
Abstract
Epigenetic modifications such as DNA methylation are both cell type and developmental age specific. Here, we show that the immunological maturation of blood cell types influences DNA methylation changes from naive cord blood to fully functional adult blood. Lymphoid cells in adult blood showed more variability than in cord blood suggesting an antigen-dependent maturation of DNA methylation in lymphoid cells throughout the lifespan. Fewer DNA methylation changes between cord and adult blood were observed in myeloid cells, particularly in monocytes, which demonstrated the least number of DNA methylation changes between cord and adult blood. We also noted differences in epigenetic ages by immune cell types within the same individuals, specifically in cord blood where monocytes were epigenetically oldest compared to the other cell types. In addition, we provide a publicly available resource to the community as an R Shiny web application to interactively explore epigenetic patterns between naive cord white blood cells and fully functional adult white blood cells for six immune cell types.
Similar content being viewed by others


Introduction
One of the main established roles for DNA methylation (DNAm) is in development, where it contributes to the functional maturation, lineage commitment, and fate of cells1. This has two important implications; the first is that DNAm within a given cell type will change over time as cells differentiate and their function develops2. The second is that different types of terminally differentiated cells will have very distinct DNAm profiles3,4. Due to this important role of DNAm in the process of sculpting and maintaining tissues and cell types, close assessment of cell type-specific changes over the lifespan can increase our understanding of developmental processes, primarily by identifying specific genes or genomic regions that dynamically change with age within a cell type. This is of great interest to human biology and perhaps best exemplified in whole blood, a commonly investigated heterogeneous tissue that comprises multiple cell types from myeloid and lymphoid lineages and where the development of the immune system in early life is linked to long-term health outcomes. Therefore, a comprehensive investigation of the changes in DNAm profiles from birth to adulthood may provide biological insights into how the immune system matures at the basic level of epigenetics and gene regulation. One commonly used non-invasive source of fetal tissue to investigate immune system changes in early life is umbilical cord blood. Because it is easy to collect from the umbilical cord post-delivery and cord blood banking is now commonly performed in several hospitals, many epigenetic studies have assessed DNAm in relatively large numbers of cord blood samples5,6.
In the context of cellular composition, while—with one notable exception (nRBCs)—the cellular constituents are largely similar, the proportions of immune cell types in cord blood are very distinct from adult blood. Specifically, cord blood has higher absolute white blood cell (WBC) counts relative to adult blood, with a higher number of natural killer (NK) cells, monocytes, regulatory T cells (Tregs), and naive T cells contributing to this distinction7,8. Further, cord blood also contains a much greater abundance of nRBCs, which declines rapidly after birth, whereas adult RBCs are enucleated. Cord blood also expresses unique proteins such as fetal hemoglobin9, as well as functionally distinct myeloid and lymphoid cells10,11. These distinct functions reflect the greater reliance on innate immunity in newborns, as adaptive immune cells require exposure to pathogens in order to mature and generate a functional memory12,13. Thus, one might expect that innate immune cells such as granulocytes, monocytes, and NK cells would be more similar over development than adaptive immune cells like B and T cells. However, this relationship is complex, with differences observed even in the absolute count and the function of innate immune cells between newborns and adults, indicating that the functionality of specific innate cell types also changes over development13,14,15.
These biologically meaningful differences in function are likely to be reflected at least in part in DNAm changes over developmental time. Beyond the obvious importance of understanding the dynamics of immune cell development at the fundamental level, this may, in turn, pose significant challenges in analyzing high dimensional DNAm data, as existing computational tools designed for adult whole blood may not perform optimally in whole blood obtained from newborns or children. A common example of this is DNAm-based cell type deconvolution algorithms, which is one of the major tools used to account for inter-individual differences in cell type composition in mixed tissue samples such as whole blood when more direct measures are not available16,17,18,19. Failing to account for these inter-individual differences in cell type composition can lead to both false positive and false negative results in epigenome-wide association studies (EWAS), and therefore, accurate implementation of this tool in a developmental context is essential2,20. Perhaps not surprisingly, deconvolution algorithms designed using adult references perform poorly on cord blood data21,22,23,24,25. Cord blood-specific algorithms have been published and are now widely used, yet some cord blood cell types remain difficult to predict despite optimization21,23,26,27,28. While accounting for the variation in cell type proportions is essential in DNAm research, it is also important to recognize that certain cellular changes are integral to the biology of some phenotypes with functional consequences27. Therefore, aside from considering cell types as merely a confounding variable in epigenetic analyses, it may be useful in specific settings to investigate the cell type proportions unique to the phenotype of interest, which will offer additional biological insights.
To refine cellular delineations and improve our current understanding of the biological differences in each cell type as they mature from an epigenetic perspective, we have compared DNAm profiles of purified major leukocyte subsets from cord blood and adult blood. We found that the molecular differences between cord and adult blood cell types reflect patterns observed during functional maturation of the immune system, and we provide a publicly available R Shiny web application to explore these findings interactively. Our work strongly suggested that age-associated DNAm differences in blood must be incorporated into the design of methods and analyses developed specifically for DNAm investigations.
Results
Publicly available DNA methylation data from isolated cord and adult WBC types showed differences in clustering between myeloid and lymphoid lineages
We hypothesized that differences in DNAm between cord and adult blood cell types might reveal biologically relevant epigenetic changes that occur during immune system maturation. To test this, we compared molecular patterns of DNAm in the adult and cord blood reference data sets3,21,23,27,28,29. In order to take advantage of as many samples as possible, we combined four previously published cord blood data sets and compared them to two publicly available adult blood data sets (Table 1). All data sets were generated from sorted blood, either flow cytometry (De Goede, Gervin, and Reinius) or magnetic sorting (Bakulski, Lin, and Salas), and were a mixture of samples that were either measured on the 450 K (Bakulski, De Goede, and Gervin) or the 850 K platform (Lin and Salas). The methodological differences in array type and sample collection methods occurred in both cord and adult data sets so we do not expect these differences will severely impact our comparisons. The six data sets were combined and processed together as described in Methods.
