
Abstract
Our study elucidates the role of FOXP1 in chemoresistance in small cell lung cancer(SCLC). FOXP1 enhances chemoresistance by regulating SP8 expression through its super-enhancer (SP8-SE), with SP8 mediating resistance via the homologous recombination repair (HRR) pathway. We also discovered that FOXP1 forms punctate nuclear structures indicative of liquid-liquid phase separation, crucial for its transcriptional regulation. Targeting the FOXP1-SP8-HR axis with BRD4 and PARP inhibitors showed synergistic effects in reducing tumor growth in vitro and in patient-derived xenograft models. These findings identify FOXP1 as a critical mediator and marker of chemoresistance in SCLC, providing a foundation for developing targeted therapies to overcome this resistance.
Similar content being viewed by others
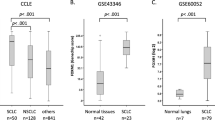
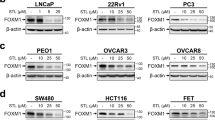

Introduction
Small cell lung cancer1 is a highly aggressive and fast-growing form of lung cancer characterized by the rapid proliferation of small cells with a neuroendocrine phenotype2. The standard treatment approach includes chemotherapy, often combined with radiation therapy, as surgery is generally not feasible in advanced cases2,3,4. Despite initial responses to treatment, SCLC frequently develops resistance, leading to relapse and limited long-term survival rates5,6. Novel targeted therapies and immunotherapies are being actively investigated to improve outcomes for SCLC patients7,8. However, chemoresistance is still an unsolved problem in clinical practice6.
In cancer, transcription factors play significant roles in the dysregulation of gene expression and the development and progression of the disease9. More critically, transcription factors also play an important role in chemoresistance in SCLC. The current mainstream view divides SCLC into different groups classified by differences in the expression of transcription factors ASCL1 and NEUROD1. Among them, different groups have different sensitivity to drugs10,11. As striking as this classification is another family of genes that are also transcription factors: the MYC homolog classification12,13. The Myc family presents interesting mutually exclusive expressions in SCLC, and different expressions present different therapeutic vulnerabilities14,15.
The impact of phase separation extends to the modulation of transcriptional activity, exerting control over DNA accessibility and promoting interactions between regulatory elements and the transcriptional apparatus16,17,18. Notably, phase separation has also been implicated in drug resistance in tumors, as it can disrupt drug sensitivity by promoting transcriptional irregularities in tumorous cells19,20. In addition, phase separation can also directly affect the distribution of drugs in tumors21,22,23. However, the role of phase separation in SCLC has not been reported, nor has the role of phase separation in the transcription factors associated with chemotherapy resistance in SCLC.
In the present investigation, our study reveals that FOXP1 facilitates phase separation by interacting with the super-enhancer SP8-SE, forming a transcription complex that fosters the expression of target genes. Concurrently, our findings demonstrate that the target gene SP8 mediates chemoresistance in SCLC by facilitating homologous recombination repair. This elucidation of the molecular mechanism enhances our understanding of the underlying principles governing the cooperative effects of epigenetic and PARP inhibition in combating drug resistance in SCLC by patient-derived xenograft. Furthermore, it offers a new therapeutic strategy with potential clinical utility.
Results
FOXP1 overexpression in small cell lung cancer is associated with chemotherapy resistance
In our previous study, using H3K27Ac pull-down chromatin immunoprecipitation sequencing and MEME-ChIP analysis, we identified three key master transcription factors-SP1, FOXP1, and IRF1-in the chemoresistant small cell lung cancer (SCLC) cell line H69AR24. We analyzed the expression of these factors in samples from relapsed and treatment-naïve patients using published data. FOXP1 expression was significantly elevated in relapsed samples13, while SP1 and IRF1 showed no significant differences (Fig. 1A). This suggests a potential link between FOXP1 and chemotherapy resistance in SCLC.

A Relative mRNA expression of three transcription factors identified as master transcription factors in chemoresistance cell line H69AR from patient-derived xenografts of naive (n = 9) and relapse (n = 10) SCLC patients. B Relative mRNA expression of FOXP1 in four pairs of chemotherapy-sensitive to induced chemoresistance patient-derived xenografts of SCLC patients (n = 5). C In the GDSC data, the expression level of queried the RNA-seq of FOXP1 in 63 SCLC cell lines was positively correlated with the level of cisplatin resistance in the public dataset GSE151904. D Endogenous expression of FOXP1 in eight different SCLC cell lines. E Immunohistochemistry was used to detect the expression of FOXP1 in normal lung tissue, adjacent tissues (PLCT) and small cell lung cancer tissue of the same patient in clinicopathological samples. Scale bar, 100 μm. F Kaplan–Meier analysis of the overall survival in the 62 paraffin embedding SCLC tissues based on FOXP1. Hazard ratio = 1.938 (Student t-test). G CCK8 was used to detect the change of IC50 for cisplatin and etoposide after knocking down FOXP1 expression in H69AR and H446DDP, respectively. H Flow cytometry was used to detect the effect on apoptosis exposed to cisplatin and etoposide after knocking down FOXP1 in H69AR and H446DDP. I The difference of apoptosis between each group was analyzed (n = 3). J Cell cycle was detected after interference with FOXP1 expression exposed to cisplatin(left) and etoposide(right). K A statistical of the cell cycle in in H69AR and H446DDP. n = 3.
We further validated this using a patient-derived xenograft model of induced chemoresistance in SCLC. FOXP1 expression was significantly higher in the induced resistant group compared to the sensitive group (Fig. 1B). Analysis of RNA-seq data from 63 SCLC cell lines (dataset GSE151904) and cisplatin IC50 values from the GDSC public drug database showed a significant positive correlation between FOXP1 expression and cisplatin IC50 (Fig. 1C)25. Western blot analysis confirmed elevated FOXP1 in resistant lines H69AR, H446DDP, and the relatively insensitive line H1339 (Fig. 1D).
To explore the clinical significance of FOXP1 in SCLC, we conducted qRT-PCR and immunohistochemical analyses on tumor samples from 62 SCLC patients. High FOXP1 protein expression, mainly in the nuclei of SCLC cells, was observed (Fig. 1E). Kaplan–Meier survival analysis indicated that high FOXP1 expression was associated with poorer survival (Fig. 1F).
To determine FOXP1’s role in chemotherapy resistance, we assessed chemosensitivity changes following FOXP1 knockdown in H69AR and H446DDP cells(Fig. S1A, B). Knockdown significantly decreased IC50 values for chemotherapy drugs (Fig. 1G) and increased apoptotic cell populations upon chemotherapy exposure(Fig. 1H, I). FOXP1 downregulation led to significant G2 phase cell accumulation in cisplatin-treated cells and accelerated S to G2 phase transition in etoposide-16 treated cells, resulting in G2 phase arrest and reduced S phase cell numbers (Fig. 1J, K). we also done the overexpression of FOXP1 in parent sensitive cell lines H69 and H446(Fig. S1C, D), the opposite change happens(Fig. S1E–G).
These results highlight FOXP1’s critical role in chemotherapy resistance in SCLC, suggesting it as both a marker and mediator of chemoresistance, with potential for therapeutic targeting.
