
Abstract
Anti-Müllerian Hormone (AMH), secreted by granulosa cells (GCs) of preantral follicles, regulates early-stage follicle growth by inhibiting the activation of primordial follicles. While GCs facilitate follicle development and ovulation through estrogen and progesterone secretion, the role of AMH in sheep GCs and its impact on follicle development remains unclear. Here, we show that exogenous AMH inhibits primordial follicle development utilizing in vitro culture of fetal sheep ovarian cortex model. Knockdown of AMH in sheep GCs increases cell proliferation, decreases apoptosis, and elevates estradiol (E2) secretion, while overexpression has the opposite effects. RNA-seq analysis suggests that AMH modulates genes involved in cell cycle regulation and MAPK signaling. Notably, AMH downregulates estrogen receptor β (ESR2) expression and suppresses FSH-induced upregulation of aromatase (CYP19A1). These findings demonstrate AMH regulates GCs function through multiple pathways, offering insights into sheep fertility and potential targets to enhance reproductive efficiency.
Similar content being viewed by others
Introduction
AMH is specifically expressed in ovarian granulosa cells and testicular Sertoli cells, playing a crucial role in regulating sex differentiation and gonadal function1,2. In fetal male animals, AMH drives the degeneration of the Müllerian duct, which leads to the development of the uterus, fallopian tubes, and the upper part of the vagina in females3. In female animals, the number of primordial follicles is determined from the fetal stage, is fixed and non-renewable, and serves as the source of developing follicles and oocytes throughout the reproductive process4. AMH is secreted by the granulosa cells of preantral and small antral follicles, not by primordial follicles. Therefore, it cannot directly represent the number of follicles in the ovaries, just represent the number of growing follicles recruited from the primordial pool5. As a marker of ovarian reserve, AMH can only indirectly reflect the quantity of remaining oocytes in the ovaries6. The development process of primordial follicles into primary follicles is called the activation of primordial follicles7, which ultimately becomes a mature follicle that can be ovulated. AMH suppresses the activation of primordial follicles8,9,10, inhibits the responsiveness of follicles to FSH, and regulates FSH-dependent preantral follicle growth11, therefore, preventing the selection of multiple follicles12. In the context of ovarian granulosa cell function, previous studies have demonstrated that in mice, AMH and INHA could act synergistically to inhibit the synthesis and secretion of steroid hormones, thereby significantly enhancing fertility13. Conversely, in chicken ovarian granulosa cells, AMH does not appear to regulate follicle development by directly affecting steroidogenesis markers, FSHR, or BMPR2 at the corresponding follicle stage14. In bovine, AMH signaling plays a crucial role in regulating granulosa cell proliferation and preventing premature differentiation in granulosa cells from 5–8 mm follicles before follicle selection occurs15. AMH is a reliable marker for ovarian reserve in sheep. In both Norduz ewes and Romanov ewes, serum AMH levels remain stable across different phases of the sexual cycle, including anestrus, estrus, and diestrus, and are not influenced by age or body condition score (BCS)16,17. In Santa Ines ewe lambs, AMH levels remain stable despite parasite infection and variations in diet protein levels18. A single blood sampling can reliably assess peripheral AMH levels. However, some studies have shown that serum AMH levels in sheep are not stable before puberty. In Aragonesa sheep, serum AMH levels are not stable until the age of 6 months19. In Romanov sheep, higher AMH levels are associated with larger litter sizes16. However, there are differences among breeds. For example, high-fertility Booroola ewes carrying the FecB mutation exhibit low AMH levels2. AMH also correlates positively with the number of ovarian follicles ≤3 mm in diameter (AFC)20. Recent studies have shown that extending FSH treatment improves embryo yield in low-AMH ewes but not in high-AMH ewes, suggesting AMH as a potential biomarker for embryo yield in sheep multi-follicle prediction21. However, both AMH and AFC (antral follicle count) have limitations in predicting fertility. Given that fertility is a multifactorial trait, a comprehensive evaluation integrating AMH, AFC, and other reproductive indicators—such as progesterone and estrogen levels—is recommended for more accurate fertility prediction. These findings underscore the complexity of AMH’s role across different species. However, the role of AMH in sheep granulosa cells remains unclear and the underlying mechanisms require further investigation.
