
Abstract
Soluble PD-1 is a dissociated form of membrane PD-1 broadly present in cancer, infections, or autoimmune diseases. However, the clinical significance of sPD-1 remains controversial due to the uncertainty of its isoforms, origin, and production mechanism. Here, using antibodies specifically binding to the intracellular domain of PD-1, we identified two sPD-1 isoforms in cancers at the protein level: FL sPD-1 containing both the extra- and intracellular domains of PD-1, and ECD sPD-1 containing only the extracellular fragment. Subsequently, we tracked their origin and found that in tumor models, both sPD-1 isoforms were generated by activated CD8 T cells highly expressing membrane PD-1. However, ECD sPD-1 was released from live PD-1+T cells by matrix metalloproteinases, while FL sPD-1 production was accompanied by PD-1+T cell death. Therefore, only ECD sPD-1 predicts effective immune response and better tumor outcome. Our study distinguished sPD-1 isoforms and highlighted ECD sPD-1 as a prognostic biomarker in cancer.
Similar content being viewed by others


Introduction
Programmed death-1 (PD-1) is a crucial inhibitor of an immune response. Full-length (FL) PD-1 is a typical type I transmembrane protein consisting of an extracellular domain (ECD), a transmembrane region (TM), and an intracellular domain (ICD)1. A soluble form of PD-1 (soluble PD-1, sPD-1) has been reported in diseases, including cancers2,3, inflammation4,5, and autoimmune disorders6,7. Given the promising application of anti-PD-1 therapy in malignant tumors, the clinical significance of sPD-1 in cancer has been explored. Some studies showed that high levels of sPD-1 in cancers were associated with aggressive tumor growth and advanced stages8,9,10 while others observed the opposite or no correlation11,12. Likewise, when tumors were treated with targeted therapies or immunotherapies, the correlation between sPD-1 increase and therapeutic response was not conclusive13,14,15,16,17. Further elucidation of sPD-1 isoforms, origin, and production mechanism would help to clarify these questions.
In contrast to extensive clinical association studies, there are fewer reports on the isoforms and production mechanisms of sPD-1. Although five alternatively spliced PD-1 mRNA transcripts were observed in naïve peripheral blood mononuclear cells (PBMCs)18, these variants were not confirmed at the protein level. Furthermore, most sPD-1 examinations utilized antibodies that bind to the ECD fragment of PD-114,15,16; however, no soluble ECD PD-1 transcript was identified, suggesting that mRNA splicing may not be the only mechanism for sPD-1 production. In contrast, some studies have reported that the amount of sPD-1 was positively correlated with the level of membrane PD-1 (mPD-1) on T cells5,17,19, implying that sPD-1 may be directly derived from expressed PD-1. PD-1 is expressed on multiple cells, including T, B, and NK cells20,21,22,23,24. On T cells, PD-1 can be upregulated upon antigen priming1,25,26, and to avoid overactivation, it generally decreases when the immune response is completed. Persistent PD-1 expression results in T-cell exhaustion and dysfunction, leading to immune abnormalities and disease27. Recent studies have shown that the downregulation of PD-1 also occurs at the protein level28,29, which may result in sPD-1 production; however, further investigation is warranted to address this.
Herein, we started with a meta-analysis of sPD-1 in cancers. Then, two enzyme-linked immunosorbent assay (ELISA) systems were developed to distinguish different isoforms of sPD-1 at the protein level, and their cellular origin and production mechanism were explored by multiple murine models. Finally, the correlation of sPD-1 with cancer prognosis was assessed. Our findings might offer a deeper understanding of T cell activation and immune response in cancer.
Results
A meta-analysis of sPD-1 level and cancer prognosis
Overall, 158 studies were obtained by bibliographic retrieval; after excluding duplicates, non-original articles, and irrelevant biomarkers to sPD-1, 30 articles covering various cancers (hepatocellular carcinoma, gastric cancer, esophageal cancer, melanoma, non-small cell lung cancer, pancreatic cancer, diffuse large B-cell lymphoma, and gastrointestinal stromal tumor) were included in our meta-analysis.
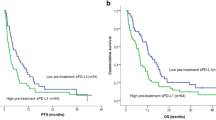
Initially, we examined sPD-1 levels in untreated cancer cases. The association between sPD-1 level and overall survival (OS) (Fig. 1a) or disease-/progression-free survival (DFS/PFS) (Fig. 1b) was pooled and analyzed. Results showed that the median hazard ratios (HRs) (high- vs. low-sPD-1 group) were 1.00 (p = 0.04, I2 = 43%) for OS and 1.27 (p < 0.01, I2 = 79%) for DFS/PFS by the random-effects model, meaning that no association was observed between the baseline sPD-1 and cancer outcomes. Additionally, both the p-value (p < 0.05) and I2 value (I2 > 25%) indicated high heterogeneity among the analyzed studies, which might contribute to the negative result. Further qualitative meta-analysis revealed no significant differences in the number of poor events between baseline high and low sPD-1 levels, the pooled risk ratio (RR) was 0.75 (95% CI: 0.53, 1.06, p < 0.01, I2 = 90%) (Supplementary Fig. 1a). A similar result was obtained by quantitative analysis (pooled RR = −0.22, 95% CI: −1.02, 0.59, p < 0.01, I2 = 96%) (Supplementary Fig. 1b).

Forest plots for OS (a, c) or DFS/PFS (b, d) with sPD-1 levels of untreated patients (a, b) or patients treated with immunotherapy or targeted therapies (c, d).
We next analyzed sPD-1 in cancers treated with immunotherapy (PD-1/PD-L1 inhibitors) or targeted therapies (erlotinib). Surprisingly, a positive association was found between increased sPD-1 upon treatment and improved OS (pooled HR = 0.34, 95% CI: 0.20, 0.60, p = 0.20, I2 = 37%, Fig. 1c), as well as DFS/PFS (pooled HR = 0.37, 95% CI: 0.21, 0.63, p = 0.88, I2 = 0%, Fig. 1d). A similar result was obtained from quantitative analysis (Supplementary Fig. 1c). In summary, our meta-analysis revealed that high sPD-1 levels did not directly correlate with long survival in untreated patients with cancer. However, increased sPD-1 levels following immunotherapy or targeted therapies were associated with a favorable cancer prognosis.