Given that our combined set of samples (cord and adult blood) represent different stages of development with cell types belonging to different lineages, we first sought to assess the drivers of DNAm variation in our data. Unsupervised hierarchical clustering on the 389,901 CpGs showed that samples grouped first by myeloid (granulocytes, monocytes) versus lymphoid (B, T, NK cells) lineage. Interestingly, within myeloid cells, samples clustered next by age, and finally by specific cell type, whereas within lymphoid cells, they clustered by cell type and then by age (Fig. 1). A small group of adult lymphocytes was the most distinct group, followed by nRBCs. In cord blood samples, CD4 T and CD8 T cells clustered in one large group, paired primarily by individual as opposed to cell type. To quantify the fit within clusters, we performed a silhouette analysis. Clustering by cell type and age resulted in better silhouette scores than clustering by cell type alone for all cell types except NK cells (Supplementary Fig. 1). CD4 T adult and CD8 T cord and adult blood cells showed particularly higher silhouette scores when clustered by both cell type and age, indicating that within those two cell types, cord and adult blood cells were particularly distinct.

Unsupervised clustering via dendrogram of beta values on 389,901 CpGs from n = 348 samples. Samples clustered first by lineage (pink = myeloid, pale blue = lymphoid), then by age (black = adult, gray = cord), and then by specific cell type (color scale below).
Adult lymphoid cells demonstrated higher variability compared to their corresponding cord blood cell types
We next sought to determine high-level patterns of variability in DNAm between cord and adult WBCs using a principal component analysis (PCA). We examined the first six principal components (PCs), accounting for >80% of the variance and which separated the different cell types. The first PC, accounting for 37% of the variance, separated the myeloid and lymphoid lineages (t-test p < 1 × 10−16), with distinct clustering of cord and adult blood samples in the second PC (t test p < 1 × 10−16), accounting for 13% of the variance (Fig. 2, Supplementary Data 1). It is important to note that this PC may reflect both age-associated and batch differences between the samples, though given that the cord samples represent four different batches, and the adult samples two batches, the batch effects appear to be smaller than the differences between cord and adult blood. When we specifically visualized the effect of the batch by color coding the samples based on batch, we still observed clustering by cell type with no apparent influence of batch effects (Supplementary Fig. 2). Overall, myeloid cell types clustered more closely than lymphoid cell types across PCs, perhaps reflecting a relatively functional and lineage proximity. These findings were consistent with the results of our unsupervised hierarchical clustering analysis. Next, we visually examined the spread of PC scores within a cell type, as an indication of how similar the samples within a cell type are relative to one another. Across both top PCs, adult lymphoid cell types showed greater variability compared to myeloid cell types (Fig. 2). The variability within adult lymphoid cells was also higher than their corresponding cord blood cell types in these first PCs.

The first six principal components of the combined cord and adult blood cell type data (n = 348), where circles are adult samples and triangles are cord blood samples, colors as specified on the legend, and percent of variance indicated on the relevant axis.
To quantify the observed differences in variability between cord and adult blood observed by PCA, we examined the number of variable sites within each cell type. We defined a variable site as having a beta value standard deviation greater than 0.05 (Fig. 3, Supplementary Data 2–4). Notably, nRBCs exhibited a large number of variable sites (77,386). All cell types except granulocytes showed more variable sites in adults than in cord blood. Specifically, B cells, CD8 T cells, and NK cells showed considerably more variability than granulocytes and monocytes. It is difficult to infer whether the larger number of variable sites in cord than adult blood cell types is a function of sample size differences, as we had more cord samples than adults across all cell types.

All cell types demonstrated significant DNAm differences between cord and adult blood, with lymphoid cell types demonstrating more than myeloid cells
To identify distinct cell type DNAm differences between cord and adult blood that drove the separation of samples observed in the hierarchical clustering and PCA, we next performed EWAS analyses between cord and adult blood in the six common cell types (CD4 T, CD8 T, NK, B, granulocytes and monocytes). As expected, a substantial proportion of CpGs were differentially methylated between cord and adult blood at an unadjusted p value of 9 × 10−8 and a mean DNAm difference of 10% (Fig. 4, Supplementary Fig. 3, Supplementary Data 5–10). One complication of the combined data sets was that some sets had total granulocytes, some purified neutrophils only, and one had both total granulocytes and purified neutrophils. While neutrophils are the predominant group in granulocytes, we ensured that the methylomes between granulocytes and the purified neutrophils were comparable by first testing DNAm differences between them. We used the Reinius reference containing both granulocytes and neutrophils from the same individuals (GSE35069) and identified no differentially methylated sites at the thresholds stated above (Supplementary Fig. 4). This led us to combine the neutrophils in the Salas reference (GSE110554) along with the granulocytes in the Reinius reference (GSE35069) and subsequent analyses were performed with the newly aggregated adult granulocyte group. In the purified cell types, 5010 CpGs were differentially methylated between cord and adult samples in all six types, and 11,641 and 1741 CpGs were lymphoid or myeloid specific, respectively (Fig. 4).

We performed an EWAS comparing cord to adult samples for each cell type, retaining sites with a p value < 9 × 10−8 and a mean DNA methylation difference >10% (visualized in Supplementary Fig. 3). UpSet plot indicating overlaps between significantly differentially methylated CpGs between cord and adult blood in the six cell types, with the number of those which are mQTLs in cord, adult, or both cord and adult indicated using greyscale bars.
Because CpGs, which are located within 400 bps of each other typically exhibit correlated DNAm states and have been shown to function as a co-methylated biologically regulated unit, we sought to identify whether our differentially methylated sites in the six cell types were contained in co-methylated regions (CMRs), using the CoMeBack algorithm30. Depending upon the cell type, we identified 31.5–43.8% of the differentially methylated sites in cord blood, and 46.3–52.66% of the differentially methylated sites in adult blood were contained in a CMR. Irrespective of the cell type, the majority of the identified CMRs were 2-CpG CMRs, though individual CMRs as large as 53-CpGs in NK cells were also identified. Further, ~30% of differentially methylated sites across all the cell types were within a CMR in both cord and adult blood samples.