AMH is a member of TGFβ family which shares similar structural and mechanism. AMH signals via specific receptor AMHR2 in tandem with type I receptors (ALK2/ACVR1, ALK3/BMPR1A, ALK6/BMPR1B) and activation of BMP R-Smads 1, 5, and 822,23. This results in SMAD1, 5, and 8 binding to SMAD4 and relocating the complex to the nucleus in AMH target cells24. Additionally, AMH is capable of modulating microRNA expression, thereby promoting the silencing of downstream target genes in mouse ovaries and regulating follicular development and neuroendocrine through FSH25. AMH expression in the ovaries of sheep, cattle, buffalo, mice, and humans begins in primary follicles, with the highest expression levels observed in preantral and small antral follicles, showing a significant decrease as the follicle diameter increases13. In recent years, AMH has been detected in organs other than the ovaries, including the hypothalamus, pituitary gland, uterus and breast. AMH exerts its effects across the Hypothalamic-Pituitary-Gonadal (HPG) axis. AMH positive feedback regulates GnRH/LH secretion. AMH can act on the hypothalamus and increase the activity of GnRH neurons in the hypothalamus. GnRH signal acts on the adenohypophysis to positively regulate LH secretion26. In Leydig cells, AMH controls LH-mediated androgen production27. PCOS patients are characterized by serum AMH levels 2–4 times higher than normal people28. In PCOS patients, altered GnRH pulse frequency from the hypothalamus results in elevated serum LH and reduced FSH, increasing the LH/FSH ratio. This disrupts follicular development, slows small follicle growth, enhances antral follicle recruitment, and stimulates granulosa cells to secrete excess AMH29,30. These findings indicate that AMH may regulate the development of sheep follicles via the hypothalamic-pituitary-ovarian (HPO) axis, which is closely associated with prolificacy. Further investigation is needed to understand the underlying mechanisms in sheep.
This study presents a systematic investigation of the role of AMH in sheep ovarian function and its regulatory impact on granulosa cells, utilizing both in vitro ovarian cortex cultures and granulosa cell models. Our findings reveal that AMH significantly inhibits the development of primordial follicles and affects the proliferation, apoptosis, and hormone synthesis of granulosa cells through multiple signaling pathways. These results not only deepen our comprehension of AMH’s function in the sheep ovary but also identify potential therapeutic targets for enhancing reproductive efficiency in sheep. It also provides a theoretical basis for improving the litter size and economic benefits of sheep in production practice.
Results
AMH inclusion in vitro inhibits primordial follicle growth in ovarian cortical cultures
In gonadal tissues, AMH is highly expressed in the ovary, and AMHR2 in the testis (Fig. 1A). In Hu sheep ovaries, AMH and AMHR2 exhibit an initial upregulation followed by downregulation across developmental stages, peaking at postnatal Day 90 (D90) and Day 0 (D0), respectively (Fig. 1B). Furthermore, the AMH was more highly expressed in follicles <3 mm than in follicles 3–5 mm and >5 mm (Fig. 1C, D).

A Relative mRNA expression of AMH and AMHR2 in different tissues of Hu sheep. B Relative mRNA expression of AMH and AMHR2 in Hu sheep ovary at different ages. C Relative mRNA expression of AMH and AMHR2 in different size follicles (<3 mm, 3–5 mm, and >5 mm). D Relative protein expression of AMH in different size follicles (<3 mm, 3–5 mm, and >5 mm). E Comparative Ovarian Imaging at 80 and 120 Days of Fetal sheep. F HE staining of fresh ovarian tissue from fetal Hu sheep at 80 Days. G–J HE staining of each treatment group after 15 days of in vitro culture of fetal sheep ovarian cortical pieces at 80 Days. K HE staining result of fresh ovarian tissue from fetal Hu sheep at 120 Days. L–O HE staining results of each treatment group after 15 days of in vitro culture of fetal sheep ovarian cortical pieces at 120 Days, Scale = 25 μm. P Relative mRNA expression levels of AMH, FSHR, HSD3B, and CYP19A1 in each treatment group after 15 days of in vitro culture of ovarian cortices from three-month-old Hu sheep. The values with different labeling a-f mean significantly differences (P-value < 0.05). n = 3 biologically independent samples/animals/independent experiments. The same as follows.
