
Abstract
As the world of agrochemicals is entering a race for efficient and safe modalities, there is an urgent and specific need for entirely new modes of action. Proteolysis-targeting chimeras (PROTACs) recruit naturally occurring E3 ubiquitin ligases to induce potent and selective degradation of protein targets via the ubiquitin-proteasome system (UPS). Herein, we demonstrate the degradative abilities of the insect VHL (von Hippel-Lindau) E3 ligase, making it the first PROTAC-ready ligase for agriculture applications. In doing so, we developed VHL-recruiting PROTACs capable of degrading fall armyworm (Spodoptera frugiperda) sfBRD3 protein with potencies as high as >80% in Sf9 cells and >60% in larvae. We also successfully designed, optimized, and tested PROTACs that significantly degraded sfWDS protein in both cells and whole organisms. This proof-of-concept study pioneers the use of PROTACs for agricultural applications and establishes this modality as a promising, disruptive alternative to traditional small molecule inhibitors.
Similar content being viewed by others
Introduction
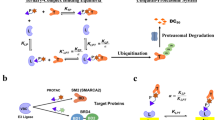
Targeted protein degradation (TPD) is revolutionizing the field of drug discovery, offering a unique approach to target and eliminate disease-causing proteins1. This family of modalities exploits different cellular mechanisms for recycling proteins, such as the ubiquitin-proteasome system (UPS), lysosomal pathways, and autophagy2,3,4. Among these, proteolysis-targeting chimeras (PROTACs) have emerged as particularly powerful and versatile tools, capable of targeting a wide array of proteins, including those previously deemed “undruggable”5. PROTACs are hetero-bifunctional small molecules that operate by recruiting a naturally occurring E3 ubiquitin ligase to a target protein of interest (POI), thereby facilitating its ubiquitination and subsequent degradation by the UPS2,6,7. Rationally-designed PROTACs possess several distinct advantages over traditional small molecule inhibitors, including the ability to degrade rather than merely inhibit their targets, the potential to target proteins lacking enzymatic activity (e.g., scaffolding proteins and transcription factors), and the capacity to overcome resistance mechanisms through the complete removal of target proteins from the cell8,9,10. Furthermore, the PROTAC mode of action is catalytic (i.e., one single PROTAC molecule can degrade multiple proteins via the iterative formation and dissociation of ternary complexes) and thus effective at relatively low concentrations, potentially reducing the risk of off-target effects and overall toxicity11,12,13,14. The current wave of PROTACs entering the clinic in human health is exclusively driven by two workhorse E3 ligases: Cereblon (CRBN), and to a lesser extent, the von-Hippel-Lindau tumor suppressor (VHL)5. Thus, discovering and validating additional PROTAC-ready E3 ligases is an industry-enabling, ongoing effort.
Amidst the healthcare TPD revolution, the agricultural sector (Ag) stands on the cusp of a similar paradigm shift. Crop protection chemicals have been foundational to the advancement of modern agriculture, playing a crucial role in safeguarding crops from pests, diseases, and weeds, thereby significantly enhancing food production and security15,16. In the face of increased pesticide resistance, stricter environmental regulations, and an industry-wide paucity of new active ingredients, there is an urgent need for innovative crop protection solutions to meet the agricultural demands of a growing population17,18,19. Intriguingly, the UPS machinery is functionally conserved across eukaryotes, underscoring the potentially broad utility of PROTACs in agriculture20,21. However, while the compendium of knowledge regarding E3 ligases across kingdoms is growing22,23, the feasibility of PROTACs in agriculture remains completely unexplored.
Herein, we pioneer the use of small molecule PROTACs in insects, demonstrating the degradation of endogenous proteins in cells and larvae of fall armyworm (S. frugiperda), an agricultural pest of over 350 host plants that is responsible for crop losses valued in the tens of billions of dollars annually worldwide24,25. Specifically, we establish the PROTAC-mediated degradative abilities of the S. frugiperda VHL (sfVHL) E3 ligase and its recruitment via a small molecule ligand. After identifying S. frugiperda homologs of proteins known to be degraded by VHL-based PROTACs in human cells, we show that sfVHL can be recruited by PROTACs to efficiently degrade two target POIs. UPS inhibitors and E3-negative PROTAC controls successfully rescued that degradation, proving the UPS-mediated mechanism of action. Finally, further optimized PROTACs show significantly improved degradation potency, demonstrating the opportunity for tuning and achieving high protein reduction in critical pest insects via VHL-recruiting PROTACs.
The PROTAC modality represents a highly promising alternative to traditional occupancy-based small molecule inhibitors in Ag. This proof-of-concept study validates the PROTAC technology beyond mammalian cells, and as such marks the first step towards making rationally designed PROTAC molecules a chemistry of the future for agrochemicals.
Results
Identification and characterization of S. frugiperda homologs of hsVHL, hsBRD4 and hsWDR5
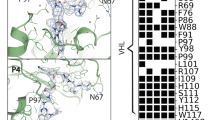
A BlastP26 search targeting the S. frugiperda proteome (GCF_023101765.2) was performed using the human VHL (hsVHL, Uniprot ID P40337), hsBRD4 (O60885, Bromodomain-containing protein 4) and hsWDR5 (P61964, WD repeat domain 5 protein) sequences as query templates. Four homologs of the VHL beta domain were identified, however only one, XP_035451646.1 (sfVHL), is predicted to contain the alpha helical bundle required to interact with Elongin-B and, thus, be a competent E3 ligase. Comparison of the amino acid sequences between hsVHL and sfVHL revealed only 29% sequence identity (Supplementary Fig. 1). Despite the low sequence identity, a threading model of sfVHL, based on hsVHL, shows that the majority of the VHL ligand binding site residues are conserved, except for three significant amino acid variations adjacent to the binding pocket (Fig. 1, Table 1). In the search for hsBRD4 homologs, we identified four isoforms in S. frugiperda, with the longest sequence hit (XP_035429406.2) annotated as bromodomain-containing protein-3 (sfBRD3). We noted residues 369-474 of sfBRD3 as the second of the two bromodomains which has the highest homology, both globally (72% identical) and within the binding site, to the second bromodomain of human BRD4 (residues 349-455). This is critical as hsBRD4 has been extensively degraded using PROTACs recruiting VHL27,28,29. This domain (bromodomain 2, BD2) is referenced throughout as sfBRD3(BD2). Finally, one unambiguous homolog (XP_035429818.1) was found for human WDR5 with 93% sequence identity, referred herein as sfWDS for its homology to the Drosophila melanogaster protein will die slowly30.

Structural comparison of VHL binding site in human and Spodoptera frugiperda. Binding site of the VHL binder of the PROTAC MZ1 bound to hsVHL (PDB 5T35, left) and sfVHL (threading model, right). Binding site residues that differ between the two orthologs are labeled and colored in pink.
Human ligands bind to recombinant S. frugiperda proteins
To assess if the putative S. frugiperda proteins bind known small molecule ligands, we recombinantly expressed proteins from E. coli. First, we co-expressed sfVHL with putative S. frugiperda elongins B and C (elonginB: XP_035440746.1, elonginC: XP_035451917.1) and purified the three-protein complex via affinity chromatography (the complex is referred to as sfVBC). Further, we purified bromodomain 2 (amino acids 356-474) of sfBRD3 as well as another version with an N-terminal Maltose-binding protein (MBP) tag (MBP-sfBRD3(BD2)). Finally, sfWDS was expressed as an N-term truncate to improve solubility (amino acids 33-346) with an N-terminal MBP tag (cleaved for biochemical studies) and purified via affinity chromatography.
