
Abstract
Parastagonospora nodorum is necrotrophic fungal pathogen of wheat with significant genomic resources. Population-level pangenome data for 173 isolates, of which 156 were from Western Australia (WA) and 17 were international, were examined for overall genomic diversity and effector gene content. A heterothallic core population occurred across all regions of WA, with asexually-reproducing clonal clusters in dryer northern regions. High potential for SNP diversity in the form of repeat-induced point mutation (RIP)-like transitions, was observed across the genome, suggesting widespread ‘RIP-leakage’ from transposon-rich repetitive sequences into non-repetitive regions. The strong potential for RIP-like mutations was balanced by negative selection against non-synonymous SNPs, that was observed within protein-coding regions. Protein isoform profiles of known effector loci (SnToxA, SnTox1, SnTox3, SnTox267, and SnTox5) indicated low-levels of non-synonymous and high-levels of silent RIP-like mutations. Effector predictions identified 186 candidate secreted predicted effector proteins (CSEPs), 69 of which had functional annotations and included confirmed effectors. Pangenome-based effector isoform profiles across WA were distinct from global isolates and were conserved relative to population structure, and may enable new approaches for monitoring crop disease pathotypes.
Similar content being viewed by others


Introduction
Parastagonospora nodorum is a necrotrophic fungal pathogen causing septoria nodorum blotch (SNB) of wheat (Triticum spp.)1 leading to significant yield losses2. P. nodorum is primarily spread by infected seed, infested debris or by wind-dispersed sexual ascospores. Secondary infections can occur when water-splash spreads asexual pycnidiospores to higher leaves and glumes, causing further necrotic patches and crop loss. P. nodorum is observed to be highly diverse in the field3,4, and appears to regularly reproduce sexually5,6,7. This suggests that P. nodorum populations have a high capacity for adaptation, with potential for selective pressures to be quickly overcome by extant diversity.
P. nodorum infection relies on necrotrophic effector proteins (NEs), which are secreted into the host and cause disease symptoms upon recognition by cognate host susceptibility (S)-receptors8. Five NEs have been characterised (SnToxA9, SnTox110 SnTox311, SnTox26712 and SnTox513) and additional NE interactions have been proposed14,15,16,17,18,19,20,21. An additional ceratoplatanin-like effector homolog that is broadly conserved across plant-pathogenic fungi (SnodProt1) has also been characterised in P. nodorum22,23. Currently identified effectors have led to the deployment of resistant wheat cultivars24. Quantitative trait loci (QTL) that are associated with disease-resistance indicate additional effectors, which if characterised can provide further crop improvement. However, epistatic interactions of SnTox1 and SnTox267 over SnTox316,25 indicate that combined interactions between multiple effectors may be complex and may vary under different conditions. Reliable markers for host S-genes and an improved understanding of NE epistatic interactions are important for ongoing disease-resistance breeding. These advancements in crop-protection rely on the prior discovery of NEs26 and upon accumulating genomic and bioinformatic resources27 which have enabled effector discovery across multiple pathogen species28.
P. nodorum was among the first fungal species for which a reference genome sequence was generated (Western Australian (WA) isolate Sn15)29, and the first species of the class Dothideomycetes that comprises several important cereal pathogens30,31. Since its initial genome analysis, the Sn15 isolate has become an important reference and model for cereal necrotrophs1, accumulating significant bioinformatic resources over time, including transcriptomic29,32,33,34,35, proteomic35,36, and metabolomic37,38,39,40 datasets. Chromosome-scale reference genome assemblies have been generated for four isolates: the Australian Sn15 isolate and 3 USA-derived isolates: LDN03-Sn4, Sn2000 and the avirulent/Agropyron-isolated Sn79-108734,41.
The study of effector content and of other genomic features that may contribute to the virulence of P. nodorum is ongoing. A ~ 400 kb accessory chromosome, typically designated chromosome 23 (or AC23) is absent from Sn79-108742 and is highly mutated34,41. Regions high in RIP-like mutations and AT-rich sequences were observed around repeat-rich stretches of AC23 and sub-telomeric regions of other chromosomes34,41,43. Candidate secreted effector-like proteins (CSEPs) have been predicted based on an ensemble of features including predicted secretion signals, sequence-based or structural homology to known effectors, positive selection, presence-absence variation (PAV), genomic location (including: G:C content, distance to telomeres, and proximity to transposable elements), genome-wide association34,41,43,44, and predictive models trained on the physicochemical properties of known fungal effectors45,46. For the Australian reference isolate Sn15, CSEP predictions have been combined with additional supporting experimental and bioinformatic indicators, including: in planta gene expression33, predicted lateral gene transfers with other cereal-pathogens (https://effectordb.com), and priority-ranking based on aggregation of multiple prediction types41,43.
Decreasing costs of genome sequencing over the last decade has progressively shifted focus from the study of solitary reference isolates to comparative genomics at increasingly larger scales. Three pangenomic comparative studies of P. nodorum have been conducted on regional scales, including isolates from Iran, Finland, Sweden, Switzerland, South Africa, the USA, and Australia41,43,44,47. Iran appeared to be the most genetically heterogeneous region, reflecting a longer history of host co-evolution during the early domestication of wheat in the fertile crescent48. Positive selection pressures and presence-absence variation (PAV) have been observed for effector loci and for accessory sequences with potential roles in virulence43. Pangenome-based surveys of fungicide-resistance adaptations have been performed across Australia, Iran, South Africa, Switzerland, and the USA47, indicating higher incidences of azole resistance in Switzerland. A pangenomic survey of isolates infecting Spring, Winter and Durum wheat across the USA44 identified 2 sub-populations corresponding to geographic regions and host wheat lines. Presence of effector loci was variable, with SnToxA, SnTox1 and SnTox3 being absent in 37%, 5% and 41% of US isolates respectively, and SnToxA being mostly absent in one sub-population. Collectively, these studies highlight the regional profiles of pathogenicity factors in P. nodorum and the emerging diagnostic potential of pangenomic surveys.
Genomic diversity of P. nodorum in Western Australia (WA) was initially surveyed using 28 simple sequence repeat (SSR) markers versus 55 WA isolates collected over a period of 44 years, and contrasted to 23 French and US isolates49. This prior study indicated two core admixed sub-population groups in WA, and at least three homogeneous groups that were restricted both geographically and temporally. Population shifts between these groups over time appeared to correlate with the historical preference for different wheat cultivars, and was prominent from 2013 when mass adoption of the SnToxA-insensitive “Mace” comprised up to 70% of areas sown50. Although overall disease-resistance among wheat cultivars may have increased over time, recently sampled isolates from emergent clusters were also reportedly more aggressive49. In this study, we have generated pangenome resources corresponding to this prior survey (Fig. 1, Supplementary Data 1). In corroboration with previous findings, we observed the WA P. nodorum population was separated into a core population and a handful of small, homogeneous sub-population groups. We generated a panel of orthologous genes that represent the observed gene content across the P. nodorum pangenome, and have used this panel to predict effector candidates, and note the subtle influence of repeat-induced point mutations (RIP) upon the evolution of this model cereal necrotroph. As effectors are the key determinants in necrotrophic interactions with host sensitivity loci8, we mined the P. nodorum pangenome for protein isoforms of known effectors, and report on isoform diversity between isolates sampled across Western Australian wheat-growing regions49 and a representative panel of international isolates43.