Ovarian cortical pieces were obtained from mid-pregnancy (80 days) and post-pregnancy (120 days) fetal Hu sheep (Fig. 1E) were cultured for 15 days under AMH, FSH, or a combination of both. Mid-pregnancy fetal ovaries are predominantly primordial follicles (Fig. 1F), while post-pregnancy ovaries are primarily composed of primary follicles (Fig. 1K). In 80-day fetal sheep ovarian cortical pieces, AMH treatment maintained flat granulosa cells around follicles, unlike the cubic morphology observed in control, FSH, and AMH + FSH groups (Fig. 1G-J). In the 120-day fetal sheep ovarian cortical pieces, granulosa cell layers were minimal in both AMH and AMH + FSH treated groups, with all cells forming monolayers (Fig. 1L-O). In cultures of three-month-old Hu sheep ovarian cortical pieces, the expression of FSHR, HSD3B, and CYP19A1 gene significantly reduced by AMH treatment, but this effect was reversed by co-treatment with FSH (Fig. 1P).
AMH suppresses Hu sheep follicular GCs proliferation in vitro
To further confirm the regulatory effect of AMH on GCs growth, we transfected AMH siRNA sequence (SI-AMH) and overexpression vector (OE-AMH) into in vitro cultured primary GCs (Fig. 2A). Granulosa cells were identified by immunofluorescence using FSHR. AMH was expressed in both the cytoplasm and nucleus, while the receptors AMHR2 and BMPR1B were primarily localized in the cytoplasm (Fig. 2B). Compared to GCs transfected with control siRNA (NC), transfection with AMH-specific siRNA (SI-AMH 332) resulted in the most significant reduction in both AMH mRNA expression (Fig. 2C) and protein expression (Fig. 2E). Knockdown of AMH significantly increased cell viability, promoted cell proliferation, and decreased cell apoptosis. In contrast, transfection of GCs with an AMH overexpression vector increased AMH mRNA (Fig. 2D) and protein levels (Fig. 2F), suppressed cell viability, inhibited cell proliferation, and increased cell apoptosis (Fig. 2G-I).

A Bright field images of primary GCs cultures from Hu sheep, taken from the first day to the third day, Scale = 100 μm. B Fluorescence staining of FSHR, AMH, AMHR2, and BMPR1B (red), and nuclei were counterstained with DAPI (blue), Scale = 100 μm. C–F AMH mRNA and protein expression levels in GCs following AMH knockdown and overexpression. G GCs viability was detected using CCK-8, n = 3 for each group. H GCs proliferation was detected using EdU, Scale = 100 μm. I The ratio of apoptosis was determined using annexin V-based flow cytometry. * and ** means significantly differences (P-value < 0.05). The same as follows.
We further detected the expression of mRNA and proteins associated with proliferation and apoptosis in cultured GCs. Following transfected with AMH siRNA and overexpression vector, quantitative PCR (qPCR) showed a downregulation of BAX and the BAX/BCL2 ratio, along with an upregulation of BCL2 and PCNA after AMH knockdown (Fig. 3A-D). Consistent with these findings, Western blot analysis demonstrated decreased levels of PARP and the BAX/BCL2 ratio, as well as increased levels of BCL2 and PCNA after AMH knockdown (Fig. 3I–N). Furthermore, the overexpression of AMH resulted in an upregulation of BAX and the BAX/BCL2 ratio, alongside a downregulation of PCNA, as shown by qPCR analysis in Fig. 3E–H. Similarly, western blot analysis revealed increased levels of PARP and the BAX/BCL2 ratio, as well as decreased levels of PCNA following AMH overexpression (Fig. 3O–T).

A–H BAX, BCL2, BAX/BCL2, PCNA mRNA expression levels in GCs following AMH knockdown and overexpression. I–N BAX, BCL2, PARP, PCNA protein expression levels in GCs following AMH knockdown. O–T BAX, BCL2, PARP, PCNA protein expression levels in GCs following AMH overexpression.