Differentially methylated CpG sites between cord and adult blood cell types showed cell type and lineage-specific enrichment in biological processes and genomic feature locations
To further understand the biological processes and underlying function associated with genes annotated to CpGs that showed differential methylation between cord and adult blood cell types, we conducted a Gene Ontology (GO) enrichment analysis using the missMethyl R package (Supplementary Data 11–16). Applying an FDR cutoff of <0.01 and a minimum of 100 genes per GO term, we first restricted the enrichment analysis to genes associated with differentially methylated CpGs between cord and adult blood cell types specifically within the lymphoid lineage. This revealed 181 unique biological processes, including regulation of the immune effector process, regulation of T cell activation, negative regulation of immune responses, and ion homeostasis. Similarly, when we focused the GO analysis on the set of genes associated with differentially methylated CpGs between cord and adult blood cell types within the myeloid lineage, we found 114 unique biological processes, such as leukocyte differentiation, immune response-activating signal transduction, cellular response to cytokine stimulus, positive regulation of immune responses, and antigen receptor-mediated signaling pathway. Further, considering that chromatin states provide critical insights into the influence of DNAm patterns, we also performed an enrichment analysis of chromatin states underlying our identified CpG associations (Supplementary Data 17–22). Differentially methylated CpGs between cord and adult blood cell types, in both lymphoid and myeloid lineage, were enriched in weak repressed polycomb, repressed polycomb, bivalent enhancer, bivalent TSS, active enhancer, and weak enhancer. However, CpGs associated with lymphoid cell types were also enriched in quiescent/low chromatin state, and myeloid cell types showed enrichment in genic enhancers.
Given that the cord and adult blood samples in the current study were not from the same individuals, this may result in potential false positives due to genetic differences. While we did not have genotyping data available on the samples, we estimated genetic effects on the identified DNAm signatures by taking advantage of the publicly available ARIES data set and examined the overlap with CpGs previously identified as being associated with genotype (mQTLs) in cord and adult blood in the ARIES data set6. In the individual lymphoid cell types, 7.8-12.2% of the differentially methylated CpGs between cord and adult blood were mQTLs (Fig. 4). Interestingly, the myeloid cell types showed an increased number of mQTLs, where >15% of the differentially methylated CpGs in granulocytes and in monocytes were mQTLs.
We next examined the genomic feature locations of the differentially methylated sites between cord and adult in all cell types by mapping each CpG to CpG island class (HC: high-density CpG island, LC: low-density CpG island, IC: intermediate-density CpG island) and genomic features. In B cells, more CpGs mapped to high-density (HC) islands than intermediate-density islands (IC), while all other cell types showed the opposite pattern. Statistically significant differences between the cell types were observed for the enrichment of CpG island status (HC, IC, ICshore, and LC, Cochran-Mantel p < 2.2 × 10−16) and the six genomic regions (1st exon, 3’UTR, 5′UTR, gene body, TSS200, and TSS1500, Cochran-Mantel p < 2.2 × 10−16) investigated (Supplementary Fig. 5).
Epigenetic clock-specific differences in epigenetic age estimates were observed across cord and adult blood cell types
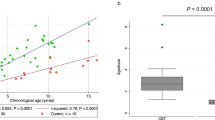
Given that some of the DNAm differences between cord and adult cell types may reflect age-associated variation potentially captured by epigenetic clocks, we examined the lists of CpG sites that were significantly differentially methylated across all cell types, as well as those specific to the myeloid or lymphoid lineage, for enrichment in the pan-tissue adult epigenetic clock sites25. 29/5010, 12/11,641, and 4/1741 sites that were differentially methylated between cord and adult in all cell types, lymphoid, and myeloid cells, respectively, were also in the epigenetic clock (hypergeometric p values 0.006, 0.005, 0.005, respectively, with 100,000 permutations). Owing to the differing aspects of development and aging captured by the epigenetic clocks trained on birth tissues such as cord blood and adult samples such as whole blood, we examined whether the cell types in the two tissues showed concordance in the epigenetic age estimates calculated using these DNAm-based clocks. Further, we assessed whether specific cell types showed higher or lower epigenetic age compared to the other cell types. We opted to test the two cord blood gestational epigenetic clocks specifically designed for samples from the 450 K array, the Knight and Bohlin clocks, and three commonly used first-generation adult epigenetic clocks, the Horvath pan-tissue clock, the Hannum clock, and the SkinBlood clock. Two cord blood data sets and one adult blood data set had reported chronological ages, and we calculated epigenetic age acceleration in weeks of gestational age for cord blood, or years of chronological age for adult blood as the residuals extracted from a linear model of predicted epigenetic age regressed on reported chronological age (Fig. 5, top, Supplementary Data 23)25. The other two cord blood data sets and one adult data set did not have known ages, but as the samples were matched across cell types, we were able to still assess mean differences in epigenetic age (Fig. 5, bottom, Supplementary Data 23). Overall, epigenetic age and epigenetic age acceleration showed similar patterns across data sets, though the patterns differed between cord blood and adult samples and between the clocks. In cord blood, for both epigenetic age and epigenetic age acceleration using the Bohlin clock, monocytes were epigenetically oldest, followed by B cells, while NK, CD4 T, CD8 T, granulocytes, and nRBC were relatively younger in terms of epigenetic age (p value < 2 × 10−16). For the Knight clock, monocytes were also epigenetically oldest, followed by granulocytes whereas B cells were the youngest (p value < 2 × 10−16). The remaining cell types, NK, CD4 T, CD8 T, and nRBCs, showed intermediate predicted gestational ages. B cells and granulocytes displayed the largest differences in both the Bohlin and Knight clock estimates (Fig. 5), which were statistically significant for both gestational epigenetic age acceleration (Bohlin p < 2 × 10−16, Knight p < 2 × 10−16) and gestational epigenetic age (Bohlin p < 2 × 10−16, Knight p < 2 × 10−16) in cord blood. Similarly, in adults, epigenetic age acceleration was significantly different between the cell types (Horvath p = 0.003, Hannum p < 3.39 × 10−12, SkinBlood p < 9.79 × 10−9), with B cells, granulocytes, and CD8 T cells demonstrating the greatest differences between the three adult epigenetic clocks. When examining epigenetic ages between matched samples, only the Hannum epigenetic clock showed specific cell type differences (p = 0.0377), notably underestimating the epigenetic age of CD8 T cells compared to the other cell types. In contrast, both the Horvath pan tissue and the SkinBlood epigenetic clocks did not exhibit differences in epigenetic ages by cell type (Horvath p = 0.771, SkinBlood p = 0.694).