Locations across Western Australia (dark grey) where Parastagonospora nodorum isolates were sourced for whole-genome sequencing.
Results
Phylogeny and structure of the Western Australian P. nodorum population
Mean genome size across the pangenome was 37.8 Mb per isolate (Supplementary Data 2) with an average of 18,392 annotations per isolate (Supplementary Data 3). There was an average of 6% repetitive DNA, comprised of 3% LTR retrotransposons, 2% DNA transposons, and 1% MITEs (Supplementary Data 4). There were 1,340,429 SNP variant sites detected across the pangenome relative to the Sn15 reference (Fig. 2), with RIP-like C:G↔T:A mutations comprising 78% of SNPs (Fig. 3). However SNPeffect analysis vs Sn15 annotations indicated that only 136,860 RIP-like (10.2%) and 78,401 (5.8%) non-RIP-like SNPs caused non-synonymous amino-acid changes (Fig. 3). For effector loci present in Sn15 (SnToxA, SnTox1, SnTox3 and SnTox267), 33% of RIP-like SNPs corresponded to non-synonymous changes and 73% to synonymous changes (Supplementary Table 1). Filtering of sequence variants relative to the Sn15 reference isolate produced 6787 bi-allelic, conserved SNPs occurring in ≥95% of isolates. A phylogenetic tree and sub-population groups predicted using this data indicated 6 groups, with Iranian isolates strongly associated with group 3, and US and European isolates assigned to groups 3 and 4 (Figs. 2 and 4, Supplementary Fig. 1). The majority of WA isolates were assigned to group 4 representing the core WA population (equivalent to groups 1 and 2 from a previous SSR-based study49). However a handful of phylogenetically-similar and regionally-proximal clades corresponded to other groups (1, 2, 5 and 6), which were also indicated in the previous study. Isolates assigned to these groups were typically collected from, but not exclusively representative of, the northern Geraldton region (Fig. 4). Interestingly, the Sn15 reference isolate was assigned to group 2 and is not a typical representative of the core WA population (group 4).

Rings proceeding inwards represent: Sn15 chromosomes from 1 to 23 (black/grey, accessory chromosome = red), with labels indicating the locations of 4 effector loci present in Sn15; G:C content (grey); gene density (green); Predector (effector-likelihood) score (green/red); repetitive DNA density (red); composite RIP index (CRI) (green/red); SNP site density (blue = total, yellow = (RIP-like) transition mutations, red=non-synonymous RIP-like transitions; presence-absence variation (PAV) relative to all isolates (red). The geographic region, predicted phylogeny, and population grouping of isolates are indicated alongside corresponding PAV tracks.

SNP mutation sites were categorised (middle) into RIP-like (C↔T or A↔G SNPs) and Other/non-RIP-like (not C↔T or A↔G SNPs) and by their predicted effects on protein-coding genes (right).

SNP-derived phylogeny (left) of Western Australian and internationally-sampled P. nodorum isolates (see legend), shows: isolates (branch labels); sampling year; sub-population groups (green) from this study (right) and a previous study (left)49; mating-type loci (blue); and the presence of effector loci (red-middle) and effector protein isoforms (red-right, from left-to-right: highest to lowest frequency). An alternate version with overlaid branch lengths is presented in Supplementary Fig. 1.
Effector protein isoform profiles were consistent with phylogeny
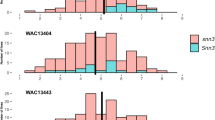
The presence of known necrotrophic effector (NE) loci SnToxA (represented by Parastagonospora nodorum ortholog group (SNOO) SNOO_16571A), SnTox1 (SNOO_20078A), SnTox3 (SNOO_08981A), SnTox267 (SNOO_14493A) and SnTox5 (SNOO_50320) was ubiquitous across WA, with the majority of isolates possessing all 5 NE loci (Fig. 4). Infrequently, SnToxA, SnTox1, SnTox3 and SnTox5 loci were absent, although this presence-absence variation was more common among international isolates and rare among WA isolates. Notably, SnTox5 was consistently absent from sub-population group 2 which included the Sn15 reference isolate, yet absence of SnTox5 was not observed among international isolates. At the protein isoform level, NE profiles of WA isolates were distinct from international isolates. Across WA, dominant isoforms and less frequent secondary isoforms were observed, and additional isoforms were rare. NE Isoform profiles also tended to conform to the predicted phylogenetic structure (Fig. 4).
Leveraging comparative pangenomics and function for prediction of effector candidates
There were 34,381 clusters of orthologs predicted across the P. nodorum pangenome, with 14,050 (40.9%) core groups present in all isolates, 11,470 (33.3%) variable (accessory) groups and 8861 (25.8%) singleton groups (Supplementary Figs. 2, 3, Supplementary Data 5). Rarefaction analysis of ortholog group presence across all isolates indicated this dataset represents a ‘closed’ pangenome51 (Supplementary Fig. 4). After functional annotation, there were 19,465 groups (56.6%) remaining with no informative matches. Based on dN/dS branch site tests, 5294 groups (15.4%) were under positive selection. Accessory orthogroups tended to be closer to repeat and telomere regions, with lower dN/dS and higher FYKIN:GAP ratios52 that would indicate relative increase in diversifying selection driven by RIP mutations (Supplementary Fig. 3, Supplementary Data 6). Singleton orthogroups tended to have slightly higher Predector scores that may indicate effector-like properties. Accessory orthogroups appeared to be enriched in several functions including cell death, membrane transport, regulation of transcription and DNA replication. Singleton orthogroups were also enriched in functional annotations related to protein repeats, protein-protein interactions, ubiquitinilation and viral replication (Supplementary Data 6).
Prediction of candidate secreted effector proteins (CSEPs, see methods) resulted in 186 orthogroups, of which 69 (37.1%) had functional information, and 17 (9.1%) were under positive selection (Supplementary Data 7). The 69 functionally-annotated CSEPs included ortholog groups corresponding to 6 known P. nodorum NE loci SnToxA, SnTox1, SnTox3, SnodProt122,23, SnTox5 and SnTox267 at predicted ranks 2, 3, 13, 27, 42 and 43 respectively (Supplementary Data 8). Other groups were homologous to several effector loci identified in other plant-pathogen species, including MoCDIP453, MoAAT54, FgXYLA55,56, CfTom157,58, MoSPD5/MoBas459,60, and Mycgr3G3810561 (Table 1, Supplementary Data 8).
