
Abstract
The lineage commitment and differentiation of mesenchymal stem cells (MSCs) play a crucial role in bone homeostasis. MAPK7 (Mitogen-activated protein kinase 7), a member of MAPK family, controls cell differentiation, proliferation and survival. However, the specific role of Mapk7 in regulating osteogenic and adipogenic differentiation of MSCs remains to be determined. In this study, depletion of Mapk7 in MSCs by crossing Prx1-Cre mice to Mapk7flox/flox resulted in severe low bone mass and accumulation of fat in bone marrow exhibiting osteoporosis (OP) in mice. Mapk7 promoted osteogenic differentiation and inhibited adipogenic differentiation of MSCs after knocking out and over-expressing Mapk7 in vitro. Mechanistically, Mapk7 activated Wnt/β-catenin signaling by phosphorylating Lrp6 at Ser1490, which ultimately enhanced osteogenesis and suppressed adipogenesis of MSCs. This is of great clinical and scientific significance for understanding biological function of Mapk7 and developing potential therapeutic targets for treatment of MSCs differentiation imbalance related bone diseases, such as, osteoporosis.
Similar content being viewed by others


Introduction
Mesenchymal stem cells (MSCs) are a class of mesodermal derived stem cells that served as a pool of multipotent progenitors, which have the potential to differentiate exclusively into either osteogenesis or adipogenesis lineages, isolated from a variety of adult or fetal tissues1,2,3,4,5,6. As a common progenitor of adipocytes and osteoblasts, the tightly controlled lineage commitment of MSCs play a critical role in the maintenance of bone homeostasis7. With age and estrogen deficiency, MSCs preferentially differentiate into adipocytes, resulting in increasing in bone marrow fat and progressive bone loss, which lead to a series of complications such as increased bone fragility and fracture8,9. Therefore, searching for key factors that promote osteogenic differentiation of MSCs while inhibiting adipogenic differentiation is a potential endeavor for OP.
Recent findings suggested that many transcription factors, signaling pathways, epigenetic regulation, and other factors played key roles in regulating the lineage commitment of MSCs, and alteration of these factors contributed to the imbalance of osteogenic and adipogenic differentiation of MSCs resulting OP10,11. Decades of in-depth studies have demonstrated that mitogen-activated protein kinases (MAPKs), Wingless-Type MMTV Integration Site Family (Wnt)/β-catenin signaling, bone morphogenetic protein (BMP) pathway, and Notch signaling exhibit dual regulatory functions in modulating MSCs differentiation10,11. This modulation occurs through targeting downstream transcription factors like runt-related transcription factor 2 (Runx2), osterix (Osx), and peroxisome proliferative activated receptor, gamma (Pparγ), as well as through intricate cross-talk among these signaling pathways10,11. Nonetheless, the mechanisms that govern the imbalance between osteogenic and adipogenic differentiation of MSCs are exceptionally intricate, and numerous pivotal regulators remain undefined, necessitating additional exploration and comprehensive research.
MAPK (ERK) family, containing ERK1 (also known as MAPK3), ERK2 (MAPK1), c-Jun N-terminal kinase (MAPK8/9), and p38 (MAPK14), are crucial in skeletal development and bone mass maintenance, mediating the effects of various extracellular ligands, such as BMPs, WNTs, and parathyroid hormone (PTH), which impact osteogenic potential of MSCs12,13,14. As one of the less studied and structurally unique members of the MAPK family, MAPK7, also known as ERK5, contains a distinctive C-terminal extension, which encodes two proline-rich domains and incorporates a signal for nuclear localization15. Mouse genetic studies have revealed its crucial role in heart function via its deletion in the heart, resulting in embryonic lethality15. Thus, utilizing specific tool mice to investigate its function in particular tissues was a viable solution. Previous research revealed that MAPK7 was a novel causative gene for adolescent scoliosis (AIS), playing a crucial role in the development of cartilage within the skeletal system; Deletion MAPK7 impaired osteogenic differentiation potential of human bone marrow-derived MSCs (hBMSCs)16. Besides, deletion Mapk7 in chondrocytes (Col2a1-Cre; Mapk7flox/flox) impaired endochondral bone formation within the limbs along with bone mass loss17. Chondrocytes were terminally differentiated cells17. Therefore, we hypothesize that Mapk7 may play a role in differentiation in earlier differentiated cells, such as MSCs, and influence skeletal phenotype. Moreover, imbalance between osteogenic and adipogenic differentiation of MSCs was one of the pathological mechanisms of OP11,17,18,19. However, there is currently a lack of direct evidence regarding the impact of Mapk7 deficiency, in MSCs fate commitment, as well as its effect on OP. Therefore, elucidating the role of Mapk7 in MSCs fate transition and its downstream molecular mechanism contributes to refining our understanding of the functions of MAPK family members in maintaining bone homeostasis, as well as provides new insights for related bone disease therapies in the future.
To address the questions above, we crossed Prx1-Cre mice with Mapk7flox/flox mice to delete Mapk7 in MSCs and conducted further mechanistic studies using primary MSCs and C3H10T1/2 MSCs. Our results demonstrated that Prx1-Cre; Mapk7flox/flox mice exhibited bone loss and increased bone marrow adiposity. Further investigation showed deletion Mapk7 decreased phosphorylation of Lrp6 at Ser1490 and inhibited Wnt/β-catenin signaling, thereby suppressing osteogenic differentiation and enhancing adipogenic differentiation of MSCs.
Methods
Animals
Mapk7flox/flox mouse strains were previously described in our study20. Prx1-Cre mouse strain were gifted from Bai lab21. To generate Prx1-Cre; Mapk7flox/flox (conditional knock out, CKO) and Mapk7flox/flox (wild type, WT) mice, we used Mapk7flox/flox strain mice to cross with Prx1-Cre mice. All mice were maintained in an environment with carefully regulated conditions, including temperature (20–26 °C), humidity (40–70%), and lighting. The animal experiments were conducted at the Laboratory Animal Center of Sun Yat-sen University, approved and regulated by the Committee for the Management and Use of Laboratory Animals of Sun Yat-sen University (Approval No. SYSU-IACUC-2022-001823), and all experimental procedures were carried out in compliance with relevant regulations. All efforts were made to minimize animal usage, manipulation, and pain. Mice genotyping was performed by PCR using DNA Extraction Kit and PCR reaction kit (B40015, Selleck, China). The primers used were listed in Supplementary Table 1.
Cell lines and reagents
L Wnt-3A cells (CL-0375, Procell, China) were genetically modified mouse subcutaneous connective tissue cells to stably secrete Wnt3a protein to active Wnt/βcatenin signaling. L Wnt-3A were cultured in DMEM medium (C11995599BT, Gibco, USA) containing 10% fetal bovine serum (FBS) (A5669701, Gibco, USA). L Wnt-3A was used to co-culture with MSCs in a trans-well plate (TCS016024, JET, China). C3H10T1/2 MSCs were gifted from Wang lab22. CHIR99021 (10 nM; HY-10182A, MCE) was an activator of Wnt/β-catenin signaling. We added CHIR99021 in the induction differentiation medium during the differentiation of MSCs to assess the role of Wnt/β-catenin signaling in Mapk7 regulation in osteogenesis and adipogenesis of MSCs.
Primary MSCs isolation
Primary mouse MSCs were isolated via flushing bone marrow out of the long bones, digesting the bone chips with collagenase type II, deprivation of the released cells, and culturing the digested bone fragments, out of which fibroblast-like cells migrate and grow in the defined medium as previous described23. In detail, we isolated primary MSCs by aspirating long bones with a syringe and subsequently digesting bone fragments with type II collagenase (Sigma, V900892-100MG, USA) in a shaker at 37 °C for 2 h. Subsequently, the supernatant was discarded through a centrifuge, and the cellular precipitate, along with bone fragments were collected and re-suspended in α-MEM medium supplemented with 10% FBS. The suspension was cultured for a period of 1–3 days until a sufficient number of large and clonal cell clusters which could be visually confirmed under a microscope. Once the cells reached 80–90% confluence, they were subjected to 0.25% trypsin–EDTA (25200-056, Gibco, USA) digestion, with each digestion step controlled to within 2 min at room temperature. Cells within the passage range of 3–8 were utilized for the procedure.