Two cord data sets (de Goede and Gervin, A) and one adult data set (Salas, B) had known gestational or chronological ages, respectively, and thus we calculated epigenetic age acceleration in days (cord) or years (adult) for each sample (n = 348). Two cord (Bakulski and Lin, C) and one adult (Reinius, D), did not have chronological ages, but did have matched samples from each participant, and so we compared epigenetic gestational age (cord) or age (adult) across cell types. In boxplots, the center thick line represents the median, the outer box ranges are the 25th and 75th percentiles, and the whiskers represent the farthest value no further than 1.5 times the IQR. Outliers are not displayed as part of boxplot since points are shown.
A publicly accessible interactive web-based tool will allow researchers to query epigenome-wide differences between cord and adult blood cell types
Overall, our results provide a comprehensive map of DNAm differences in immune cell types between naive cord blood and fully functional adult blood. To facilitate community access to this information, we created an interactive web application (CoADD – Cord blood and Adult blood DNAm Differences, https://rasiimwe.shinyapps.io/CoADD_KoborLab/). This R application allows searching by CpG IDs based on Illumina’s 450 K array, importantly including CpG sites that are differentially methylated between cord and adult blood in at least one cell type. We built CoADD to allow researchers to explore epigenetic patterns of DNAm in both adult and cord blood samples when DNAm data is not available for either or both tissues. By inputting a single CpG site/a list of CpG sites in .csv format, researchers can easily cross-reference if the identified differentially methylated CpG sites (hits) in their respective study exhibit differential DNAm patterns between cord and adult blood in a specific cell type or across the six immune cell types, thus facilitating the interpretation of DNAm results.
Discussion
Here, we have explored intrinsic biological differences in DNAm between cord and adult blood cell types, in order to better understand how changes in DNAm reflect immune cell maturation across cell types. It has been previously shown that DNAm is highly variable with age, but age-related effects on individual cell types are yet to be rigorously explored2,20,25. Using five data sets comprised of purified cell types from cord or adult blood, we examined differences in broad patterns of DNAm, variability, and cell type-specific age-related changes in DNAm as well as epigenetic age across cell types and age.
Given the impact of immunological maturation of WBC types after birth29,31, we noted marked molecular differences between cord and adult blood methylomes which could influence analysis of whole blood in these tissues. While not yet documented, it was perhaps not surprising that DNAm patterns from cord and adult cell types were visually distinct in both unsupervised hierarchical clustering and PCA analysis. Further, the number of variable sites within cell types was highly different between cord and adult blood samples. While PCA showed variable DNAm patterns in all cell types between cord and adult blood, this was accentuated in lymphoid compared to myeloid lineage cells. We hypothesize that overall, lymphoid cells are more variable in adults than in cord blood due to the functional maturation of lymphoid cells postnatally, in contrast to myeloid cells, whose function matures earlier during fetal development. For example, when a naive B-cell encounters a foreign antigen, it activates and differentiates into an antibody-producing memory or an effector B cell. Similarly, activation of a resting, naive T cell relies on the recognition of an antigen bound to an antigen-presenting cell. Because lymphoid cells are activated by antigens and can utilize their immunologic memory to generate a heightened immune response upon a re-encounter of the same antigen32,33,34, it is reasonable to assume that an increase in DNAm variability within lymphoid cells may reflect an age-related increasing proportion of differentiated T and B cells due to antigen exposure over lifespan11,31,35. T cells in umbilical cord blood are predominantly naive36,37 and show reduced expression of proteins that facilitate activation of T cells38,39 whereas adult T cells are antigen-experienced and thus, exhibit increased functional immune responses.
Further, CD4 T and CD8 T cells clustered differently in cord blood than in adult blood. In adults, CD4 T and CD8 T cells clustered separately, whereas in cord blood they clustered by individual rather than by cell type. This implied that the influence of genotypic variation within our study population likely outweighed the influence of cell type on DNAm patterns of CD4 T and CD8 T cells in cord blood. These results suggested that neonatal CD4 T and CD8 T cells were highly similar at the DNAm level, which may reflect a relative lack of functional differentiation between these two cell lineages prior to antigen exposure. This may also partly explain why CD8 T cell proportions have proved the most difficult cell type to predict in cord blood26.
Both variability and EWAS results demonstrated that DNAm differences between cord and adult blood were distinct between cell types. Adult cell types were more variable than in cord blood, with CD8 T, B, and CD4 T cells showing the largest differences. EWAS within cell types identified thousands of DNAm differences between cord and adult blood, but the specific number of differentially methylated CpGs varied across cell types, with CD8 T showing the largest number of differentially methylated sites and monocytes the least. Additionally, there were many more sites that differed between cord and adult blood and were more common between the four lymphoid cell types than between the myeloid cells. This is partly reflective of the fact that there were far fewer changes between cord and adult blood in myeloid cells as a whole, perhaps once again because their immunological function is already mature at birth.
Given that cord and adult blood cell types are involved in maintaining immune homeostasis, we observed the expected enrichment of varying biological processes related to immune activation and regulation across both lymphoid and myeloid DNAm associations. However, processes linked with negative regulation of immune responses, such as apoptotic pathways, were uniquely enriched in lymphoid lineage cell types. In contrast, processes underlying positive regulation of immune responses, such as activation of enzyme activity, were overrepresented in myeloid cell types, delineating the distinct biological roles of these DNAm associations based on cell type lineage. While our GO analyses revealed enrichment of both distinct and shared pathways in the developmentally age-related CpGs associations, the ChromHMM findings predominantly showed enrichment of common chromatin states underlying the differential DNAm patterns, regardless of cell type lineage. This may suggest that similar chromatin features underlying the age-associated molecular changes in DNAm may drive the regulation of key immune responses during development.