FOXP1 promotes chemoresistance in SCLC by regulating SP8 expression
Results from MEME analysis suggest that FOXP1 plays a critical role in H69AR by regulating associated super-enhancers. To explore FOXP1’s involvement in chemoresistance in SCLC, we performed and analyzed ATAC-seq and identified FOXP1 binding sites in the super-enhancer region and promoter of the SP8 gene (Figs. 2A and S2A), naming this super-enhancer SP8-SE. ChIP-qPCR validated these binding sites (Fig. 2B). FOXP1 knockdown and overexpression experiments confirmed that FOXP1 positively regulates SP8 at both RNA and protein levels (Fig. 2C, D), while also regulates general H3K27Ac expression. This indicates that FOXP1 exerts an important regulation of the whole transcription in SCLC while regulating the specific gene SP8.

A The location of SP8-SE, SP8, and component enhancers. B ChIP-qPCR assays were performed to detect the binding between FOXP1 and SP8 promoter and enhancer. C qPCR was detected mRNA of SP8 after knocking down FOXP1 expression in H69AR and H446DDP, respectively. (n = 3). D Western blot was detected SP8 and H3K27Ac after knocking down FOXP1 expression in H69AR and H446DDP, respectively. E Volcanic map of DEGs between H69AR and H69 cells. DEGs: differentially expressed genes. F H69AR and H446DDP were exposed to cisplatin and etoposide with their IC50 values, respectively, after interference of SP8 expression (n = 3). G CCK8 was used to detect the IC50 value of cisplatin and etoposide after FOXP1 was knocked down the while SP8 was overexpressed (n = 3). H Relative Luciferase activity of different components of SP8-SE (n = 3). I Western blot was detected the protein expression between H69AR WT and SP8-SE knockout. J CCK8 was detected between H69AR WT and SP8-SE knockout (n = 3).
Combined H3K27Ac and RNA-seq analyses showed significantly higher SP8 expression in chemoresistant cell lines compared to sensitive ones (Fig. 2E), suggesting that FOXP1 promotes chemoresistance by regulating SP8-SE. Interfering with SP8 expression(Fig. S2B, C) increased chemosensitivity (Fig. 2F). Overexpression of SP8 partially restored resistance in FOXP1-knockdown cells (Fig. 2G). These results proved that SP8 is the downstream molecule of FOXP1 and exerts the chemoresistance function.
The SP8-SE was divided into four active components based on H3K27Ac peak values. Dual-luciferase reporter assays revealed that the E3 component exhibited the highest activity (Fig. 2H). CRISPR-Cas9 knockout of the E3 component in resistant cell line significantly reduced SP8 expression and the IC50 value for chemotherapy drugs, confirming the regulatory role of this component (Fig. 2I, J).
These findings indicate that FOXP1 promotes chemoresistance by regulating SP8 expression through SP8-SE. these transcription elements are vital roles in chemoresistance SCLC.
SP8 mediates chemotherapy resistance in SCLC through homologous recombination repair pathway
To investigate the mechanism through which SP8 contributes to chemotherapy resistance in SCLC, we performed RNA transcriptome sequencing following SP8 knockdown in the H69AR cell line. Transcriptome analysis revealed the involvement of the PALB2 gene and the homologous recombination repair (HRR) pathway (Fig. 3A–C). PALB2 (Partner and Localizer of BRCA2) operates in the HRR pathway by interacting with BRCA1 and BRCA2, facilitating SCLC cell survival after chemotherapy-induced DNA damage.

A–C RNA expression difference analysis was performed after interfering SP8 expression in H69AR cells, n = 3. Volcanoplot and KEGG analysis and biological process analysis were performed on the results of RNA-sequencing analysis. D After interfering with SP8 expression in H69AR, H446DDP, respectively, western blot was used to detect the protein expression level of PALB2. E H69AR and H446DDP were exposed to cisplatin and etoposide with their IC50 values, respectively, after interference of SP8 expression, and the protein levels of PALB2 and RAD51 were measured by Western blot. F H69AR and H446DDP were exposed to cisplatin with their IC50 values, and the effects of interference with SP8 expression on the degree of trailing were observed by alkaline comet assay. Scale bar, 10 μm. G A statistical of the percentage of tailing DNA and γH2AX intracellular density. H H69AR was exposed to cisplatin and etoposide with IC50 value, and the change of γH2AX intracellular density after interference of SP8 expression was observed by immunofluorescence. Scale bar, 10 μm. I H69AR was exposed to cisplatin with their IC50 values, and the homologous recombination repair was observed by transferring into I-SCEl and DR-report plasmids. J H69AR was exposed to Talazoparib of their IC50 value, and the homologous recombination repair was observed by transferring into I-SCEl and DR-report plasmids.
To confirm the association between SP8 and the HRR pathway, we knocked down SP8 in two resistant cell lines. Western blot results showed a significant decrease in PALB2 and HRR-related protein RAD51 levels in the knockdown group, with or without chemotherapy drug exposure (Fig. 3D, E). This indicates that SP8 positively regulates PALB2 and HRR protein expression.
The comet assay revealed increased nuclear damage in cells with SP8 knockdown upon chemotherapy drug exposure, as shown by more pronounced comet tails compared to the control group (Fig. 3F, G). Immunofluorescence staining of γH2AX further confirmed exacerbated DNA damage with SP8 knockdown, suggesting SP8’s role in DNA damage repair (Figs. 3H and S3B, C).
To specifically determine SP8’s role in the HRR pathway of DNA damage repair, we used the DR-GFP homologous recombination reporter plasmid for flow cytometry analysis. SP8 downregulation significantly reduced the population of cells reporting homologous recombination (Figs. 3I and S3D). Additionally, using the PARP inhibitor Talazoparib to selectively inhibit DNA damage repair, we found that SP8 overexpression partially restored HR efficiency (Figs. 3J and S3E).
Furthermore, Co-immunoprecipitation (Co-IP) experiments revealed direct interactions between SP8, PALB2, and RAD51 (Fig. S3G), meanwhile, PALB2 can slowdown the degradation of SP8 (Fig. S3H), providing strong evidence that SP8 is directly involved in the HRR pathway by forming a complex with these key HRR proteins.
Interestingly, we also observed that SP8 degrades more rapidly when cisplatin is added to cell cultures(Fig. S3F). This suggests that cisplatin treatment may destabilize SP8, potentially disrupting its role in the HRR pathway. This degradation might be a cellular response to limit the repair of chemotherapy-induced DNA damage, thereby enhancing the drug’s cytotoxicity. However, in chemoresistant cells, the remaining SP8 might still be sufficient to support HRR, thus contributing to resistance.
These results conclusively demonstrate that SP8 mediates chemotherapy resistance in SCLC through its involvement in the homologous recombination repair pathway.
FOXP1 exists in a phase-separated form in chemoresistant SCLC
Building on previous findings, we aimed to uncover the structural basis of how the transcription factor FOXP1 contributes to chemotherapy resistance in SCLC. Transcription factors are known to cluster around super-enhancers and activate transcription through interactions with nucleic acids, exhibiting properties of liquid-liquid phase separation (LLPS).