Discussion
Previously the structure of the WA P. nodorum population was assessed with SSR markers from which 5 sub-population groups were predicted49. Two of these groups were proposed to represent a gradual change over time in the core population in response to wheat cultivar use, while the remaining homogeneous clusters may be clonally-expanded populations. In this pangenome-based study 6 sub-population groups were predicted, with a core WA group (group 4) and geographically-restricted clonal groups. The ratio of mating-type loci in the core population was close to 1:1 indicating heterothallic meiotic potential, in line with previous reports from WA5,6 and elsewhere62. In contrast, the clonal sub-groups only had a single mating type and were thus asexual (Fig. 4), with 1 exception. A single WA isolate (group 6) and a single Iranian isolate both appeared to match both mating-type loci (Fig. 4). This may potentially indicate contamination of those samples where more than one isolate has been sequenced, or alternatively this can indicate a spontaneous shift to homothallism which may occur rarely. The clonal sub-groups exhibited a similar proportion of RIP-like SNP mutations relative to the core population (~80%) with the exception of group 2 (96%), which notably contains the reference isolate Sn15 and consistently lacked the Tox5 locus (Fig. 4, Supplementary Data 9). Clonal sub-groups were also primarily collected from the northern Geraldton region, which is relatively hotter with less rainfall63. High temperatures have been negatively correlated with P. nodorum disease load64. Conversely rainfall and splash dispersal have been associated with higher disease loads1,64, and rain impacts may also promote airborne dispersal over longer distances65. The combination of these climatic factors may have contributed to the homogeneity across this region. Furthermore, the phylogeny of the core-group did not indicate strong association with geographic regions. Long-range wind dispersal of sexual ascospores has been reported in WA5 and dispersal by infected seed is also a possibility66,67,68. Speculatively, the population structure of P. nodorum may be less dependent on geographic distance, when compared to influences of climatic and anthropic factors.
Presence of necrotrophic effector loci that correspond to cognate host sensitivity receptor loci is a useful predictor of the outcome of P. nodorum infection26. Previously, discrete sets of effector candidates were predicted for two US sub-populations44, highlighting the importance of region-specific analysis. In this study focused on the Western Australian wheat belt region, conserved effector isoform profiles for effector loci SnToxA, SnTox1, SnTox3, SnTox267 and SnTox5 generally conformed to phylogenetic structure (Fig. 4). Despite the extreme genome plasticity of fungal genomes52,69,70,71 and the unsurprisingly high levels of RIP-like mutations observed across the P. nodorum pangenome (Figs. 2 and 3), relatively little effector protein isoform diversity was observed across WA (Fig. 4). Effector loci of Sn15 are located at or near telomeres, which are hotspots for TEs, SNPs, intrachromosomal recombinations, duplications, and positive selection34,42,69,70,72. Yet, only a strongly dominant isoform and an infrequent secondary isoform were observed, and if present additional isoforms were extremely rare. RIP appears to be a strong driving force causing many DNA-level mutations across the entire landscape of the P. nodorum pan-genome, presumably due to “RIP-leakage”73 which is frequently observed in the Pezizomycotina52 and extends up to (at least) 4-5 Kb from a RIP-targeted repeat74,75. Although the majority of protein-coding genes of P. nodorum are within 2-3 Kb of their nearest repeat (Supplementary Fig. 3), RIP-leakage in P. nodorum is balanced by strong selection against mutations causing amino acid changes, even for necrotrophic effector loci. The ratio of RIP-like to non-RIP mutations for all loci (78401:136860 = 0.57) was the same as that observed for known effectors (9:18 = 0.57, Supplementary Table 1). The pathogenic fitness of biotrophs and hemibiotrophs52,76 can benefit from RIP-driven pseudogenisation of effector or other PAMP-producing loci, however this does not typically apply to a necrotroph like P. nodorum. Consequently, these observations suggest that most RIP mutations altering protein-coding gene regions are strongly selected against, to avoid deleterious losses of function.
Pathogen pangenomics has the potential to enable affordable genome-based crop disease surveillance tailored to local regions41,44,77. This study focusses on a population of the wheat pathogen Parastagonospora nodorum from the Western Australian wheat belt region. The collective bioinformatic resources for P. nodorum pathogen have significantly improved over time, including the development of approaches to pangenomic analysis at regional scales. By aggregating multiple predictive methods and data sources, a stringent set of 69 candidate effectors has been generated that may guide experiment-validation and discovery of effectors. Alternate reproductive modes were also observed in some regions, highlighting the potential need for differential disease management under altered population growth conditions. At the genome-level we observed high potential for adaptability, indicated by widespread RIP-like mutations that appeared to drive heterogeneity at the DNA level. Counter-intuitively, there were relatively few mutations retained at the protein isoform level, even within necrotrophic effector loci typically associated with mutation hotspots. In P. nodorum and potentially other necrotrophs, the majority of RIP-driven heterogeneity may be purged by strong selection against non-synonymous mutations, resulting in relative homogeneity across its ‘pan-proteome’. Regardless, there is encouraging potential to extend pangenome-based insights and the effector isoform profiling approaches described here to future plant pathology applications. The reduction of total gene content and SNP-level diversity down to simplified isoform profiles could be used as an alternative to traditional and haplotype-based pathotyping48,78, and GWAS approaches testing for SNPs associated with cultivar susceptibility12,13,44. In this manner, despite the vast potential for DNA mutation observed for most fungal pathogen genomes, future effector studies that use isoform profiling may be less prone to RIP-related errors.
Materials and methods
Whole genome sequencing of Western Australian P. nodorum isolates
Genomic DNA of 141P. nodorum isolates49 sampled across the Western Australian wheat-belt region (Fig. 1, Supplementary Data 1) were extracted79 and sequenced by the Australian Genome Research Facility (Melbourne, Australia) (Illumina HiSeq2500, TruSeq PCR-free, 125 bp paired end (PE), 600 bp insert size) [NCBI BioProject: PRJNA612761]. Genomic DNA of 17 new isolates and 2 repeated isolates (14FG141 and Mur_S3 from the previous 141) were extracted with the Qiagen DNeasy Plant Mini kit (Venlo, Netherlands. Catalogue ID: 69104) and sequenced by Novogene (Beijing, China) (Illumina HiSeq2500, TruSeq PCR-free, 150 bp PE, 350 bp insert size). Data from prior studies was also used, including draft genomes of 15 international P. nodorum isolates43 [NCBI BioProject: PRJNA476481]; and chromosome-scale genome assemblies for Western Australian reference isolate Sn1541 [NCBI Assembly: GCA_016801405.1], and; US isolates LDN03-Sn4 [NCBI Assembly: GCA_002267005.1], Sn2000 [NCBI Assembly: GCA_002267045.1] and Sn79-1087 [NCBI Assembly: GCA_002267025.1] [NCBI BioProject: PRJNA398070]34.
Reads were trimmed with CutAdapt v1.18 (2 passes, 3 trims/pass, terminal Phred score >2, average Phred score ≥5, length ≥50)80 and BBduk v38.38 (read kmer coverage 0.7)81 versus UniVec (https://www.ncbi.nlm.nih.gov/tools/vecscreen/univec/) and PhiX (NCBI RefSeq: NC_001422.1)82. Sample contamination was checked with Kraken v2.0.783 versus NCBI Refseq (bacteria, archaea, protozoa, virus, and fungi: downloaded: 2019-03-16), and human GRCh3884, as well as to 4 reference P. nodorum genomes as a positive set34,41. Insert size and completeness was assessed by alignment to Sn15, LDN03-Sn4, Sn2000 and Sn79-1087 genomes with BBmap v38.3881 and quality control statistics were assessed with FastQC v0.11.8 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), Bbmap, Samtools85, and MultiQC86 witihn the qcflow pipeline87.