We then assessed whether literature-published ligands used as starting scaffolds for previously reported hsBRD4-hsVHL and hsWDR5-hsVHL PROTACs could maintain binding affinity towards their insect homologs. sfVHL ligands were based on the hydroxyproline binder commonly used in publicly available VHL PROTACs. We first tested ligands based on human VHL binders (compounds 1, 2 and 3, Fig. 2, Supplementary Fig. 2, Table 2). These ligands displaced a Fluorescein Amidite (FAM)-labeled VHL peptide tracer (compound 4, FAM-DEALA-Hyp-YIPD) in a ligand displacement (LD) binding screen with affinities in the low- to mid-nanomolar range (Table 2). Their tight binding affinity (KD) to sfVBC was also confirmed by surface plasmon resonance (SPR): 210, 26, and 270 nM respectively.

a Ligand displacement trace of GST-sfVBC for binding of compounds 3 (cobalt, sfVBC vestigial), 18 (green, PROTAC), and 21 (bronze, E3 neg. PROTAC). b Ligand displacement trace for sfWDS binding of compounds 13 (cobalt, sfWDS vestigial), 18 (green, PROTAC), and 20 (bronze, E3 neg. PROTAC. c SPR sensorgrams for binding of 18 (PROTAC) to sfVBC. d SPR sensorgrams for binding of 18 (PROTAC) to sfWDS. e SPR sensorgrams for ternary complex formed by 18 with sfVBC immobilized on the surface. f AlphaLISA trace of sfVBC(BIO)/sfWDS-His6 ternary formation for 18 (green, PROTAC), and 20 (bronze, E3 neg. PROTAC) and sfVBC/sfWDS vestigials combined (cobalt, 3, sfVBC, 13 sfBRD3). g anti-His6 western blot of IVQ reaction containing, E1, E2, sfVBC, sfWDS, Fl-Ubq, ATP, and PROTAC (18). h anti-His6 western blot of IVQ reaction containing, E1, E2, sfVBC, sfWDS, Fl-Ubq, ATP, and E3 neg. PROTAC (20). i Chemical structures of compounds 18 and 20. Error bars on affinity and ternary plots represent standard error of the mean from n = 3 experimental replicates.
Initial sfBRD3 binders were based off three well-documented hsBRD4 binders: triazolodiazepine (JQ1)31, tetrahydroquinoline (I-BET)32, and dihydro-pyrrolopyridinone33. Binding affinity of these ligands towards sfBRD3(BD2) was measured by SPR. Ligand compounds 5, 6, and 7 demonstrated excellent SPR binding affinities (KD) to sfBRD3(BD2) of 290, 1.2, and 6.8 nM respectively (Fig. 3, Supplementary Fig. 2, Table 3). Based on these results, we synthesized the corresponding “vestigial” compounds (8, 9 and 10, defined as the ligands with the first few atoms of the PROTAC linker covalently linked at a well-chosen attachment point) and a JQ-1-based fluorophore tool for LD (11). This step is essential to verify that the linker attachment at a particular position of those ligands (which defines a PROTAC “exit vector”) does not affect nor prevent protein binding. These vestigials also showed very tight affinity towards sfBRD3(BD2) with LD Kis of 240, 260, and 220 nM and SPR KDs of 70, 0.9, and 23 nM respectively, suggesting the linker placement was clearly viable to enable PROTACs with conserved binding affinity towards the POI (Fig. 3, Supplementary Fig. 2, Table 3). Moreover, sfWDS ligands were derived from well characterized hsWDR5-hsVHL PROTACs, such as compound 12 (MS67)34. The vestigial compounds 13 and 14 displaced a FAM-labeled peptide tracer (compound 15) with excellent Ki values of 110 and 170 nM, and SPR KDs of 11 and 47 nM respectively, suggesting these ligands as excellent starting points for PROTACs.

a Ligand displacement trace of GST-sfVBC for binding of compounds 1 (cobalt, sfVBC vestigial), 21 (green, PROTAC), and 25 (bronze, E3 neg. PROTAC). b Ligand displacement trace for MBP-sfBRD3(BD2) binding of compounds 8(cobalt, sfBRD3 vestigial), 21 (green, PROTAC), and 25 (bronze, E3 neg. PROTAC. c SPR sensograms for binding of 21 (PROTAC) to sfVBC. d SPR sensograms for binding of 21 (PROTAC) to sfBRD3(BD2). e SPR sensograms for ternary complex formed for 21 with sfVBC immobilized on the surface. f AlphaLISA trace of ternary formation of sfVBC(BIO)/MBP-sfBRD3(BD2) for 21 (green, PROTAC), and 25 (bronze, E3 neg. PROTAC) and sfVBC/sfBRD3 vestigials combined (cobalt, 1, sfVBC, 8, sfBRD3). g anti-His6 western blot of IVQ reaction containing, E1, E2, sfVBC, MBP-sfBRD3(BD2), Fl-Ubq, ATP, and PROTAC (21). h anti-His6 western blot of IVQ reaction containing, E1, E2, sfVBC, MBP-sfBRD3(BD2), Fl-Ubq, ATP, and E3 neg. PROTAC (25). i Chemical structures of compounds 21 and 25. Error bars on affinity and ternary plots represent standard error of the mean from n = 3 experimental replicates.
Human WDR5 PROTACs show good affinity but fail to induce significant sfWDS degradation in cell
With all ligand and vestigial binding affinities confirmed to sfVBC, sfBRD3(BD2), and sfWDS, we turned our interest to characterizing the in vitro and in cell efficacy of the known PROTACs based on these same ligands. Our interest in identifying potential bifunctional sfWDS degraders first led us to examine well-characterized hsWDR5-hsVHL degraders such as MS67 (compound 12) and MS33 (compound 16)34. These two PROTACs have been reported to degrade WDR5 in MV4-11 cells with DC50 = 3.7 nM (Dmax = 94%) and DC50 = 260 nM (Dmax = 71%), respectively34. MS67 afforded good binary affinity to sfWDS (SPR KD = 14 nM, LD Ki = 130 nM) and sfVBC (SPR KD = 140 nM, LD Ki = 57 nM, Table 2, Supplementary Fig. 3) and was able to induce ternary complex formation between sfWDS and sfVBC in SPR (KD,ternary = 290 nM, t1/2 = 13 s, Supplementary Table 2). We then tested MS67 in an in vitro ubiquitination (IVQ) cell free system; polyubiquitination of sfWDS was observed in a dose-dependent manner (Supplementary Fig. 4). However, the PROTAC failed to prompt degradation of sfWDS in our cell-based targeted LCMS degradation assay. PROTAC MS33 (16) also achieved good binding affinity for sfWDS (SPR KD = 29 nM, LD Ki = 67 nM) and sfVBC (SPR KD = 37 nM, LD Ki = 3.4 nM) and was able to induce ternary complex formation between sfWDS and sfVBC in both SPR and AlphaLISA (AL) assays (SPR KD,ternary = 130 nM, t1/2 = 18 s; AL KD,app = 940 nM, Table 2, Supplementary Table 2). Like MS67, MS33 induced ubiquitination of sfWDS in the IVQ system, but degradation of sfWDS was still not observed in our cellular degradation assay. While both PROTACs facilitate ternary complex formation, the interaction exhibits negative cooperativity (α < 1;KD,binary < KD,ternary) and short complex half-lives. The transient ternary complex is sufficient to induce in vitro ubiquitination, but insufficient in inducing degradation in the cellular environment.