Our observations revealed a distinct punctate distribution pattern of FOXP1 within the nuclei of resistant cell lines and patient-derived xenograft (PDX) samples, in contrast to the diffuse distribution seen in sensitive samples (Fig. 4A, B). This distinct pattern indicates that FOXP1 undergoes phase separation in resistant samples. To explore this further, we used CRISPR/Cas9 to insert a GFP tag into the FOXP1 gene and performed immunofluorescence microscopy. FOXP1 showed a punctate nuclear distribution in live cells (Fig. 4C). Adding 5% 1,6-hexanediol for 120 seconds significantly disrupted these foci (Fig. 4D), suggesting they are maintained by weak hydrophobic interactions. Fluorescence recovery after photobleaching (FRAP) confirmed the liquid nature of the EGFP-FOXP1 foci (Fig. 4E, F). Three-dimensional reconstruction demonstrated that FOXP1 exists as spherical droplets within the nucleus (Figs. 4G, and S4A).

A Endogenous expression of FOXP1 in two pairs of chemotherapy-sensitive or chemoresistance cell lines subjected to immunofluorescence (IF) analyses. Scale bar, 10 μm. B Endogenous expression of FOXP1 in PDX2-chemotherapy-sensitive or resistant samples subjected to immunofluorescence (IF) analyses. Scale bar, 100 μm. C Knock in EGFP-tag of FOXP1 into H69AR and H446DDP cells, and subjected to IF analyses. Representative images are shown. Scale bar, 10 μm. D 5% 1,6-hexanediol (Hex) disrupted EGFP-FOXP1 foci formation. Scale bars, 10 μm. E Representative images of the FRAP analysis of EGFP-FOXP1 are shown. The white boxes highlight the foci undergoing targeted bleaching. The indicated region was chosen randomly under live-cell observation and photobleached to 75% of the original intensity. Scale bar, 10 μm. F Quantitation of FRAP results of EGFP-FOXP1 puncta (n = 3). The FRAP recovery curve was generated by averaging the signals of 3 droplets per group with similar sizes after photobleaching. The relative fluorescence intensity of each droplet prior to photobleaching was set to 100%. Time 0 refers to the time point right at the start of photobleaching. Data are presented as mean ± SD (n = 3). G 3D Z-stack scanning of EGFP-FOXP1 foci was constructed. Rendered 3D shapes of a punctum with panels showing the Y axes. Scale bar, 10 μm. H Representative picture of droplet fusion observed under confocal glass, Scale bar, 2.5 μm. I Representative images of FRAP of the droplets in vitro, Scale bar, 10 μm, J Quantitation of FRAP results of EGFP-FOXP1 puncta outside the cells (n = 3). The FRAP recovery curve was generated by averaging the signals of 3 droplets per group with similar sizes after photobleaching. The relative fluorescence intensity of each droplet prior to photobleaching was set to 100%. Time 0 refers to the time point right at the start of photobleaching. Data are presented as mean ± SD (n = 3).
Disrupting cell structure showed that EGFP-FOXP1 molecules remained liquid-like in the medium, albeit with reduced fluidity (Fig. 4H, J). Direct microscopic observation revealed the flow and fusion of fluorescent proteins (Fig. 4J). Additionally, we expressed and purified His-tagged EGFP-FOXP1 recombinant protein in Escherichia coli (Fig. S1E). At a concentration of 20 mM, EGFP-FOXP1 formed droplet-like foci in a buffer containing 10% PEG-8000, with FRAP confirming rapid molecular exchange, verifying the liquid-like structure of FOXP1 in vitro (Fig. S4B).
These findings suggest that phase separation could be fundamental to FOXP1’s function in chemoresistant SCLC. The ability of FOXP1 to form phase-separated condensates may underpin its role in transcriptional regulation and contribute to the cellular mechanisms driving resistance.
The phase-separated structure of FOXP1 is intrinsic to its transcriptional function
To verify the connection between FOXP1’s phase-separated structure and its role in forming transcription factor-super enhancer complexes, we examined its colocalization with BRD4 protein and histone markers H3K27Ac and H3K4me1. In two resistant cell lines, endogenous FOXP1’s punctate structures significantly colocalized with endogenous BRD4, H3K27Ac, and H3K4me1 within the nucleus (Fig. 5A). Similar colocalization trends were observed in primary cells from chemoresistant PDX tissues (Fig. 5B). PLA assay also confirmed that a connection between FOXP1’s structure and its transcriptional role(Fig. S4D). These findings suggest a potential link between FOXP1’s phase-separated structure and its transcriptional function.

A, B Co-localization of H3K27Ac, H3K4me1, BRD4, and FOXP1 were assessed using immunofluorescence (IF) in H69AR cells and PDX2-resistant using antibodies against each one. Nuclei were stained with DAPI. Fields were observed using confocal microscopy. Scale bar, 10 μm. An enlargement of the boxed punctum is shown in the insert. C IDRs of FOXP1 were analyzed using PONDR. D PrD-like sequences were analyzed by http://plaac.wi.mit.edu/. E The diagram of FOXP1 mutants. F Living cell imaging of in different mutants in H69AR cells. Twenty-four hours after plasmid transfection. Scale bar, 10 μm. G The foci number of all cells observed in living cell imaging was counted. H Luciferase report assay was used to detect the effect of different fragments on SP8 promoter and enhancer activity in H69AR cells. I ChIP-qPCR assays were performed to detect the H3K27Ac and H3K4me1 binding on SP8 promoter and enhancer. J Curve of FRAP assay on different mutants’ condensate in H69AR cells, n = 3. K After the treatment of EGFP-FOXP1 with ARV-771 with IC50 value, the representative images of the fluorescence bleaching recovery experiment were carried out, and the fluorescence recovery efficiency was calculated to draw the curve of the fluorescence bleaching recovery experiment. n = 3. L Promoter and enhancer activity was tested by luciferase assays after adding ARV-771 with IC50 value.
Previous studies indicate that intrinsically disordered regions (IDRs) and prion-like domains (PRDs) are crucial for protein phase separation. Using prediction tools, we identified three IDRs (Fig. 5C) and two PRDs (Fig. 5D) in the FOXP1 amino acid sequence, all independent of the forkhead box domain. We constructed deletion mutants of these five sequences fused with EGFP and transfected them into SCLC cell lines (Fig. 5E). Compared to the wild-type FOXP1 vector, different deletion mutants showed varying degrees of reduction in the number of condensates (Fig. 5F, G), and the FRAP decreased, with the most significant reduction observed in the IDR1 and IDR2 regions (Fig. S4C, Fig. 5H, K). Deleting the PRD1/PRD2 regions alone did not affect the proportion of cells exhibiting LLPS. These results indicate that the deletion of IDR1 and IDR2 regions leads to reduced fluidity of the FOXP1 protein.
Further experiments showed that deletions in IDR1 and IDR2 resulted in reduced promoter luciferase activity and decreased enrichment of histones at binding sites (Fig. 5H). These results demonstrate that IDR1 and IDR2 are essential regions for FOXP1’s function as a transcription factor.