Finally, one surprising finding we observed was the difference in epigenetic age estimates among WBC types isolated from the same individuals, though differences in proportions of immune cell subtypes, primarily between naive and activated T and NK cells, have been previously shown to influence epigenetic age prediction in adult blood40. Similarly, cell type differences in gestational epigenetic age between four immune cell types, including T cells, granulocytes, monocytes, and nRBCs, have been previously observed in cord blood, with unique patterns noted in preterm and term cell populations41. While limited studies have assessed the epigenetic age profiles independently in cord and adult blood, a comparison of patterns between them has not been explored. Here, we observed differences in epigenetic aging profiles of the immune cell types in cord and adult blood. Specifically, in cord blood, using both Bohlin and Knight clocks, both gestational age acceleration calculated for those participants with known gestational age and gestational epigenetic age for those whose gestational age was unknown showed the same pattern: monocytes were estimated to have the highest epigenetic gestational ages of all cell types, as was previously reported in adult blood42. Unlike the majority of our analysis, there was no clear myeloid or lymphoid pattern in the cell types with higher or lower epigenetic gestational age estimates. In adult blood samples, epigenetic age acceleration was different between the cell types in the three adult epigenetic clocks, though, the patterns were less clear compared to those observed in cord blood, perhaps due to lower sample size and warrants further investigation.
One important limitation of this analysis is that the granulocytes are composed of neutrophils (>95%), eosinophils, and basophils. While the percentage of neutrophils was very high, we cannot be entirely certain that the difference in composition of granulocyte samples was not influencing our results. However, using the Reinius reference that contained both granulocytes and purified neutrophils from the same individuals, we investigated DNAm differences between the two cell types and found the DNAm profile of the granulocytes and the purified neutrophils were largely similar, suggesting the influence of eosinophils and basophils on DNAm patterns of granulocytes was minimal. A second issue of importance is the differences in sorting strategies between cord and adult blood samples. All data sets in the current study utilized either a magnetic-activated cell sorting method for cell separation and/or confirmed enrichment of cell types by fluorescence-activated cell sorting technology. Importantly, cord blood samples in the study were selectively chosen based on purity estimates26. Although these methods are recognized as gold standards for the isolation of cell populations, discrepancies in cell yield, viability, purity, and processing time exist between these two techniques43. Further, both methods are antibody-based and rely on cell surface markers, which can represent significant challenges for cell types that do not exhibit unique cell surface markers. Dependence on cell surface markers may also restrict cell separation to only those markers for which antibodies are readily available. Based on these considerations, while cell sorting strategies may likely contribute to some of the DNAm differences observed across the cell types in cord and adult blood samples, the opposing pattern observed in CD4 T and CD8 T cells would clearly suggest otherwise. Specifically, CD8 T cells showed the greatest differences between cord and adult blood samples both in overall variability and in the number of variable CpG sites. These cells were isolated from the same staining panel and using the same sorting strategy, with only the CD4 T or CD8 T cell surface markers being different. Thus, we believe that there is very limited, if any, evidence that there was any difference in purity between them, yet we still observed differences in variability. It is also possible that using antibodies for sorting resulted in stimulation of cells, particularly antigen-responsive lymphoid cells. This could contribute to some of the variability within a cell type, but as it would have been applied similarly to cord and adult blood cells, it would have affected DNAm patterns in both cord and adult blood.
Aside from implications of cell sorting strategies on DNAm differences, genetic influences typically account for a substantial proportion of DNAm variation within a tissue44,45,46. Interestingly, in the individual lymphoid cell types, ~7.8−12.2% of the differentially methylated CpGs between cord and adult blood overlapped with cord blood mQTLs, suggesting that the proportion of apparent differences between cord and adult blood cells are largely contributed by genetic differences between the cord and adult samples. In myeloid cells it was much higher, up to 19% in monocytes and granulocytes, suggesting that a greater proportion of apparent DNAm differences between cord and adult blood types were influenced by genetic variation. This implies much fewer DNAm changes occur in monocytes with age compared to other lymphoid cell types, which is also consistent with previous reports and our assessment of DNAm variability47. This may partly explain why, even after recent improvements to cord blood cell type prediction via deconvolution, monocytes remain one of the most difficult cell types to predict26. Additionally, we acknowledge that the cord and adult blood samples were obtained from different data sets and different individuals, which means that both genetic differences and batch effects might inflate our estimate of age-specific DNAm differences. While we did not have available genotyping data to address genetic variation, we assessed mQTL enrichment of our age-specific findings using a publicly available ARIES (ALSPAC) mQTL resource. We were unable to correct batch effects using tools such as ComBat, as either the batch variables were confounded by age and/or the distribution of samples across the chips was unbalanced. This imbalance was an unavoidable consequence of extracting varying subsets of samples from publicly available GEO data sets. Although it was not possible to specifically remove these effects, we thoroughly examined our data for potential batch influences. Plotting the top principal components and color-coding the samples by batch/study showed no substantial batch effects. Additionally, we used a stringent p value cutoff (9 × 10− 8), recommended for studies on the larger EPIC/850 K array which we adopted in our 450 K data, and a high effect size (∆β > 0.10/10%) cutoff to enrich our findings for biological effects, ensure greater stringency and minimize the residual influence of batch. Importantly, since there is no expectation that batch effects would be cell type-specific, the comparisons between cell types should be equally affected by batch effects and thus not bias the interpretation of the findings.
Both the immune system and the methylome exhibit sex-specific patterns, which may change during development. Unfortunately, due to the minimal or complete lack of representation of females in the adult blood GEO data sets, we were unable to broadly test for sex-specific effects. However, the uneven number of sexes across our data sets, particularly between the cord and adult blood data sets, raised the possibility of sex being a potential confounding variable in our differential DNAm analysis. This then offered us the opportunity to address these issues, using a more targeted approach. Specifically, we leveraged previously published lists of sex-specific autosomal CpGs in cord and adult blood and cross-referenced these lists with the differentially methylated CpGs between cord and adult blood cell types identified in the current study. We noted an overlap of only 1–7% between our differentially methylated CpGs and the sex-specific CpGs, depending on the study examined48,49,50,51,52, suggesting that sex had a limited influence on our identified DNAm associations.