PROTAC linker optimization leads to sfWDS degradation
While MS67 and MS33 showed tight binary/ternary affinities and IVQ signals, cellular degradation was not observed at all, leading us to focus on linker composition and optimization to improve sfWDS-sfVBC PROTACs for in cell degradation. Linkers were iteratively modified to shorten their lengths, increase their rigidity and dihedral constraints, and decrease the number of rotatable bonds. The bifunctional degrader compound 17 was first reported in the patent literature as one in a set of compounds “able to degrade WDR5 in MV4-11 and/or MIAPACA2 cells.”35 We measured that 17 achieved good binary affinity for sfWDS (SPR KD = 95 nM, LD Ki = 330 nM) and sfVBC (SPR KD = 93 nM, LD Ki = 64 nM, Table 2, Supplementary Table 2), comparable to MS67 and MS33. However, compound 17 formed a more robust ternary complex by SPR (KD = 61 nM, t1/2 = 41 s) compared to the other PROTACs. Interestingly, this improved ternary complex stability translated into weak but observable degradation of sfWDS (DC50 = 3.2 µM, Dmax = 37%). Through several iterative rounds of linker optimization, we designed the PROTACs 18 and 19, that afforded excellent binding affinity for sfWDS (SPR KD = 10 and 9.1 nM, LD Ki = 94 and 150 nM, respectively) as well as very good affinity for sfVBC (SPR KD = 48 nM and 44 nM, LD Ki = 59 and 31 nM, respectively, Table 2, Supplementary Fig. 3). In our SPR and AlphaLISA (AL) biochemical assays, both 18 and 19 demonstrated ternary complex formation (Table 3, Supplementary Fig. 2). Analysis of the binding constants from SPR revealed compound 18 induced ternary complex formation with KD,ternary = 13 nM, t1/2 = 270 s and α = 3.8, where the cooperativity (α) is a measure of the fold-improvement in affinity of the ternary complex over the binary interaction. Compound 19 showed a similar response with KD,ternary = 16 nM, t1/2 = 160 s and α = 2.8. Importantly, both molecules formed productive ternary complexes, inducing robust ubiquitination of sfWDS in our IVQ assays (Fig. 2, Supplementary Fig. 4). We also showed that both sfWDS and sfVBC were required for ubiquitination confirming involvement of both proteins in ubiquitin transfer. Furthermore, the corresponding E3 negative compound 20 which showed no binding to sfVBC due to a stereochemical modification on the sfVBC ligand (sfVBC SPR KD and LD Ki no binding observed, Table 2) abolished ubiquitination of sfWDS, further confirming sfVBC involvement (Supplementary Fig. 5).
We used our targeted LCMS assays36,37 to monitor the endogenous protein abundance changes of the targets resulting from treatment with compounds identified by in vitro assays. Both compounds 18 and 19 (10 μM) promoted robust degradation of sfWDS in Sf9 cells, degrading the protein in the 64-67% range (Fig. 4). Follow up control experiments employing the proteasome inhibitor bortezomib and cotreatment with either the target binder 13 or the E3 binder 3, were shown to “rescue” (i.e., abolish) the PROTAC-induced sfWDS degradation, suggesting a proteasome mediated event as expected (Fig. 4). Dose-response experiments also indicated the high potency of those compounds with DC50 of ~50 nM (Fig. 4), while time-course studies at the DC50 showed consistent degradation at 4 h and 24 h time. Additionally, treatment with the E3 negative compound 20 increased the abundance of sfWDS, indicating a dependance on full sfVHL engagement to achieve degradation (Fig. 4). To better understand the role of cellular permeability of these PROTACs in degradation of sfWDS, compound distribution assays were performed, revealing that PROTACs 18 and 19, which showed cellular degradation, had robust intracellular concentrations (with 32% total PROTAC recovery detected in cells vs media for 18 and 14% in cells for 19). PROTAC 16 was also present in cells (29% PROTAC in cells) at similar cellular distribution to PROTACs 18 and 19 but did not show degradation. PROTAC 12, which also did not demonstrate in cell degradation, was only detected at very low intracellular levels, suggesting that limited permeability may contribute to the lack of activity for this compound. Finally, global proteomics analysis performed in Sf9 cells treated with compounds at 10 µM for 4 h revealed selective degradation of sfWDS, with minimal off-target effects, supporting the conclusion of on-target activity (Supplementary Fig. 6).

a Sf9 cells (1e6) were treated with DMSO or compound (1 μM) +/- bortezomib (10μM) for 24 h and sfBRD3 abundance was measured by targeted MS assays. b Dose response of sfBRD3 degradation. Sf9 cells were treated with DMSO, 21 (black) or 25 (red) for 24 h. sfBRD3 abundance was measured by targeted MS. c In vivo degradation of sfBRD3. Spodoptera frugiperda larvae (L5) were injected with 0.1 nmol/mg sfBRD3 degrader and protein abundance was monitored by targeted MS. d Sf9 cells (1e6) were treated with DMSO or compound (10 μM) +/- bortezomib (10μM) for 24 h and sfWDS abundance was measured by targeted MS. e Dose response of sfWDS degradation. Sf9 cells were treated with DMSO, 18 (black) or 20 (red) for 24 h. sfWDS abundance was measured by targeted MS. f In vivo degradation of sfWDS. Spodoptera frugiperda larvae (L5) were injected with 0.1 nmol/mg sfWDS degrader and protein abundance was monitored by targeted MS assay. Bar graphs show averages +/- sem, (n = 4 for Sf9 cell studies, n = 6 biological replicates in larvae studies). Significance is expressed as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 as compared to the DMSO control in an unpaired two-tailed t-test. Dose-response curves are fitted with an Inhibitor vs. response, variable slope (4 parameter) curve using GraphPad Prism version 10.2.0. for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com.
PROTACs based on multiple sfBRD3 ligands induce significant sfBRD3 degradation in cell
With sfWDS in cell degradation vastly improved, we turned to determining whether hsBRD4-hsVHL PROTACs could degrade sfBRD3. Three distinct bromodomain chemical scaffolds (8- JQ1, 9- dihydro-pyrrolopyridinone, and 10- I-BET) were shown to exhibit tight binding affinity to sfBRD3(BD2). Utilizing these three binders in concert with sfVHL ligands 1 and 3, early PROTAC designs were centered around PEG- and alkyl-based linkers, eventually transitioning to more rigid linkers. The subsequent pairing of the three sfBRD3 binders with 1 and a selection of rigid linkers culminated in the synthesis of PROTACs 21-24. Note that 21, 22 and 23 all share the common rigid 1-(3-azetidinyl)piperazine linker, whereas 24 possesses an alternative rigid biphenyl linker.
Potent binary binding affinities to sfBRD3(BD2) for all four PROTACs were obtained, indicating a reasonable incorporation of the sfBRD3 POI binder (Table 3, Fig. 3, Supplementary Fig. 7). Interestingly, the binary binding affinities to sfVBC for the PROTACs 23 (SPR KD = 70 nM, LD Ki = 45 nM), 22 (SPR KD = 120 nM, LD Ki = 81 nM), and 21 (SPR KD = 91 nM, LD Ki = 77 nM, Fig. 3, Supplementary Fig. 7) were better than that of the ligand 1 on which they are based (SPR KD = 210 nM, LD Ki = 370 nM, Table 3). Considering the PROTAC 24 (SPR KD = 850 nM, LD Ki = 430 nM) did not show such improvement, we concluded there is a beneficial contribution to the sfVBC binding affinity by the 1-(3-azetidinyl)piperazine linker. Additionally, 23 demonstrated a 10-fold tighter affinity (KD = 1.2 nM) to sfBRD3(BD2) compared to 24 (KD = 12 nM) despite employing the same ligand (9) further indicating the important role of the linker in PROTAC design.