Two isoforms of sPD-1 protein existed in plasma from individuals with tumor
To clarify the clinical significance of sPD-1 levels in treated and untreated cancers, we first examined isoforms of sPD-1 that may have different clinical implications. We selected two pairs of anti-human PD-1 antibodies (Abs) to distinguish different sPD-1 isoforms at the protein level by ELISA (Supplementary Fig. 2a). Clone D4W2J specifically binds to the ICD of human PD-1, and clone J105 binds to the ECD (Fig. 2a). ELISA-#1 using them as capture and detection Abs could effectively detect FL sPD-1 (Fig. 2b). Likewise, another two ECD-binding Abs—clone MIH4 and polyclonal BAF1086 (Fig. 2c), which were previously reported as an efficient couple to detect sPD-1, were used in ELISA-#2. ELISA-#2 could detect both the ECD and FL form of sPD-1 (Fig. 2d). Each sample was examined by both of the two ELISA assays, ELISA-#1 used ECD-hIgG fusion protein as a standard and generated standard curve-I, and ELISA-#2 used ECD-hIgG and ΔFL-hIgG (ΔFL is FL PD-1 without TM region) and generated standard curve-II and -III, respectively. Further, Abs doses in ELISAs were adjusted, so that both ECD and FL sPD-1 could be detected with high and comparable sensitivity (Fig. 2b, d). Detailed calculations of plasma sPD-1 were performed as follows: if sample S1 is positive in ELISA-#2 with OD450nm = RS1, but negative in ELISA-#1, then S1 contains only ECD sPD-1, and the content could be calculated by standard curve-II in ELISA-#2. If sample S2 is positive in both ELISA-#1 (OD450nm = RS2-1) and -#2 (OD450nm = RS2-2), we will first calculate the FL sPD-1 amount through standard curve-I in ELISA-#1, then obtain RS2-3 by reversely calculating the standard curve-III. RS2-2 = RS2-3 means only FL sPD-1 was contained in S2; otherwise, RS2-2 > RS2-3 means that both FL and ECD sPD-1 were contained therein, the OD450nm of ECD sPD-1 in S2 is obtained by subtracting RS2-3 from RS2-2, and the amount could be calculated by standard curve-II. In this way, sPD-1 in plasma samples could be analyzed qualitatively and quantitatively through the two ELISAs.

a ELISA validation of anti-human PD-1 clones D4W2J and J105 used in ELISA-#1. Fusion proteins CD80-hIgG, ECD-hIgG, ICD-hIgG, or ΔFL-hIgG were coated into 96-well plate at 1 μg/ml; D4W2J or J105 was used as detection Abs. CD80-hIgG was negative control. b Standard curves of PD-1 ΔFL (blue line) and ECD (orange line) fusion proteins in ELISA-#1, OD45 nm ≤ 0.5 area was magnified. c ELISA validation of anti-human PD-1 clones MIH4 and BAF1086 used in ELISA-#2; coated antigens and ELISA procedures were the same as in (a). d Standard curves of PD-1 ΔFL (blue line) and ECD (orange line) fusion proteins in ELISA-#2; OD45 nm ≤ 0.5 area was magnified. e Level of ECD (orange) and FL sPD-1 (blue) in ten patients with NSCLC. Plasma samples were diluted 10 folds with PBS, and examined by ELISA-#1 and -#2. f Generation procedures and g binding specificity of monoclonal CH30; splenocytes from murine PD-1 ICD-mIgG immunized PD-1 knockout (KO) mice were electrofused into SP2/0 cells. CH30 was screened and confirmed by ELISA. p < 0.0001. h Distribution of ECD and FL sPD-1 in B16 tumor model, and representative mice single- (1#, 2#) or double-positive (8#) for the two isoforms. B16 melanoma cells were injected subcutaneously into 10 mice at 3 × 105 cells/mouse. Bars indicate mean ± SD.
Subsequently, we examined sPD-1 levels in patients with cancer. Plasma from 10 treatment-naïve patients diagnosed with non-small cell lung cancer (NSCLC) (Supplementary Table 1 cohort #1) were collected. As shown, both FL and ECD sPD-1 could be detected in plasma from patients with cancer (Fig. 2e), samples #3 and #5 were double-positive for both isoforms, while some plasma samples were single-positive, e.g., only FL sPD-1 appeared in samples #7 and #10, and only ECD sPD-1 appeared in sample #8, indicating the independent existence of these two sPD-1 isoforms.
To facilitate mechanistic studies, a monoclonal Ab (mAb) specifically binding to the ICD of murine PD-1 (Fig. 2f, g), named CH30, was generated, and two similar sandwich ELISA systems were developed (Supplementary Fig. 2b). Murine tumor models were established for sPD-1 detection. The results showed that, despite being sparse, sPD-1 in B16 melanoma-bearing mice also had two isoforms, FL and ECD sPD-1. Similarly, some plasma samples were double-positive for the two isoforms, e.g., the plasma from mouse #8 collected on day 10 and 14, and the one from mouse #7 on day 14. However, the two isoforms were not consistently found together; plasma from different mice (e.g., mouse #1 and #2) or collected at different time points (e.g., samples from mouse #8 on day 10 and 17) were single-positive for either FL or ECD sPD-1 (Fig. 2h), indicating the independence of these two sPD-1 isoforms. Similar findings were observed from the MC38 tumor model (Supplementary Fig. 2c). Collectively, these findings demonstrated the existence of FL and ECD sPD-1 proteins in tumor-bearing mice and patients, suggesting a complex relationship between sPD-1 and tumor progression or prognosis.
sPD-1 was produced by activated lymphocytes
Next, we attempted to track the cellular origin of sPD-1. B16 melanoma models were established in immunodeficient rag1-KO (NOD-Rag1em1Smoc) and wild-type (WT) C57BL/6 (B6) mice. To enhance the immune response, anti-4-1BB mAb30 (4-1BB agonist, clone 2A, made in-house) was administrated. Total sPD-1 was the sum of ECD and FL sPD-1. As shown, plasma sPD-1 was detected in B16-inoculated WT mice; mice receiving no tumor injection or merely anti-4-1BB had no sPD-1, suggesting that antigens and immune activation might be required for sPD-1 production. Furthermore, compared to the ctrl IgG, the anti-4-1BB agonist significantly elevated sPD-1 level (p < 0.0001), from 20 ng/ml on average to ~120 ng/ml on day 14, indicating that immune promotion could facilitate sPD-1 production. However, in the rag1-KO group, no sPD-1 production was detected from either ctrl IgG or the anti-4-1BB group, demonstrating that sPD-1 production requires lymphocytes (Fig. 3a). Additionally, to confirm our observation, we employed a concanavalin A (ConA) activation model with rag1-KO and WT B6 mice. Similarly, sPD-1 was produced in WT mice after ConA stimulation, with anti-4-1BB also elevating its level significantly (p < 0.0001). In contrast, there was no sPD-1 production in rag1-KO mice (Supplementary Fig. 3a). Collectively, lymphocytes and immune activation are necessary for sPD-1 production.

a Total sPD-1 from B16 inoculated WT or rag1-KO mice treated with ctrl IgG or anti-4-1BB; 3 × 105 B16 melanoma cells were injected per mouse at day 0, ctrl IgG or anti-4-1BB was administered at 200 μg/mouse on day 3, control groups were anti-4-1BB-injected or blank mice, n = 10. b Time course of the ConA/LPS model, and change in total sPD-1 in ConA (n = 5) or LPS (n = 4) models post CD8, CD4, or CD20 depletion. c Gating strategy and percentages (d) of mPD-1 expression on B, CD4, or CD8 T cells. Blood from the anti-4-1BB/B16 model was collected and assayed regularly by flow cytometry. n = 10. Bars indicate mean ± SEM.