Flow cytometry
To identify the characteristics of primary MSCs, we detected cell surface markers, such as: CD105-phycoerythrin (PE) (12047, Biolegend, USA), CD29-PE (102207, Biolegend, USA), Sca1-fluorescein isothiocyanate (FITC) (108105, Biolegend, USA), CD45-FITC (103107, Biolegend, USA), CD34-PE (119107, Biolegend, USA) using flow cytometers (CytoFLEX, Beckman Coulter, USA) and analyzing data via FlowJo10.8.1 software.
MSCs differentiation and staining
The differentiation potential of MSCs was further verified by evaluating the multilineage differentiation capabilities in passages 3–6. In details, primary MSCs were cultured in osteogenic/adipogenic induction medium (AIM/OIM) (MUXMX-90031/MUXMX-90031, Cyagen, China). Alkaline phosphatase (ALP) staining was conducted for up to the seventh day of incubation in OIM, as previously described24. Additionally, ARS/ORO staining was performed until visible calcium nodules or oil droplets were observed under a microscope. Calcium nodules were dissolved in DMSO and measured at 405 nm for quantitative analysis. Lipid droplets were extracted using isopropanol, and measured at 510 nm for quantitative analysis. For differentiation to chondrocytes, 1% (vol/vol) Insulin–Transferrin–Selenium (ITS) (41400045, Gibco, USA) was added in α-MEM medium (C12571500BT, Gibco, USA) with 10% (vol/vol) FBS to culture MSCs for 21 days. After the end of chondrogenic differentiation, chondrocytes were stained using alcian blue (ALCB-10001, Cyagen, China) after fixed with 4% PFA (G1101, Servicebio, China).
Real-time quantitative PCR (RT-qPCR) analysis
Total RNA was extracted from cells or tissue using TRIzol reagent (15596-026, Invitrogen, USA) based on manufacturer’s protocol. Reverse transcription was conducted using 1–0.5 μg of RNA and cDNA Synthesis Mix (TSK322S, Tsingke, China) as per the manufacturer's instructions. For RT-qPCR, ArtiCanATM SYBR qPCR Mix (TSE501, Tsingke, China) was employed following manufacturer’s guidelines. GAPDH was utilized as an internal control, and the 2−ΔΔCt method was applied to assess relative gene expression. The primer sequences used for RT-qPCR is provided in Table 1.
Bone tissue imaging and immunological detection
Mouse femur was obtained with removal of muscle tissue, fixed in 4% PFA at 4 °C overnight and stored in 70% ethanol, allowing for direct micro-quantitative computed tomography (μCT) scanning using a resolution of 10 μm with a voltage of 70 kV and a current of 200 μA on a μCT scanner (SkyScan1276, Bruker, Germany). Bone parameters including bone volume per tissue volume (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N), and the trabecular separation (Tb.Sp) were calculated25, quantifying the trabecular region beneath the growth plate as previously described20. For tibia to be sectioned and stained, decalcification in EDTA for several weeks was conducted, followed by hematoxylin–eosin (H&E) and immunohistochemical (IHC) staining as necessary, rabbit anti-OSX (ab209484, Abcam, IHC: 1:50), mouse anti-OPN (ab69498, Abcam, IHC: 1:50), tartrate-resistant acid phosphatase (TRAP) staining was performed to calculate bone resorption parameter: osteoclast number (N.Oc/BS), which was performed in line with TRAP assay kit procedure (387A-1KT, Sigma, USA).
Dynamic histomorphometry was performed using calcein (C0004-5G, TCI, Japan) (10 mg/kg body weight) and alizarin red S (A3882, Sigma, USA) (20 mg/kg body weight) were injected subcutaneously 12 and 2 days before sacrifice in the 9-week-old mice, respectively. Femur were embedded in methyl methacrylate (M813511-500 ml, Macklin, China), and approximately 100-mm-thick horizontal sections of distal metaphyses and midshaft regions were prepared. Calcein and alizarin red labeling was visualized using a fluorescence microscope (DS-U3, Nikon, Japan) with an excitation wavelength of 450–490 nm and a 510–560 nm band-pass filter, respectively, for clarifying bone formation parameters: mineral apposition rate (MAR) and bone formation rate as bone surface reference (BFR/BS)25.
Western blot (WB) and immunoprecipitation (IP)
Primary MSCs, C3H10T1/2 MSCs were harvested and lysed with RIPA buffer containing 1% phosphatase inhibitor cocktail and protease inhibitor cocktail (HY-K0023-1 mL and HY-K0010-1 mL, MCE, USA) on ice for 20 min. Nuclear lysate of primary MSCs was extracted using Nuclear and Cytoplasmic Protein Extraction Kit (P0028, Beyotime, China) according to operating rules. The lysate was centrifuged at 12,000×g for 15 min at 4 °C. The resulting immunoprecipitation (IP) complexes were prepared using corresponding antibodies according to the instructions provided by DynabeadsTM Protein G (10004D, Thermo Fisher, USA). The complexes were washed and heat denatured, followed by separation of the proteins using SDS–PAGE and transfer onto 0.22 μm PVDF membranes (3010040001, Roche, Germany). After blocking for 1 h at room temperature, antibody–antigen immunohybridization was performed, and gray-scale detection was carried out using the enhanced chemi-luminescence (ECL) detection kit (WBKLS0500, Millipore, USA). GAPDH was used as an internal reference protein. The primary antibodies used were as follows: mouse anti-MAPK7 (3372s, Cell Signaling Technology, WB: 1:200), mouse anti-GAPDH (60004-1-Ig, Proteintech, WB: 1:2000), rabbit anti-RUNX2 (12556, Cell Signaling Technology, WB: 1:1000); mouse anti-OPN (ab69498, Abcam, WB: 1:1000), rabbit non-phospho (Active) anti-β-catenin (19807T, Cell Signaling Technology, WB:1:1000), GSK3β (A11731, ABclonal, WB:1:1000), p-GSK3β (AP1088, ABclonal, WB:1:1000), Phospho-LRP6-S1490 (AP1191, ABclonal, WB:1:1000), LRP6 (A6134, ABclonal, WB:1:1000), MEF2C (10056-1-AP, Proteintech, WB: 1:1000), Phospho-MEF2C (ab78888, Abcam, WB: 1:1000).
Immunofluorescence (IF) staining
Cells were fixed with 4% PFA for 30 min, permeabilized with 0.1% Triton X-100 (V900502, Sigma, USA) for 5 min, and subsequently blocked in 1% BSA (V900933-100G, Sigma, USA) diluted with PBS for 30 min. After three times washing with PBS, cells were incubated with primary anti-β-catenin (D10A8) XP antibody (8480T, Cell Signaling Technology,1:50) overnight at 4 °C. Following another three washes in PBS, cells were incubated with anti-rabbit IgG Alexa Fluor 555 (44135, Cell Signaling Technology, IF:1:1000) for 1 h. Finally, Anti-fading Mounting Medium with DAPI (S2110, Solarbio, China) was used to visualize nuclear fluorescence. The samples and captured images were observed using a fluorescent microscope (Axio Imager.Z2, Zeiss, Germany).
RNA-sequencing (RNA-seq) analysis
We isolated MSCs from 3 weeks CKO mice (n = 3) and WT littermates (n = 3). Total RNA of non-induction MSCs in passage 3 was extracted using TRIzol regent (15596-026, Invitrogen, USA). Libraries were prepared using TruSeq Stranded mRNA LTSample Prep Kit (Illumina, USA) and sequenced on the Illumina sequencing platform according to the manufacturer’s instructions. Genes exhibiting a fold change of 2 and P-value < 0.05 were identified as significantly differentially expressed. GO and KEGG pathway enrichment of differentially expressed genes were analyzed using online tool of DAVID Bioinformatics Resources 202121.