Examination of the ternary affinity data for all PROTACs revealed that 21 (KD,ternary = 8.9 nM, t1/2 = 250 sec., α = 10) afforded the most stable ternary complex between sfBRD3(BD2) and sfVBC. Interestingly, while 24 (KD,ternary = 25 nM, t1/2 = 32 sec., α = 33) demonstrated a greater cooperativity, its respective ternary complex affinity was weaker with a much shorter complex half-life that is more comparable to 23 (KD,ternary = 79 nM, t1/2 = 20 sec., α = 0.9) and 22 (KD,ternary = 32 nM, t1/2 = 50 sec., α = 3.7, Supplementary Fig. 7, Table 3, Supplementary Table 3). Subsequently, all four PROTACs showed robust ubiquitination of MBP-sfBRD3(BD2) in IVQ testing (Supplementary Fig. 8). Like sfWDS, excluding either MBP-sfBRD3(BD2) or sfVBC abolished ubiquitination, confirming the involvement of both proteins in ubiquitin transfer (Supplementary Fig. 8). Further, the E3 negative controls 25 (sfVBC SPR KD and LD Ki no binding observed, Fig. 3, Table 3) and 26 (sfVBC SPR KD and LD Ki no binding observed, Table 3) corresponding to PROTACs 21 and 22 also suppressed ubiquitination, further confirming sfVHL involvement (Supplementary Fig. 8).
Similarly to sfWDS, Sf9 cell lines were utilized to assess the functionality of the PROTACS toward sfBRD3. We observed robust degradation of sfBRD3 in a compound specific manner: treatment with 21 and 24 at 1 μM reduced the sfBRD3 protein abundance by >85% after 24h treatment, whereas 23 (1 µM) and 22 (1 µM) led to sfBRD3 degradation levels as high as 70% and 74%, respectively (Fig. 4, Supplementary Fig. 9). Dose-response experiments with these compounds showed their high potency, with DC50 values of ~5 nM (Fig. 4, Supplementary Fig. 9), while time-course studies performed at the DC50 demonstrated an increase of degradation over 24 h. Furthermore, treatment with E3 negative compounds 25 and 26 (1 µM) showed no degradation of sfBRD3 and some modest increases in protein abundance, again indicating sfVHL involvement in the observed degradation. A modest increase in sfBRD3 was also observed with cotreatment of the target binder 8 and no degradation was observed in the presence of the E3 binder 1 or in the presence of the UPS inhibitor bortezomib (Fig. 4). Lastly, target selectivity was assessed using global proteomics in the same manner as described for sfWDS, with less than 1% of identified proteins decreasing significantly (Supplementary Fig. 6) indicating sfBRD3 specific degradation.
PROTACs induce degradation of sfBRD3 and sfWDS in whole fall armyworm larvae
With UPS-dependent degradation confirmed in cell for both sfWDS and sfBRD3, we then tested POI degradation with these PROTACs in vivo. In vivo degradation was assessed using targeted LCMS for both sfBRD3 and sfWDS. Several tested PROTACs showed significant levels of POI degradation. Phenotypic observation of larvae showed no changes to size or behavior, nor any signs of generalized toxicity 24 h after injection with the compounds tested. Specifically, compound 21 exhibited a substantial 68% degradation of sfBRD3, while compounds 22 and 23 showed comparatively lower levels at 29% and 35% respectively; importantly, 24 did not demonstrate any degradation. Rationalizing on possibilities why 23 degrades but 24 did not, with the only difference being the 1-(3-azetidinyl)piperazine in 23 replaced by the biphenyl moiety in 24, we refer to our reasoning that incorporation of the hydrophobic biphenyl group results in a compound with poor physicochemical properties (calculated LogP = 8.4 and measured kinetic solubility in Sf9 media <1.56 µM)—these properties promote poor solubility, compound aggregation and consequently reduce exposure at the drug target. In contrast, the basic 1-(3-azetidinyl)piperazine in 23 offers more favorable properties (calculated LogP = 4.6 and measured kinetic solubility in Sf9 media of 120 µM, Supplementary Table 4), leading to enhanced overall characteristics and greater degradation exposure. Treatment of fall armyworm larvae with sfBRD3-sfVHL E3-negative PROTACs 25 and 26 led to an increase in measurable sfBRD3 peptide levels of 8% and 20% with respect to the DMSO control (Fig. 4c). Regarding sfWDS, degradation was observed by targeted MS for sfWDS PROTACs 18 and 19 at 35% and 22% respectively, but not for compound 17, consistent with the trends observed in cell-based data. For sfWDS, the sfWDS-sfVHL E3-negative PROTAC 20 displayed a 14% increase in total sfWDS levels in larvae, which is in full agreement with results seen for sfBRD3 E3-negative and POI-vestigial compounds, again suggesting sfVHL involvement in the observed in vivo degradation. (Fig. 4f)
Discussion
Achieving protein degradation using sfVHL for two different POIs in Sf9 cells and in fall armyworm larvae makes (i) sfVHL the first PROTAC-ready E3 ligase for agricultural applications, (ii) the PROTACs disclosed herein the first proven PROTACs to degrade a POI in a non-mammalian cell and organism, and (iii) the PROTAC technology a validated modality to tackle the grand challenges of Ag, especially when it comes to next-generation pesticides.
This study established sfVHL as a PROTAC-ready ligase in fall armyworm. sfVHL shares only 29% sequence identity to the human variant but maintains binding affinity to the ligands designed for PROTACs that use hsVHL. Despite this, published hsVHL-hsWDR5 PROTACs showed very little to no degradation for sfWDS and only the linker-focused chemical modifications made in this study were able to facilitate sfVHL-induced sfWDS degradation in fall armyworm. Improvements in sfWDS degradation were driven by the cheminformatics-fueled, Oerth-proprietary PROTAC linkerology, where transitioning from the short linker in 12 to the longer-chain linkers in 16 and 17, followed by using more rigid linkers in compounds 18 and 19 resulted in the most effective degradation profiles. This highlights the importance of integrating the right linker length, rigidity, and conformational space, all along with bringing the POI and the E3 ligase together to form a ternary complex in the correct geometry with the appropriate exit vector on both sides of the PROTAC. The linkers introduced in these PROTACs show markedly enhanced biochemical measurements and in cell/in vivo efficacy. The relationship between in vitro PROTAC affinity and in vivo degradation, while poorly understood, can be discussed in terms of the ternary cooperativity, which describes the improvement in affinity of the ternary complex (the PROTAC in complex with the E3 ligase and the POI) over the separate binary interaction (the PROTAC with the E3 ligase alone and the POI alone). Notably, PROTACs 18 and 19 that demonstrated the longest ternary complex half-life ( > 150 s) and greatest cooperativity were the only two PROTACs in that series to show significant in vivo degradation. Further, the published PROTACs MS33 (16) and MS67 (12) with much shorter half-lives and lower cooperativity did not show degradation in cells.