Rag1-KO mice are deficient in CD4 T, CD8 T, and B cells. To further identify the sPD-1-producing cells, we depleted each cell subset (Supplementary Fig. 3b) and examined sPD-1 production in the ConA activation model. In control mice without any cell depletion, plasma sPD-1 increased to 98.5 ng/ml on day 10 after ConA and anti-4-1BB stimulation. However, it was totally absent in CD8 T cell-depleted and B cell-depleted mice. Although CD4 T cell depletion also resulted in a significant sPD-1 decrease (p < 0.0001), it still remained 47.4 ng/ml on day 10 (Fig. 3b, Supplementary Fig. 3c). These results indicated that CD8 T and B cells were required for ConA-induced sPD-1 production. A lipopolysaccharide (LPS) model was also established; results revealed that all three cell depletions decreased sPD-1 levels significantly (p < 0.0001) on day 7; however, on day 10, only CD8 T depletion resulted in a significant reduction of sPD-1 (p < 0.0001), while CD4 T or B cell depletion made no difference. These results suggest that CD8 T depletion abolished sPD-1 production completely, while CD4 T and B cell depletion only delayed production (Fig. 3b, Supplementary Fig. 3d, e). Collectively, both T and B cells were involved in sPD-1 production, and CD8 T seems particularly indispensable.
We proceeded to analyze the expression of mPD-1 in the B16 tumor model. Flow cytometry revealed that B cells lacked mPD-1 expression throughout the immune response (Fig. 3c); the percentage of mPD-1+/B cells remained not more than 2% in both the anti-4-1BB and ctrl IgG groups. However, mPD-1 expression was upregulated in T cells. In detail, the percentage of mPD-1+/CD4 T cells was 4–9% in control mice, with anti-4-1BB slightly increasing the percentage to 5–12%. In contrast, anti-4-1BB significantly promoted mPD-1 expression on CD8 T cells (p < 0.0001); the mean positive percentage was 30% on day 14 (Fig. 3d), suggesting that activated CD8 T might be the cellular origin of sPD-1 production.
ECD sPD-1 required CD8 T cells expressing PD-1 and matrix metalloproteinase (MMP)
We tried to figure out how PD-1-expressing T cells produced sPD-1. A lymphoma cell line—EL4—was cultured in complete RMPI 1640 medium, and the supernatant was collected daily for ELISAs. Results showed that sPD-1 was detected in the supernatant, and its amount gradually increased over time; in contrast, blank medium without EL4 had no sPD-1 (Fig. 4a). Additionally, when sPD-1 isoforms were distinguished, we found that the ECD fragment was the sole component of sPD-1 at the first 3 days, FL sPD-1 emerged on day 4 and continually increased till all cells died of nutrient deficiencies or excessive cell density (Fig. 4b). Further flow cytometry assays demonstrated that EL4 cells constitutively expressed a high level of mPD-1 (Fig. 4c), and they began to die after 3 days of proliferation (Fig. 4d, e). These results demonstrated that EL4 cells could produce ECD sPD-1 upon survival and release FL sPD-1 upon death. Similar results (Supplementary Fig. 4a) were obtained from EG7, another lymphoma stably expressing mPD-1. We also checked a human-originated lymphoma cell line—Jurkat. Jurkat had neither mPD-1 expression nor sPD-1 production in regular culture. However, its mPD-1 was obviously elevated upon phytohemagglutinins (PHA) stimulation (Supplementary Fig. 4b); correspondingly, ECD sPD-1 also increased, and FL sPD-1 only appeared when the activated Jurkat cells died (Supplementary Fig. 4c). Collectively, these results suggest that mPD-1 could turn into sPD-1, and the specific sPD-1 isoforms depend on the viability of mPD-1+cells.

a Total sPD-1 levels in the supernatant from continual EL4 culture, EL4 cells were plated at 0.3 × 105 cells/well into 48-well plates, and the supernatant was collected from 3 wells per day for 7 days. n = 3. b Dynamic increases in ECD and FL sPD-1 in the EL4 culture, n = 3. c mPD-1 expression on EL4 cells by flow cytometry. d Cell numbers and e death marker staining of EL4 cells at different time points. EL4 cells were cultured as in (a). f Changes in ECD and FL sPD-1 production upon ilomastat treatment; ilomastat was 2-fold diluted from 40 μM. n = 3. g MMP activity of ECD sPD-1-positive plasma (ECD+), FL sPD-1-positive but ECD sPD-1-negative plasma (FL+ECD−), or FL sPD-1 and ECD sPD-1 double-negative plasma (ECD−FL−) in the ConA and LPS models. Bars indicate mean ± SD, ns p > 0.05.
Soluble forms of immune checkpoint molecules are generally cleaved by MMPs. To check whether this applied to sPD-1, we added an MMP inhibitor, named ilomastat, into EL4 cell culture; results showed that ECD sPD-1 production was dose-dependently blocked by ilomastat (Fig. 4f). Besides, no FL sPD-1 production and cell death (Supplementary Fig. 4d) were detected during ilomastat treatment, indicating that ECD sPD-1 was reduced upon MMP inhibition but was not converted to FL sPD-1. We further checked MMP activity in all plasma from the above ConA and LPS models; varying degrees of MMP activity were detected in ECD sPD-1-positive plasma; however, only negligible MMP activity was found in plasma from the FL sPD-1 single-positive or sPD-1 negative group (Fig. 4g). Collectively, sPD-1 production derives from mPD-1; ECD sPD-1 was cleaved from live mPD-1+T cells by MMPs, while FL sPD-1 was released from dead mPD-1+T cells.
FL and ECD sPD-1 reflect different viability of CD8 T cells
We next addressed the sPD-1 production mechanism in a tumor model. sPD-1 isoforms from anti-4-1BB-treated B16 tumors were distinguished by ELISAs; both ECD and FL sPD-1 were detected. The ECD fragment was the main component of sPD-1 in 7/10 mice (termed as ECD sPD-1+ mice), while FL sPD-1 was the main isoform in the other 3/10 mice (FL sPD-1+ mice) (Fig. 5a). Flow cytometry revealed that mPD-1 was upregulated in both groups but did not significantly differ between them (Fig. 5b). Further dynamic assays showed that mPD-1 all declined post sPD-1 release. FL sPD-1 production was also accompanied by a dramatic reduction of mPD-1+CD8+T cells; the percentage of CD8 T cells in lymphocytes decreased from 21.4% to 1.86%. In comparison, CD8 T cell percentage was maintained when only ECD sPD-1 was produced (Fig. 5c), suggesting that FL sPD-1 is released from dead mPD-1+CD8+T cells, while ECD sPD-1 is released from live cells.

a Levels of ECD and FL sPD-1 in ECD or FL sPD-1-positive mice from anti-4-1BB-treated B16 tumor model. n = 10. b Percentage of mPD-1 expression on CD8 T cells from FL (n = 3) or ECD sPD-1 (n = 7) positive or sPD-1 negative (n = 7) mice. Blood cells were stained with anti-PD-1 and analyzed by flow cytometry. c Representative plots of mPD-1 and CD8 T cell changes pre and post-ECD or FL sPD-1 production. Lymphocytes were first gated FSC-SSC as Fig. 3c, then CD8 T cells were gated as CD3+CD4−T cell, and PD-1+ was gated on CD8 T cells. d Changes of FL and ECD sPD-1 in CD8 T competent or depleted ConA model, CD8 T depletion mAbs were administrated on day 7 post-ConA stimulation, CD8 T competent mice had no CD8 T depletion. n = 5. Bars indicate mean ± SD, ns p > 0.05.