Molecular docking
The three-dimensional (3D) structures of LRP5/LRP6 were generated using Alphafold2 based on the amino acid sequence obtained from the UniProt website (www.uniprot.org/) through multiple sequence alignment. Obtained the 3D structures of ERK5 (PDB: 4ZSG) from the Protein Data Bank (PDB). The protein structure was visualized using PyMOL 2.5.32 software26. We used PyMOL software to preprocess the protein structures (the removal of water molecules, hydrogen atoms, and extraneous protein regions). Subsequently, the RosettaDock software was employed to dock the target protein (ERK5) with ligand proteins (predicted LRP5 and LRP6), and calculating the docking energy within a negative value is interpreted as indicating the potential for spontaneous binding between the two proteins.
Plasmid and transfection
The single guide RNA (sgRNA) sequences for knocking out Mapk7 using CRISPR-Cas9 were listed in Supplementary Table 2. The negative control was an empty vector plasmid (Lenti-V2). Besides, the TEY motif dominant-negative form of MAPK7 (DN-MAPK7), wild-type form of full-length MAPK7 (WT-MAPK7), as well as constitutively active form of MEK5 (CA-MEK5) were constructed as described previously20. Plasmids encoding constitutive activation of Lrp6 at serine 1490 (CA-Lrp6) and mutant Lrp6 at serine 1490 (Mut-Lrp6) were constructed separately (Genecard, China). Primary MSCs or C3H10T1/2 MSCs were plated at a density of 2 × 105 cells per well in a 24-well plate overnight and transfected with plasmids using Lipofectamine 3000 (L3000-015, Invitrogen, USA). Cells were cultured in the appropriate medium to induce differentiation for various time periods.
Statistical analysis
Results were presented as mean ± standard deviation (SD). Besides, the error bars in the bar chart showed the corresponding SD values, and statistical analysis was conducted using GraphPad Prism 8 software. Statistical significance was determined through one-way analysis of variance, two-way analysis of variance, or Student’s t-tests, with values of p < 0.05 considered statistically significant.
Results
The expression of Mapk7 positively correlated with osteogenic differentiation of MSCs and negatively correlated with adipogenic differentiation
To explore the functions of Mapk7 in MSCs, we first surveyed the expression of Mapk7 in bone, fat, and other various mouse tissues. As shown in Fig. 1A, Mapk7 was highly expressed in lung, kidney, brain, and bone marrow, and was less expressed in adipose tissue, heart, and colon, especially perirenal fat and inguinal fat. Subsequently, Mapk7 expression was evaluated in mouse femur, and IHC results showed that Mapk7 was highly expressed in osteoblasts and at a relatively low level in bone marrow adipocytes (Fig. 1B).

A The expression of Mapk7 in heart, liver, spleen, lung, kidney, brain, skeletal muscle, bone marrow, stomach, perirenal fat, inguinal fat, testis, epididymal fat, and colon of 3-week-old mice. RNA expression in fat, inguinal fat, testis, epididymal fat, and colon (GAPDH as an internal reference gene, n = 3). B IHC results of Mapk7 in the femur of 9-week-old mice, BM represents bone marrow, TB represents trabecular bone, MA represents bone marrow adipocytes, red arrows mark trabecular surface osteoblasts, scale bar:50 μm (left), 20 μm (right). C Mouse primary MSCs derived from the femur in 3-week-old mice were stained for ALP and ARS on 0, 3, 7, 10, and 14 days of OS induction, scale bar: 500 μm. D After OS induction, western blot was performed to detect the expression of Mapk7 and osteogenesis marker genes Runx2 and Opn, respectively, with GAPDH as an internal reference. E Pearson correlation analysis between Mapk7/GAPDH and Opn/GAPDH, n = 3. F Oil red O staining was performed on 0, 3, 7, 10, and 14 days of AD induction in MSCs, scale bar: 100 μm. G After AD induction, WB was performed to detect the expression of Mapk7 and adipogenesis marker genes Pparγ and Fabp4 in MSCs, respectively. β-actin was used as an internal reference gene. H Pearson correlation analysis between the relative grayscale ratio of Mapk7/GAPDH and OD values of Oil Red O, n = 3.
To observe the expression pattern of Mapk7 during MSCs differentiation, we examined Mapk7 expression during primary MSCs differentiation. As shown in Fig. 1C, ALP staining of primary MSCs was enhanced with prolonged osteogenic induction time, indicating successful differentiation. Calcium nodules appeared on day 7 of induction and increased in size and number until day 14. Mapk7 expression elevated after osteogenic induction and peaked at day 14 (Fig. 1D). In contrast, Mapk7 was significantly reduced during adipogenesis of MSCs (Fig. 1F and G). In addition, the expression levels of adipocyte markers, such as Pparγ and fatty acid binding protein 4 (Fabp4), were elevated when primary MSCs were differentiated into adipocytes, indicating effective differentiation of adipocytes. Pearson correlation analysis revealed that Mapk7 expression was positively correlated with osteogenic differentiation (Fig. 1E), while it was negatively correlated with the absorbance value of ORO staining during adipogenic differentiation of MSCs (Fig. 1H). Results above suggested that the expression of Mapk7 positively correlated with the osteogenic differentiation of MSCs and negatively correlated with adipogenic differentiation.
Mapk7 positively regulated osteogenic differentiation and negatively modulated adipogenic differentiation of C3H10T1/2 MSCs
C3H10T1/2 MSCs are immortalized mesenchymal stem cells derived from mouse embryos possessing the general characteristics of MSCs27,28. To clarify the influence of Mapk7 on osteogenic (OS) and adipogenic (AD) differentiation capacities of MSCs, we employed CRISPR-Cas9 to silence Mapk7 (Supplementary Fig. 1A). After OS induction for 3 days, we observed reduced in osteogenesis-related genes Runx2 and Opn, as well as markedly diminished ALP and mineralization ability in Mapk7-knockout C3H10T1/2 cells (Fig. 2A–D). In contrast, Fabp4 and Pparγ, were considerably up-regulated, and lipid droplet formation was enhanced following Mapk7 depletion in C3H10T1/2 MSCs (Fig. 2E–H). Moreover, we overexpressed Mapk7 (OE-Mapk7) in C3H10T1/2 cells (Supplementary Fig. 1B) and performed OS and AD induction, respectively. Results showed the protein and mRNA levels of osteogenesis-related genes (Runx2, Opn) in OE-Mapk7 C3H10T1/2 were significantly up-regulated after 3 days of OS induction (Fig. 2I, J). In addition, the ALP as well as mineralization ability of C3H10T1/2 cells were also significantly enhanced in OE-Mapk7 compared with the vector group (lv242) (Fig. 2K, L). Moreover, overexpression of Mapk7 significantly reduced protein and RNA expression of adipogenesis-related genes (Fabp4, Pparγ) and inhibited the formation of lipid droplets in C3H10T1/2 MSCs (Fig. 2M–P). The above experiments indicated that overexpression of Mapk7 in C3H10T1/2 MSCs enhanced osteogenic differentiation and decreased adipogenic differentiation in MSCs.