sfVHL-mediated in vivo degradation of sfBRD3 was achieved through three distinct chemical scaffolds. The PROTACs incorporating these respective scaffolds afforded notable differences in ternary cooperativity and half-life as well as in vivo degradation of sfBRD3. PROTACs 21 and 24 produced strong ternary complex affinities (KD,ternary < 50 nM) and cooperativities (α > 10) but quite dissimilar half-lives (250 s and 32 s, respectively). Similar to the trend observed for WDS degradation, only PROTAC 21 with the longer ternary half-life demonstrated significant in vivo degradation. Moreover, 21 (MW = 1038 g/mol) afforded excellent measured solubility ( > 200 µM in the sf9 cell culture media) with improved physicochemical properties such as calculated logP <5, H-bond donors <5, and a reduced number of only 15 rotatable bonds. Interestingly, PROTACs 22 and 23 led to in vivo degradation despite having relatively short ternary complex half-lives ( < 50 sec) and weak cooperativities. Furthermore, PROTAC 24, which differs from PROTAC 23 in linker composition only, failed to demonstrate significant in vivo degradation despite producing a ternary complex affinity (KD,ternary < 100 nM) and an in cell degradation profile (Dmax > 70%) that is comparable to PROTAC 23. These results, along with those of sfWDS, begin to uncover the complex stack of non-linear structure-activity relationships between long-lived ternary complexes and in vivo degradation efficacy in fall armyworm. While these parameters are important, they are likely not the sole determining factors in governing the relationships between in vitro affinity and in cell/in vivo degradation. It is still not fully understood what other critical factors mainly drive differences in degradation efficiency but bioavailability, solubility, formulation, and PROTAC metabolism are among the top contenders. The PROTACs presented herein possess physicochemical properties within range of the “beyond rule of 5” definition38: for example, both compounds 16 (MS33) and 19 have high molecular weight (1209 and 1312 g/mol respectively), high LogP ( > 7), and low calculated and experimental solubility ( < 1 µM). It is interesting to note that compound 19, a confirmed degrader of sfWDS, has a lower number of H-bond donors (4 instead of 6) and a much lower number of rotatable bonds (18 instead of 25, Supplementary Table 4). Obviously, their physicochemical properties require further optimization, along with formulation efforts, to enable viable insecticidal products based on VHL-based PROTACs. Continued monitoring of the intricate relationships between in vitro, in cell, and in vivo function and physiochemical properties will be important to help guide future PROTAC design and optimization campaigns. Importantly, the two targets sfWDS and sfBRD3 considered for this study were solely chosen as proof-of-concept and are not necessarily intended as insecticidal targets per se. Future studies will involve PROTACs designed to degrade known targets with higher insecticidal potential as well as previously untapped pesticidal targets considered undruggable with traditional small molecule inhibitors. Moreover, insect-specific ligase recruiters, appropriate formulations, and their associated insecticidal phenotypes will be pursued.
The VHL ligase is a promising E3 ligase for PROTACs in insects. Validated with two different POIs in this study, it could indicate a level of promiscuity and broad diversity of proteins to be potentially PROTAC accessible via this ligase, as seen for hsVHL in pharmaceutical research. Due to the human-insect conservation, ongoing efforts to develop insect-specific VHL binders enabling the development of VHL-based PROTAC degraders that can only degrade an insect POI and be completely inactive in a human cell are critical to ensure the viability of that ligase in future insecticidal products based on VHL PROTACs. With approximately three hundred E3 ligases currently known or predicted in insects, our success in translating and optimizing VHL-based PROTACs to insects suggests the opportunity to harness other E3 ligases. Numerous other PROTACable E3 ligases in humans are also present in insects39,40, such as cereblon (XP_035442906.1 with only 30% overall sequence identity but a very well conserved binding site). Despite the broad conservation of some of those E3 ligases, species-specific responses to modulators of mammalian cereblon41 could suggest the potential to develop tools that are insect-specific. Additionally, E3 ligases specific to the metazoan kingdom and, perhaps, narrow taxa within the kingdom, as suggested by bioinformatic analyses42 may allow for the design of highly targeted modulators for specific ranges of pest species. The E3 ligase space in insects is relatively unexplored compared to humans43 and plants44. Therefore, further efforts into ligase discovery and validation have the potential to catalyze the development of a broad set of PROTAC-ready E3 ligases for insect control.
Even though PROTACs are synthetic molecules, they capitalize on the cell’s natural protein recycling machinery to achieve the degradation of a well-defined POI and as such, represent a disruptive modality for small molecules in Ag. While other crop protection modalities such as RNA interference (RNAi) and peptides (typically referred to as biologics) offer innovative alternatives, they also suffer from distinct challenges. RNAi, targeting gene silencing, is precise but limited by delivery issues and environmental degradation45. Peptides, on the other hand, provide targeted action against pests but struggle with stability and higher production costs46. PROTACs have the potential to combine the best of both worlds, balancing kingdom of life specificity with the tunable stability and bioavailability of small molecule chemistries. On top of lower potential field application rates due to their catalytic effect, PROTACs also offer the unique possibility of overcoming pesticide resistance through their ability to degrade rather than inhibit target proteins. Indeed, several pharmaceutical studies have demonstrated that PROTACs can overcome drug resistance caused by either target protein overexpression or point mutations that render typical chemical inhibitors ineffective10. Target site resistance is one of the most common manifestations of pesticide resistance in agriculture47,48, so enabling the rescue of existing modes of action via PROTACs would be extremely valuable. Finally, PROTACs offer the exciting prospect of targeting proteins (e.g., transcription factors, structural proteins) that are not currently approachable with traditional small molecule inhibitors. Considering the relatively small number of currently utilized targets in crop protection and the slow pace of identifying additional modes of action18,49, expanding the target list with PROTACs will be a welcome addition to the crop protection toolbox and a true enabler for crop efficiency applications. This proof-of-concept study establishes PROTAC molecules as the modality of choice for conceiving the next-generation of rationally-designed agrochemicals.
Methods
Ethical Statement
The authors confirm that the research in this study complies with all relevant ethical regulations.
Chemical Synthesis
A list of final compounds is compiled in Supplementary Fig. 1. Details regarding the preparation and characterization of all aforementioned compounds are available in the Compound Synthesis section of the Supplementary Information.
Homology Modeling
3D atomic models of the Spodoptera frugiperda homologs were constructed using the homology model tool in Schrodinger Maestro (version 13.6). PDB 5T3529, was used as the structural template for both sfVHL (chain D) and sfBRD3 (chain A). The structural template for sfWDS was chain D of PDB 7JTP34. Homology models were constructed in the presence of ligand.
Expression and purification of proteins
Genes encoding Spodoptera frugiperda proteins VHL (sfVHL XP_035451646.1), elongins B and C (sfElonginB: XP_035440746.1, sfElonginC: XP_035451917.1), WDS (sfWDS XP_035429818.1), and BRD3 (sfBRD3 XP_035429406.2) were synthesized at Genscript and codon optimized for expression in E. coli and cloned into respective backbones for E. coli overexpression. Plasmids were transformed into E. coli BL21 (DE3) or E. coli BL21 (DE3) pLysS (Invitrogen) for protein expression. Genes encoding Homo sapiens CUL2 (hsCUL2 NP_001185706.1and RBX1 (hsRBX1 NP_055063.1) were codon optimized for expression in S. frugiperda Sf9 cells, synthesized, cloned into pFastBac1 vectors, transformed into DH10bac cells, and finally into baculovirus using the Bac-to-Bac method at Genscript.
For all E. coli expressed proteins cultures were grown at 37 °C in either Luria Broth or Terrific Broth to an OD600 ~ 0.6, cooled on ice, then induced with 500 µM IPTG for expression overnight at 16 °C. Cells were harvested by centrifugation at 4500 x g. Cell pellet was resuspended 50 mL wash buffer (50 mM HEPES, 300 mM NaCl, 5 mM imidazole, 10% glycerol, pH 7.5) and lysed using sonication, 1 sec “on” time 3 sec “off” time, 33% amplitude. 3 min total “on” time. Cell debris was pelleted at 35,000 x g. Proteins were purified by Immobilized Metal Affinity Chromatography (IMAC) and size exclusion steps. Detailed purification protocols are included in the Supplementary Information.
For hsCUL2 and hsRBX1, stocks of baculovirus infected insect cells (BIICs) were produced and stored in the vapor phase of liquid nitrogen. For protein expression, BIIC aliquots were diluted into Sf9 cells in suspension culture. The Sf9 insect cells were isolated from Spodoptera frugiperda cell line IPLB-Sf-21-AE. The Sf9 cells used in in cell degradation assays were obtained from Expression Systems, LLC. Infection and protein expression were allowed to proceed for 90 h at 27 °C, at which point cells were harvested by centrifugation. Cell pellets were gently resuspended in Sf9 lysis buffer (50 mM HEPES, pH 7.5, 200 mM NaCl, 1 mM TCEP, and complete protease inhibitor cocktail (Sigma)). Proteins were purified by IMAC, size exclusion, and ion exchange chromatography steps. Detailed expression and purification protocols are included in the Supplementary Information.