RNA-seq and DEG analysis of AMH knockdown and overexpression in GCs
To delineate the mechanisms regulated by AMH that alter granulosa cell function, we constructed and sequenced twelve cDNA libraries from transfected GCs, with three libraries each from the SI-AMH, NC, OE-AMH, and OE groups. Analysis of the RNA-seq dataset showed that the granulosa cells of the four different treatment groups were distributed in different quadrants, as exemplified by principal component analysis (PCA) (Fig. 4A). The TPM method was employed to assess gene expression levels, using a fold-change of > |1.5| and a P-value of < 0.05 as the screening criteria. A total of 1486 differentially expressed genes (DEGs) were identified between the NC and SI-AMH groups, comprising 541 upregulated and 945 downregulated DEGs (Fig. 4B). Compared to the OE and OE-AMH groups, a total of 324 DEGs were identified, including 263 that were upregulated and 61 that were downregulated (Fig. 4C). Heatmap analysis showed distinct gene expression patterns between SI-AMH and NC, as well as between OE-AMH and OE (Fig. 4D). The groups of knockdown and overexpression shared 37 common genes (Fig. 4E). GO and KEGG analyses of DEGs from AMH knockdown showed enrichment in biological processes such as regulation of cell cycle and developmental process modulation, with affected pathways included the cell cycle, MAPK, and FOXO (Fig. 4F, H). In contrast, GO and KEGG analyses of DEGs from AMH overexpression enrichment in protein binding, with affected pathways included NOD-like receptor and RIG-I-like receptor (Fig. 4G, I). Many of these processes and pathways were highly related with GCs growth and follicular development.

A PCA analysis between samples. B Volcano plot displays DEGs with a P-value < 0.05 between the NC and SI-AMH groups, and the OE and OE-AMH groups. C The expression patterns of DEGs are illustrated in a cluster map that compares SI-AMH with NC and OE-AMH with OE. (P-value < 0.05; log2 fold change > |1.5 | , analyzed using DESeq2). D Heatmap showing the gene expression pattern between the SI-AMH and NC groups, and the OE-AMH and OE groups. Red indicates the maximum value within a row, while blue represents the minimum value. E Venn analysis. F–I GO and KEGG enrichment results (P < 0.05) of all DEGs between the SI-AMH and NC groups, and the OE-AMH and OE groups.
AMH regulats cell cycle and inhibits steroid hormone synthesis of GCs
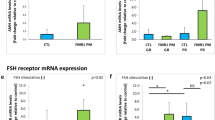
The knockdown of AMH resulted in a significant downregulation of most genes associated with the cell cycle. Conversely, the overexpression of AMH led to the upregulation of the majority of cell cycle-related genes (Fig. 5A). The qPCR results showed that the mRNA levels of CCND1, CCNE2, CCNB3, CCNA2, P53, and CDK6 significantly decreased after AMH knockdown and increased after AMH overexpression (Fig. 5B). Western blot analysis showed that the protein levels of CDK6 and CCND1 significantly decreased after AMH knockdown and increased after AMH overexpression (Fig. 5C, D). Flow cytometric analysis showed that AMH knockdown decreased the number of S phase cells, whereas AMH overexpression had no effect (Fig. 5E). Heatmap analysis showed that genes related to the estrogen signaling pathway, including FSHR, ESR2, and MAPK13, were upregulated after AMH knockdown. However, following the overexpression of AMH, the ESR2 and FSHR genes were downregulated in certain samples (Fig. 5F). The qPCR results showed that the mRNA levels of 3BHSD, 17BHSD, STAR, CYP11A1, CYP17A1, and CYP19A1 significantly increased after AMH knockdown, while they decreased after AMH overexpression (Fig. 5G). Western blot analysis showed that the protein levels of CYP19A1 significantly increased after AMH knockdown and decreased after AMH overexpression (Fig. 5H). The ELISA results showed that E2 secretion levels significantly increased after AMH interference and significantly decreased after AMH overexpression (Fig. 5I).

A Expression heatmap of DEGs in cell cycle signaling pathway. B CCND1, CCNE2, CCNB3, CCNA2, P53, and CDK6 mRNA expression levels in GCs following AMH knockdown and overexpression. C, D CDK6, CCND1 protein expression levels in GCs following AMH knockdown and overexpression. E Effects of knockdown and overexpression AMH gene on cell cycle progression in GCs. F Expression heatmap of DEGs related to the estrogen signaling pathway. G 3BHSD, 17BHSD, STAR, CYP11A1, CYP17A1, and CYP19A1 mRNA expression levels in GCs following AMH knockdown and overexpression. H CYP19A1 protein expression levels in GCs following AMH knockdown and overexpression. I E2 secretion levels were detected by ELISA following AMH knockdown and overexpression.