A Proteins were collected after OS differentiation for 3 days to detect the expression of Mapk7 and osteogenic marker gene Runx2, following knocking out of Mapk7 in C3H10T1/2 MSCs, Lenti-V2 was the control group, with GAPDH as an internal reference gene. B After knocking out of Mapk7 in C3H10T1/2, RNA was collected after OS differentiation for 3 days to detect the RNA level of Mapk7 and osteogenic marker genes Runx2 and Opn, with GAPDH as an internal reference gene, n = 3, ***p < 0.005, ****p < 0.001. C Following OS differentiation, ALP and ARS staining were performed, respectively. D ARS nodules were solubilized with DMSO to detect OD values (405 nm), n = 3, ****p < 0.001. E After AD differentiation for 3 days, proteins were collected to detect the expression of Mapk7 and adipogenesis-related gene Fabp4, with β-actin as an internal reference gene. F After AD differentiation for 3 days, RNA was collected to detect the expression of Mapk7 and adipogenesis marker genes (Fabp4 and Pparγ), and β-actin was used as an internal reference gene, n = 3, ***p < 0.005, ****p < 0.001. G ORO staining of AD differentiation, scale bars: 100 μm. H Lipid droplets were solubilized with isopropanol to detect the OD value (510 nm), n = 3, ***p < 0.005. I After OS differentiation, proteins were collected for western blot to detect protein expression of Mapk7 and osteogenic marker genes Runx2 and Opn, with GAPDH as an internal reference gene. (J) After OS differentiation was performed for 3 days, RNA was collected for qPCR to detect the expression levels of Mapk7 and osteogenic marker genes (Runx2, Opn and Alpl), with GAPDH as internal reference gene, n = 3, ***p < 0.005, ****p < 0.001. K ALP and ARS staining. L ARS positive nodules were solubilized with DMSO, and OD values (405 nm) were detected, n = 3, *p < 0.05. M After AD differentiation was performed for 3 days, the expression of Mapk7 and adipogenesis marker protein (Pparγ and Fabp4) were detected, β-actin was used as an internal control. N After overexpression of Mapk7 in C3H10T1/2, AD differentiation was performed for 3 days, RNA was collected for qPCR, and expression of Mapk7 and adipogenesis marker genes (Fabp4 and Pparγ) were detected, β-actin as an internal reference gene, n = 3, ***p < 0.005, ****p < 0.001. O After AD differentiation 14 days, ORO staining was performed, scale bar: 500 μm. P Lipid droplets were solubilized with isopropanol, and OD value (510 nm) was detected, n = 3, **p < 0.01.
MSCs specific (Prx1-Cre) deletion of Mapk7 led to decreased bone mass and increased bone marrow adipose tissue in mice
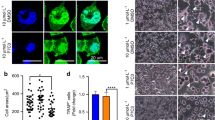
To investigate the function of Mapk7 in MSC differentiation, we constructed Prx1-Cre; Mapk7flox/flox mice (conditional knockout, CKO) by crossing Mapk7flox/flox mice (wild type, WT) with Prx1-Cre mice which mainly targeted at MSCs using CRISPR-Cas9 technology and Cre-loxp system (Supplementary Fig. 2A)20,21. Using specific primers, we conducted genotype identification on mouse tails (Supplementary Fig. 2B). Besides, Prx1-Cre; Mapk7flox/+, Mapk7flox/flox, and Mapk7flox/+ showed no significant differences in appearance, body length or skeletal phenotypes (Supplementary Fig. 2C and D). After extracting long bone MSCs, WB and qPCR were performed to confirm the deletion efficiency of Mapk7 (Supplementary Fig. 2E and F). Upon birth, the CKO mice demonstrated robust survival rates and maintained normal fertility. Regardless of gender, CKO mice exhibited dwarfism and a decrease in body weight (Supplementary Fig. 2G and H). Interestingly, μCT analysis of femurs from both control littermates (WT) and knockout littermates (CKO) postnatal was performed to compare the changes in bone-related elements in the long bones (Fig. 3A). Parameters such as BV/TV, Tb.Th, Tb.N, and Tb.Sp also indicated a substantial decrease in bone mass, indicating severe osteoporosis in CKO mice compared with 2-month-old WT littermates (Fig. 3B). Besides, the bone loss in CKO mice exhibited no sex bias (about 1:1). Furthermore, the results of Ct. Th indicated that knockout of Mapk7 in MSCs did not significantly affect cortical bone thickness (Supplementary Fig. 2I). Furthermore, Mapk7 CKO mice (5-month-old) exhibited a slower mineral apposition rate (MAR) and bone formation rate (BFR) compared to WT group (Fig. 3C and D). HE staining revealed a significant reduction in the number of tibial trabeculae in CKO group compared to WT group (Fig. 3E). Additionally, there is a significant decrease of Ob.S/Bs and N.Ob/B.Pm compared with CKO in expression of osteopontin (Opn), which is a marker of osteogenic cell maturation and differentiation, and osx (an osteogenic transcription factors) (Fig. 3F)18. Quantitative analysis of fat cells showed that the fat cell number of CKO increased compared to control WT littermates in the bone marrow cavity (Fig. 3G–I). In addition, we examined the osteoclastic activity of CKO mice. TRAP-positive osteoclastic activity was significantly enhanced in CKO littermates (Supplementary Fig. 3A and B). Subsequently, after we co-cultured MSCs with osteoclast precursor cells, the supernatant of CKO MSCs significantly promoted the multi-nuclear fusion ability of osteoclast precursor cells (Supplementary Fig. 3C). In conclusion, the skeletal manifestations of Mapk7 CKO mice resembled the pathological phenotypes of OP with bone loss and increased bone marrow adiposity.

A and B μCT maps (A) and quantitative results (B) of scan-related bone parameters in 2-month-old Mapk7 knockout mice and WT littermates regards of gender, n = 4, *p < 0.05, **p < 0.01, ***p < 0.005, scale bar: 2 mm. C Representative image of double labeling with Calcein-Alizarin red from in cancellous bone regions from proximal femur sections in 9-week-old mice, scale bar: 5 μm. D The dynamic bone histomorphometry indexes of 9-week-old mice, including MAR and BFR/BS, n = 3, ***p < 0.005. E HE staining of tibiae of 3-week-old mouse, scale bar: 20 μm. F IHC results of osteogenic marker protein Opn and Osx in tibiae of 3-week-old mice and the quantitative of bone surface covered by osteoblasts Ob.S/Bs (%), the number of osteoblasts per mm endocortical bone surface (N. Ob/B.Pm,/mm), n = 3,**p < 0.01, scale bar: 50 μm. G HE staining of tibiae from 2-month-old mice, yellow arrows show adipocytes, scale bar: 20 μm. H and I IHC of adipose tissue marker protein Fabp4 and relative area of adipocytes in the distal marrow per tissue area (n = 3); ***p < 0.005, scale bar: 50 μm.
Mapk7 deficiency promoted adipogenic differentiation and inhibited osteogenic differentiation of primary MSCs
The accumulation of adipocytes and reduced bone formation in skeleton of Prx1-Cre; Mapk7flox/flox mice prompted us to further validate the role of Mapk7 in regulating the differentiation of primary MSCs. Firstly, the flow cytometry results demonstrated that the isolated cells from mouse compact bone were positive for mesenchymal markers CD29 and CD105, as well as the stem cell marker Sca-1, but negative for hematopoietic markers CD34 and CD4529 (Fig. 4A). Further differentiation experiments revealed that primary MSCs by enzymatic digestion could readily differentiate into osteoblasts, adipocytes, and chondrocytes (Fig. 4B). Therefore, we isolated MSCs from WT and CKO mice to verify osteogenic and adipogenic potentials in vitro. Results showed that compared with WT group, the expression of Runx2 and Opn of CKO MSCs was down-regulated (Fig. 4C, D). Likewise, ALP and ARS staining decreased in CKO MSCs (Fig. 4E), which indicated that Mapk7 knockdown resulted in reduced osteogenesis of MSCs. Next, we investigated the role of Mapk7 in adipogenesis of MSCs. Consistently, expression of adipogenesis genes (Fabp4 and Pparγ) and ORO staining were both significantly enhanced in CKO compared with WT MSCs (Fig. 4F–H), indicating that Mapk7 deletion enhanced adipogenesis of MSCs. Collectively, above results further clarified that Mapk7 regulated the balance of fate commitment of MSCs by inhibiting adipogenesis and enhancing osteogenesis.