SPR binary affinity assessment
All measurements obtained for binary interaction SPR experiments were collected using a Biacore 8 K instrument (Cytiva) at 25 °C. Protein immobilization was completed in 10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05% P20 (Cytiva BR100671). Protein was immobilized on a carboxymethylated dextran surface with nitrilotriacetic acid (Cytiva BR100532) using a combined His-tag capture (Cytiva 28995043) and direct amination coupling approach (Cytiva BR100633). The protein was immobilized on flow cell 2, while flow cell 1 was activated and deactivated to be used as a reference surface. Binary interaction analysis of compounds to sfVBC, sfWDS, and sfBRD3 was completed in 10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05% P20, 1 mM DTT, and 2% DMSO (Sigma Aldrich 276855). Binary interaction analysis of sfWDS PROTACs to sfVBC was performed in 10 mM HEPES, pH 7.4, 300 mM NaCl, 0.05% P20, 250 mM Sucrose (Sigma Aldrich S5016), 5 mg/mL BSA (Sigma Aldrich A9418), 1 mM DTT, and 2% DMSO. Binary interaction analysis of sfBRD3 PROTACs to sfVBC was completed in 10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05% P20, 1 mg/mL carboxymethyl dextran sodium salt (Sigma Aldrich 86524), 1 mM DTT, and 2% DMSO. A 12-point dose-response curve, including a blank control, was created by diluting compounds in three-fold steps in 384-well plates (Greiner Bio-One 781271). Compound binding was measured using multi-cycle analysis at a flow rate of 30 µL per minute, with an association phase of 60 sec and a dissociation phase of 180 sec. Reference-subtracted and solvent-corrected data were analyzed using the Biacore Insight Evaluation Software (Cytiva) using multi-cycle kinetic evaluation. Sensorgrams were fit to the 1:1 kinetic binding model.
SPR ternary affinity assessment
All measurements obtained for ternary interaction SPR experiments were collected using a Biacore 8 K instrument (Cytiva) at 25 °C. Protein immobilization was performed in 10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05% P20 (Cytiva BR100671). sfVBC was immobilized on a carboxymethylated dextran surface with nitrilotriacetic acid (Cytiva BR100532) using a combined His-tag capture (Cytiva 28995043) and direct amination coupling approach (Cytiva BR100633). The protein was immobilized on flow cell 2, while flow cell 1 was activated and deactivated to be used as a reference surface. Buffer additives were included to mitigate non-specific binding between the ternary protein in solution and the sensor surface. Ternary interaction analysis of sfWDS PROTACs to sfVBC was performed in 10 mM HEPES, pH 7.4, 300 mM NaCl, 0.05% P20, 250 mM Sucrose (Sigma Aldrich S5016), 5 mg/mL BSA (Sigma Aldrich A9418), 1 mM DTT, and 2% DMSO. Ternary interaction analysis of sfBRD3 PROTACs to sfVBC was completed in 10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05% P20, 1 mg/mL carboxymethyl dextran sodium salt (Sigma Aldrich 86524), 1 mM DTT, and 2% DMSO. A 6-point dose-response curve was created by diluting compounds in the presence of POI (sfBRD3 or sfWDS) in four-fold steps in 384-well plates (Greiner Bio-One 781271). Ternary titrations were designed such that the POI concentration is greater than the PROTAC concentration and greater than 10x the binary KD50. Ternary complex binding was measured using single-cycle analysis at a flow rate of 30 µL per minute, with association phases of 75 sec and a dissociation phase of 900 sec. Reference-subtracted and solvent-corrected data were analyzed using the Biacore Insight Evaluation Software (Cytiva) using single-cycle kinetic evaluation. Sensorgrams were fit to the 1:1 kinetic binding model.
LD binary affinity assessment
All LD affinity measurements were performed in LD buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 0.05% TWEEN-20, 1 mM DTT, and 2% DMSO). The affinity of all fluorescent tracer compounds was determined based on the fluorescence polarization difference between free and bound compound, read in a fluorescence plate reader (Biotek Synergy 2). For LD assays, protein (sfVBC at 20 nM, sfWDS at 200 nM, or sfBRD3(BD2) at 100 nM) and appropriate tracer (10 nM of compound 4 for sfVBC, 20 nM of compounds 15 or 11 for WDS or BRD3(BD2)) were preincubated for 30 min, shielded from light. Unlabeled compounds of interest were diluted from 10 mM stocks to 200 μM in LD buffer and titrated in 15 μL volumes in 384-well black microplates (Corning 3820) for 16-point dose curves. Protein + tracer solution was added to all wells to bring the final volume to 30 μL and initiate binding. The displacement of the fluorophore tracer was monitored by changes to fluorescence polarization after a 45-minute incubation. All LD assays were performed with a minimum of n = 3 experimental replicates. Affinity values were determined from global fits using competitive binding and displacement equations in GraphPad Prism, with standard error reported.
AlphaLISA ternary complex assessment
All AL ternary complex measurements were performed in 1× PPI buffer (Revvity). All AL experiments were performed in 384-well white OptiPlates (Revvity). AlphaLISA signal was ready in a Biotek Synergy2 plate reader, and all reactions contained 30 μg/mL of both acceptor and donor beads (Revvity). All compounds were diluted from 10 mM stocks in 100% DMSO. A biotinylated avi-tagged form of sfElongin B was used to form sfVBbioC. 3x working stocks of sfVBbioC + sfWDS-His6 or sfVBbioC + MBP-sfBRD3(BD2) were prepared in PPI buffer at 105 nM. Compounds of interest were diluted to a 3x working stock of 100 μM in PPI buffer and titrated at 10 μL volume down wells in the 384-well OptiPlates in a 16-point series. Following this, 10 μL of E3 ubiquitin ligase and target protein solutions were added to all wells, and mixtures were incubated at room temperature for 30 min. After this incubation, 10 μL of pre-mixed AlphaLISA donor and acceptor beads were added to wells, bringing the final volume to 30 μL. For all sfVBbioC and sfBRD3(BD2) mixtures, streptavidin donor and MBP acceptor beads were added. For all sfVBbioC and sfWDS-His6 mixtures, streptavidin donor and His6 acceptor beads were added. Plates were sealed to prevent evaporation and incubated an additional 30 min, shielded from light, before Alpha response signals were measured. All AL assays were performed with a minimum of n = 3 experimental replicates. A global fit of the AL data in GraphPad Prism was used to determine ternary complex formation KDapp values, with standard error reported.