We also used BRD4 inhibitors to target this complex. BRD4 is a well-known transcriptional coactivator and epigenetic reader of acetylated chromatin. The inhibitors exhibited global and concentration-dependent inhibition of transcription levels, and the levels of transcription activation markers H3K27Ac and H3K4me1 decreased. Six hours post treatment with ARV-771, FRAP significantly decreased (Fig. 5K, L), indicating reduced protein fluidity and confirming the disruption of FOXP1 condensates. Dual-luciferase reporter assays further confirmed the reduction in FOXP1’s transcriptional capacity (Fig. 5L). These findings suggest that FOXP1 condensates possess transcriptional activity and that, in its phase-separated form, FOXP1 interacts with transcription-related proteins to perform its function as a transcription factor.
As described before, when we knocked out the E3 component of SP8-SE, the phase-separation degree of FOXP1 decreased obviously (Fig. S4F, G). it suggests that the interaction with specific enhancer regions is crucial for the formation and maintenance of FOXP1 condensates. This also indicates that FOXP1’s ability to phase-separate is at least partially dependent on its binding to certain enhancers, while further corroborating this by showing that without the enhancer binding, FOXP1’s phase-separation and, consequently, its transcriptional activity are compromised.
Overall, these findings demonstrate that the phase-separated structure of FOXP1 is intrinsic to its function as a transcription factor, with specific regions (IDR1 and IDR2) being critical for its transcriptional activity.
The phase-separated structure is also the basis for FOXP1’s role in chemoresistance
We conducted experiments overexpressing full-length EGFP-FOXP1 in the sensitive parent cell lines H69 and H446, selectively deleting various fragments of the FOXP1 protein. Compared to the EGFP-negative control group (EGFP-NC), the IC50 of the FOXP1-EGFP overexpression group significantly increased after exposure to chemotherapy drugs (Fig. 6A), while the apoptosis rate decreased (Fig. 6B–D). Notably, in the sensitive cell lines, deletion of IDR1 or IDR2 did not lead to a statistically significant increase in IC50 values or a decrease in apoptosis rate. This aligns with the previously confirmed transcription-related sequences, suggesting that FOXP1 acts as a transcription factor involved in SCLC resistance, regulated by phase separation.

A CCK8 was used to detect the change of IC50 after transferring full length and different mutant EGFP-FOXP1 in H446 and H69 cells, respectively, n = 3. B–D Changes in apoptosis after overexpression of full length and different mutant and EGFP-FOXP1 in H446 and H69 cells, respectively, n = 3. E Endogenous expression of FOXP1 subjected to IF analyses of H69AR when different concentrations of cisplatin/etoposide were added from top to bottom. Scale bar, 10 μm. F The immunofluorescence results of FOXP1 exposed to different concentrations of chemotherapy. G Western blot detected changes in FOXP1 protein levels in H69AR cells exposed to different concentrations of chemotherapy drugs. H Recombinant plasmid expression of FOXP1 subjected to IF analyses of H69AR when different concentrations of cisplatin were added from top to bottom. Scale bar, 10 μm. I The immunofluorescence results of EGFP-FOXP1 exposed to different concentrations of cisplatin were quantitatively analyzed. n = 5. J The foci size of EGFP-FOXP1 condensate was quantitatively analyzed. n = 20. K Changes in mRNA of SP8 after the stimulation of chemotherapy drugs in H69AR. L After the treatment of EGFP-FOXP1 with cisplatin and etoposide with IC50 value, the representative images of the fluorescence bleaching recovery experiment were carried out, and the fluorescence recovery efficiency was calculated to draw the curve of the fluorescence bleaching recovery experiment. n = 3 Scale bar, 10 μm.
Previous studies have provided insights into the non-random and selective DNA platination induced by small molecule drugs like cisplatin within tumors. This phenomenon is closely related to the condensates formed by super enhancers and transcription factors, significantly impacting the degree of phase separation. We exposed cells to varying concentrations of chemotherapy drugs, particularly within a defined range. With increasing drug concentration, FOXP1’s punctate distribution within the nucleus gradually intensified (Figs. 6E and S5B). This was accompanied by increased fluorescence intensity and enlarged discrete spots (Fig. 6F). Importantly, this distribution change was not accompanied by an upregulation in FOXP1 protein expression levels (Fig. 6G).
Next, we placed live cells transfected with EGFP-FOXP1 under the same concentration gradient of chemotherapy drugs. Subsequently, we observed a marked enhancement in the liquid-like structures and increased fluorescence intensity (Fig. 6H, I). Correspondingly, FRAP experiments showed increased fluidity of EGFP-FOXP1 under chemotherapy drug stimulation (Figs. 6L and S5C). It indicates that FOXP1’s phase-separated structure is dynamically responsive to chemotherapeutic stress. This heightened phase separation suggests that FOXP1 may be actively involved in the cellular response to cisplatin treatment. The increased phase separation of FOXP1 under cisplatin stress likely enhances its role in transcriptional regulation, contributing to the cell’s adaptive mechanisms for chemoresistance. The use of BRD4-targeting inhibitors reduces the binding of FOXP1 to transcription-related proteins, as demonstrated by the PLA experiment (Fig. S5D, E). Consistent with earlier results, under such drug stimulation, SP8 RNA levels also increased, indicating enhanced transcriptional activity of FOXP1 (Fig. 6H, K). In summary, these findings underscore the critical role of FOXP1 phase separation in enhancing transcriptional regulation and promoting chemoresistance in SCLC, providing potential targets for therapeutic intervention.
Targeting BRD4 and PARP in chemotherapy-resistant SCLC: a synergistic approach
Targeting BRD4 has proven to be an effective strategy against SCLC26. Our findings indicate that BRD4 inhibitors can suppress the transcription of target genes by disrupting phase segregation. Among the two chemotherapy-resistant cell lines, H69AR and H446DDP, ARV-771 exhibited the highest inhibitory efficiency on SCLC growth in vitro, as determined by IC50 assays, aligning with BRD4’s inhibitory potency (Figs. S4E, S5D).
In SCLC, the homologous recombination (HR) pathway is frequently mutated. Our previous results suggest that FOXP1 regulates SP8 transcription through phase separation, and that the protein encoded by SP8 is directly involved in homologous recombination repair. We wanted to look at the role of SP8 in the antitumor effects of BRD4 inhibitors and PARP inhibitors, respectively. We transplanted drug-resistant cells of control group and cells of SP8 knockdown group subcutaneously in nude mice, respectively, and treated ARV-771 and Talazoparib at the same time point to observe the tumor response of the two groups to the drug27,28,29,30. We found that tumors that were knocking down SP8 were more responsive to both drugs. And the effect was better than EP chemotherapy group (Fig. 7A–D). IC50 assays revealed a synergistic effect between ARV-771 and Talazoparib in vitro, further supporting the notion that SP8 serves as a pivotal target in the treatment strategy (Fig. 7E, G).