A Immunophenotypic characterization of CD45, CD34, CD29, Sca1 and CD105. B Osteogenic, adipogenic, and chondrogenic differentiation assays of mouse primary MSCs derived from femur and tibia in 3-week-old mice, scale bar: 650 μm (upper); 50 μm (middle). C The expression of Mapk7, osteogenic marker protein (Runx2) was detected 3 days after OS induction of primary MSCs in passage 3 derived from 3-week-old WT and littermate CKO mice. D mRNA levels of Runx2, Opn, n = 3, ****p < 0.001. E Results of ALP and ARS staining in MSCs in passage 3 after 7 and 14 days of OS induction, n = 3, ****p < 0.001. F Expression of Mapk7, adipogenic marker protein Fabp4 was detected 3 days of primary MSCs after AD induction. G mRNA levels of Fabp4 and Pparγ were detected 3 days of primary MSCs after AD induction, n = 3, ****p < 0.001. H Results of ORO staining and quantification at 14 days of AD induction of primary MSCs at passage 3 derived from WT and littermate CKO mice, scale bar: 50 μm, n = 3, *p < 0.05.
Wnt/β-catenin signaling was significantly reduced after Mapk7 deletion in MSCs
To explore the underlying mechanism of Mapk7 in the regulation of osteoblast-adipocyte lineage transition in MSCs, we extracted RNA from the same generation of non-induction MSCs in littermate WT and CKO mice for RNA-seq, and results showed that 151 genes were up-regulated and 376 genes were down-regulated in CKO compared with WT (Fig. 5A), where down-regulated genes were mainly enriched in extracellular matrix receptor interaction, cytoskeleton regulation, PI3K-Akt signaling and Wnt pathways (Fig. 5B). In recent years, numerous studies have shown that MAPK family acted as important intermediate signaling molecules to regulate Wnt signaling activity which regulates cell fate determination and tissue homeostasis10,30. Since Wnt signaling consists of both β-catenin-dependent and β-catenin-independent pathways, previous researches have indicated that ERK could potentiate non-canonical Wnt cascades, such as Wnt/PCP signaling20. However, Nagano et al. confirmed that enhancing Erk phosphorylation could activate classical Wnt signaling to regulate bone formation in the appendicular skeleton10. Gene set enrichment analysis (GSEA) results showed that Wnt signaling was significantly enriched in Mapk7-deficient MSCs (Fig. 5C). Subsequently, we examined the expression of active (non-phospho-) β-catenin, an effector molecule of classical Wnt pathway. Immunofluorescence results showed active (non-phospho-) β-catenin decreased in CKO tibia compared to WT tibia (3-weeks-old mice) (Fig. 5D). Besides, translocation of β-catenin into nucleus was reduced in CKO MSCs (Fig. 5E). In addition, active β-catenin were significantly reduced in CKO MSCs compared to WT in both total protein extract and nuclear protein extract (Fig. 5F), and deletion of Mapk7 resulted a significant decrease in the expression of canonical Wnt target genes (C-Myc and Cyclin D1) (Fig. 5G), accompanied by an elevation in active β-catenin, which was also consistent with the reduced of Runx2, an osteogenic-related target factor of canonical Wnt signaling30,31. The above results suggested that Mapk7 deletion in MSCs inhibited Wnt/β-catenin signaling activity.

A Differentially expressed genes after RNA-seq of RNA from primary MSCs isolated from 3-week-old. WT and littermate CKO mice at passage 3. B and C Results of KEGG enrichment (B) and GSEA analysis of differential genes (C). D Representing active β-catenin IF images of tibia at 3-weeks old mice. Dotted lines mark different locations, BM bone marrow, TB trabecular bone, M muscle, CB cortical bone, GP growth plate; scale bars: 100 μm. E IF results of total β-catenin in MSCs at passage 4 co-cultured with or without L-Wnt3A cells via a trans-well device, scale bar: 50 μm. F Detection of Mapk7 and classical Wnt pathway effector molecular activity, active β-catenin expression in total protein extracts and nuclear protein extracts from mouse MSCs at passage 4, GAPDH and Lamin B1 were used as internal reference genes for total and nuclear proteins, respectively. G Detection of the RNA level of β-catenin target genes C-Myc and CyclinD1, with GAPDH as an internal reference gene, n = 3, *p < 0.05.
Wnt/β-catenin pathway activator, CHIR-99021 rescued Mapk7 knockdown-induced attenuated osteogenesis and increased adipogenesis in MSCs in vitro
To further confirm that Wnt/β-catenin signaling was a critical pathway for Mapk7-mediated differentiation of MSCs, we introduced an activator for classical Wnt/β-catenin signaling, CHIR-9902127. After OS differentiation, calcium nodules in CKO MSCs were notably increased after CHIR-99021 (10 μM) treatment compared with the DMSO group (Fig. 6A and B). In contrast, ORO results also showed that the adipogenic capacity of CKO MSCs was inhibited by CHIR-99021 (Fig. 6C and D). Besides, Runx2 was significantly up-regulated after the CHIR-99021 application (Fig. 6E). At the same time, expression of active β-catenin in CKO MSCs did appear to be down-regulated and increased after the CHIR-99021 application (Fig. 6E), reflecting that CHIR-99021 could indeed activate Wnt/β-catenin signaling. Conversely, CHIR-99021 decreased Fabp4 expression in CKO MSCs (Fig. 6G). Moreover, CHIR-99021 activation of Wnt signaling rescued the expression of osteogenic genes (Runx2, Alpl) in CKO MSCs (Fig. 6F). Likewise, the levels of adipogenesis-related genes (Cebpα, Fabp4) were both partially rescued in CKO group after activation of Wnt/β-catenin signaling (Fig. 6H). Taken together, CHIR-99021 ameliorated the imbalance of osteogenic and adipogenic differentiation in MSCs caused by Mapk7 deletion through activation of Wnt/β-catenin pathway, which further identified the indispensable role of Wnt/β-catenin signaling in Mapk7-deficiency mediated imbalance between osteogenesis and adipogenesis of MSCs.

A ARS staining for CHIR-99021 (10 μM) treatment to primary MSCs at passage 4 after OS induction for 14 days, scale bar: 150 μm. B Quantitative of ARS staining, n = 3, ***p < 0.005. C ORO staining for CHIR-99021 (10 μM) treatment to primary MSCs at passage 4 after OS induction, scale bar: 200 μm. D Quantitative of ORO staining, n = 3, ***p < 0.005. E Protein expression of Mapk7, osteogenesis-related genes (Runx2), and β-catenin was detected after OS induction of primary MSCs for 3 days with CHIR-99021 (10 μM) treatment. F Application of CHIR-99021 (10 μM) to MSCs at passage 4 after OS induction for 3 days, qPCR was performed to detect the levels of osteogenesis-related genes (Runx2, Alpl), n = 3, *p < 0.05; **p < 0.01. G Primary MSCs at passage 4 of WT/CKO mice were treated with CHIR-99021 (10 μM) for 3 days after AD induction, and Western blot was performed to detect the expression of Mapk7, adipogenesis-related gene (Fabp4) and active β-catenin, β-actin was used as an internal control. H Adipogenesis of primary MSCs at passage 4 was induced for 3 days along with CHIR-99021 (10 μM) treatment, then qPCR was performed to detect the mRNA levels of lipid formation-related genes (Fabp4, Cebpα), n = 3, ***p < 0.005.
Mapk7 enhanced osteogenesis and inhibited adipogenesis of MSCs through phosphorylation of Lrp6 at Ser1490
MAPK7 is one of the kinases of the MAPK family which phosphorylates a range of substrates essential for osteogenesis28. Therefore, we co-transfected MEK5 constitutive activated (CA-MEK5), wild-type MAPK7-WT and MAPK7 dominant negative plasmids (DN-MAPK7)20 in C3H10T1/2 to investigate whether Mapk7 regulated osteogenic and adipogenic differentiation in MSCs through phosphorylation function (Fig. 7A). After MAPK7 was persistently activated by MEK5, osteogenic genes were significantly up-regulated and adipogenic genes were significantly down-regulated, while the expression of osteogenesis and adipogenesis-related genes were all reverted after MAPK7 mutation (Fig. 7B and C), indicating that phosphorylation function of Mapk7 was involved in regulating osteogenic and adipogenic differentiation in MSCs.