In vitro ubiquitination (IVQ)
E2 was purchased from R&D biosytems (Homo sapiens UbcH5a (hsUBcH5a), R&D E2-616-100). All other proteins were purified internally as described in protein purification section. All reaction components were added in a master mix at 2× final concentration, with the exception of buffer (1× concentration: 25 mM HEPES, pH 7.5, 125 mM NaCl, 2 mM MgCl2, 1 mM DTT (fresh)). Buffer was added from a 10× stock followed by 35 µM Fluorescein-Ubiquitin, 200 nM sfUBA1 (E1), 400 nM E2, 1.5 µM MBP-sfBRD3(BD2)-His6, 300 nM sfVBC, 300 nM hsCUL2/hsRBX1, and 4 mM ATP/MgCl2 solution (pH 7.0). PROTACs were diluted to 2× final concentration in 1× buffer with 0.25% DMSO. Six µL of protein master mix was added to a 384 well PCR plate followed by 6 µL of compound dilutions. The plate was spun at 500 × g for 5 min to mix reaction, sealed, and incubated at room temperature for 2 h. After the incubation 12 µL of 2× loading dye (NuPAGE™ LDS Sample Buffer) was added to each reaction and spun at 500 × g. A total of 8 µL of each reaction was loaded into an SDS gel (Invitrogen Bolt™ 4-12% Bis-Tris) for analysis by in gel fluorescence of Fluorescein-ubiquitin (488 channel, Licor Odyssey M). SDS-Page gels were then transferred to nitrocellulose using an Invitrogen iblot3. Membranes were blocked with blocking buffer (StartingBlock™ Blocking Buffer), then incubated in primary antibody (Invitrogen PA1-983B, 1:1000 in blocking buffer) overnight at 4 °C. Membrane was washed three times with TBST (TBS with Tween, Thermo Scientific), incubated in secondary antibody (IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody, 1:10,000 in blocking buffer) for 1 h, washed three times with TBST, then visualized to detect His6 tagged proteins (800 channel, Licor Odyssey M).
Sf9 cell degradation assays
In cell protein abundance measurements were carried out on peptide digests from treated Sf9 cells (the same as used for recombinant protein expression). Briefly, Sf9 cells were cultured in ESF921 media (Expression Systems LLC). The culture was divided into a 1e6 cell aliquots and treated with compounds for 24 h. Protein was isolated from the individual cultures in Tris SDS buffer. The samples were precipitated with Acetone and resolubilized with Urea for reduction/alkylation and digestion using Trypsin/LysC (Promega). The resulting peptide mixtures were cleaned with mixed mode cation exchange (MCX) (Waters) and combined with custom stable isotope peptide standards (Genscript) matching the target sequences as internal reference standards. All target specific abundance data was collected using a Shimdazu Nexera LC coupled to an LC-8050 triple quadrupole mass spectrometer operated in MRM mode. Data processing was carried out using Skyline software51. Global proteomics samples were all prepared in the same manner and 0.5 µg/sample was analyzed using a Thermo Scientific Orbitrap Astral Mass Spectrometer coupled to a Vanquish Neo UHPLC system. Representative spectra and fragmentation for sfBRD3 and sfWDS targeted data, along with detailed sample isolation and instrument settings for both global and targeted methods are represented in Supplementary Figs 10–13 and additional information about the protocol is available in the Supplementary information.
In vivo experiments with Fall Armyworm
Fall Armyworm (FAW) larvae were obtained from Benzon Research and maintained in an incubator at 25 °C with 60-70% humidity for 24 h prior to experimentation. Fifth instar (L5) larvae were weighed and evenly allocated across treatment groups; subsequent sample processing and data collection were performed in a blinded manner, with samples assigned random numerical identifiers and processed in this randomized order. Although no formal sample size calculation was performed, 6 larvae per group were used consistently for experiments and no samples were excluded for statistical analysis. All in vivo experiments were performed independently at least twice to confirm reproducibility. Each larva received an injection of either 0.1nmol/mg PROTAC or DMSO vehicle, unless otherwise stated, before being returned to the incubator for either 6 or 24 h until dissection. Carcasses and fat bodies were extracted from dissected larvae, homogenized with 1.4 mm ceramic beads through three cycles of 1700 rpm shaking with 15-second intervals in PBS containing HALT protease inhibitor. Lysates underwent centrifugation, and the protein concentration of the soluble fraction was determined via BCA assay. Subsequently, 1 mg of protein was acetone-precipitated, solubilized in 8 M urea, reduced with DTT, alkylated with iodoacetamide, and digested overnight with Trypsin/LysC. The resulting peptides were quantified using the Pierce peptide assay, with 150 μg/sample undergoing separation through plate-based reverse-phase fractionation, followed by additional purification by plate based MCX. Approximately 2 μg of each sample was subjected to targeted MS analysis using a Shimadzu QQQ. MS Instrumentation details and data processing of in vivo samples was carried out in the same manner as previously described for in cell experiments.
Statistics and Reproducibility
The peptide intensity values obtained from the mass spectrometer are normalized by taking the ratio of sample peptide to isotopic peptide intensity in each well. That ratio is divided by the sample’s total peptide quantity to remove the effect of total protein differences between samples. Finally, replicates of each treatment are converted to a percentage by dividing by the average of DMSO treated samples and multiplying by 100. The percentages are used in statistical analysis of in cell and in-organism degradation data with GraphPad Prism using an unpaired t-test algorithm. Error bars on plots represent±sem. Ligand displacement affinity curves are fitted using the One site—Fit Ki algorithm in GraphPad Prism. Cellular dose response curves are fitted using the Inhibitor vs. response (4-parameter) algorithm in GraphPad Prism. All GraphPad Prism data was analyzed in version 10.2.0. for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com. Global proteomic data were analyzed using the software packages DIANN v1.8, Direct LFQ and Perseus, additional details available in supplementary materials52,53,54. Regarding experimental and technical replicates, “n =” refers to the number of biological replicates unless otherwise specified. The degradation results in sf9 cell and Spodoptera frugiperda experiments are representative of results obtained from at least three independent experiments on the compounds presented in this publication. All ligand displacement affinity, SPR affinity, and ternary complex results were produced from at least three experimental replicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All source data generated in this study are provided in the Supplementary Information and Supplementary Data files. The raw data and DIA_NN search files for comparative proteomics described in Supplementary Fig. 6 have been deposited to the ProteomeXchange Consortium via the PRIDE55 partner repository with the dataset identifier PXD062088.
References
Bondeson, D. P. & Crews, C. M. Targeted Protein Degradation by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 57, 107–123 (2017).
Sakamoto, K. M. et al. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 98, 8554–8559 (2001).
Békés, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 (2022).
Zhao, L., Zhao, J., Zhong, K., Tong, A. & Jia, D. Targeted protein degradation: mechanisms, strategies and application. Signal Transduct. Target. Ther. 7, 113 (2022).
Schapira, M., Calabrese, M. F., Bullock, A. N. & Crews, C. M. Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discov. 18, 949–963 (2019).
Ciulli, A. & Hsia, O. PROTAC degraders: mechanism, recent advances, and future challenges. in Protein Homeostasis in Drug Discovery (eds. Kostic, M. & Jones, L. H.) 317–356 (Wiley, 2022). https://doi.org/10.1002/9781119774198.ch9.
Samarasinghe, K. T. G. & Crews, C. M. Targeted protein degradation: A promise for undruggable proteins. Cell Chem. Biol. 28, 934–951 (2021).
Burslem, G. M. et al. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem. Biol. 25, 67–77.e3 (2018).
Gao, H. et al. FAK-targeting PROTAC as a chemical tool for the investigation of non-enzymatic FAK function in mice. Protein Cell 11, 534–539 (2020).
Burke, M. R., Smith, A. R. & Zheng, G. Overcoming Cancer Drug Resistance Utilizing PROTAC Technology. Front. Cell Dev. Biol. 10, 872729 (2022).
Bondeson, D. P. et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11, 611–617 (2015).
Mares, A. et al. Extended pharmacodynamic responses observed upon PROTAC-mediated degradation of RIPK2. Commun. Biol. 3, 140 (2020).
Cromm, P. M. & Crews, C. M. Targeted protein degradation: from chemical biology to drug discovery. Cell Chem. Biol. 24, 1181–1190 (2017).
Chirnomas, D., Hornberger, K. R. & Crews, C. M. Protein degraders enter the clinic — a new approach to cancer therapy. Nat. Rev. Clin. Oncol. 20, 265–278 (2023).
Oerke, E. -C. Crop losses to pests. J. Agric. Sci. 144, 31–43 (2006).