Variant calling relative to the P. nodorum Sn15 reference isolate
Reads were aligned to the Sn15 ref. 41 with bwa mem 0.7.17-r119888 and outputs were converted to aligned BAM format with GATK 4.2.6.1 (MarkIlluminaAdapters, MarkDuplicates, MergeBamAlignment -CREATE_INDEX -ADD_MATE_CIGAR)89. Sequence variants relative to Sn15 were generated in gVCF format with GATK HaplotypeCaller (-ERC GVCF –minimum-mapping-quality 20 –min-base-quality-score 20 -G StandardAnnotation -G AS_StandardAnnotation -G StandardHCAnnotation) and isolates were genotyped with GATK CombineGVCFs and GenotypeGVCFs, filtering variants with GATK VariantFiltration for SNPs (QD < 2, QUAL < 30, SOR > 3, FS > 60, MQ < 40, MQRankSum < −12.5, ReadPosRankSum < −8) and InDels (QD < 2, QUAL < 30, FS > 200, ReadPosRankSum < −20). Filtered variants resulting in non-synonymous or nonsense mutations relative to Sn15 gene annotations were identified with SnpEff90.
To predict population structure groups, VCFs were converted to PLINK bed format using PLINK v1.90b7, used as input to fastSTRUCTURE v1.091. Initially, for K = 1–12, twelve independent runs were performed with default parameters and the function “chooseK.py” was used to select the optimal run. To predict phylogeny, ≤2 bi-allelic and conserved (≤5% missing data) SNPs were randomly selected within 5 kbp increments with BCFtools92 (view --max-alleles 2 -e ‘F_MISSING < = 0.05; +prune -l 0.9 -w 5000 bp -n1 -N rand) and used to predict a phylogenetic tree with IQTree v2.0.3 (-bb 1000 -alrt 1000)93. The SNP-derived phylogenetic tree was visualised with iTOL v594 alongside geographic location, mating-type genes, FastSTRUCTURE-based population groups, previously published SSR-marker-derived population groups49, and pathogenicity effector profiles.
Previous studies have established repeat-induced point mutation (RIP) in P. nodorum41 and broadly across many other fungal species52 have a strong bias for mutation of CpA to TpA dinucleotides. Therefore, bi-allelic SNP variants which were comprised of either “C” and “T” allele pairs, or the reverse complement “A” and “G”, were designated “RIP-like” for subsequent analysis. SNP variants relative to Sn15 were also used to calculate Composite RIP Index (CRI)95.
De novo genome assembly of Western Australian P. nodorum isolates
Overlapping read pairs were merged with BBmerge v38.3896 (strict = t k = 62 rem = 50 ecctadpole = t) and combined with unmerged pairs for de novo genome assembly with Spades v3.13.097 (--careful --cov-cutoff auto). Mitochondrial genomes (mtDNAs) were assembled with Novoplasty v2.7.298, seeded with the Sn15 mtDNA [NCBI RefSeq: EU053989.1]29 (k = 31-81, selected for min. contigs with assembly size=47-52 Kb) (Supplementary Data 2) (via mitoflow v.1099). Nuclear assemblies were filtered for mtDNA with minimap2 (git commit 371bc95)100 (≥95% coverage, median depth > = 99.2% total depth). Assembly quality was assessed with Quast v5.0.2101, bbtools v38.3881, and KAT v2.4.2102 (via postasm v1.0103). Genome assemblies were aligned to Sn15 [NCBI Assembly: GCA_016801405.1]41 with nucmer v4.0.0beta2 (--maxmatch)34,104. Mean coverage within non-overlapping 50 Kb windows was calculated with BEDTools v2.28.0105 and visualised with circlize106.
Annotation of DNA repeats and non-protein coding gene features
DNA repeats were predicted using a combination of tools (Supplementary Data 4): EAHelitron (git commit c4c3dca)107, LTRharvest108, LTRdigest (genometools v1.5.10)109, MiteFinder (git commit 833754b)110, RepeatModeler v1.0.11111, and RepeatMasker v4.0.9p2112 (-species “Parastagonospora nodorum”). Putative transposable element (TE) protein-coding regions were predicted with MMSeqs2 v9-d36de113 versus selected Pfam families, GyDB families114, and a custom MSA database sourced from TransposonPSI (http://transposonpsi.sourceforge.net/) and LTR_retriever115 (via PanTE v1.0116).
Predicted TE sequences from EAHelitron, MiteFinder, RepeatModeler, and MMSeqs protein finding were clustered with VSEARCH v2.14.1117 (--cluster_fast combined.fasta --id 0.90 --weak_id 0.7 --iddef 0 --qmask dust), filtered for >=4 copies in >=20% isolates, aligned with DECIPHER v2.10.0118, classified into subtypes with RepeatModeler (RepeatClassifier), and mapped to each isolate assembly with RepeatMasker. Non-coding rRNA and tRNA features were predicted with RNAmmer v1.2119 and tRNAscan-SE v 2.0.3120. Genome assemblies were soft-masked with TE and non-coding RNA features with BEDTools105.
Annotation of protein-coding genes
Data supporting gene annotation in the P. nodorum pangenome was derived from multiple sources. Previous annotations for Sn1541, LDN03-Sn4, and Sn7934 were mapped to all assemblies with Spaln v2.3.3 (-KP -LS -M3 -O0 -Q7 -ya1 -yX -yL20 -XG20000)121. Fungal proteins from UniRef50 (release 2019_08, downloaded: 2019-10-29, taxonomy = “Fungi [4751]” AND identity = 0.5) were aligned with Exonerate v2.4.0 (--querytype protein --targettype dna --model protein2genome --refine region --percent 70 --score 100 --geneseed 250 --bestn 2 --minintron 5 --maxintron 15000 --showtargetgff yes --showalignment no --showvulgar no)122 with pre-filtering using MMSeqs2 (-e 0.00001 --min-length 10 --comp-bias-corr 1 --split-mode 1 --max-seqs 50 --mask 0 --orf-start-mode 1). RNAseq reads for Sn15 in vitro and 3 days post infection on wheat leaves33 [GEO: GSE150493; SRA: SRX8337774-SRX8337777, SRX8337782-SRX8337785] were de novo assembled into transcripts using Trinity v2.8.4 (--jaccard_clip --SS_lib_type FR)123. RNAseq reads were also aligned to all assemblies with STAR v2.7.0e124 and assembled into transcripts with StringTie v1.3.6 (--fr -m 150)125. Assembled transcripts were aligned to genomes using Spaln v2.3.3 (-LS -O0 -Q7 -S3 -yX -ya1 -Tphaenodo -yS -XG 20000 -yL20)121, and GMAP v2019-05-12126.