A Schematic illustration of MAPK7-WT and the DN-MAPK7 mutation. B and C Expression of Mapk7, p-Mapk7, osteogenesis-related genes, and adipogenesis-related genes (Pparγ, Fabp4) were detected after 3 days of OS (B) and AD (C) induction in C3H10T1/2 cells transfected with the corresponding plasmid, respectively, and GAPDH and β-actin were used as internal reference genes. D The expression of key molecules of Wnt/β-catenin signaling in C3H10T1/2 cells after transfected with the corresponding plasmid, and GAPDH was used as an internal reference gene. E p-Lrp6 staining of 2-month-old mouse tibiae using IHC, yellow arrowheads mark the positive region of p-Lrp6 expression. scale bar: 50 μm. F IP assay of Lrp6 and p-Mapk7 interactions in C3H10T1/2 cells. G C3H10T1/2 MSCs were transfected with different ratios of MAPK7-WT: CA-MEK5 to detect the expression of p-Mapk7, p-Lrp6 (Ser1490), and GAPDH as an internal reference gene. H Primary MSCs at passage 4 were transfected with constitutive activation Lrp6 at Ser1490 plasmid (CA-Lrp6) and inactivation mutation plasmid (Mu-Lrp6) and were co-cultured with L-Wnt3A cells, whose p-Lrp6 (Ser1490)/ osteogenesis-related genes (Opn and Runx2) were detected after 3 days of osteogenic induction. I ARS staining of primary passage 4 MSCs which were co-cultured with L-Wnt3A cells under the condition of OS induction for 14 days, scale bar: 500 μm. J Quantitative results of ARS staining, n = 3, **p < 0.01. K Expression levels of p-Lrp6 (Ser1490), Lrp6, and adipogenesis-related genes (Pparγ and Fabp4) were tested in passage 4 MSCs, which were transfected with CA-Lrp6 and Mu-Lrp6, co-cultured with L-Wnt3A cells after 3 days of AD induction. L ORO staining after 14 days of AD induction under co-culture conditions of passage 4 MSCs and L-Wnt3A, scale bar: 500 μm. M Quantitative results of ORO, n = 3, **p < 0.01.
Since Mapk7 deletion resulted in reduced nucleation of β-catenin, we further investigated whether Mapk7 regulated the activity of key molecules in Wnt/β-catenin signaling. It is known that in the absence of Wnt stimulation, β-catenin can be constitutively phosphorylated by glycogen synthase kinase-3β (Gsk3β), leading to ubiquitination and proteasome-dependent degradation of β-catenin32. Furthermore, members of the LRP family, including LRP4, LRP5, and LRP6, are all single-pass trans-membrane proteins containing multiple phosphorylation sites32,33,34. Considering that LRP4 facilitated sclerostin-mediated inhibition of Wnt signaling, while LRP5/6, in conjunction with Frizzled receptors regulated β-catenin stability, and functional mutations in LRP6 were associated with the occurrence of OP32,33,34, we conducted molecular docking between MAPK7 (ERK5) and LRP5/ LRP6, respectively. The results revealed the docking energy between LRP6 and ERK5 was −7.9 kJ/mol, which was lower than the minimum docking energy observed for LRP5 (Supplementary Fig. 4A). This suggested a stronger binding potential between ERK5 and LRP6. Furthermore, the phosphorylation levels of Lrp6 and Gsk3β were significantly decreased after MAPK7 mutation (Fig. 7D). Meanwhile, p-Lrp6 levels in CKO tibiae were also significantly lower than in WT group via IHC (Fig. 7E). Previous study has shown that ERK directly induced Gsk3β T43 phosphorylation, which in turn regulated β-catenin activity33, so we need to clarify whether Gsk3β was involved in the regulatory process in our system. Therefore, to verify whether Mapk7 phosphorylated Gsk3β/Lrp6 by binding them, we found that p-Mapk7 could co-precipitate with Lrp6 by IP (Fig. 7F), while there was no co-precipitation with Gsk3β (Supplementary Fig. 5A), indicating that p-Mapk7 interacted with Lrp6. And as the phosphorylation level of Mapk7 increased, the phosphorylation level of Lrp6 at Ser1490 also gradually increased (Fig. 7G).
Given that the phosphorylation level of Lrp6 at Ser1490 was activated by Mapk7, therefore, we further investigated whether the phosphorylation of Lrp6 at Ser1490 by Mapk7 was involved in MSCs osteogenic and adipogenic differentiation. Activation of classical Wnt/β-catenin pathway was initiated by the binding of Wnt proteins to cell surface receptors11, we utilized a co-culture system of L-Wnt3A and MSCs to activate Wnt/β-catenin signaling in MSCs. Besides, the transfection of CA-Lrp6 under activation of Wnt3a significantly enhanced the level of p-Lrp6 (Ser1490) in CKO MSCs (Fig. 7H). Moreover, activation of Lrp6 at Ser1490 significantly enhanced the level of osteogenic markers (Opn and Runx2) in CKO MSCs (Fig. 7H). As expected, ARS staining was also consistent with WB (Fig. 7I, J). Similarly, under AD differentiation conditions, activation of Lrp6 at Ser1490 significantly reduced the level of adipogenesis protein (Pparγ and Fabp4) and improved the accumulation of lipid droplets in CKO MSCs (Fig. 7K–M). In conclusion, the results above suggested that the activating effect of Mapk7 on Lrp6 Ser1490 was essential for enhancing osteogenesis and inhibiting adipogenesis of MSCs.
Discussion
In this study, we initially demonstrated that deletion of Mapk7 in MSCs led to a significant loss in bone mass accompanied by an increase in bone marrow adiposity. Furthermore, overexpression and deletion experiments showed that Mapk7 played a crucial role in promoting osteogenic differentiation in MSCs while simultaneously inhibiting adipogenesis. Mechanistically, the findings revealed that Mapk7 could phosphorylate Ser1490 of LRP6, promoting the nuclear translocation of β-catenin, and thereby regulating bone mass and bone marrow adiposity. The findings further broaden the understanding of the crosstalk between the MAPK family member, Mapk7, and Wnt/β-catenin signaling in the differentiation of MSCs.
MAPK7, through its phosphorylation function, regulated cellular functions in a wide variety of tissues35,36. The present findings underscored the importance of the phosphorylation function of MAPK7 (ERK5) phosphorylation in promoting osteogenic differentiation while inhibiting adipogenic differentiation in MSCs. Adam et al.37. demonstrated that knockdown of ERK5 or the use of ERK5 inhibitor XMD8-92 to suppress ERK5 phosphorylation inhibited osteogenic differentiation in human MSCs, aligning with our findings and further verifying the crucial regulatory role of Mapk7 phosphorylation in MSC osteogenic differentiation. Prior research has shown that MSCs could be traced using markers such as Prx1, Dermo1, LepR, and Nestin38,39,40. Our study revealed that the ablation of Mapk7 in MSCs reduced bone mass in mouse long bones, with further confirmation in C3H10T1/2 MSCs that the lack of Mapk7 inhibited osteogenic differentiation. However, the deletion of Mapk7 in LepR+ MSCs enhanced osteogenic differentiation in MSCs41. One explanation was that Prx1 was expressed predominantly in mesenchymal progenitors during embryonic development. This suggested that Prx1+ MSCs might be present much earlier in development than LepR+ MSCs. LepR might thus represent an MSC population that was more associated with osteogenic and osteocytic generative activity. This also suggested that Mapk7 exhibited functional heterogeneity in different cell types. Furthermore, in the physiological microenvironment of bone marrow, bone formation increased while fat accumulation decreased35. In our study, Prx1-Cre; Mapk7flox/flox mice exhibited decreased osteogenesis accompanied by abnormally increased adipose tissue in the marrow. Our in vitro gain- and loss-of-function studies also confirmed that Mapk7 inhibited adipogenic differentiation in MSCs. Similarly, the deletion of Mapk7 in LepR+ MSCs resulted in abnormal increases in bone marrow adiposity41. LepR was primarily a marker of MSCs in adults, whereas Prx1 was pressed largely by MSCs during embryonic development35,36, suggesting that the absence of Mapk7 at different developmental stages led to increased marrow adipose tissue.