A, B Subcutaneous tumor experiments were performed with or without interference with SP8 expression in H69AR (n = 5). Divide mice into control group(PBS), chemotherapy group(Etoposide 10 mg/kg + Cisplatin 3 mg/kg,3 times a week, intraperitoneal injection), single-drug ARV-771 group(10 mg/kg, intraperitoneal injection), and single-drug Talazoparib group (0.5 mg/kg, everyday, Oral Administration). C Tumor size was measured and subcutaneous tumor growth curve was drawn. D Tumor weight was measured. E After H69AR was exposed to the gradient combination of Talazoparib and ARV-771, cell activity was measured by CCK8 to calculate cell survival rate, and heat map was drawn. F H69AR and H446DDP were exposed to the control group, ARV771 group with IC50 concentration, Talazoparib group, and the two-drug combination group, respectively, to detect the changes of the PALB2, SP8, PARP, and H2X protein levels. G The synergistic index of the two drugs calculated by Synergyfinder+. H PDX2-R was used to divide patients into control group, single-drug ARV-771 group(10 mg/kg, intraperitoneal injection), single-drug Talazoparib group(0.5 mg/kg, everyday, Oral Administration) and double-drug combination group. I Tumor size was measured and subcutaneous tumor growth curve was drawn, n = 4. J Tumor weight was measured. K The quantified result of positive SP8 cells percentage in PDX subcutaneous tumors detected by immunohistochemistry. L Schematic representation of the hypothetical structure of FOXP1 droplet and the combination therapy. Created in BioRender. Tang, Y. (2025) https://BioRender.com/x83d522.
The results of Wb suggest that ARV-771 itself has the function of damaging DNA while inhibiting PARP process. When combined with PARP inhibitors, the DNA damage was more severe, providing further evidence for the effect of combination therapy (Fig. 7F). To validate these findings, we utilized a patient-derived xenograft (PDX) model of SCLC for drug testing. Results showed that the combination treatment group exhibited the strongest tumor inhibition effect (Fig. 7H–J). Immunohistochemical analysis of subcutaneous tumors reconfirmed that FOXP1 protein levels remained unaffected by BRD4 inhibitors, whereas SP8 expression was inhibited, consistent with our previous results (Fig. 7K).
These findings suggest that the combination of BRD4 inhibitors and PARP inhibitors offers a promising therapeutic strategy for overcoming chemoresistance in SCLC by targeting the FOXP1-SP8-HR pathway (Fig. 7J).
Discussion
SCLC is a tumor of epigenetic disorder, and the role of various epigenetic molecules has been gradually discovered31,32,33. This study explored the epigenetic effects of transcription factors and super-enhancers on drug resistance in SCLC. Master transcription factors activate the expression of specific genes by binding to super-enhancers, which perform specific functions in specific cells. Among them, transcription factors and super-enhancers in cancer have been shown to bind to each other through a phase separation mechanism and promote tumor development22,34.
In this study, we found that in chemoresistant cells, the degree of FOXP1 phase separation to form condensate increased with the stimulation of chemotherapy drugs. FOXP1 was reporter to influence the chemotherapy sensitivity35. Immunocoprecipitation, fluorescence co-localization and PLA experiments confirmed that FOXP1 condensate and transcription-related proteins form complex, and the destruction of transcription protein BRD4 can also disrupt the fluidity of FOXP1 condensate, and the deletion of IDR fragments corresponding to FOXP1 can disrupt the formation of condensate and affect the transcriptional function of FOXP1. Subsequent studies confirmed that FOXP1 plays a transcriptional role on DNA fragments by binding in the form of homologous dimers. The main functional segment is the leucine zipper fragment, which almost coincides with the predicted IDR2 fragment36. The deletion of IDR1 fragment has the greatest effect on the condensate. Most of this fragment is an unidentified protein disorder. Notably, the polyQ sequence has been shown to be involved in protein aggregation in the FOX protein family37. This sequence is located 70–107 in FOXP138, independent of PRD-1/2 in IDR1. This, combined with our experimental results, suggests that polyQ sequence really plays the role of LLPS in IDR1 sequence. Consistent with this, deletion of IDR1 had the greatest effect on FOXP1 transcription and drug resistance.
We found that the target gene SP8 was involved in the homologous recombination repair pathway by transcriptome sequencing. At the same time, we demonstrated that PALB2 and SP8 can influence protein stability with each other. We hypothesize that SP8 protein is recruited to participate in the homologous recombination repair pathway during DNA damage, which needs further investigation.
Based on phase isolation results and target genes’ involvement in homologous recombination repair pathways, we propose a combination therapy that combines BETi and PARP inhibitors to treat chemotherapy-resistant SCLC. Previous studies have used this method in SCLC, but there have been no reports of chemoresistance in SCLC12,39,40. And in this study, the combination therapy is based on a new mechanism. It provides a new treatment opportunity for SCLC resistant to chemotherapy. However, this study does not rule out whether other mechanisms are involved in the effectiveness of this joint scheme. Further research is needed6,41,42.
Overall, this study is the first to validate the presence of phase isolation of the transcription factor FOXP1 in SCLC and demonstrate that it is associated with chemotherapy resistance and mediates a downstream homologous recombination repair pathway. The transcription factor SP8 was first found to be involved in the homologous recombination repair pathway of SCLC resistance. On this basis, we propose the possibility of PARP+BETi combination therapy, which provides a new strategy for the treatment of SCLC after chemotherapy resistance.
Materials and methods
Cell culture
HEK293T cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, USA). Human small-cell lung cancer (SCLC) cell lines, including H69, H69AR, and H446, were sourced from the American Type Culture Collection (ATCC). The drug-resistant subline H446DDP was generated by exposing H446 cells to gradually increasing concentrations of cisplatin (DDP) up to 0.5 µg/mL over a 12-month period and subsequently maintained in a complete medium supplemented with DDP. Cell line authentication was confirmed through short tandem repeat (STR) profiling, and polymerase chain reaction (PCR) screening verified the absence of mycoplasma contamination.
Immunofluorescence (IF) microscopy
Cells grown on confocal dishes were fixed in 4% paraformaldehyde for 20 min, followed by permeabilization with 0.5% Triton X-100 in PBS for 10 min. After blocking, confocal dishes were incubated overnight with diluted primary antibodies at 4 °C. Following several washes with 0.1% Triton X-100/PBS buffer, confocal dishes were incubated with Alexa Fluor 488-conjugated anti-rabbit or Alexa Fluor 594-conjugated anti-mouse secondary antibodies for 1 h. DAPI was used to stain nuclei. Images were captured using a confocal microscope(Nikon, ECLIPSE Ti2-E) with a 63× oil objective lens. Quantitative analysis was conducted utilizing the ImageJ software.
Protein expression and purification
Rossetta’s component cells were transferred into a plasmid heat shock ice bath, a 30 μl coated LB plate (including 50 μg/ml Kan) was incubated at 37 °C for 14 h, and the monoclones were selected for activation culture. The bacteria were transferred to 5 ml LB liquid medium (containing 50 μg/ml Kan and 34 μg/ml Chl) and cultured to 0.6–0.8 OD600, and then IPTG with concentration of 1 mM was added to induce expression at 37 °C for 4 h, and then the bacteria were collected. SDS-PAGE analysis of bacterial systems was performed before and after induction. Fusion proteins are purified on Ni-Sepharose® columns according to the manufacturer’s instructions (Beyotime).