Overall, DNAm profiling using microarrays is a powerful approach to capture DNAm variation associated with cell types; however, quantifying the methylome with the newer microarrays will facilitate broader epigenome-wide coverage than the 450 K array. Additionally, measuring other molecular features of cord and adult blood cell types, including other epigenomic, proteomic, and transcriptomic profiles, will refine existing cellular definitions and will provide a complete picture of functional changes in cell types as the immune system matures.
Conclusions
The exciting potential of epigenetic profiling of cord blood as a marker of in utero environmental exposure should be balanced by an understanding of the unique cellular properties of that tissue. Based on our results, it is evident that leukocytes of different lineages mature differently in utero and after birth resulting in different DNAm between cord and adult cell types. These appeared to be primarily driven by lymphocytes, which have very different DNAm profiles in cord blood compared to adults, mirroring their acquisition of immunological memory postnatally upon antigen exposure. These findings suggest an important role of DNAm in immune cell maturation or maintenance of a matured state and indicate why DNAm-based tools that are generated in adult samples should be applied to other fetal samples like cord blood with caution. Further, our work also showed age prediction by epigenetic clocks differs by immune cell types, a finding observed in both cord and adult blood, and requires further investigations to understand the biological processes that contribute to these cell type differences in epigenetic clock estimations. The rich molecular information generated from the current study is made available to the community through a user-friendly R Shiny web application – CoADD. This interactive tool will allow users to visualize and compare DNAm patterns between adult blood and cord blood and thus aid in the interpretation of DNAm findings in the context of immune cell type differences.
Methods
Datasets merging
The two adult data sets, here named by the first author of the respective papers: Reinius reference and Salas reference, were run on two Illumina Infinium Human Methylation BeadChip platforms. While the underlying probe chemistry to measure DNAm across these platforms remains the same, the Illumina HumanMethylation450 BeadChip (450 K) quantifies DNAm at 485,512 CpGs, and Illumina MethylationEPIC BeadChip (850 K) measures 866,091 CpGs on the human genome. Both the data sets are available through Bioconductor: FlowSorted.Blood.450k (http://bioconductor.org/packages/release/data/experiment/html/FlowSorted.Blood.450k.html) and FlowSorted.Blood.EPIC (http://bioconductor.riken.jp/packages/3.8/data/experiment/html/FlowSorted.Blood.EPIC.html).
Similarly, the four umbilical cord blood data sets (de Goede reference, Bakulski reference, Gervin reference, and Lin reference) consisted of three 450 K data sets and one 850 K DNAm data set from the Lin reference. The R package FlowSorted.CordBloodCombined.450 K available through Bioconductor (https://bioconductor.org/packages/devel/data/experiment/html/FlowSorted.CordBloodCombined.450k.html) was used to obtain the cord blood data sets. Finally, combineArrays() in R minfi package53 was used to combine the adult and cord blood data set, leaving only the 404,779 CpGs present on both the 450 K and 850 K arrays for preprocessing.
Data preprocessing
A battery of checks was conducted to determine sample identity and quality. The control probes on the array were used to assess technical parameters including array staining, extension, hybridization, target removal, specificity, and bisulfite conversion. All the adult and cord blood samples passed the control probes check and the detectOutlier function in R lumi package54 did not identify any outliers. None of the samples exhibit more than 1% detection p values over the background. Further, nRBCs have been shown to physically interact with other immune cell types, including T and B cells during cell sorting, which may result in substantial contamination impacting DNAm readouts28. Therefore, we checked nRBC contamination of purified hematopoietic cells based on measurements at eight CpG sites which were expected to exhibit less methylation in nRBC compared to other cell types (magnitude of DNAm difference >0.50). At these CpGs, we observed the expected patterns and confirmed no nRBC contamination (Supplementary Fig. 6, Supplementary Data 24). Hierarchical clustering based on the beta values of the X and Y chromosomes was used to confirm the reported sex.
Background correction with dye-bias normalization on the merged DNAm data was done using preprocessNoob() in R minfi package, followed by BMIQ normalization to account for probe type differences on the array. Next probe filtering was performed to remove the SNP probes, XY probes, polymorphic probes, and cross-hybridizing probes55. Finally, probes that exhibit a bad detection p value > 0.01 in 5% of the samples were eliminated, leaving 389,901 probes for further analyses.
Unsupervised dendrogram and PCA of cord and adult blood samples
The dendrogram was generated using complete linkage of a Euclidean distance matrix of samples based on methylation beta values including all three of these FACS-sorted cell type data sets, with samples colored by cell type, age, and myeloid vs lymphoid lineage. Silhouette analysis used the same distance matrix, clustered by cell type or cell type and age. We performed PCA on the same data set using the prcomp()in R. For the number of variable sites, we counted the number of sites with SD > 0.05 in beta value in each cell type by age. As DNAm beta values are heteroscedastic, M values are log-transformed beta values that avoid the typical statistical problems associated with heteroscedasticity.
Epigenome-wide association study comparing cord to adult blood cells
EWAS analysis was performed on the adult and our sorted cord blood data sets3,21,23,27,28,29. We applied the R limma package on the M values with a categorical variable of cord vs adult and no other covariates to normalized data, using a p value cutoff of 9 × 10−8 and a mean absolute DNAm difference of 0.1 (10%) to define a significant CpG site56.
Co-methylated region-based calling and mQTL identification
CMRs were identified in a cell type-specific way in both cord blood and adult blood using the R CoMeBack package (https://bitbucket.org/flopflip/comeback/src/master/)30. We constructed the CMRs based on the default settings and a 0.3 spearman correlation cutoff. For the mQTL identification to investigate the genetic influence on the differentially methylated sites, we accessed the published ARIES (ALSPAC) mQTL data set via http://www.mqtldb.org/.