The study showed that Mapk7 deletion in MSCs resulted in short stature at birth, consistent with previous research28. The elongation of the limbs and long bones occurs through endochondral ossification. Researchers have found that specific knockout of Mapk7 in chondrocytes (Col2al-Cre; Mapk7floxflox) inhibited hypertrophic differentiation of growth plate chondrocytes in mice42. Since chondrocytes originate from mesenchymal cells43, the short-stature phenotype observed in CKO mice in this study might have been due to the absence of Mapk7 in MSCs. Additionally, MSC-mediated bone formation and osteoclast-mediated bone resorption jointly maintain bone homeostasis. Interestingly, the CKO mice in the study exhibited enhanced osteoclast activity in long bones. Previous reports have shown that the deletion of Mapk7 in osteoclast lineage precursors led to increased osteoclast activity, resulting in increased bone resorption and reduced bone mass in postnatal Nkx3.1-Cre; Mapk7flox/flox mice13. However, Prx1 was not a marker of osteoclast lineage precursors39. Therefore, we co-cultured bone marrow-derived macrophages (BMMs) with MSCs and found that BMMs co-cultured with CKO MSCs exhibited enhanced osteoclast activity. Previous studies have reported that MSCs acted as secretory cells influencing the activities of surrounding cells, with MSCs-derived extracellular vesicles (MSC-EVs) loaded with microRNAs regulating osteoclast activity44. This suggested that CKO MSCs might affect the osteoclast activity of BMMs through paracrine signaling. Another possible explanation is that, following Mapk7 knockout, the tendency of MSCs toward adipogenic differentiation promoted their ability to enhance osteoclast activity in BMMs. Recent research by Fan et al. indicated that in the absence of Pth1r in MSCs, bone marrow adipocytes served as one of the sources of receptor activators of nuclear factor-κB ligand (RANKL), affecting osteoclast activity43. Therefore, elucidation of the differentiation potential of MSCs toward adipocytes following Mapk7 deletion and its impact on osteoclast-inducing factors such as RANKL in regulating osteoclast activity will be an intriguing area for further research. In conclusion, our findings suggested that Mapk7 regulated cell differentiation and fate across different cell types, to collectively maintain bone homeostasis.
MAPK7, as a member of the conserved MAPK family, transmits downstream signals through the phosphorylation of various substrates20,45. ERK5, rather than ERK1/2, phosphorylated FAK at Ser910 to modulate cell migration46. The findings of the present study further extended the known functions of MAPK7 to include the promotion of β-catenin nuclear translocation in MSCs. It was shown that the osteogenic and adipogenic phenotypes of MSCs were dependent on Mapk7 phosphorylation of Lrp6 at Ser1490, and the activation of TEY motif in the kinase domain played a crucial role in this process. In addition, another family member of ERK, ERK1/2, has been previously demonstrated to bind to LRP6 to regulate Wnt/β-catenin signaling activity, serving as a regulatory factor for MSC differentiation although the site of interaction between ERK1/2 and LRP6 was not investigated3,10. However, the TEY motif in the kinase domain was relatively conserved in the MAPK family. Therefore, we speculated that LRP6 might be a common substrate of MAPK family members with the TEY motif. Besides, we have previously shown that Mapk7 participates in vertebral development by regulating MEF2C/PTEN/AKT signaling to modulate chondrocyte hypertrophic differentiation43. Therefore, considering that MEF2C represents as a classic substrate in the MAPK cascade15,47, we overexpressed MEF2C to assess its potential in reversing the imbalance between osteogenic and adipogenic differentiation in MSCs due to Mapk7 deficiency. Nevertheless, our findings indicated that MEF2C was unable to compensate for changes resulting from the absence of Mapk7 (Supplementary Fig. 6A and B), hinting at the existence of Mapk7 functional heterogeneity across different cell types.
In all, our study established a connection between MSC fate decisions and protein kinases, demonstrating that Mapk7 was a vital regulator of MSC differentiation through Lrp6 phosphorylation. Clarifying the relationship between Mapk7 and the Wnt/β-catenin pathway may provide new insights for future research on targeted therapy for diseases associated with MSC differentiation, such as OP. Due to its expression in multiple tissues and the significant roles of MAPK7, a systemic treatment that targets MAPK7 directly would pose considerable risks. It has been reported that transplantation of genetically modified MSCs stimulated osteogenic differentiation and new bone formation to suppress age-related OP-like phenotypes48. Besides, clinical trials using MSC injection for the treatment of various diseases, such as osteoarthritis, are currently underway49. Therefore, our findings may provide therapeutic targets for OP and preliminary laboratory data for future clinical treatment of severe OP, for instance, the use of intramedullary injections of genetically modified MSCs.
Data availability
All data of RNA-seq have been deposited to the NCBI GeneExpression Omnibus (GSE255596). The source data behind the graphs in the paper was exhibited in Supplementary Data 1. Uncropped and unedited blot images were exhibited in Supplementary Fig. 7. Plasmids first constructed in this study have been deposited to the Addgene (ID:234364 and 234365).
References
Deng, P. et al. Loss of KDM4B exacerbates bone–fat imbalance and mesenchymal stromal cell exhaustion in skeletal aging. Cell Stem Cell 28, 1057–1073 (2021).
Zhang, L. et al. Hedgehog signaling controls bone homeostasis by regulating osteogenic/adipogenic fate of skeletal stem/progenitor cells in mice. J. Bone Miner. Res. 37, 559–576 (2022).
Tian, L. et al. A novel Sprouty4-ERK1/2-Wnt/β-catenin regulatory loop in marrow stromal progenitor cells controls osteogenic and adipogenic differentiation. Metabolism 105, 154189 (2020).
Lee, K. S. et al. Extracellular vesicles from adipose tissue-derived stem cells alleviate osteoporosis through osteoprotegerin and miR-21-5p. J. Extracell. Vesicles 10, e12152 (2021).
Liu, Z. Z. et al. Autophagy receptor OPTN (optineurin) regulates mesenchymal stem cell fate and bone–fat balance during aging by clearing FABP3. Autophagy 17, 2766–2782 (2021).
Olona, A. et al. Adipoclast: a multinucleated fat-eating macrophage. BMC Biol. 19, 246 (2021).
Chen, Q. et al. Fate decision of mesenchymal stem cells: adipocytes or osteoblasts? Cell Death Differ. 23, 1128–1139 (2016).
Basisty, N. et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18, e3000599 (2020).
During, A. Osteoporosis: a role for lipids. Biochimie 178, 49–55 (2020).
Nagano, K. et al. R-spondin 3 deletion induces Erk phosphorylation to enhance Wnt signaling and promote bone formation in the appendicular skeleton. Elife 11, e84171 (2022).
Tian, L. et al. A novel Sprouty4-ERK1/2-Wnt/beta-catenin regulatory loop in marrow stromal progenitor cells controls osteogenic and adipogenic differentiation. Metabolism 105, 154189 (2020).
Cong, Q. et al. p38a MAPK regulates lineage commitment and OPG synthesis of bone marrow stromal cells to prevent bone loss under physiological and pathological conditions. Stem Cell Rep. 6, 566–578 (2016).