The mechanism of AMH regulating the function of GCs by inhibiting ESR2/ p38-MAPK signaling pathway
We treated GCs with 0, 2, 10, and 50 IU/mL of FSH for 24 h. The CCK8 results showed that the cell viability was the highest after treatment with 10 IU/mL of FSH. Additionally, both qPCR and Western blot results showed that the mRNA and protein levels of PCNA were the highest after treatment with 10 IU/mL of FSH (Fig. 6A-C). The Western blot results showed that the expression level of AMH decreases after treatment with 10 IU/mL of FSH. However, this decrease is rescued when FSH is co-treated with overexpress AMH (OE-AMH). At the same time, the expression level of CYP19A1 increases after FSH treatment; however, it decreases when co-treated with OE-AMH (Fig. 6D). KEGG enrichment analysis showed that AMH regulate granulosa cell function through the MAPK signaling pathway. Western blot results showed that the p-p38/p38 ratios increased after AMH knockdown; conversely, these ratios decreased after AMH overexpression (Fig. 6E-F). PPI (Protein-Protein Interaction Networks) analysis showed an interaction between AMH and ESR2 (Fig. 6G). After AMH knockdown, the expression level of ESR2 significantly increased (Fig. 6H). When GCs were treated with the ESR2 agonist DPN (20 nM) and the ESR2 inhibitor PHTPP (20 nM), qPCR results showed that AMH expression decreased after DPN treatment and increased after PHTPP treatment (Fig. 6I). Western blot results showed that the protein level of ESR2 increased after DPN treatment, but it is suppressed when treated with both DPN and OE-AMH (Fig. 6J). However, the combined treatment with PHTPP and SI-AMH did not significantly affect the expression level of ESR2 (Fig. 6K). There is evidence of negative feedback regulation between AMH and ESR2, where AMH inhibits the expression of ESR2, thereby reducing the effects of estrogen. The above results showed that AMH influences the function of granulosa cells by inhibiting the ESR2/p38 MAPK pathway.

A GCs were treated with FSH for 24 h, and cell viability was detected using CCK8, n = 3. B, C The mRNA and protein expression levels of PCNA were detected after the treatment of GCs with FSH. D The protein expression levels of AMH and CYP19A1 were detected after the treatment of GCs with FSH or co-treatment with OE-AMH. E, F The protein expression levels of p-38 and p-p38 in GCs following AMH knockdown and overexpression. G Protein interaction network of DEGs. H The mRNA expression levels of ESR2 in GCs following AMH knockdown and overexpression. I The mRNA expression levels of AMH in GCs following treatment with DPN (ESR2 agonists, 20 nM) or PHTPP (ESR2 inhibitors, 20 nM) for 24 h. J The protein expression levels of ESR2 in GCs following treatment with DPN or co-treatment with OE-AMH. K The protein expression levels of ESR2 in GCs following treatment with PHTPP or co-treatment with SI-AMH.
Discussion
This study systematically investigates the role of AMH in sheep ovaries and its regulatory effects on granulosa cells through in vitro ovarian cortex culture and sheep granulosa cell models. Initially, our in vitro ovarian culture experiments demonstrated that the exogenous addition of AMH protein inhibited the development of primordial follicles. Subsequently, we performed interference and overexpression experiments of the AMH gene in sheep granulosa cells, revealing that the expression levels of the AMH gene significantly modulate critical physiological processes, including cell proliferation, cell cycle progression, and hormonal secretion in granulosa cells. Notably, AMH overexpression was found to suppress granulosa cell proliferation and enhance apoptosis, while concurrently reducing the biosynthesis of estradiol (E2). RNA-seq suggested that AMH potentially exerts its regulatory effects on granulosa cell function and estrogenic activity by modulating the p38 mitogen-activated protein kinase (MAPK)/estrogen receptor 2 (ESR2) signaling pathway. Furthermore, AMH was observed to diminish the regulatory influence of FSH on granulosa cells. Collectively, these findings elucidate the crucial role of AMH in the ovine ovary and offer perspectives on the mechanisms underlying AMH’s involvement in reproductive health and disease pathology.