To confirm the correlation of cell death with FL sPD-1 production, we altered the timing of CD8 T cell clearance in the ConA model. In CD8 T cell competent mice, ECD and FL sPD-1 both increased after ConA and anti-4-1BB injection, with ECD sPD-1 levels significantly higher than the FL ones on day 10. Unlike the CD8 T cell depletion in Fig. 3b, where CD8 T cells were completely cleared before ConA stimulation, and neither ECD nor FL sPD-1one was produced. We depleted CD8 T cells at day 7 post-ConA and anti-4-1BB administration; it turned out that FL sPD-1 level was increased on day 10 (Fig. 5d). In contrast, ECD sPD-1 disappeared in the subsequent plasma samples, consistent with the reduction of CD8 T cells upon depletion (Supplementary Fig. 5a). A similar correlation of sPD-1 production with PD-1+T cell viability was observed in vitro when OT-1 T cells were activated with OVA257-264 peptide (Supplementary Fig. 5b, c). In summary, our results further addressed the different mechanisms of ECD and FL sPD-1 production.
ECD sPD-1 rather than FL sPD-1 predicted an effective antitumor immune response
Based on the above results, we explored the association between sPD-1 production and tumor prognosis. First is the untreated tumor, owing to the low frequency of sPD-1 from untreated tumors, we expanded the sample size to 30 in the B16 tumor model and analyzed the correlation between tumor growth and sPD-1 production. ELISAs revealed that 4 of the 30 B16-implanted mice were ECD sPD-1+, with 2 producing ECD sPD-1 only and another two also having detectable but lower FL sPD-1 levels (29.0 and 33.4 ng/ml of ECD sPD-1 vs. 17.9 and 18.7 ng/ml of FL sPD-1 on day 17) (Fig. 6a, b). Further tumor size monitoring revealed that these four ECD sPD-1+ mice had significantly slower tumor growth than the remaining 26 mice (p < 0.0001) (Fig. 6c). Besides, three of 26 ECD sPD-1− mice produced FL sPD-1 (29.2 ng/ml on average); however, tumors in the three mice grew comparably to those in sPD-1− mice (Fig. 6b, c). Our results suggest that high levels of ECD sPD-1 but not FL sPD-1 indicate delayed tumor growth.

a Levels of ECD and b FL sPD-1 in the B16 tumor model, mice were injected with 3 × 105 cells/mouse at day 0, and blood was collected at the indicated time points. The gray line for sPD-1 negative mice represents the mean of all sPD-1− mice, colored lines represent individual values. n = 30. c Tumor growth of ECD sPD-1+, FL sPD-1+, and sPD-1 negative mice. d B16 tumor growth in C57BL/6 mice treated with anti-4-1BB. Mice were implanted with B16 cells and treated with anti-4-1BB (n = 15) or ctrl IgG (n = 10). Among the 4-1BB-treated mice, 5 were FL sPD-1+, and 10 were ECD sPD-1+. e sPD-1 levels and f tumor growth in radiation-treated B16 models. B16 tumors were implanted at day 0 and locally radiated with 15 Gy x-ray at day 7. n = 10. Bars indicate mean ± SEM, ns p > 0.05.
Next are the treated tumors; sPD-1 from the anti-4-1BB/B16 tumor model was monitored, and the results showed that tumor growth in ECD sPD-1+ mice was significantly slower than that in the FL sPD-1+ group (p < 0.0001), which had no apparent difference with the control (Fig. 6d). Similar results were obtained from the anti-PD-L1-treated MC38 tumor model (Supplementary Fig. 6a, b). Besides immunotherapy, we also tried local radiotherapy; pre-implanted B16 tumors received 15 Gy radiation, and sPD-1 production was examined regularly. ELISAs revealed that both ECD and FL sPD-1 production was promoted upon radiation (Fig. 6e); however, only ECD sPD-1 indicated significant tumor regression (p < 0.0001), while tumor growth in the FL sPD-1+ group was similar to that in the control group (Fig. 6f). Collectively, these results suggest that ECD sPD-1 increase indicates better tumor prognosis upon treatment.
Indicative role of ECD sPD-1 in clinical cancers
Finally, we validated our findings in patients with cancers. A total of 66 patients—26 with lung cancer, 27 with gastric cancer, and 13 with esophageal cancer—were enrolled (Supplementary Table 1, cohort #2). Patients were grouped into early-stage (I–IIIa, n = 30) and late-stage (IIIb–IV, n = 36) groups according to their histological pathology, and 20 healthy donors were enrolled as control (HC). Plasma was collected at diagnosis when patients had not yet received antitumor therapy. We first used MIH4 and BAF1086 to detect sPD-1 and analyzed its clinical association. Results showed that the sPD-1 levels in patients with cancer were significantly greater than those in healthy donors; however, no significant difference was detected between patients with early-stage cancer and those with late-stage cancer (Fig. 7a). In contrast, when the two sPD-1 isoforms were distinguished, we found that the ECD fragment was the main component of sPD-1 from early-stage patients, while FL sPD-1 tended to be produced by late-stage patients. Therefore, ECD sPD-1 level was significantly higher in early-stage patients than in late-stage patients (p = 0.0152) (Fig. 7b), suggesting that a high ECD sPD-1 level is also associated with the slow progression of cancers.

a Levels of total and b ECD sPD-1 from patients with early (n = 30) or late-stage (n = 36) cancers, 20 healthy donors were enrolled as control (HC). c–f Changes of ECD and FL sPD-1 levels post immunotherapy, targeted therapy or combo, responders, n = 88, non-responders, n = 9. Bars indicate mean ± SD, ns p > 0.05.
Lastly, we examined sPD-1 production in treated cancers. Patients with advanced cancers (lung cancer, gastric cancer, esophageal cancer) were treated with immunotherapy (anti-PD-1, anti-PD-L1), targeted therapy (targeted EGFR, VEGF, or VEGFR), or combo (Supplementary Table 2), and blood was collected at baseline and post-treatment (18–54 days, median 31 days). Among the enrolled 97 patients, nine patients experienced tumor progression, metastasis, or relapse during the follow-up (9–12 months), thus considered as non-responders, and 88 had regressed or stable tumors, thus as responders. ELISAs revealed that compared to that at baseline, plasma collected post-treatment had significantly higher sPD-1 levels (p < 0.0001), with 30.9% (30/97) patients having elevated ECD sPD-1 and 19.6% (19/97) having elevated levels of FL sPD-1 upon therapies (Fig. 7c). Moreover, almost all (29/30) patients with elevated ECD sPD-1 responded, especially those with elevation greater than 100 ng/ml (Fig. 7d), suggesting that ECD sPD-1 increase may predict response to therapies. In contrast, although FL sPD-1 also increased significantly upon treatment (p = 0.0136), its increases were observed in both responders (63.2%) and non-responders (36.8%) (Fig. 7e, f). Besides, seven out of 9 non-responders had elevated FL sPD-1, whereas only 1 case of ECD sPD-1 increased (Fig. 7f), suggesting that FL sPD-1 increase is not prognostic of tumor prognosis post-treatment. Collectively, these results indicate that elevated ECD sPD-1 predicts a better prognosis after single or combined immunotherapy/targeted therapy, whereas FL sPD-1 does not.