Loveridge, C. J. et al. Analysis of Nkx3.1: Cre-driven Erk5 deletion reveals a profound spinal deformity which is linked to increased osteoclast activity. Sci. Rep. 7, 13241 (2017).
Schroyer, A. L., Stimes, N. W., Abi Saab, W. F. & Chadee, D. N. MLK3 phosphorylation by ERK1/2 is required for oxidative stress-induced invasion of colorectal cancer cells. Oncogene 37, 1031–1040 (2018).
Paudel, R., Fusi, L. & Schmidt, M. The MEK5/ERK5 pathway in health and disease. Int. J. Mol. Sci. 22, 7594 (2021).
Zhou, T. et al. Mutant MAPK7-Induced Idiopathic Scoliosis is Linked to Impaired Osteogenesis. Cell. Physiol. Biochem.48, 880–890 (2018).
Zhao, X. et al. ZBP1 (DAI/DLM-1) promotes osteogenic differentiation while inhibiting adipogenic differentiation in mesenchymal stem cells through a positive feedback loop of Wnt/beta-catenin signaling. Bone Res. 8, 12 (2020).
Suo, J. et al. The RNA-binding protein Musashi2 governs osteoblast-adipocyte lineage commitment by suppressing PPARgamma signaling. Bone Res. 10, 31 (2022).
Yu, L., Xie, M., Zhang, F., Wan, C. & Yao, X. TM9SF4 is a novel regulator in lineage commitment of bone marrow mesenchymal stem cells to either osteoblasts or adipocytes. Stem Cell Res. Ther. 12, 573 (2021).
Yang, X. et al. Conditional ablation of MAPK7 expression in chondrocytes impairs endochondral bone formation in limbs and adaptation of chondrocytes to hypoxia. Cell Biosci. 10, 103 (2020).
Gong, Y. et al. Vangl2 limits chaperone-mediated autophagy to balance osteogenic differentiation in mesenchymal stem cells. Dev. Cell 56, 2103–2120 (2021).
Cao, H. et al. PDGF-BB prevents destructive repair and promotes reparative osteogenesis of steroid-associated osteonecrosis of the femoral head in rabbits. Bone 167, 116645 (2023).
Yin, T. The stem cell niches in bone. J. Clin. Investig. 116, 1195–1201 (2006).
Li, C. et al. M13, an anthraquinone compound isolated from Morinda officinalis promotes the osteogenic differentiation of MSCs by targeting Wnt/β-catenin signaling. Phytomedicine 108, 154542 (2023).
Parfitt, A. M. et al. Bone histomorphometry: standardization of nomenclature, symbols, and units,. J. Bone Mineral Res. 2, 595–610 (1987).
Li, J. et al. Genetic detection of two novel LRP5 pathogenic variants in patients with familial exudative vitreoretinopathy. BMC Ophthalmol. 23, 489 (2023).
Cai, Z. et al. Directed differentiation of human induced pluripotent stem cells to heart valve cells. Circulation 149, 1435–1456 (2024).
Iezaki, T. et al. The MAPK Erk5 is necessary for proper skeletogenesis involving a Smurf-Smad-Sox9 molecular axis. Development 145, dev164004 (2018).
Hoover, M. Y. et al. Purification and functional characterization of novel human skeletal stem cell lineages. Nat. Protoc. 18, 2256–2282 (2023).
Nusse, R. & Clevers, H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017).
Almalki, S. G. & Agrawal, D. K. Key transcription factors in the differentiation of mesenchymal stem cells. Differentiation 92, 41–51 (2016).
Gajos-Michniewicz, A. & Czyz, M. WNT signaling in melanoma. Int. J. Mol. Sci. 21, 4852 (2020).
Clevers, H. & Nusse, R. Wnt/beta-catenin signaling and disease. Cell 149, 1192–1205 (2012).
Laudes, M. Role of WNT signalling in the determination of human mesenchymal stem cells into preadipocytes. J. Mol. Endocrinol. 46, R65–R72 (2011).
Fan, Y. et al. Parathyroid hormone directs bone marrow mesenchymal cell fate. Cell Metab. 25, 661–672 (2017).
Cong, Q. et al. p38α MAPK regulates lineage commitment and OPG synthesis of bone marrow stromal cells to prevent bone loss under physiological and pathological conditions. Stem Cell Rep. 6, 566–578 (2016).
Adam, C. et al. The MEK5/ERK5 mitogen-activated protein kinase cascade is an effector pathway of bone-sustaining bisphosphonates that regulates osteogenic differentiation and mineralization. Bone 111, 49–58 (2018).
Li, Q., Xu, R., Lei, K. & Yuan, Q. Insights into skeletal stem cells. Bone Res. 10, 61 (2022).
Liu, H. et al. Prrx1 marks stem cells for bone, white adipose tissue and dermis in adult mice. Nat. Genet. 54, 1946–1958 (2022).
Logan, M. et al. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80 (2002).
Horie, T. et al. Erk5 in bone marrow mesenchymal stem cells regulates bone homeostasis by preventing osteogenesis in adulthood. Stem Cells 40, 411–422 (2022).
Yang, R. et al. 1,25-Dihydroxyvitamin D protects against age-related osteoporosis by a novel VDR-Ezh2-p16 signal axis. Aging Cell 19, e13095 (2020).
Wu, C. et al. Mapk7 deletion in chondrocytes causes vertebral defects by reducing MEF2C/PTEN/AKT signaling. Genes Dis. 11, 964–977 (2023).
KS, L. et al. Extracellular vesicles from adipose tissue-derived stem cells alleviate osteoporosis through osteoprotegerin and miR-21-5p. J. Extracell. Vesicles 10, e12152 (2021).
Horie, T. et al. Erk5 in bone marrow mesenchymal stem cells regulates bone homeostasis by preventing osteogenesis in adulthood. Stem Cells (Dayton, OH) 40, 411–422 (2022).
Jiang, W. et al. Extracellular signal regulated kinase 5 promotes cell migration, invasion and lung metastasis in a FAK-dependent manner. Protein Cell 11, 825–845 (2020).
Nishimoto, S. & Nishida, E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 7, 782–786 (2006).
Lee, D. S. et al. NFI-C regulates osteoblast differentiation via control of osterix expression. Stem Cells 32, 2467–2479 (2014).
Copp, G., Robb, K. P. & Viswanathan, S. Culture-expanded mesenchymal stromal cell therapy: does it work in knee osteoarthritis? A pathway to clinical success. Cell. Mol. Immunol. 20, 626–650 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 82472388, 82172376, and 82330074). Guangzhou Science and Technology Project (No. 2023B03J0137). Guangdong Province Basic and Applied Basic Research Fund Project (No. 2023A1515010307).
Author information
Authors and Affiliations
Contributions
Chuan Li, Jiahui Long, and Shuqing Chen: Methodology, data curation, writing-original draft. Liru Tian and Ya Xiao: Methodology and software. Shulin Chen and Deying Su: Investigation. Baolin Zhang: Statistical analyses. Peiqiang Su, Zhiheng Liao, and Caixia Xu: Conceptualization, supervision, writing-review & editing, funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The animal experiments for this study were conducted at the Laboratory Animal Center of Sun Yat-sen University, approved and regulated by the Committee for the Management and Use of Laboratory Animals of Sun Yat-sen University (Approval No. SYSU-IACUC-2022-001823), and all experimental procedures were carried out in compliance with relevant regulations.
Peer review
Peer review information
Communications Biology thanks Gretl Hendrickx and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Martina Rauner and Joao Valente. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, C., Long, J., Chen, S. et al. Mapk7 enhances osteogenesis and suppresses adipogenesis by activating Lrp6/β-catenin signaling axis in mesenchymal stem cells. Commun Biol 8, 310 (2025). https://doi.org/10.1038/s42003-025-07765-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07765-x
This article is cited by
-
Activation of GLP-1 receptors enhances the osteogenic differentiation process of STRO-1-positive BMSCs
Molecular Biology Reports (2025)