Savary, S. et al. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 3, 430–439 (2019).
United Nations. World Population Prospects 2019: Highlights. (UN, 2019). https://doi.org/10.18356/13bf5476-en.
Duke, S. O. Why have no new herbicide modes of action appeared in recent years?. Pest Manag. Sci. 68, 505–512 (2012).
Gould, F., Brown, Z. S. & Kuzma, J. Wicked evolution: Can we address the sociobiological dilemma of pesticide resistance?. Science 360, 728–732 (2018).
Zuin, A., Isasa, M. & Crosas, B. Ubiquitin signaling: extreme conservation as a source of diversity. Cells 3, 690–701 (2014).
Zheng, N. & Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 86, 129–157 (2017).
Shu, K. & Yang, W. E3 Ubiquitin Ligases: Ubiquitous Actors in Plant Development and Abiotic Stress Responses. Plant Cell Physiol. 58, 1461–1476 (2017).
Finley, D., Ulrich, H. D., Sommer, T. & Kaiser, P. The Ubiquitin–Proteasome System of Saccharomyces cerevisiae. Genetics 192, 319–360 (2012).
Montezano, D. G. et al. Host Plants of Spodoptera frugiperda (Lepidoptera: Noctuidae) in the Americas. Afr. Entomol. 26, 286–300 (2018).
Overton, K. et al. Global crop impacts, yield losses and action thresholds for fall armyworm (Spodoptera frugiperda): A review. Crop Prot. 145, 105641 (2021).
Johnson, M. et al. NCBI BLAST: a better web interface. Nucleic Acids Res 36, W5–W9 (2008).
Krieger, J. et al. Systematic potency and property assessment of VHL ligands and implications on PROTAC design. ChemMedChem 18, e202200615 (2023).
Testa, A., Hughes, S. J., Lucas, X., Wright, J. E. & Ciulli, A. Structure-based design of a macrocyclic PROTAC. Angew. Chem. Int. Ed. 59, 1727–1734 (2020).
Gadd, M. S. et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 13, 514–521 (2017).
Hollmann, M., Simmerl, E., Schäfer, U. & Schäfer, M. A. The essential Drosophila melanogaster gene wds (will die slowly) codes for a WD-repeat protein with seven repeats. Mol. Genet. Genomics MGG 268, 425–433 (2002).
Filippakopoulos, P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010).
Nicodeme, E. et al. Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123 (2010).
Hewings, D. S. et al. Progress in the development and application of small molecule inhibitors of bromodomain–acetyl-lysine interactions. J. Med. Chem. 55, 9393–9413 (2012).
Yu, X. et al. A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci. Transl. Med. 13, eabj1578 (2021).
Jin, J., Gang, W.,Jing, l., Xufen, Y. & Dongxu, L. Wd40 repeat domain protein 5 (wdr5) degradation/disruption compounds and methods of use. (2019).
Lange, V., Picotti, P., Domon, B. & Aebersold, R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 4, 222 (2008).
LC-MS/MS in Proteomics. vol. 658 (Humana Press, Totowa, NJ, 2010).
Hornberger, K. R. & Araujo, E. M. V. Physicochemical property determinants of oral absorption for PROTAC protein degraders. J. Med. Chem. 66, 8281–8287 (2023).
Wakabayashi, S. et al. Ohgata, the single drosophila ortholog of human cereblon, regulates insulin signaling-dependent organismic growth. J. Biol. Chem. 291, 25120–25132 (2016).
Lane, D. P. et al. The Mdm2 and p53 genes are conserved in the Arachnids. Cell Cycle 9, 748–754 (2010).
Matyskiela, M. E. et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol. 14, 981–987 (2018).
Choy, A. et al. Decoding the ubiquitin-mediated pathway of arthropod disease vectors. PLoS ONE 8, e78077 (2013).
Medvar, B., Raghuram, V., Pisitkun, T., Sarkar, A. & Knepper, M. A. Comprehensive database of human E3 ubiquitin ligases: application to aquaporin-2 regulation. Physiol. Genomics 48, 502–512 (2016).
Gingerich, D. J., Hellmann, H., Christians, M. J. & Stone, S. L. Editorial: structure, function, and evolution of E3 ligases and targets. Front. Plant Sci. 12, 767281 (2021).
Bramlett, M., Plaetinck, G. & Maienfisch, P. RNA-based biocontrols—a new paradigm in crop protection. Engineering 6, 522–527 (2020).
Gressel, J. Perspective: It is time to consider new ways to attack unpesticidable (undruggable) target sites by designing peptide pesticides. Pest Manag. Sci. 78, 2108–2112 (2022).
Powles, S. B. & Yu, Q. Evolution in action: plants resistant to herbicides. Annu. Rev. Plant Biol. 61, 317–347 (2010).
Hawkins, N. J., Bass, C., Dixon, A. & Neve, P. The evolutionary origins of pesticide resistance. Biol. Rev. 94, 135–155 (2019).
Sparks, T. C., Wessels, F. J., Lorsbach, B. A., Nugent, B. M. & Watson, G. B. The new age of insecticide discovery-the crop protection industry and the impact of natural products. Pestic. Biochem. Physiol. 161, 12–22 (2019).
Roy, M. J. et al. SPR-Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate. ACS Chem. Biol. 14, 361–368 (2019).
Pino, L. K. et al. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 39, 229–244 (2020).
Demichev, V. et al. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Ammar, C., Schessner, J. P., Willems, S., Michaelis, A. C. & Mann, M. Accurate label-free quantification by directLFQ to compare unlimited numbers of proteomes. Mol. Cell. Proteom. 22, 100581 (2023).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Perez-Riverol, Y. et al. The PRIDE database at 20 years: 2025 update. Nucleic Acids Res. 53, D543–D553 (2025).
Acknowledgements
We would like to thank all our Bayer and Arvinas colleagues for in-depth feedback and suggestions regarding this research study. We also gratefully thank all other present or former Oerthlings for their fruitful input and discussions.
Author information
Authors and Affiliations
Contributions
D.J.S., K.R.P., and D.F. conducted bioinformatics analysis and molecular modeling. H.D., J.C., J.S., and D.F. designed the PROTAC molecules. G.L.M., B.E.W., J.P., M.S., and E.D.S. conducted the biochemical assays. G.L.M. and D.O. performed the recombinant protein expression. B.F. and S.C.B. conducted the proteomics work and all corresponding data analysis. M.L., C.C.K., and A.P. performed preliminary ligase and assay work. B.F., A.N., E.A., and S.C.B. conducted the in cell and in vivo degradation assays and all associated data analysis. P.I., A.P., C.L., S.G., J.S., S.V., K.B., and D.F. conceived, monitored, and supervised the project. G.L.M., H.D., J.C., B.F., B.E.W., S.C.B., D.J.S., P.I., E.D.S., and D.F. wrote the paper. G.L.M., H.D., B.F., B.E.W., and D.F. edited and formatted the paper. D.F. reviewed, packaged, and submitted the paper.
Corresponding author
Ethics declarations
Competing interests
Oerth Bio has filed IP (WO2024050016) on compounds related to this work. Oerth Bio is supporting the commercialization of this IP. Oerth Bio is a joint venture between Bayer CropScience AG and Arvinas. All co-authors who are current or former employees of Oerth Bio, have equity shares of Oerth Bio and/or Bayer AG and/or Arvinas.
Peer review
Peer review information
Communications Biology thanks Laura Santos and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Laura Rodríguez Pérez.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Morgan, G.L., Dickson, H., Ford, B. et al. Pioneering protein degradation for agricultural applications. Commun Biol 8, 591 (2025). https://doi.org/10.1038/s42003-025-08013-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08013-y