Western blot analysis
Adherent cells were cultured under standard conditions and harvested at 80% confluency for protein extraction. After washing with cold PBS to remove residual media, cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors. The lysates were incubated on ice for 30 min, then centrifuged at 12,000 × g for 15 min at 4 °C to remove cell debris. Protein concentration was quantified using the BCA assay. Equal amounts of protein were resolved by SDS-PAGE and transferred onto PVDF membranes. After blocking with 5% non-fat milk or BSA, membranes were incubated overnight at 4 °C with primary antibodies, followed by HRP-conjugated secondary antibodies. Protein detection was performed using enhanced chemiluminescence (ECL), and images were acquired using a gel documentation system.
Immunoprecipitation (IP)
Immunoprecipitation was carried out using the Thermo Scientific Pierce™ Cross-linked Magnetic IP/Co-IP Kit. Microbeads were prewashed with 1× coupling buffer, followed by incubation with protein A/G magnetic beads and the target antibody for 15 min. After three washes with coupling buffer, the antibodies were cross-linked to the microbeads using DSS for 30 min. The beads were then subjected to three elution buffer washes and two additional washes with washing buffer. Prepared microbeads and cell lysates were incubated overnight at 4 °C. The following day, microbeads were washed twice with washing buffer and once with purified water, and protein complexes were eluted using elution buffer.
Chromatin immunoprecipitation (ChIP) qPCR
Formaldehyde was added to the prepared cells to a final concentration of 1%, gently mixed, and incubated at room temperature for 10 min to allow chromatin cross-linking. The chromatin was then sheared by sonication on ice at 50% power, with six cycles of 20-s pulses followed by 20-second cooling intervals. After sonication, the diluted supernatant was incubated overnight at 4 °C with the appropriate antibody on a mixer, followed by the addition of 25 µL of ChIP-grade Protein A/G Magnetic Beads to each sample and further incubation at 4 °C under rotation. To reverse cross-linking, 6 µL of 5 M NaCl and 1 µL of 20 mg/mL Proteinase K were added to both Input and ChIP samples, and the reaction was incubated at 65 °C for 2 h. DNA was then purified using a DNA purification kit according to the manufacturer’s protocol, and the resulting DNA was analyzed by quantitative PCR (qPCR) with the appropriate reagents and primers following the provided instructions.
CRISPR-Cas9 for knockout and knock-in
In this study, guide RNAs for CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) were designed using the CRISPR direct design tool, which can be accessed at https://crispr.dbcls.jp/. The E3 component of SP8-SE was chosen to be knocked out. Knockout bands were detected by nucleic acid gel electrophoresis. We also knocked in green fluorescent protein (GFP)-coding sequences into the C-terminal region of the FOXP1 gene in H69AR cell line through CRISPR-Cas9-mediated homologous recombination. In order to inoculate single cells into 96-well culture plates, a flow cytometer was utilized. The clones were cultured for two weeks until visible colonies formed, after which they were digested with pancreatic enzymes and transferred to 24-well plates for continued growth. Once the cells reached approximately 80% confluency, a portion was collected for DNA extraction to enable subsequent identification and verification.
Luciferase assay
SCLC cells were plated in 24-well plates at 100,000 cells per well, co-transfected with 500 ng firefly luciferase reporter vector, 500 ng of different plasmids, and 50 ng of pRL-TK Renilla luciferase reporter vector. Cells were incubated for 48 h and the luciferase activities were assessed using the Dual-Luciferase Reporter Assay System (Promega) following the manufacturer’s protocol.
Fluorescence recovery after photobleaching (FRAP) assay
Fluorescence recovery after photobleaching (FRAP) experiments were performed using a Nikon ECLIPSE Ti2-E confocal microscope equipped with a 40× immersion objective. In vivo EGFP-FOXP1 bodies were photobleached until fluorescence intensity was reduced by 95% from the initial value. Time-lapse images were acquired over the designated recovery period, with the pre-bleaching fluorescence intensity set as the 100% reference point, and fluorescence intensity at each time point was subsequently normalized. Quantitative analysis was carried out using ImageJ software, while GraphPad Prism was used for data visualization and statistical analysis of FRAP results.
Duolink® proximity ligation assay22
Duolink®In Situ PLA® kit (DUO92101, Sigma-Aldrich) was used PLA determination. First, primary antibodies specific to the target proteins are applied, followed by secondary antibodies conjugated to oligonucleotide probes. The oligonucleotides hybridize and undergo ligation upon the addition of a ligase enzyme. The ligated probes then serve as templates for rolling circle amplification43, generating a localized DNA product. Finally, the amplified DNA is detected via fluorescence microscopy, enabling visualization of protein interactions at the single-molecule level. Fluorescence images were obtained using a 40× objective lens under a confocal scanning microscope (Nikon, ECLIPSE Ti2-E).
Comet assay
The Comet assay was performed using the Comet Assay Kit (3-well slides) (ab238544), with all solutions thoroughly chilled to 4 °C before use. Cells were digested with trypsin, resuspended in PBS, and adjusted to the appropriate cell concentration. A pre-warmed slide was first coated with 0.5% normal melting-point agarose and solidified at 4 °C, followed by the addition of 1.5% low melting-point agarose containing the cells. After solidification, slides were lysed in RIPA buffer for 2.5 h, rinsed in PBS, and placed in TBE buffer for alkaline unwinding (20 min), followed by electrophoresis at 4 °C (25 V, 30 min). The slides were then neutralized in Tris–HCl, stained with ethidium bromide under light protection, and immediately imaged under a fluorescence microscope to evaluate DNA damage. Fluorescence images were acquired using a 20× objective lens on a Nikon ECLIPSE Ti2-E confocal scanning microscope.
DR-GFP report
The DR-GFP plasmids were transferred into SCLC cells and several stably transfected cell clones by selection with puromycin (1 μg/ml). I-SceI expression plasmids containing a hemagglutinin (HA) tag (pCBASceI, Addgene #26477) or pcDNA3.1 control plasmids were transfected into SCLC cells using Lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. One day after transfection, cells were harvested and analyzed for the percentage of GFP-positive cells by means of flow cytometry (Beckman CytoFlex). Data were analyzed in FlowJo software (version 9.4.2; Treestar, Ashland, OR, USA).
Live-cell imaging
Cells were cultured in confocal dishes one day before transfection with the plasmid. Forty-eight hours after transfection, live-cell images were collected through (Nikon, ECLIPSE Ti2-E) confocal microscope (64× oil objective). 1,6-Hexanediol treatment: cells were cultured on confocal dishes, and transfected with plasmids for 48 h before observation. The culture medium was removed and re-replaced with medium containing 6% or 10% 1,6-hexanediol. The Micropoint system was directly coupled to the epifluorescence path of the (Nikon, ECLIPSE Ti2-E) confocal imaging system, and time-lapse images were captured at the indicated times.
Three-dimensional (3D) rendering
Z-Stack scanning images of cells transiently expressing EGFP-FOXP1 were acquired using a confocal microscope(Nikon, ECLIPSE Ti2-E) with a 63× oil immersion lens. A series of optical slices collected at different levels perpendicular to the optical axis (z axis) within the sample. It is exported to a computer 3D reconstruction program specially designed for processing confocal images. 3D rendering was conducted using the Nikon software and calibrated with spherical beads.