In prepubertal Sarda ewe lambs, both AFC and AMH levels can predict ovarian response to FSH stimulation, but neither reliably predicts the size of the primordial follicle pool or the number of healthy follicles31. In Romanov ewes, higher AMH levels are associated with larger litter sizes16, and barren ewes exhibit significantly lower numbers of follicles (including both antral and primordial follicles) and serum AMH levels compared to ewes with normal fertility32. However, significant differences exist among breeds. For example, high-fertility Booroola ewes carrying the FecB mutation (FecBB) exhibit low AMH levels2. This phenomenon is likely attributable to reduced BMP signaling, which may diminish the activation of the AMH promoter and lead to lower AMH gene expression levels. Consistent with this finding, our lab’s prior transcriptomic analysis of <3 mm follicles from high-prolificacy (genotype: FecBB) and low-prolificacy (genotype: FecB + ) Hu sheep revealed higher AMH gene expression in the low-prolificacy group. Similar results have been reported in previous studies on sheep, which demonstrated that AMH knockdown does not affect the recruitment of primordial follicles but accelerates the rate at which follicles progress through the gonadotropin-responsive phase. This acceleration subsequently leads to a significant increase in the number of gonadotropin-dependent follicles and the ovulatory quota33. Given these findings, our current research aims to explore how AMH affects the fertility of Hu sheep ewes and their regulatory mechanisms in ovarian follicular development and granulosa cell function, areas that remain underexplored in current research.
As a multiparous animal, the reproductive biological characteristics of the Hu sheep differ from those of uniparous animals. Therefore, the results of this study are of great significance for understanding and addressing the reproductive issues of sheep and other multiparous animals. In ovarian organ culture experiments of a newborn rat, AMH treatment for two or ten days revealed its inhibitory effect on primordial follicle assembly through modulating the developmental interaction between ovarian stroma and epithelium. This finding underscores the critical role of AMH in regulating follicle pool size, thereby influencing female fertility potential34. In line with these results, recombinant AMH at a concentration of 100 ng/ml was found to exert an inhibitory effect on the early-stage development of human ovarian follicles in vitro8. Furthermore, consistent with our observations, AMH has been shown to inhibit the activation of primordial follicles and to decelerate follicle growth in bovine ovarian cortex cultures35. These collective findings highlight the conserved role of AMH across species in the control of ovarian folliculogenesis. Single-cell sequencing of AMH-injected newborn mouse ovaries revealed that AMH suppresses early follicle development by inhibiting granulosa cell proliferation and differentiation, thereby maintaining them in a quiescent state36, a pattern that mirrors our observations. BMP6 upregulated GATA4 through the SMAD1/5/8 signaling pathway, which in turn promoted AMH expression37. Besides the TGF/SMAD signaling pathway, AMH has been shown to inhibit proliferation and induce apoptosis in breast cancer cell lines through the NF-κB signaling pathway38. BMP15 can regulate the expression of AMH by inhibiting the p38 MAPK pathway39. FSH and cAMP enhance AMH transcription through the protein kinase A and p38 MAPK signaling pathways40. Our results also revealed that AMH regulates the function of granulosa cells through the p38 MAPK signaling pathway. Most studies have demonstrated a negative correlation between AMH and FSH levels in the ovaries. In mice, AMH decreased the expression of CYP19A1 aromatase, which is mediated by FSH. Additionally, high concentrations of FSH specifically reduced the expression of AMH in preantral and early antral follicles41. Furthermore, in a human granulosa tumor cell line (KGN cells), FSH inhibits GDF9 and BMP15-induced AMH expression through HDAC2-mediated deacetylation of H3K27ac42, which is consistent with our results.
Estrogens are primarily synthesized in the growing follicles of the ovaries through the FSH-induced aromatization of androgens secreted by theca cells. Estrogens are responsible for regulating the functions of the female reproductive system by binding to nuclear estrogen receptors (ESR1 and ESR2)43. Unlike previous studies, which demonstrated that E244or ESR2, rather than ESR145, inhibits the expression of AMH and AMHR2, our results indicate that, conversely, AMH can also inhibit the expression of ESR2 in granulosa cells. Consistent with the downregulation of ESR2 expression by AMH, serum AMH levels are higher in patients with PCOS, and it has been reported that ESR2 expression is reduced in individuals with PCOS46. This suggests that the negative feedback between AMH and ESR2 may be reciprocal. AMH negatively impacts antral follicle maturation47. There is also research indicating that AMH not only inhibits the growth of PMFs but also activates FOXO3A to induce autophagy, which allows them to survive and, in turn, protects the ovarian reserve48.
In summary, our findings demonstrate that exogenous AMH binds to AMHR2 on granulosa cells, inhibiting FSH-induced aromatase expression and reducing E2 synthesis, which in turn arrests the development of small antral follicles. Moreover, endogenous AMH gene suppresses E2 activity by downregulating ESR2, leading to decreased E2 receptor binding and ultimately inhibiting granulosa cell proliferation via the p38 MAPK pathway. These results highlight the pivotal role of AMH in follicular development and its potential as a therapeutic target for fertility regulation.