Protein-coding gene annotations (Supplementary Data 3) were predicted in several stages. Initial predictions for each isolate used multiple tools: PASA2 v2.3.3 (-T --MAX_INTRON_LENGTH 15000 --ALIGNERS blat --transcibed_is_aligned_oriented --TRANSDECODER --stringent_alignment_overlap 30.0)127, GeneMark-ET (--soft_mask 100 --fungus)128, CodingQuarry v2.0 (standard and “pathogen mode”), Augustus (git commit 8b1b14a, iindependently for forward and reverse strands; --hintsFile = hints.gff3 --strand = $ --allow_hinted_splicesites=’gtag,gcag,atac,ctac’ --softmasking = on --alternatives-from-evidence = true --min_intron_len = 5)129, and GeMoMa v1.6.1 (Sn15 annotations only)130. PASA2 predictions used GMAP- and BLAT-aligned RNASeq data. Augustus predictions used GMAP alignments, STAR intron features, and Spaln protein alignments as hints. PASA2, Augustus, and CodingQuarry predictions were clustered with MMSeqs2 (90% identity, 98% reciprocal coverage) and transferred with GeMoMa. Outputs from Genemark-ET, CodingQuarry, Augustus, PASA, GeMoMa, Exonerate, Spaln protein and transcript alignments, and GMAP alignments were combined using EVidenceModeler (git commit 73350ce) (--min_intron_len 5)131. Augustus (all hints, parameters as above) was used to predict additional genes not overlapping with EVidenceModeler outputs.
Multiple steps were then taken to ensure accuracy and reliability of annotations across the pangenome. Pseudogenes were screened with AntiFam132 using HMMER v3.2.1 (--cut_ga). Annotations were considered “low confidence” if supported only by Spaln or GMAP transcript alignments, Exonerate protein alignments, or transfers of annotations between isolates performed via GeMoMa (unless derived from previously curated Sn15 annotations), or for Sn15, if supported only by the above, or Augustus. “Low-confidence” annotations overlapping annotations on either strand by more than 30% of their length were discarded. Frame/phase-shift annotation errors in outputs were corrected by mapping to all annotations of all isolates without internal stop codons, and all Pezizomycota proteins from UniRef-90 (2020-05-13; taxonomy: “Pezizomycotina [147538]”; identity:0.9’) with blastx v2.10.0 (-strand plus -max_intron_length 300 -evalue 1e−5)133. In-phase matches lacking internal stops were retained, out-of-phase matches with stops were marked as pseudogenes, and annotations with internal stops and no matches were discarded. Annotations overlapping predicted rRNA genes (≥50% length) were discarded. Annotations with exons spanning assembly gaps were split in separate annotations if ≥60 bp. Annotation completeness was evaluated with BUSCO v3 (pezizomycotina_odb9)134 with additional statistics collected with genometools v1.5.10135. For Sn15, updated annotations were compared to previous versions41 with ParsEval/AEGeAn v0.15.0136 and BEDTools (bedtools subtract -a new -b old -s -A -F 0.2).
Orthology & positive selection
Orthology relationships were predicted with Proteinortho v6.0.30 (-singles -seflblast)137 and Diamond v 2.0.8138, with alternate transcript isoforms (Sn15 only) allowed to cluster into separate ortholog groups. The prefix ‘SNOO_‘ was assigned to clusters, with a numerical suffix based on: the “SNOG” locus numbers of corresponding Sn15 annotations41, or sequential numbers starting from 50,000 if not present in Sn15. Alphabetical suffixes also indicate Sn15 isoforms present in the cluster. Representative sequences were selected from each cluster, in descending order of priority: 1) Sn15 sequence with closest to average length, 2) presence in LDN03-Sn4, 3) Sn2000, 3) Sn79, 4) random selection from closest to average length (Supplementary Data 5). CDS sequences of clusters were codon-aligned with DECIPHER v2.16.1118, gene trees were estimated with FastTree v2.1.11139, tested for positive selection with HYPHY v2.5.15140,141 (BUSTED method, p-value ≤ 0.01).
Functional analysis & effector candidate prediction
MetaEuk (v4, easypredict)142 was used to search for SnToxA (SNOO_16571A), SnTox1 (SNOO_20078A), SnTox3 (SNOO_08981A), SnTox267 (SNOO_14493A) and SnTox5 (SNOO_50320), and effector protein isoform profiles for each isolate were extracted matching regions. Functional annotation was performed versus the representative ortholog clusters with InterProScan143,144 with additional GO-terms added with PANNZER145 and eggNOG-Mapper146 (excluding “anti-slim”). Additional annotations, properties, and effector-likelihood were added with Predector v.0.1.0147 (Supplementary Data 7). Candidate secreted effector-like proteins (CSEPs) were predicted by filtering ortholog clusters for the criteria: predicted secretion, ≥2 cysteine residues, Predector score ≥2, present in ≥1 reference isolate, excluding singletons, and ≥1 functional annotation (Supplementary Data 8).
Statistics and reproducibility
Functional enrichment tests were performed using the total set of ortholog groups (n = 34,381). Fisher’s exact test (two-tailed) was applied to orthogroup counts assigned to individual functional annotations, comparing orthogroups belonging to sub-categories ‘accessory’ (n = 11,470) and ‘singleton’ (n = 8861) with those of the ‘core’ sub-category (n = 14,050). Functional annotations with p ≤ 0.05 are reported in Supplementary Data 6.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
New sequence data available from NCBI BioProject PRJNA612761. Gene annotations for isolates Sn2000, Sn4, and Sn79-1087 (https://doi.org/10.6084/m9.figshare.13340975). Data from prior studies: draft genomes of 15 international P. nodorum isolates [NCBI BioProject: PRJNA476481]; chromosome-scale genome assemblies for: 1) Western Australian reference isolate Sn15 [NCBI Assembly: GCA_016801405.1]; 2) US isolates LDN03-Sn4 [NCBI Assembly: GCA_002267005.1]; 3) Sn2000 [NCBI Assembly: GCA_002267045.1]; 4) Sn79-1087 [NCBI Assembly: GCA_002267025.1] [NCBI BioProject: PRJNA398070].
Code availability
Custom code available from: a) https://github.com/darcyabjones/qcflow87; b) https://github.com/darcyabjones/mitoflow99; c) https://github.com/darcyabjones/postasm(commit:c94c3b9)103; and d) https://github.com/darcyabjones/pante/tree/master/data/proteins116.
References
Solomon, P. S., Lowe, R. G. T., Tan, K.-C., Waters, O. D. C. & Oliver, R. P. Stagonospora nodorum: cause of stagonospora nodorum blotch of wheat. Mol. Plant Pathol. 7, 147–156 (2006).
Murray, G. M. & Brennan, J. P. Estimating disease losses to the Australian wheat industry. Austral. Plant Pathol. 38, 558–570 (2009).
McDonald, M. C., Razavi, M., Friesen, T. L., Brunner, P. C. & McDonald, B. A. Phylogenetic and population genetic analyses of Phaeosphaeria nodorum and its close relatives indicate cryptic species and an origin in the Fertile Crescent. Fungal Genet. Biol. 49, 882–895 (2012).
Stukenbrock, E. H., Banke, S. & McDonald, B. A. Global migration patterns in the fungal wheat pathogen Phaeosphaeria nodorum. Mol. Ecol. 15, 2895–2904 (2006).
Bathgate, J. A. & Loughman, R. Ascospores are a source of inoculum of Phaeosphaeria nodorum, P. avenaria f. sp. avenaria and Mycosphaerella graminicola in Western Australia. Austral. Plant Pathol. 30, 317 (2001).
Murphy, N., Loughman, R., Appels, R., Lagudah, E. & Jones, M. Genetic variability in a collection of Stagonospora nodorum isolates from Western Australia. Aust. J. Agric. Res. 51, 679–684 (2000).