Discussion
In this study, we initially conducted a meta-analysis to systematically summarize the clinical relevance of sPD-1 in cancers, and the results showed that in untreated patients, the levels of sPD-1 were not indicative of tumor progression and malignancy. However, upon treatment, increased sPD-1 production was consistently observed in responders, suggesting that sPD-1 increase positively correlated with better cancer prognosis. To elucidate these conflicting results, we developed two sandwich ELISA systems and identified two different isoforms of sPD-1 in tumors, namely FL or ECD sPD-1. Subsequently, we analyzed the dynamic production of sPD-1 during immune response and tracked its cellular origin through various murine models. High levels of the two isoforms were both associated with significant upregulation of mPD-1 expression on CD8 T cells upon antigen stimulation; however, FL sPD-1 was released from dead CD8 T cells, and ECD sPD-1 was cleaved off live cells by MMPs. Therefore, only ECD sPD-1 production was associated with effective CD8 T activation and delayed tumor growth. Our findings revealed the existence of two sPD-1 isoforms and highlighted the predictive value of ECD sPD-1 in cancer outcomes.
Numerous immune regulatory molecules, including both co-inhibitory and co-stimulatory checkpoints, have been reported to have soluble forms besides membrane-bound expression31,32. Some of these soluble forms are produced by proteolytic cleavage from surface forms, such as sLAG-333, sB7-H334, and sTim335,36, while others are translated from alternate splice variants, such as sB7-237 and sCTLA-438,39. So far, the mechanism of sPD-1 production has not been fully elucidated. The full-length human PD-1 gene comprises five exons encoding the signal peptide (exon 1), extracellular IgV domain (exon 2), transmembrane region (exon 3), and intracellular domain (exons 4 and 5)40. Four mRNA transcripts were generated upon alternative splicing18 namely, PD-1Δex2, PD-1Δex3, PD-1Δex2,3, and PD-1Δex2,3,4. Among them, PD-1Δex3 and PD-1Δex2,3 can be translated into soluble forms by splicing out exon 3. PD-1Δex2,3,4 also has no transmembrane region but is only translated into a signal peptide. Besides, no transcript-encoding soluble ECD fragment has been reported. Herein, ECD sPD-1 was detected in cell culture in vitro and in antigen stimulation models in vivo, demonstrating the existence of ECD sPD-1 at the protein level. Therefore, alternatively spliced mRNA might not be the only origin of soluble isoforms. Another reason we did not stick to mRNA variants was the observation that sPD-1 positively correlated with mPD-1. sPD-1 was produced only when mPD-1 was constitutively expressed or upregulated upon antigen stimulation. Treatments promoting mPD-1 expression correspondingly elevated sPD-1 levels. Similar associations were reported in clinical studies. Heli Vajavaara et al.17 found that serum sPD-1 from patients with high-risk diffuse large B-cell lymphoma positively correlated with the proportions of tumor-infiltrating PD-1+T cells. Chiu et al.5 and Zilber et al.19 also reported that sPD-1 from HIV-infected patients was associated with the surface expression of PD-1 on T cells. Therefore, we reasoned that sPD-1 was probably derived from mPD-1.
Theoretically, PD-1 has three soluble forms at the protein level: ECD fragment, ICD fragment, or FL fragment containing both ECD and ICD domains. The first two originate from a single FL PD-1 protein and thus appear in equal quantities. Therefore, the challenge lies in distinguishing ECD sPD-1 from the FL isoform. In this study, through different ELISAs, two isoforms of sPD-1—ECD and FL sPD-1—were identified. ECD sPD-1 production was blocked by MMP inhibition, indicating that the ECD form was cleaved off the cell membrane by MMPs. This is similar to the way some immune checkpoint molecules produce soluble ECD fragments. ADAM7 and ADAM10 are two critical transmembrane metalloproteases. sTim-335 was cleaved by ADAM10, and sLAG-333 was regulated by ADAM10 and AMAD17. The specific MMP required for ECD sPD-1 production warrants further study. We also observed FL sPD-1 production along with PD-1+T cell death; this is not surprising, because all cell components, including membrane-bound proteins, would be released when cells die8,41. Moreover, when lymphoma cells or activated T cells begin to die, ECD sPD-1 production begins to decline, but FL sPD-1 increases; and vice versa, mice producing high levels of ECD sPD-1 generally have low FL sPD-1 levels. This exclusive pattern further validates the sPD-1 production mechanism that sPD-1 derives from mPD-1, and isoforms were determined by the viability of PD-1+ cells.
Distinguishing sPD-1 isoforms at the protein level may help elucidate the clinical significance of sPD-1 in cancer. The two isoforms represent opposite statuses of mPD-1+CD8+ T cells: ECD sPD-1 is released from live mPD-1+CD8+T cells, indicating the survival of effector T cells and mPD-1 downregulation; in contrast, FL sPD-1 is generated following the death of mPD-1+CD8+ T cells, indicating the collapse of immune activation. Therefore, a mixed assay of total sPD-1 in tumors inevitably leads to an unclear conclusion. Consistently, neither our meta-analysis for untreated cancers nor the total sPD-1 results from untreated patients with cancers demonstrated a clear correlation of sPD-1 with tumor outcomes. In contrast, when using ECD sPD-1 as the only readout, the association between sPD-1 and tumor malignancy became explicit. Regarding treated cancers, our findings showed that ECD sPD-1 could predict effective responses in treatment groups. High ECD sPD-1 was produced only when CD8 T cells were highly activated, and mPD-1 expression was significantly upregulated. Notably, CD8 T cells remained alive when ECD sPD-1 was released. These are general features when an effective immune response occurs following therapies42,43. No increase in sPD-1 or only FL sPD-1 increase indicated that CD8 T cells either had low/no mPD-1 expression or died after activation, both resulting in no response. Therefore, ECD sPD-1 elevation necessarily predicts response to treatments. By analyzing the sPD-1 isoforms separately, we provided a more comprehensive approach to clarify the clinical significance of sPD-1 in cancers and found that ECD sPD-1, but not FL sPD-1, is associated with a better response and tumor prognosis.
Our study also has some limitations. Although we demonstrate that both isoforms are derived from mPD-1, the precise mechanism that determines whether ECD or FL sPD-1 will be produced remains unclear. Experiments are going on to identify which and how MMP is activated and to cut off ECD sPD-1. Besides, sPD-1 is also present in autoimmune diseases and infections; distributions of ECD and FL sPD-1 in these patients need further study.
PD-1 plays a crucial role in regulating the entire course of T-cell response, including T-cell priming, expansion, function, and exhaustion44. The two isoforms of sPD-1 identified in the present study directly reflect mPD-1 regulation and antigen-specific CD8 T-cell survival. Therefore, the strength and duration of the immune response could be evaluated by qualifying and quantifying circulating sPD-1. Our study offers new insights into how to assess and further manipulate antitumor immune responses.
Methods
Meta-analysis
The meta-analysis in this study was conducted under the guideline of the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement. On December 12, 2024, a systematic search for literature from PubMed and Web of Science via the search terms: ((“prognosis”[Title/Abstract] OR “outcome”[Title/Abstract]) AND ((“soluble pd 1”[Title/Abstract] OR “soluble pdcd1”[Title/Abstract]) AND ((“cancer”[Title/ Abstract] OR “tumor”[Title/Abstract] OR “carcinoma”[Title/Abstract]) was conducted. Studies in which HRs) or Kaplan–Meier curves or individual patient data for OS, DFS, and PFS were comprehensively recorded, were confirmed as eligible. To avoid selection bias, only studies detecting sPD-1 production by ELISA or Luminex were included. Meetings, dissertation thesis, reviews, case reports, and letters were excluded.