Antibodies information
Rabbit anti-FOXP1 mAbs were purchased from Cell Signaling Technology(FoxP1 (D35D10) XP® Rabbit mAb #4402) and Abcam (EPR4113; ab134055), Mouse anti-FOXP1 mAb were purchased from Abcam(ab32010); Rabbit anti-SP8 mAbs were purchased from Abcam(EPR26224-86; (ab302915)); Rabbit anti-PALB2 mAb were purchased from Absin(abs120051); Rabbit anti-H3K27Ac mAbs were purchased from GeneTex(AB_2885844); Mouse anti-H3K27Ac mAbs were purchased from GeneTex(AB_2888358); Rabbit anti- H3K4me1mAbs were purchased from Abmart(P37961); Mouse anti-BRD4 mAbs were purchased from Abmart(M073541); Rabbit anti-β-actin mAbs were purchased from Abcam(ab8227); Rabbit anti-PARP mAbs were purchased from Cell Signaling Technology(#9532); Rabbit anti-gamma H2AX (phospho S139) mAbs were purchased from Abcam(ab81299); Rabbit anti-RAD51 mAbs were purchased from Affinity(#DF8066); The detail of antibody dilutions can be seen in Supplementary Data 2.
RNA sequencing
Total RNA was extracted from H69AR by RNAiso Plus (Takara Bio, Dalian, China). RNA integrity was evaluated using a Bioanalyzer, ensuring only high-quality RNA was selected for library preparation. Following adapter ligation and PCR amplification, the libraries underwent sequencing on high-throughput Illumina platforms. Raw sequencing reads were subjected to quality control, aligned to the reference genome, and analyzed for gene expression, differential expression, and pathway enrichment. Additionally, the Illumina genetic algorithm was applied to optimize the pipeline for image processing and signal calibration. Gene expression levels were quantified in FPKM units, with differential expression defined as a minimum two-fold change and an absolute difference of at least 0.5 FPKM between groups.
Assay for transposase-accessible chromatin sequencing (ATAC-seq)
ATAC-seq pipeline was utilized for stringent quality control and statistical analysis of sequencing data, ensuring the reliability of accessibility profiles. Paired-end reads (150 bp) were aligned to the reference genome (human: hg38; mouse: mm10) following a standardized mapping protocol. To detect differentially accessible regions, the DiffBind R package was employed, using statistical modeling to identify significant chromatin accessibility variations between conditions.
Super-enhancer identification
This study utilized the chemoresistant H69AR cell line as the research model. The ROSE (Rank Ordering of Super-Enhancers) algorithm was employed to identify super-enhancers by first merging adjacent enhancers within a defined genomic distance. These enhancers were then ranked based on H3K27ac ChIP-seq signal intensity, and a threshold was established at the inflection point of the enhancer signal distribution to differentiate super-enhancers from typical enhancers. Finally, super-enhancers were mapped to nearby genes to infer their regulatory targets, offering insights into cell-type-specific transcriptional regulation.
Tumor xenograft experiments
All animal studies were approved by the Ethics Committee of Southern Medical University. We have complied with all relevant ethical regulations for animal use H69AR or H69 cells were resuspended in cold 0.9% sterile saline. A total of 2 × 106 H69AR or H69 cells were injected into the right flank of BALB/c nude mice (male, 20–30 g, 5 weeks old) and allowed to reach between 100 and 150 mm3 before beginning treatment. Tumors were measured at least twice a week by caliper and tumor volume was calculated using the formula: volume = length × width2/2. Drug administration was counterbalanced to ensure that each treatment condition was equally represented at different times of the day. Research involving animals was performed in compliance with the policies of the animal ethics committee of the Southern Medical University of China.
Patient-derived xenograft (PDX) Construction
In this study, four Patient-derived xenografts (PDX) models from small-cell lung cancer patients were used. B-NDG mice (NOD.CB17-Prkdcscid/IL2rgtm1/Bcgen) used were all female, 4–6 weeks old, and reared in the SPF Laboratory Animal Center laboratory. All patients signed informed consent forms and passed the review by the Ethics Committee of Southern Medical University. At least two pathologists confirmed the diagnosis of SCLC. Surgically resected or biopsy-obtained tumor samples are processed and transplanted subcutaneously into B-NDG mice. The tumors are monitored for growth, and once established, they are passaged into subsequent generations of mice to maintain tumor heterogeneity. Drug treatments interventions are administered, and tumor responses are evaluated through volume measurements, histopathology, and molecular analyses. Outcome assessment was performed by a blinded evaluator who was unaware of the group allocation. Samples were coded before analysis to ensure objectivity.
Clinical samples
A total of 74 formalin-fixed, paraffin-embedded (FFPE) tissue samples from small-cell lung cancer (SCLC) patients were collected between 2008 and 2020 at The First Affiliated Hospital of Hebei North University and Zhujiang Hospital, Southern Medical University. Informed consent was obtained from all patients prior to sample collection, and all procedures adhered to ethical regulations for human research participants. All ethical regulations relevant to human research participants were followed. The study was approved by the Ethics Committee of The First Affiliated Hospital of Hebei North University and complied with the Declaration of Helsinki guidelines. Survival analysis was conducted using the Kaplan-Meier method.
Statistics and reproducibility
Nonparametric analyses were performed using Prism 10.0 (GraphPad Software). A χ² test assessed definite correlations, while Student’s t-test and the Mann-Whitney test were used for parametric and nonparametric comparisons, respectively. Data are presented as mean ± standard error of the mean (SEM). Assumptions of normality and homogeneity of variance were tested using the Kolmogorov–Smirnov test and Levene’s test, respectively.
For nude mouse studies, biochemical and pathology analyses were conducted with n ≥ 5, while cell culture experiments included at least three independent replicates. All animal studies were randomized and conducted in a blinded manner. No samples or animals were excluded from the analysis. Pairwise comparisons were performed using Student’s t-test, with statistical significance set at P < 0.05. Statistical analyses were carried out using GraphPad Prism (Version 9.0) and IBM SPSS Statistics 22.0 (IBM Corporation).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All plasmids and CRISPR sequences are in Supplementary Data 1. Source data can be found in Supplementary Data 2 and unprocessed blots in Fig. S6. FACS gating strategies are included in Fig. S7. RNA sequencing data can be obtained via SRA datasets (PRJNA1222335) and DNA sequencing data can be obtained via (PRJNA1219381, PRJNA1224241).
References
Gaspar, L. E. et al. Small-cell lung cancer: prognostic factors and changing treatment over 15 years. Clin. Lung Cancer 13, 115–122 (2012).
Wang, Q. et al. SCLC: Epidemiology, risk factors, genetic susceptibility, molecular pathology, screening, and early detection. J. Thorac. Oncol. 18, 31–46 (2023).
Roth, B. J. et al. Randomized study of cyclophosphamide, doxorubicin, and vincristine versus etoposide and cisplatin versus alternation of these two regimens in extensive small-cell lung cancer: a phase III trial of the Southeastern Cancer Study Group. J. Clin. Oncol. 10, 282–291 (1992).