Sommerhalder, R. J., McDonald, B. A. & Zhan, J. The frequencies and spatial distribution of mating types in Stagonospora nodorum are consistent with recurring sexual reproduction. Phytopathology 96, 234–239 (2006).
Tan, K.-C., Oliver, R. P., Solomon, P. S. & Moffat, C. S. Proteinaceous necrotrophic effectors in fungal virulence. Funct. Plant Biol. 37, 907–912 (2010).
Liu, Z. et al. The Tsn1–ToxA interaction in the wheat–Stagonospora nodorum pathosystem parallels that of the wheat–tan spot system. Genome 49, 1265–1273 (2006).
Liu, Z. et al. The cysteine rich necrotrophic effector SnTox1 produced by Stagonospora nodorum triggers susceptibility of wheat lines harboring Snn1. PLOS Pathog. 8, e1002467 (2012).
Liu, Z. et al. SnTox3 acts in effector triggered susceptibility to induce disease on wheat carrying the Snn3 gene. PLOS Pathog. 5, e1000581 (2009).
Richards, J. K. et al. A triple threat: the Parastagonospora nodorum SnTox267 effector exploits three distinct host genetic factors to cause disease in wheat. N. Phytologist 233, 427–442 (2022).
Kariyawasam, G. K. et al. The Parastagonospora nodorum necrotrophic effector SnTox5 targets the wheat gene Snn5 and facilitates entry into the leaf mesophyll. N. Phytologist 233, 409–426 (2022).
Abeysekara, N. S., Friesen, T. L., Keller, B. & Faris, J. D. Identification and characterization of a novel host–toxin interaction in the wheat–Stagonospora nodorum pathosystem. Theor. Appl. Genet. 120, 117–126 (2009).
Friesen, T. L., Chu, C., Xu, S. S. & Faris, J. D. SnTox5–Snn5: a novel Stagonospora nodorum effector–wheat gene interaction and its relationship with the SnToxA–Tsn1 and SnTox3–Snn3–B1 interactions. Mol. Plant Pathol. 13, 1101–1109 (2012).
Friesen, T. L., Meinhardt, S. W. & Faris, J. D. The Stagonospora nodorum‐wheat pathosystem involves multiple proteinaceous host‐selective toxins and corresponding host sensitivity genes that interact in an inverse gene‐for‐gene manner. Plant J. 51, 681–692 (2007).
Friesen, T. L., Zhang, Z., Solomon, P. S., Oliver, R. P. & Faris, J. D. Characterization of the interaction of a novel Stagonospora nodorum host-selective toxin with a wheat susceptibility gene. Plant Physiol. 146, 682–693 (2008).
Gao, Y. et al. Identification and characterization of the SnTox6-Snn6 interaction in the Parastagonospora nodorum–wheat pathosystem. MPMI 28, 615–625 (2015).
Phan, H. T. et al. Novel sources of resistance to Septoria nodorum blotch in the Vavilov wheat collection identified by genome-wide association studies. Theor. Appl. Genet. 131, 1223–1238 (2018).
Shi, G. et al. The wheat Snn7 gene confers susceptibility on recognition of the Parastagonospora nodorum necrotrophic effector SnTox7. Plant Genome 8, plantgenome2015.2002.0007 (2015).
Zhang, Z. et al. Two putatively homoeologous wheat genes mediate recognition of SnTox3 to confer effector‐triggered susceptibility to Stagonospora nodorum. Plant J. 65, 27–38 (2011).
Hall, N., Keon, J. & Hargreaves, J. A homologue of a gene implicated in the virulence of human fungal diseases is present in a plant fungal pathogen and is expressed during infection. Physiological Mol. Plant Pathol. 55, 69–73 (1999).
Wang, Y. et al. Magnaporthe oryzae-Secreted Protein MSP1 Induces Cell Death and Elicits Defense Responses in Rice. MPMI 29, 299–312 (2016).
Tan, K.-C. et al. Sensitivity to three Parastagonospora nodorum necrotrophic effectors in current Australian wheat cultivars and the presence of further fungal effectors. Crop Pasture Sci. 65, 150–158 (2014).
Phan, H. T. et al. Differential effector gene expression underpins epistasis in a plant fungal disease. Plant J. 87, 343–354 (2016).
Vleeshouwers, V. G. A. A. & Oliver, R. P. Effectors as Tools in Disease Resistance Breeding Against Biotrophic, Hemibiotrophic, and Necrotrophic Plant Pathogens. MPMI 27, 196–206 (2014).
Jones, D. A. B., Bertazzoni, S., Turo, C. J., Syme, R. A. & Hane, J. K. Bioinformatic prediction of plant–pathogenicity effector proteins of fungi. Curr. Opin. Microbiol. 46, 43–49 (2018).
Kanja, C. & Hammond‐Kosack, K. E. Proteinaceous effector discovery and characterization in filamentous plant pathogens. Mol. Plant Pathol. 21, 1353–1376 (2020).
Hane, J. K. et al. Dothideomycete–Plant Interactions Illuminated by Genome Sequencing and EST Analysis of the Wheat Pathogen Stagonospora nodorum. Plant Cell 19, 3347–3368 (2007).
Aylward, J. et al. A plant pathology perspective of fungal genome sequencing. IMA Fungus 8, 1–15 (2017).
Ohm, R. A. et al. Diverse Lifestyles and Strategies of Plant Pathogenesis Encoded in the Genomes of Eighteen Dothideomycetes Fungi. PLOS Pathog. 8, e1003037 (2012).
Ipcho, S. V. S. et al. Transcriptome analysis of Stagonospora nodorum: gene models, effectors, metabolism and pantothenate dispensability. Mol. Plant Pathol. 13, 531–545 (2012).
Jones, D. A. B. et al. A specific fungal transcription factor controls effector gene expression and orchestrates the establishment of the necrotrophic pathogen lifestyle on wheat. Sci. Rep. 9, 1–13 (2019).
Richards, J. K., Wyatt, N. A., Liu, Z., Faris, J. D. & Friesen, T. L. Reference Quality Genome Assemblies of Three Parastagonospora nodorum Isolates Differing in Virulence on Wheat. G3 Genes Genomes Genet. 8, 393–399 (2018).
Syme, R. A. et al. Comprehensive Annotation of the Parastagonospora nodorum Reference Genome Using Next-Generation Genomics, Transcriptomics and Proteogenomics. PLOS ONE 11, e0147221 (2016).
Bringans, S. et al. Deep proteogenomics; high throughput gene validation by multidimensional liquid chromatography and mass spectrometry of proteins from the fungal wheat pathogen Stagonospora nodorum. BMC Bioinforma. 10, 301 (2009).
Chooi, Y.-H., Muria-Gonzalez, M. J. & Solomon, P. S. A genome-wide survey of the secondary metabolite biosynthesis genes in the wheat pathogen Parastagonospora nodorum. Mycology 5, 192–206 (2014).
Gummer, J. P. A., Trengove, R. D., Oliver, R. P. & Solomon, P. S. Dissecting the role of G-protein signalling in primary metabolism in the wheat pathogen Stagonospora nodorum. Microbiology 159, 1972–1985 (2013).
Lowe, R. G. T. et al. A metabolomic approach to dissecting osmotic stress in the wheat pathogen Stagonospora nodorum. Fungal Genet. Biol. 45, 1479–1486 (2008).