The estimates of the HRs for PFS, DFS, and OS were obtained from the studies. For Kaplan–Meier curve, the Web Plot Digitizer tool (https://automeris.io/WebPlotDigitizer/) was applied to estimate data from the figures and extract the proportion of patients who survived at each point between low and high levels of sPD-1. The natural logarithms of the HRs and standard errors were calculated according to the method described previously45. Common-effects and random-effects models were chosen to evaluate the pooled estimated HRs and the impact of sPD-1 on the patients’ outcomes. Forest plots were generated to display the heterogeneity and the weights between different studies. The plot was generated using R software (v.4.2.2) package “meta” (v.6.2.1)46 through Hiplot Pro (https://hiplot.com.cn/), a comprehensive web service for biomedical data analysis and visualization.
Human plasma
Three cohorts of patients with cancers were enrolled, cohort#1 and #2 had not received treatment, cohort#1 included 10 patients with NSCLC, and cohort#2 comprised 66 patients with lung cancer, gastric cancer, or esophageal cancer. Cohort#3 had the same three cancer types with cohor#2, cohort#3 included 97 patients who were treated with immunotherapy, targeted therapy, or combo. Table S1 and S2 shows the detailed patient information. All ethical regulations relevant to human research participants were followed. The study protocol (No. 2021-118) was approved by the Institutional Review Boards of Fujian Medical University, and informed consents were obtained. Human peripheral blood was collected through venipuncture, and plasma was isolated through centrifugation and stored at −80 °C.
Mouse strains and cell lines
C57BL/6 J and C57BL/6-rag-1−/− (rag1-KO) were procured from the Nanjing Biomedical Research Institute of Nanjing University (Nanjing, China). OT-1 mice and PD-1 KO mice were bred in-house. T cells from OT-1 mice have a transgenic T cell receptor that specifically recognizes ovalbumin peptide residues 257–264 (OVA257–264, also called OVA8). We have complied with all relevant ethical regulations for animal use. All mice were housed in specific pathogen-free facilities and treated according to ethical approvals received from the Institutional Animal Care and Use Committee of Fujian Medical University. Experiments were conducted using female mice aged 6–8 weeks, each mouse was considered to be an experimental unit. Mice were randomly divided into treatment or control groups using the standard = RAND() function in Microsoft Excel. For each group, a cage was selected randomly from the pool of all cages.
Murine B16 melanoma and MC38 colorectal tumor cell lines were purchased from ATCC (Manassas, VA, USA). Lymphoma cell lines EL4, EG7, and Jurkat were lab-retained and had been authenticated using short tandem repeat DNA profiling. All cell lines were cultured in complete medium (RPMI 1640 containing 10% fetal bovine serum and 100 μg/ml penicillin/streptomycin, Sigma) and tested to ensure that they were free of mycoplasma using a Myco-Blue Mycoplasma Detector (Cat. #D101-02, Vazyme Biotech, Nanjing, China).
Preparation of recombinant PD-1 fragments
Three fragments of human/murine PD-1, including the signal peptide (SP), ECD, and ICD, were amplified via PCR. Subsequently, through overlapping, the SP was combined with the ICD, and the ECD was combined with the ICD to create the ΔFL (FL PD-1 without the TM). Subsequently, ECD, ICD, and ΔFL were inserted into plasmids containing the Fc fragment of murine IgG2a or human IgG1 to generate the corresponding fusion proteins.
The constructed plasmids were transiently transfected into HEK-293T cells, and the resulting fusion protein was purified using a Hi trap Protein A affinity column for subsequent experiments.
CH30 generation
The murine SPIC fusion protein was used to produce the monoclonal antibody CH30. Briefly, 100 μg of murine PD-1 ICD-mIgG fusion protein in 500 μL of PBS was thoroughly mixed with an equal volume of adjuvant. PD-1 KO mice were immunized with a mixture of 25 μg of PD-1 in four places. Mice underwent four immunizations in total; initially, ICD-mIgG protein was mixed with complete Freund’s adjuvant, and the other three were mixed with incomplete Freund’s adjuvant. Three weeks after the final immunization, the mice were intraperitoneally (i.p.) injected with 200 μg of ICD-mIgG fusion protein; five days later, splenocytes were obtained for electrofusion with the myeloma sp2/0 cell line. Positive clones were screened using ELISA, and at least five rounds of subcloning were conducted to obtain the monoclonal hybridoma CH30.
ELISA detection of sPD-1
Qualitative and quantitative analyses of sPD-1 in mouse or human plasma were performed using two ELISA systems. Antibodies used in ELISAs are listed in Supplementary Fig. 1a, and all ELISAs were performed using the same procedures. Briefly, capture antibody was coated into a 96-well assay plate at 4 °C for 18 h, the plate was washed for 3 times and blocked with 5% BSA at 37 °C for 1 h. Subsequently, prediluted plasma (10×folds for human plasma and 5×folds for mice plasma) with PBS was added, and the plates were placed at 37 °C for 2 h. After 3 washes, biotinylated detection antibody was added and incubated at 37 °C for 1 h. Then, the plate was washed 3 times and incubated with streptavidin-conjugated HRP (#405210, Biolegend) for 45 min. Finally, the substrate, TMB, was loaded and incubated for 15 min, and the reaction was stopped with H2SO4, after which the OD450nm was read. Detailed calculation of sPD-1 isoform levels has been explained in Fig. 1a–d and in the results.
Murine tumor model and treatment
For the untreated tumor model, B16 cells were injected intradermally into the right flank of C57BL/6 WT mice at 3 × 105 cells/mouse. In the immunotherapy model, B16 cells were injected at 3 × 105 cells/mouse into C57BL/6 WT or rag-1 KO mice. Anti-4-1BB or control IgG was administered intraperitoneally at 200 μg/mouse 3 days post tumor implantation. MC38 cells were injected at 6 × 105 cells/mouse into C57BL/6 mice at day 0 and anti-PD-L1 (clone 10B4) was administrated at day 3. In the radiation model, B16 was implanted as above; 7 days later, mice were first anesthetized and then shielded with a lead plate, except the established tumor, and irradiated with 15 Gy x-rays in RS-2000 Biological Irradiator (RadSource, Canada). Retro-orbital bleeds were collected from each mouse at the indicated time points. Blood was centrifuged at 5000 rpm/min for 5 min after standing for 2 h, and the plasma was obtained and stored for sPD-1 detection. Tumor sizes were measured with digital calipers every 3–4 days and were calculated using the formula (l + w)/2, where l and w represent the larger and smaller dimensions, respectively. For humane treatment, the mice were classified as dead once the tumors reached 15 mm in size.
In vivo activation model
For the ConA model, 20 mg/kg ConA (#C0412, Sigma, Darmstadt, Germany) was intravenously injected into C57BL/6 WT mice, and then anti-4-1BB or control rat IgG was intraperitoneally administered on the same day (n = 5). To deplete cell subsets, anti-CD20 (clone SA271G2), anti-CD4 (clone GK1.5), and anti-CD8 (clone 53.67) depletion antibodies or blank PBS was intraperitoneally injected at −7 and −3 days prior to the injection at 200 μg/mouse. On day 0, ConA and anti-4-1BB were injected into all cell-depleted mice at the same volume as above. Delayed CD8 T depletion was achieved by injecting anti-CD8-depleting antibodies 7 days post ConA and 4-1BB treatment. For the LPS model, 15 μg/mouse was intraperitoneally injected, and an anti-4-1BB or cell subset depletion agent was administrated similarly as in the ConA model. Venous blood was obtained regularly for sPD-1 detection.