Schmittel, A. et al. A randomized phase II trial of irinotecan plus carboplatin versus etoposide plus carboplatin treatment in patients with extended disease small-cell lung cancer. Ann. Oncol. 17, 663–667 (2006).
Rossi, A. et al. Carboplatin- or cisplatin-based chemotherapy in first-line treatment of small-cell lung cancer: the COCIS meta-analysis of individual patient data. J. Clin. Oncol. 30, 1692–1698 (2012).
Herzog, B. H., Devarakonda, S. & Govindan, R. Overcoming chemotherapy resistance in SCLC. J. Thorac. Oncol. 16, 2002–2015 (2021).
Petty, W. J & Paz-Ares, L Emerging strategies for the treatment of small cell lung cancer: a review. JAMA Oncol. 9, 419–429 (2023).
Nicholson, A. G. et al. The International Association for the Study of Lung Cancer Lung Cancer Staging Project: proposals for the revision of the clinical and pathologic staging of small cell lung cancer in the forthcoming eighth edition of the TNM classification for lung cancer. J. Thorac. Oncol. 11, 300–311 (2016).
Mansour, M. R. et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346, 1373–1377 (2014).
Costanzo, F. et al. Promoters of ASCL1- and NEUROD1-dependent genes are specific targets of lurbinectedin in SCLC cells. EMBO Mol. Med. 14, e14841 (2022).
Qu, S. et al. Molecular subtypes of primary SCLC tumors and their associations with neuroendocrine and therapeutic markers. J. Thorac. Oncol. 17, 141–153 (2022).
Grunblatt, E. et al. MYCN drives chemoresistance in small cell lung cancer while USP7 inhibition can restore chemosensitivity. Genes Dev. 34, 1210–1226 (2020).
Drapkin, B. J. et al. Genomic and functional fidelity of small cell lung cancer patient-derived xenografts. Cancer Discov. 8, 600–615 (2018).
Mollaoglu, G. et al. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell 31, 270–285 (2017).
Huang, F. et al. Guanosine triphosphate links MYC-dependent metabolic and ribosome programs in small-cell lung cancer. J. Clin. Investig. 131, e139929 (2021).
Zhang, S. et al. NONO enhances mRNA processing of super-enhancer-associated GATA2 and HAND2 genes in neuroblastoma. EMBO Rep. 24, e54977 (2023).
Guo, Y. E. et al. Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572, 543–548 (2019).
Petronilho, E. C. et al. Oncogenic p53 triggers amyloid aggregation of p63 and p73 liquid droplets. Commun. Chem. 7, 207 (2024).
Yu, M. et al. Interferon-γ induces tumor resistance to anti-PD-1 immunotherapy by promoting YAP phase separation. Mol. Cell 81, 1216–1230.e1219 (2021).
Lu, B. et al. Pharmacological inhibition of core regulatory circuitry liquid-liquid phase separation suppresses metastasis and chemoresistance in osteosarcoma. Adv. Sci. 8, e2101895 (2021).
Xie, J. et al. Targeting androgen receptor phase separation to overcome antiandrogen resistance. Nat. Chem. Biol. 18, 1341–1350 (2022).
Klein, I. A. et al. Partitioning of cancer therapeutics in nuclear condensates. Science 368, 1386–1392 (2020).
Mehta, S. & Zhang, J. Liquid–liquid phase separation drives cellular function and dysfunction in cancer. Nat. Rev. Cancer 22, 239–252 (2022).
Bao, J. et al. Integrated high-throughput analysis identifies super enhancers associated with chemoresistance in SCLC. BMC Med. Genomics 12, 67 (2019).
Gay, C. M. et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 39, 346–360.e347 (2021).
Lenhart, R. et al. Sensitivity of small cell lung cancer to BET inhibition is mediated by regulation of ASCL1 gene expression. Mol. Cancer Ther. 14, 2167–2174 (2015).
Zengerle, M., Chan, K. H. & Ciulli, A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 10, 1770–1777 (2015).
Bondeson, D. P. et al. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 25, 78–87.e75 (2018).
Turk, A. A. & Wisinski, K. B. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer 124, 2498–2506 (2018).
Jain, A., Barge, A. & Parris, C. N. Combination strategies with PARP inhibitors in BRCA-mutated triple-negative breast cancer: overcoming resistance mechanisms. Oncogene 44, 193–207 (2025).
Cao, J. et al. DNA methylation-mediated repression of miR-886-3p predicts poor outcome of human small cell lung cancer. Cancer Res 73, 3326–3335 (2013).
Fang, S. et al. H3K27me3 induces multidrug resistance in small cell lung cancer by affecting HOXA1 DNA methylation via regulation of the lncRNA HOTAIR. Ann. Transl. Med 6, 440 (2018).
Zheng, Y., Wang, Z., Wei, S., Liu, Z. & Chen, G. Epigenetic silencing of chemokine CCL2 represses macrophage infiltration to potentiate tumor development in small cell lung cancer. Cancer Lett. 499, 148–163 (2021).
Silva, J. L. et al. Targeting biomolecular condensation and protein aggregation against cancer. Chem. Rev. 123, 9094–9138 (2023).
Levavasseur, F. et al. FOXP1 regulates oxidative stress, SIRT1 expression, and resistance to chemotherapies in acute myeloid leukemia cells. Blood Adv. 7, 3265–3275 (2023).
Banham, A. H. et al. The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Res. 61, 8820–8829 (2001).
Vaglietti, S. et al. PolyQ length-based molecular encoding of vocalization frequency in FOXP2. iScience 26, 108036 (2023).
Wang, B., Lin, D., Li, C. & Tucker, P. Multiple domains define the expression and regulatory properties of Foxp1 forkhead transcriptional repressors. J. Biol. Chem. 278, 24259–24268 (2003).
Belkina, A. C. & Denis, G. V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 12, 465–477 (2012).
Filippakopoulos, P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010).
Shi, J. & Vakoc, C. R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 54, 728–736 (2014).
Yokoyama, Y. et al. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. 76, 6320–6330 (2016).
Zanconato, F. et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 24, 1599–1610 (2018).
Acknowledgements
The work described in this article was supported by grants from the National Natural Science Foundation of China (Nos. 81972809 and 82172769) and also was partially supported by GuangDong Basic and Applied Basic Research Foundation (2023A1515220261, 2022A1515012400) from Natural Science Foundation of Guangdong Province China.
Author information
Authors and Affiliations
Contributions
Y Tang, Y Niu, and L Guo conceptualized the study and designed the experiments. Y. Chen and X. Zhou contributed to methodology and visualization. Y. Hu, L. Sun, and Y. Xiong were responsible for data collection and statistical analysis. Y. Xu, Q. Wang, and Y. Wang supervised the project and provided funding support. All authors contributed to manuscript writing and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Jerson Silva and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary handling editors: Silvia Belluti and Kaliya Georgieva.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, Y., Niu, Y., Chen, Y. et al. Targeting FOXP1 phase separation in small cell lung cancer mechanisms of chemotherapy resistance. Commun Biol 8, 431 (2025). https://doi.org/10.1038/s42003-025-07804-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07804-7
This article is cited by
-
Biomolecular condensates in lung cancer: from molecular mechanisms to therapeutic targeting
Cell Death Discovery (2025)