Gene ontology and ChromHMM enrichment analysis
Enrichment analysis of biological processes for the genes associated with the differentially methylated CpGs between cord blood and adult blood cell types was conducted using the gometh() embedded in the R missMethyl package. In addition, enrichment analysis of chromatin states, generated using ChromHMM57, was performed using the relevant cell type-specific ChromHMM maps.
Epigenetic age analysis
We calculated the epigenetic age of adult samples and the epigenetic gestational age of cord samples independently, as previously published25,58. For adult samples, we used the online tool available at http://dnamage.genetics.ucla.edu/submit, following the recommended guidelines. For cord blood samples, we used the publicly available package at https://zenodo.org/record/60498. We next divided the samples based on whether we knew the participants chronological or gestational ages. For those with known chronological or gestational age, we calculated age acceleration as the residuals of linear regression with epigenetic age as the dependent and chronological age as the independent variable (epigenetic age~chronological age). For those without known age, we used calculated epigenetic age on its own, but as samples were mostly matched across cell types, average epigenetic age across samples should be comparable. In all cases, we performed an ANOVA to determine whether epigenetic age acceleration or epigenetic age differed between cell types within cord or adult samples.
Statistics and reproducibility
All statistical analyses performed in the study were conducted in R programming environment. Specific R packages and their associated functions have been provided in the relevant method sections described above. The R code is publicly available in Github and through Zenodo https://doi.org/10.5281/zenodo.1467148259, whereas the source data which was used to conduct the analyses and create the figures is accessible as Supplementary Data in the form of excel sheets.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
As mentioned in the Methods, the data sets used in the current study were downloaded through the Bioconductor R packages: FlowSorted.Blood.450 K, FlowSorted.Blood.EPIC and FlowSorted.CordBloodCombined.450K. Source data for the figures can be found in the Supplementary Data.
Code availability
The R code used to conduct the statistical analyses described in the study is publicly available in Kobor lab Github https://github.com/kobor-lab/Cord_AdultBlood and through Zenodo https://doi.org/10.5281/zenodo.14671482.
Abbreviations
- CMR:
-
Co-methylated region
- DNAm:
-
DNA methylation
- EWAS:
-
Epigenome-wide association studies
- FACS:
-
Fluorescence-activated cell sorting
- mQTL:
-
Methylation quantitative trait loci
- NK:
-
Natural killer
- nRBCs:
-
Nucleated red blood cells
- PCA:
-
Principal component analysis
- PC:
-
Principal components
- Tregs:
-
Regulatory T cells
- WBC:
-
White blood cell
- 450 K:
-
Illumina infinium methylation450K beadchip
References
Smith, Z. D. & Meissner, A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204–220 (2013).
Jones, M. J., Goodman, S. J. & Kobor, M. S. DNA methylation and healthy human aging. Aging Cell 14, 924–932 (2015).
Reinius, L. E. et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 7, e41361 (2012).
Ziller, M. J. et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481 (2013).
Joubert, B. R. et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am. J. Hum. Genet. 98, 680–696 (2016).
Gaunt, T. R. et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 17, 61 (2016).
Prabhu, S. B. et al. Comparison of human neonatal and adult blood leukocyte subset composition phenotypes. PLoS One 11, e0162242 (2016).
Beck, R. & Lam-Po-Tang, P. R. Comparison of cord blood and adult blood lymphocyte normal ranges: a possible explanation for decreased severity of graft versus host disease after cord blood transplantation. Immunol. Cell Biol. 72, 440–444 (1994).
Hermansen, M. C. Nucleated red blood cells in the fetus and newborn. Arch. Dis. Child Fetal Neonatal Ed. 84, 211F–215F (2001).
Kan, B., Razzaghian, H. R. & Lavoie, P. M. An immunological perspective on neonatal sepsis. Trends Mol. Med. 22, 290–302 (2016).
van den Heuvel, D. et al. Effects of nongenetic factors on immune cell dynamics in early childhood: the Generation R Study. J. Allergy Clin. Immunol. 139, 1923–1934.e17 (2017).
Holt, P. G. & Jones, C. A. The development of the immune system during pregnancy and early life. Allergy 55, 688–697 (2000).
Quinello, C. et al. Phenotypic differences in leucocyte populations among healthy preterm and full-term newborns. Scand. J. Immunol. 80, 57–70 (2014).
Kollmann, T. R., Levy, O., Montgomery, R. R. & Goriely, S. Innate immune function by Toll-like receptors: distinct responses in newborns and the elderly. Immunity 37, 771–783 (2012).
Netea, M. G. Training innate immunity: the changing concept of immunological memory in innate host defence. Eur. J. Clin. Investig. 43, 881–884 (2013).
Shen-Orr, S. S. et al. Cell type-specific gene expression differences in complex tissues. Nat. Methods 7, 287–289 (2010).
Lam, L. L. et al. Factors underlying variable DNA methylation in a human community cohort. Proc. Natl. Acad. Sci. USA 109, 17253–17260 (2012).
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86 (2012).
Koestler, D. C. et al. Blood-based profiles of DNA methylation predict the underlying distribution of cell types: a validation analysis. Epigenetics8, 816–826 (2013).
Jaffe, A. E. & Irizarry, R. A. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 15, R31 (2014).
Gervin, K. et al. Cell type specific DNA methylation in cord blood: a 450K-reference data set and cell count-based validation of estimated cell type composition. Epigenetics 11, 690–698 (2016).
Yousefi, P. et al. Estimation of blood cellular heterogeneity in newborns and children for epigenome-wide association studies. Environ. Mol. Mutagen. 56, 751–758 (2015).
Bakulski, K. M. et al. DNA methylation of cord blood cell types: Applications for mixed cell birth studies. Epigenetics 11, 354–62 (2016).
Koch, C. M. & Wagner, W. Epigenetic-aging-signature to determine age in different tissues. Aging 3, 1018–1027 (2011).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Gervin, K. et al. Systematic evaluation and validation of reference and library selection methods for deconvolution of cord blood DNA methylation data. Clin. Epigenetics 11, 125 (2019).