In vitro lymphoma culture
EL4 or EG7 lymphoma was cultured in a complete RMPI 1640 medium with an initial concentration of 0.3 × 105 cells/well in a 48-well plate. All wells were loaded with cells on the first day, supernatant was collected from three new wells every day, and cells were assayed with flow cytometry after death marker staining. For MMP inhibition, EL4 cells were loaded into a 48-well plate at 1.5 × 105 cells/well, and ilomastat was 2 × diluted from 40 μM. Next, after a 24 h incubation, the supernatant was collected from the ilomastat-treated or blank wells for sPD-1 detection. Jurkat cells were stimulated with 1.5 μg/ml PHA for 48 h; after collecting the supernatant, cell death was induced with H2O for further sPD-1 examination.
OT-1 T cell activation
For OT-1 T cell activation, splenocytes were collected from OT-I mice and processed to single-cell suspensions at 1 × 106 cells/ml. Subsequently, the splenocytes were plated into a 48-well plate at 0.5 ml/well and stimulated with the OVA257-264 peptide (1 μM). The supernatant was collected every 2 days for sPD-1 detection, and cells from the same wells were preserved for flow cytometry. Cells from three wells were collected from each group each time. The remaining cells were replenished with fresh medium without any peptide or antibody and cultured for 10 days.
Flow cytometry
Blood cells were blocked with anti-mouse CD16/32 (#MFCR00, clone FCR-4G8, Fisher Scientific) after erythrocyte lysis and stained with antibodies against mouse CD3 (#48-0032-82, clone 17A2, Invitrogen, CA, USA), CD19 (#11-0191-82, clone MB19-1, Invitrogen), CD8 (#53-0081-82, clone 53-6.7, Invitrogen), PD-1 (#135206, Biolegend; #53-9969-42, ThermoFisher), and matched isotype controls. Lymphoma cells were stained with anti-PD-1 and death markers. OT-1 T cells were stained with CD3, CD8, or PD-1. All samples were run on a BD FACSVerse™ flow cytometer, and the data were analyzed using FlowJo software (BD, NJ, USA).
Statistics and reproducibility
All experiments were conducted three times. The group sizes (n) and applied statistical tests are indicated in each figure legend. The data are presented as the mean ± standard error of the mean or standard deviation. GraphPad Prism Version 8.0.1 (GraphPad Software CA, USA) was used to generate plots and conduct additional statistical analyses. The significant differences between the two groups were assessed using a two-tailed unpaired t-test, and a two-way analysis of variance was used to determine the statistical significance between groups.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The original data generated in the study are included in the article. The source data for graphs are provided in Supplementary Data 1. Materials, additional data, and protocols described within the manuscript are available from the corresponding author upon reasonable request.
References
Ishida, Y., Agata, Y., Shibahara, K. & Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J.11, 3887–3895 (1992).
Shreya, M. et al. Serum biomarker panel for diagnosis and prognosis of pancreatic ductal adenocarcinomas. Front. Oncol. 11, https://doi.org/10.3389/fonc.2021.708963 (2021).
Corinna, U. et al. Cellular and soluble immune checkpoint signaling forms PD-L1 and PD-1 in renal tumor tissue and in blood. Cancer Immunol. Immunther. 71, https://doi.org/10.1007/s00262-022-03166-9 (2022).
Hsiang-Yun, C. et al. Circulating programmed death-1 as a marker for sustained high hepatitis B viral load and risk of hepatocellular carcinoma. PLoS ONE 9, https://doi.org/10.1371/journal.pone.0095870 (2014).
Chris, Y. C. et al. Soluble immune checkpoints as correlates for HIV persistence and T cell function in people with HIV on antiretroviral therapy. Front. Immunol. 14, https://doi.org/10.3389/fimmu.2023.1123342 (2023).
Greisen, S. R. et al. Increased soluble programmed death-1 (sPD-1) is associated with disease activity and radiographic progression in early rheumatoid arthritis. Scand. J. Rheumatol. 43, https://doi.org/10.3109/03009742.2013.823517 (2013).
Bing, W. et al. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J. Immunol. 177, https://doi.org/10.4049/jimmunol.177.12.8844 (2006).
Shin, H. et al. Prognostic impact of serum soluble PD-1 and ADV score for living donor liver transplantation in patients with previously untreated hepatocellular carcinoma. Ann. Surg. Treat. Res. 102, https://doi.org/10.4174/astr.2022.102.1.46 (2022).
Ying, P. et al. A comprehensive profiling of soluble immune checkpoints from the sera of patients with non-small cell lung cancer. J. Clin. Lab. Anal. 36, https://doi.org/10.1002/jcla.24224 (2022).
Anna, P. et al. Programmed death-1 receptor (PD-1) as a potential prognosis biomarker for ovarian cancer patients. Cancer Manag. Res. 12, https://doi.org/10.2147/cmar.S263010 (2020).
Byeong-Gon, N. et al. Absence of association between pretransplant serum soluble programmed death protein-1 level and prognosis following living donor liver transplantation in patients with hepatocellular carcinoma. Medicine (Baltimore) 100, https://doi.org/10.1097/md.0000000000025640 (2021).
Klara, D. et al. Serum biomarker panel diagnostics in pancreatic ductal adenocarcinoma: the clinical utility of soluble interleukins, IFN-γ, TNF-α and PD-1/PD-L1 in comparison to established serum tumor markers. J. Cancer Res. Clin. Oncol. 149, https://doi.org/10.1007/s00432-022-04112-z (2022).
Manuela, T. M. et al. Predictive value of soluble PD-1, PD-L1, VEGFA, CD40 ligand and CD44 for nivolumab therapy in advanced non-small cell lung cancer: a case-control study. Cancers (Basel) 12, https://doi.org/10.3390/cancers12020473 (2020).
Steffen Filskov, S., Christina, D., Britta, W., Boe Sandahl, S. & Peter, M. J. L. C. Increase in soluble PD-1 is associated with prolonged survival in patients with advanced EGFR-mutated non-small cell lung cancer treated with erlotinib. Lung Cancer 100, https://doi.org/10.1016/j.lungcan.2016.08.001 (2016).
Jesper Geert, P. et al. Increased soluble PD-1 predicts response to nivolumab plus ipilimumab in melanoma. Cancers (Basel) 14, https://doi.org/10.3390/cancers14143342 (2022).
Ryotaro, O. et al. Increased plasma soluble PD-1 concentration correlates with disease progression in patients with cancer treated with anti-PD-1 antibodies. Biomedicines 9, https://doi.org/10.3390/biomedicines9121929 (2021).
Heli, V. et al. Soluble PD-1 but not PD-L1 levels predict poor outcome in patients with high-risk diffuse large B-cell lymphoma. Cancers (Basel) 13, https://doi.org/10.3390/cancers13030398 (2021).
Christian, N., Line, O.-L., Torben, B., Steffen, H. & Søren T, L. J. C. I. Alternative splice variants of the human PD-1 gene. Cell. Immunol. 235, https://doi.org/10.1016/j.cellimm.2005.07.007 (2005).