Muria-Gonzalez, M. J. et al. Volatile Molecules Secreted by the Wheat Pathogen Parastagonospora nodorum Are Involved in Development and Phytotoxicity. Front. Microbiol. 11 https://doi.org/10.3389/fmicb.2020.00466 (2020).
Bertazzoni, S., Jones, D. A., Phan, H. T., Tan, K.-C. & Hane, J. K. Chromosome-level genome assembly and manually-curated proteome of model necrotroph Parastagonospora nodorum Sn15 reveals a genome-wide trove of candidate effector homologs, and redundancy of virulence-related functions within an accessory chromosome. BMC Genomics 22, 1–16 (2021).
Bertazzoni, S. et al. Accessories make the outfit: accessory chromosomes and other dispensable DNA regions in plant-pathogenic Fungi. MPMI 31, 779–788 (2018).
Syme, R. A. et al. Pan-Parastagonospora Comparative Genome Analysis—Effector Prediction and Genome Evolution. Genome Biol. Evol. 10, 2443–2457 (2018).
Richards, J. K. et al. Local adaptation drives the diversification of effectors in the fungal wheat pathogen Parastagonospora nodorum in the United States. PLOS Genet. 15, e1008223 (2019).
Sperschneider, J., Dodds, P. N., Gardiner, D. M., Singh, K. B. & Taylor, J. M. Improved prediction of fungal effector proteins from secretomes with EffectorP 2.0. Mol. Plant Pathol. 19, 2094–2110 (2018).
Sperschneider, J. et al. EffectorP: predicting fungal effector proteins from secretomes using machine learning. N. Phytologist 210, 743–761 (2016).
Pereira, D., McDonald, B. A. & Croll, D. The genetic architecture of emerging fungicide resistance in populations of a global wheat pathogen. bioRxiv, https://doi.org/10.1101/2020.03.26.010199 (2020).
Ghaderi, F., Sharifnabi, B., Javan‐Nikkhah, M., Brunner, P. C. & McDonald, B. A. SnToxA, SnTox1, and SnTox3 originated in Parastagonospora nodorum in the Fertile Crescent. Plant Pathol, ppa.13233 https://doi.org/10.1111/ppa.13233 (2020).
Phan, H. T. T. et al. Low Amplitude Boom-and-Bust Cycles Define the Septoria nodorum Blotch Interaction. Front. Plant Sci. 10 https://doi.org/10.3389/fpls.2019.01785 (2020).
Trainor, G., Zaicou-Kunesch, C., Curry, J., Shackley, B. & Nicol, D. 2019 Wheat variety sowing guide for Western Australia (Department of Primary Industries and Regional Development, 2018).
Medini, D., Donati, C., Tettelin, H., Masignani, V. & Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594 (2005).
Testa, A. C., Oliver, R. P. & Hane, J. K. OcculterCut: a comprehensive survey of AT-rich regions in fungal genomes. Genome Biol. Evol. 8, 2044–2064 (2016).
Chen, S. et al. Identification and Characterization of In planta–Expressed Secreted Effector Proteins from Magnaporthe oryzae That Induce Cell Death in Rice. MPMI 26, 191–202 (2013).
Guo, M. et al. The bZIP transcription factor MoAP1 mediates the oxidative stress response and is critical for pathogenicity of the rice blast fungus Magnaporthe oryzae. PLOS Pathog. 7, e1001302 (2011).
Pollet, A., Beliën, T., Fierens, K., Delcour, J. A. & Courtin, C. M. Fusarium graminearum xylanases show different functional stabilities, substrate specificities and inhibition sensitivities. Enzym. Microb. Technol. 44, 189–195 (2009).
Sperschneider, J. et al. Genome-Wide Analysis in Three Fusarium Pathogens Identifies Rapidly Evolving Chromosomes and Genes Associated with Pathogenicity. Genome Biol. Evol. 7, 1613–1627 (2015).
Ökmen, B. et al. Detoxification of α-tomatine by Cladosporium fulvum is required for full virulence on tomato. N. Phytologist 198, 1203–1214 (2013).
Pareja-Jaime, Y., Roncero, M. I. G. & Ruiz-Roldán, M. C. Tomatinase from Fusarium oxysporum f. sp. lycopersici is required for full virulence on tomato plants. MPMI 21, 728–736 (2008).
Mosquera, G., Giraldo, M. C., Khang, C. H., Coughlan, S. & Valent, B. Interaction transcriptome analysis identifies Magnaporthe oryzae BAS1-4 as biotrophy-associated secreted proteins in rice blast disease. Plant Cell 21, 1273–1290 (2009).
Sharpee, W. et al. Identification and characterization of suppressors of plant cell death (SPD) effectors from Magnaporthe oryzae. Mol. Plant Pathol. 18, 850–863 (2017).
Kettles, G. J. et al. Characterization of an antimicrobial and phytotoxic ribonuclease secreted by the fungal wheat pathogen Zymoseptoria tritici. N. Phytologist 217, 320–331 (2018).
Caten, C. & Newton, A. Variation in cultural characteristics, pathogenicity, vegetative compatibility and electrophoretic karyotype within field populations of Stagonospora nodorum. Plant Pathol. 49, 219–226 (2000).
Western Australian Government, BOM. Western Australia in 2021: wet in the west, very warm days in the north, http://www.bom.gov.au/climate/current/annual/wa/summary.shtml (2021).
Shaw, M. W., Bearchell, S. J., Fitt, B. D. L. & Fraaije, B. A. Long-term relationships between environment and abundance in wheat of Phaeosphaeria nodorum and Mycosphaerella graminicola. N. Phytologist 177, 229–238 (2008).
Kim, S., Park, H., Gruszewski, H. A., Schmale, D. G. & Jung, S. Vortex-induced dispersal of a plant pathogen by raindrop impact. PNAS 116, 4917–4922 (2019).
Bennett, R. S., Milgroom, M. G. & Bergstrom, G. C. Population Structure of Seedborne Phaeosphaeria nodorum on New York Wheat. Phytopathology 95, 300–305 (2005).
Cunfer, B. M. The Incidence of Septoria nodorum in Wheat Seed. Phytopathology 68, 832 (1978).
Cunfer, B. M. Seasonal availability of inoculum of Stagonospora nodorum in the field in the southeastern US. Cereal Res. Commun. 26, 259–263 (1998).
Croll, D., Zala, M. & McDonald, B. A. Breakage-fusion-bridge cycles and large insertions contribute to the rapid evolution of accessory chromosomes in a fungal pathogen. PLOS Genet. 9, e1003567 (2013).
Hane, J. K. et al. A novel mode of chromosomal evolution peculiar to filamentous Ascomycete fungi. Genome Biol. 12, 1–16 (2011).
McClintock, B. The stability of broken ends of chromosomes in Zea mays. Genetics 26, 234 (1941).
Hocher, A. & Taddei, A. Subtelomeres as specialized chromatin domains. BioEssays 42, 1900205 (2020).
Van de Wouw, A. P. et al. Evolution of linked avirulence effectors in Leptosphaeria maculans is affected by genomic environment and exposure to resistance genes in host plants. PLOS Pathog. 6, e1001180 (2010).