Salas, L. A. et al. An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol. 19, 64 (2018).
de Goede, O. M. et al. Nucleated red blood cells impact DNA methylation and expression analyses of cord blood hematopoietic cells. Clin. Epigenetics 7, 95 (2015).
Lin, X. et al. Cell type-specific DNA methylation in neonatal cord tissue and cord blood: a 850K-reference panel and comparison of cell types. Epigenetics 13, 941–958 (2018).
Gatev, E., Gladish, N., Mostafavi, S. & Kobor, M. S. CoMeBack: DNA methylation array data analysis for co-methylated regions. Bioinformatics 36, 2675–2683 (2020).
de Jong, E., Strunk, T., Burgner, D., Lavoie, P. M. & Currie, A. The phenotype and function of preterm infant monocytes: implications for susceptibility to infection. J. Leukoc. Biol. 102, 645–656 (2017).
Kurosaki, T., Kometani, K. & Ise, W. Memory B cells. Nat. Rev. Immunol. 15, 149–159 (2015).
Pennock, N. D. et al. T cell responses: naïve to memory and everything in between. Adv. Physiol. Educ. 37, 273–283 (2013).
Nikzad, R. et al. Human natural killer cells mediate adaptive immunity to viral antigens. Sci. Immunol. 4, eaat8116 (2019).
Sharma, A. A. et al. Hierarchical maturation of innate immune defences in very preterm neonates. Neonatology 106, 1–9 (2014).
Jacks, R. D. et al. Cell intrinsic characteristics of human cord blood naïve CD4 T cells. Immunol. Lett. 193, 51–57 (2018).
Cantó, E., Rodriguez-Sanchez, J. L. & Vidal, S. Distinctive response of naïve lymphocytes from cord blood to primary activation via TCR. J. Leukoc. Biol. 74, 998–1007 (2003).
Berthou, C. et al. Cord blood T lymphocytes lack constitutive perforin expression in contrast to adult peripheral blood T lymphocytes. Blood 85, 1540–1546 (1995).
Kaminski, B. A. et al. Reduced expression of NFAT-associated genes in UCB versus adult CD4+ T lymphocytes during primary stimulation. Blood 102, 4608–4617 (2003).
Sinke, L., Cats, D. & Heijmans, B. T. Omixer: multivariate and reproducible sample randomization to proactively counter batch effects in omics studies. Bioinformatics 37, 3051–3052 (2021).
de Goede, O. M., Lavoie, P. M. & Robinson, W. P. Cord blood hematopoietic cells from preterm infants display altered DNA methylation patterns. Clin. Epigenetics 9, 39 (2017).
Marttila, S. et al. Biological aging of different blood cell types. GeroScience https://doi.org/10.1007/s11357-024-01287-w (2024).
Sutermaster, B. A. & Darling, E. M. Considerations for high-yield, high-throughput cell enrichment: fluorescence versus magnetic sorting. Sci. Rep. 9, 227 (2019).
Chen, L. et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167, 1398–1414.e24 (2016).
Gertz, J. et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 7, e1002228 (2011).
Islam, S. A. et al. Integration of DNA methylation patterns and genetic variation in human pediatric tissues help inform EWAS design and interpretation. Epigenet. Chromatin 12, 1 (2019).
Ecker, S. et al. Genome-wide analysis of differential transcriptional and epigenetic variability across human immune cell types. Genome Biol. 18, 18 (2017).
Singmann, P. et al. Characterization of whole-genome autosomal differences of DNA methylation between men and women. Epigenet. Chromatin 8, 43 (2015).
Solomon, O. et al. Meta-analysis of epigenome-wide association studies in newborns and children show widespread sex differences in blood DNA methylation. Mutat. Res. Rev. Mutat. Res. 789, 108415 (2022).
McCartney, D. L. et al. An epigenome-wide association study of sex-specific chronological ageing. Genome Med. 12, 1 (2019).
Grant, O. A., Wang, Y., Kumari, M., Zabet, N. R. & Schalkwyk, L. Characterising sex differences of autosomal DNA methylation in whole blood using the Illumina EPIC array. Clin. Epigenetics 14, 62 (2022).
Gatev, E. et al. Autosomal sex-associated co-methylated regions predict biological sex from DNA methylation. Nucleic Acids Res. 49, 9097–9116 (2021).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Du, P., Kibbe, W. A. & Lin, S. M. lumi: a pipeline for processing Illumina microarray. Bioinformatics 24, 1547–1548 (2008).
Price, M. E. et al. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenet. Chromatin 6, 4 (2013).
Mansell, G. et al. Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC Genomics 20, 366 (2019).
Ernst, J. & Kellis, M. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012).
Bohlin, J. et al. Prediction of gestational age based on genome-wide differentially methylated regions. Genome Biol. 17, 207 (2016).
Chaini Konwar. kobor-lab/Cord_AdultBlood: Cord_Adultblood_manuscript. Zenodo http://zenodo.org/records/14671483 (2025).
Author information
Authors and Affiliations
Contributions
Conceptualization: M.S.K., M.J.J., and C.K.; biological data generation: J.L.M., H.R.R., A.M.M, O.dG., K.G., R.L., P.M.L., W.P.R., and M.S.K.; Biological data curation and processing: M.J.J., L.D., and C.K.; Data analysis: M.J.J., K.K., L.D., and C.K.; Interpretation of results: M.J.J., K.G., P.M.L., R.N., D.C.K., J.F.F., W.P.R., S.M., and C.K.; Shiny app creation: R. A. and C.K.; Manuscript review: M.J.J., C.K., R.A., L.D., H.R.R., O.de G., J.L.M., A.M.M., K.K., K.G., R.L., R.N., D.C.K., J.F.F., P.M.L., W.P.R., S.M., and M.S.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Kristen Boyle and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Ani Manichaikul and Kaliya Georgieva. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jones, M.J., Konwar, C., Asiimwe, R. et al. DNA methylation differences between cord and adult white blood cells reflect postnatal immune cell maturation. Commun Biol 8, 237 (2025). https://doi.org/10.1038/s42003-025-07661-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07661-4