Methods
Inclusion and ethics
All experimental procedures were approved by the Animal Care and Use Committee of Nanjing Agricultural University and were strictly conducted according to the animal experiment guidelines (Approval ID: SYXK 2022-0031; Approval Date: 2022-6-10). We have complied with all relevant ethical regulations for animal testing. Hu sheep used in the experiments were transported and humanely slaughtered at a government-certified slaughter facility.
Isolation and culture of ovine cortical strips
The ovaries from Hu sheep (80 and 120 days of the embryonic period, female, n = 3), as well as the ovaries of Hu sheep (three-month-old, female, n = 3), were preserved in tissue storage solution (MACS, 130-100-108) and transported to the laboratory. The ovaries were bisected with a blade, and the cortex was separated and cut into 0.5 mm³ fragments. Each sample was selected randomly. Two to three tissue fragments were fixed in 4% paraformaldehyde to serve as a fresh tissue uncultured control group for immunohistochemical analysis, while the remaining tissues were subjected to in vitro culture. The samples were divided into four groups: the basal culture medium group, the basal culture medium plus FSH (1000 ng/ml) (Vetoquinol, 439166) group, the basal culture medium plus AMH (1000 ng/ml) (MCE, HY-P74413) group, and the basal culture medium plus both AMH and FSH group. The composition of the basal medium includes 7.5 mg/ml of BSA (Sigma, B2064), 1% non-essential amino acids (NEAA) (Sigma, M7145), 1% ITS-A (insulin, transferrin, selenium, sodium pyruvate) (Gibco, 51300-044), and 1% penicillin and streptomycin, all of which were added to MEM-α (Gibco, 32571101) medium. The samples were cultured in vitro for 15 days.
Tissue collection and cell culture
Tissues (hypothalamus, pituitary, pineal gland, testis from two-year-old rams, n = 3; ovary and uterine horn from two-year-old ewes, n = 3) were collected from Hu sheep humanely slaughtered at a government-certified facility. The tissue samples were immediately stored in liquid nitrogen after dissection from the animal’s body within half an hour after death, and kept in liquid nitrogen for more than 30 min. The samples were then transferred to a − 80 °C freezer until RNA extraction.
Fresh ovaries were obtained from healthy two-year-old Hu sheep (female) at a local slaughterhouse in Taicang, Jiangsu, China (121°10’ E, 31°45’ N) during the breeding season (October to January) and were quickly transported to the laboratory within 2 h. The ovaries were then washed three times with physiological saline containing gentamicin and maintained at a constant temperature of 37 °C. Primary granulosa cells (GCs) were isolated from <3 mm follicles and cultured in DMEM/F12 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37 °C with 5% CO2. We also selected follicles of different diameters (<3 mm, 3–5 mm, and >5 mm), categorizing them into three groups. These samples were then rapidly frozen in liquid nitrogen for subsequent experiments.
Hematoxylin-eosin (HE) staining
The ovine cortical strips were embedded in paraffin and sectioned to a thickness of 5 μm. Subsequently, the samples underwent a series of processes, including dewaxing, hematoxylin and eosin staining, dehydration, permeability treatment, and sealing. Finally, the sections were observed and photographed under a microscope.
Cell transfection
Based on the sheep AMH gene sequence (NC_056058.1) in the NCBI database, Genepharma Company (Shanghai, China) was commissioned to synthesize three specific AMH siRNAs along with a control siRNA (negative control), as shown in Table 1. The AMH overexpression vector was designed and synthesized by Qinke Company (Nanjing, China). Transfection was performed according to the Lipofectamine 3000 instructions when the cell density reached 60%–70%.
RNA extractions, cDNA synthesis, and quantitative real-time PCR (qRT-PCR) analysis
After transfecting the granulosa cells (GCs) for 24 h, total RNA was extracted using TRIzol Reagent (Invitrogen, 15596026CN). The cells were lysed with TRIzol, followed by the addition of chloroform. The mixture was thoroughly vortexed and then centrifuged at 12,000 × g for 15 min at 4 °C. The aqueous phase was carefully collected, and isopropanol was added to precipitate the RNA. The mixture was inverted to mix and incubated at room temperature for 10 min, followed by centrifugation at 12,000 × g for 10 min to collect the RNA pellet. The RNA pellet was washed twice with 75% pre-chilled ethanol to remove residual salts and impurities, followed by centrifugation at 12,000 × g for 5 min at 4 °C. After air-drying for 5–10 min, the RNA pellet was dissolved in RNase-free water. The concentration and purity of the RNA were assessed using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). Samples with a concentration greater than 200 ng/μL and an A260/280 ratio between 1.8 and 2.0 were selected for subsequent experiments.