Irelan, J. T., Hagemann, A. T. & Selker, E. U. High frequency repeat-induced point mutation (RIP) is not associated with efficient recombination in Neurospora. Genetics 138, 1093–1103 (1994).
Komluski, J., Habig, M. & Stukenbrock, E. H. Repeat-Induced Point Mutation and Gene Conversion Coinciding with Heterochromatin Shape the Genome of a Plant-Pathogenic Fungus. Mbio 14, e03290–03222 (2023).
Gervais, J. et al. Different waves of effector genes with contrasted genomic location are expressed by Leptosphaeria maculans during cotyledon and stem colonization of oilseed rape. Mol. Plant Pathol. 18, 1113–1126 (2017).
Badet, T. & Croll, D. The rise and fall of genes: origins and functions of plant pathogen pangenomes. Curr. Opin. Plant Biol. 56, 65–73 (2020).
McDonald, M. C., Oliver, R. P., Friesen, T. L., Brunner, P. C. & McDonald, B. A. Global diversity and distribution of three necrotrophic effectors in Phaeosphaeria nodorum and related species. N. Phytologist 199, 241–251 (2013).
Xin, Z. & Chen, J. A high throughput DNA extraction method with high yield and quality. Plant Methods 8, 26 (2012).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Bushnell, B. BBMap sourceforge.net/projects/bbmap/ (2016).
Sanger, F. et al. The nucleotide sequence of bacteriophage φX174. J. Mol. Biol. 125, 225–246 (1978).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46 (2014).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Jones, D. A. qcflow v1.0, https://doi.org/10.5281/zenodo.14170234 (2019).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80–92 (2012).
Raj, A., Stephens, M. & Pritchard, J. K. fastSTRUCTURE: Variational Inference of Population Structure in Large SNP Data Sets. Genetics 197, 573–589 (2014).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Minh, B. Q. et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evolution 37, 1530–1534 (2020).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Van Wyk, S. et al. The RIPper, a web-based tool for genome-wide quantification of Repeat-Induced Point (RIP) mutations. PeerJ 7, e7447 (2019).
Bushnell, B., Rood, J. & Singer, E. BBMerge – Accurate paired shotgun read merging via overlap. PLOS ONE 12, e0185056 (2017).
Bankevich, A. et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Computational Biol. 19, 455–477 (2012).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18–e18 (2017).
Jones, D. A. mitoflow v1.0, https://doi.org/10.5281/zenodo.14170230 (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Mapleson, D., Garcia Accinelli, G., Kettleborough, G., Wright, J. & Clavijo, B. J. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33, 574–576 (2017).
Jones, D. A. postasm v.10, https://doi.org/10.5281/zenodo.14170275 (2019).
Marçais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLOS Computational Biol. 14, e1005944 (2018).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014).
Hu, K. et al. Helitron distribution in Brassicaceae and whole Genome Helitron density as a character for distinguishing plant species. BMC Bioinforma. 20, 354 (2019).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinforma. 9, 18 (2008).
Steinbiss, S., Willhoeft, U., Gremme, G. & Kurtz, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 37, 7002–7013 (2009).
Hu, J., Zheng, Y. & Shang, X. MiteFinderII: a novel tool to identify miniature inverted-repeat transposable elements hidden in eukaryotic genomes. BMC Med. Genomics 11, 101 (2018).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. PNAS 117, 9451–9457 (2020).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-4.0., http://www.repeatmasker.org (2013–2015).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Llorens, C. et al. The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res. 39, D70–D74 (2011).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Jones, D. A. PanTE v.10, https://doi.org/10.5281/zenodo.14170272 (2019).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Wright, E. S. DECIPHER: harnessing local sequence context to improve protein multiple sequence alignment. BMC Bioinforma. 16, 322 (2015).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57 (2016).
Iwata, H. & Gotoh, O. Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic Acids Res. 40, e161 (2012).
Guy St C, S. & Ewan, B. Automated generation of heuristics for biological sequence comparison. BMC Bioinforma. 6, 31–31 (2005).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Lomsadze, A., Burns, P. D. & Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 42, e119 (2014).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinforma. 19, 189 (2018).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Eberhardt, R. Y. et al. AntiFam: a tool to help identify spurious ORFs in protein annotation. Database 2012, https://doi.org/10.1093/database/bas003 (2012).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinforma. 10, 421 (2009).
Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evolution 35, 543–548 (2018).
Gremme, G., Steinbiss, S. & Kurtz, S. GenomeTools: A Comprehensive Software Library for Efficient Processing of Structured Genome Annotations. IEEE/ACM Trans. Computational Biol. Bioinforma. 10, 645–656 (2013).
Standage, D. S. & Brendel, V. P. ParsEval: parallel comparison and analysis of gene structure annotations. BMC Bioinforma. 13, 187 (2012).
Lechner, M. et al. Proteinortho: detection of (co-) orthologs in large-scale analysis. BMC Bioinforma. 12, 1–9 (2011).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLOS ONE 5, e9490 (2010).
Murrell, B. et al. Gene-Wide Identification of Episodic Selection. Mol. Biol. Evolution 32, 1365–1371 (2015).
Pond, S. L. K., Frost, S. D. W. & Muse, S. V. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–679 (2005).
Levy Karin, E., Mirdita, M. & Söding, J. MetaEuk—sensitive, high-throughput gene discovery, and annotation for large-scale eukaryotic metagenomics. Microbiome 8, 1–15 (2020).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Mitchell, A. L. et al. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360 (2019).
Koskinen, P., Törönen, P., Nokso-Koivisto, J. & Holm, L. PANNZER: high-throughput functional annotation of uncharacterized proteins in an error-prone environment. Bioinformatics 31, 1544–1552 (2015).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evolution 34, 2115–2122 (2017).
Jones, D. A. et al. An automated and combinative method for the predictive ranking of candidate effector proteins of fungal plant pathogens. Sci. Rep. 11, 1–13 (2021).
Acknowledgements
This project was supported by the Australian Government Grains Research and Development Corporation (GRDC) (Grant No. CUR00023) DABJ was supported by an Australian Postgraduate Award (APA) awarded by the Australian Government Dept. of Education. MH was supported by GRDC Research Scholarship (GRS) CUR2301-006RSX and by a Research Training Program (RTP) Scholarship awarded by the Australian Government Dept. of Education. This research was undertaken with the assistance of resources provided by the Pawsey Supercomputing Centre and the National Computational Infrastructure (NCI), which is supported by the Australian Government.
Author information
Authors and Affiliations
Contributions
J.K.H. and D.A.B.J. conceived the study. H.T.T.P., K.R. and K.C.T. provided materials. D.A.B.J. and J.K.H. led analysis. M.H., S.B. and A.W. contributed supporting analyses. J.K.H. provided supervision to D.A.B.J. J.K.H. and K.C.T. provided supervision to S.B. J.K.H. and H.T.T.P. provided supervision to M.H. D.A.B.J. and J.K.H. wrote the initial manuscript. J.K.H. wrote the final manuscript. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Alex Zaccaron and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: David Favero. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jones, D.A.B., Rybak, K., Hossain, M. et al. Repeat-induced point mutations driving Parastagonospora nodorum genomic diversity are balanced by selection against non-synonymous mutations. Commun Biol 7, 1614 (2024). https://doi.org/10.1038/s42003-024-07327-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-07327-7