Eva, Z. et al. Soluble plasma programmed death 1 (PD-1) and Tim-3 in primary HIV infection. AIDS 33, https://doi.org/10.1097/qad.0000000000002165 (2019).
Ila, D. et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin. Cancer Res. 25, https://doi.org/10.1158/1078-0432.Ccr-18-4142 (2019).
Weiping, Z., Jedd D, W. & Lieping, C. J. S. T. M. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 8, https://doi.org/10.1126/scitranslmed.aad7118 (2016).
Lloyd, B. et al. B-cell-specific checkpoint molecules that regulate anti-tumour immunity. Nature 619, https://doi.org/10.1038/s41586-023-06231-0 (2023).
Linda, Q. et al. The immune checkpoint PD-1 in natural killer cells: expression, function and targeting in tumour immunotherapy. Cancers (Basel) 12, https://doi.org/10.3390/cancers12113285 (2020).
Sean J. J. et al. Minimal PD-1 expression in mouse and human NK cells under diverse conditions. 130, https://doi.org/10.1172/jci133353 (2020).
Alena, G. et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. Immunology 124, https://doi.org/10.1172/jci73639 (2014).
Kenneth J, O., Hyesuk, Y., Rafi, A. & Jeremy M., B. J. J. I. NFATc1 regulates PD-1 expression upon T cell activation. J. Immunol. 181, https://doi.org/10.4049/jimmunol.181.7.4832 (2008).
Jiaxiong, T. et al. Increased PD-1+Tim-3+ exhausted T cells in bone marrow may influence the clinical outcome of patients with AML. Biomark. Res. 8, https://doi.org/10.1186/s40364-020-0185-8 (2020).
Xiangbo, M. et al. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature 564, https://doi.org/10.1038/s41586-018-0756-0 (2018).
Xiaoli, H. et al. Emerging role of ubiquitination in the regulation of PD-1/PD-L1 in cancer immunotherapy. Mol. Ther. 29, https://doi.org/10.1016/j.ymthe.2020.12.032 (2021).
Ignacio, M. et al. CD137 (4-1BB)-based cancer immunotherapy on its 25th anniversary. Cancer Discov. 13, https://doi.org/10.1158/2159-8290.Cd-22-1029 (2022).
Daqian, G., Xiang, A., Yu, Y., Zhuo, C. & Xiang, X. J. J. I. C. Soluble immune checkpoints in cancer: production, function and biological significance. J. Immunother. Cancer 6, https://doi.org/10.1186/s40425-018-0449-0 (2018).
Wei, L. et al. Soluble immune checkpoints are dysregulated in COVID-19 and heavy alcohol users with HIV infection. Front. Immnol. 13, https://doi.org/10.3389/fimmu.2022.833310 (2022).
Nianyu, L. et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 26, https://doi.org/10.1038/sj.emboj.7601520 (2007).
Guangbo, Z. et al. Soluble CD276 (B7-H3) is released from monocytes, dendritic cells and activated T cells and is detectable in normal human serum. 123, https://doi.org/10.1111/j.1365-2567.2007.02723.x (2008).
Chaojia, C. et al. Soluble Tim-3 serves as a tumor prognostic marker and therapeutic target for CD8(+) T cell exhaustion and anti-PD-1 resistance. Cell Rep. Med. 5, https://doi.org/10.1016/j.xcrm.2024.101686 (2024).
Christian, B., Xavier, T., Laurence, G. & Jean-François, G. J. B. P. Soluble TIM-3 as a biomarker of progression and therapeutic response in cancers and other of human diseases. Biochem. Pharmacol. 209, https://doi.org/10.1016/j.bcp.2023.115445 (2023).
Jeannin, P. et al. Soluble CD86 is a costimulatory molecule for human T lymphocytes. Immunity 13, https://doi.org/10.1016/s1074-7613(00)00030-3 (2000).
Frank J. W. et al. The soluble isoform of CTLA-4 as a regulator of T-cell responses. Eur. J. Immunol. 43, https://doi.org/10.1002/eji.201242529 (2013).
Oaks M. K. & Hallett K. M. Cutting edge: a soluble form of CTLA-4 in patients with autoimmune thyroid disease. J. Immunol. 164, https://doi.org/10.4049/jimmunol.164.10.5015 (2000).
Kathleen, M. M. et al. A secreted PD-L1 splice variant that covalently dimerizes and mediates immunosuppression. Cancer Immunol. Immunother. 68, https://doi.org/10.1007/s00262-018-2282-1 (2018).
Samali, A., Zhivotovsky, B. & Orrenius, S. Current concepts in cell toxicity. Curr. Protoc. Toxicol. Chapter 2, Unit 2 1, https://doi.org/10.1002/0471140856.tx0201s00 (2001).
Samuel, C. & Valerie, C. J. J. I. C. High-dimensional immune-profiling in cancer: implications for immunotherapy. J. Immunother. Cancer 8, https://doi.org/10.1136/jitc-2019-000363 (2020).
Jason, L. & Olivera J. F. J. P. T. Current modalities in cancer immunotherapy: immunomodulatory antibodies, CARs and vaccines. Pharmacol. Ther. 178, https://doi.org/10.1016/j.pharmthera.2017.03.008 (2017).
Matthew D, V., Tianxiang, Z. & Lieping, C. J. A. R. I. Resistance mechanisms to anti-PD cancer immunotherapy. Annu. Rev. Immunol. 40, https://doi.org/10.1146/annurev-immunol-070621-030155 (2022).
Jayne F, T., Lesley A, S., Davina, G., Sarah, B. & Matthew R. S. J. T. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials 8, https://doi.org/10.1186/1745-6215-8-16 (2007).
Sara, B., Gerta, R. & Guido, S. J. E. B. M. H. How to perform a meta-analysis with R: a practical tutorial. Evid. Based Mental Health 22, https://doi.org/10.1136/ebmental-2019-300117 (2019).
Acknowledgements
We thank Lieping Chen for providing the anti-4-1BB (clone 2A) and anti-B7-H1 (clone 10B4) antibodies. This work was supported by the Fujian Province Joint Funds for the Innovation of Science and Technology grant (2020Y9050), the Fujian Provincial Health Technology project (2024ZD01004), the Nature Science Foundation of the Fujian Province (2023J01105), the National Nature Science Foundation of China grant (81702828), and the Fujian Provincial Health Commission grant (2020CXA017).
Author information
Authors and Affiliations
Contributions
Ping Hou: Project administration, writing—original draft, and conceptualization; Li Hu: Writing— original draft, investigation, methodology, data curation; Junrong Zhang: Software, formal analysis; Xiaoyan Zhou: Methodology, data curation; Yonglei Xiao: Software, methodology; Lijun Li: Methodology, project administration; Qiongwen Wu: Methodology; Jing Liu: Investigation, data curation; Yuhong Lin: Writing—review and editing, validation, funding acquisition; Ling Chen: Writing—review and editing, supervision, project administration, funding acquisition, and conceptualization.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Pengfei Xu, Avijit Goswami and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Kaliya Georgieva. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hou, P., Hu, L., Zhang, J. et al. Characterization of alternative sPD-1 isoforms reveals that ECD sPD-1 signature predicts an efficient antitumor response. Commun Biol 8, 406 (2025). https://doi.org/10.1038/s42003-025-07800-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07800-x