First-strand cDNA was synthesized from 1 μg of total RNA using the HiScript III RT SuperMix for qPCR (Vazyme, R223) according to the manufacturer’s instructions. The reaction mixture included 4 μL of 5× HiScript III Buffer, 1 μL of HiScript III Enzyme Mix, 1 μL of Oligo(dT)20VN primer, and RNase-free water to a final volume of 20 μL. The reaction was performed at 42 °C for 2 min, followed by heat inactivation at 50 °C for 15 min, and 85 °C for 5 s.
qPCR reactions were performed using AceQ qPCR SYBR Green Master Mix (Vazyme, Q711-02). Each reaction was performed in a 20 μL volume containing 10 μL of Master Mix, 0.8 μL of forward and reverse primers (10 μM each), 2 μL of cDNA template, and 7.4 μL of RNase-free water. The qPCR cycling conditions included an initial denaturation at 95 °C for 30 s, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. The final step involved a melt curve analysis at 95 °C for 15 s, 60 °C for 1 min, and 95 °C for 15 s. Each sample was run in triplicate. GAPDH was used as an internal control, and the 2 − ΔΔCt method was applied to assess relative gene expression. The primer sequences used for RT-qPCR are shown in Table 2.
Western blot analysis
Proteins were extracted from ovarian follicles, and GCs were transfected for 48 h using RIPA buffer (Thermo Pierce, 89,900) combined with protease inhibitors and phosphatase inhibitors (Beyotime, P1045) in a ratio of 100:1:1 for 30 min at 4 °C. All protein samples were denatured at the same concentration. A total of 100 μg of protein solution was taken, to which 15 μL of 4x LDS Sample Buffer and 10 μL of 10x Sample Reducing Agent were added. Finally, ddH2O was added to bring the total volume to 100 μL, and the mixture was denatured at 70 °C for 10 min using a PCR instrument. Equal amounts of protein (10 µg) were resolved on 12% Bis-Tris SDS-PAGE gels (Invitrogen, Carlsbad, CA, USA) and transferred to 0.45 µm polyvinylidene fluoride (PVDF) membranes (Millipore, Burlington, MA, USA) using a wet transfer system. Membranes were blocked with 5% bovine serum albumin (BSA; Solarbio, Beijing, China) in Tris-buffered saline with 0.1% Tween-20 (TBST) for 2 h at room temperature, followed by overnight incubation at 4 °C with primary antibodies (dilution ratios listed in Table 3). After three 5-min TBST washes, membranes were incubated with HRP-conjugated goat anti-rabbit/mouse IgG secondary antibodies (Table 3) for 1 h at room temperature. Signals were detected using enhanced ECL ultra-sensitive luminescence fluid (Thermo Pierce, USA) and imaged on an Image Quant LAS 400 (Fiji film, Tokyo, Japan). The gray values of the Western blot were analyzed using ImageJ. Uncropped and unedited Western Blot images were presented as described in Supplementary Fig. 1.
Cell counting kit-8 (CCK8) and EdU assay
Cell viability was assessed using the CCK-8 Cell Counting Kit (Biosharp, BS350B). Granulosa cells (GCs) were cultured in 96-well plates (JetBiofil) and transfected with plasmids or siRNA. After 0, 12, and 24 h, 10 µL of CCK-8 reagent was added to the culture medium and incubated at 37 °C for 4 h. The absorbance was then measured at 450 nm using a microplate reader.
Cell proliferation was assessed using the kFluor555 Click-iT EdU assay (RCF041, Huilanbio BiologicalTechnology, Shanghai, China). After transfecting the GCs for 24 h, they were incubated with EdU reagent (50 µM), washed with DPBS, and fixed with 4% PFA. After being permeabilized with 0.5% Triton X-100, washed with 3% BSA, and incubated with the Click-iT reaction mixture in the dark. Following this, counterstaining was performed using Hoechst 33342, also in the dark. Finally, images were captured using a laser scanning confocal microscope (Carl Zeiss, Oberkochen, Germany).
Cell apoptosis assay
Cell apoptosis was detected using the Annexin V-FITC/PI double staining apoptosis detection kit (Keygen BioTECH, KGA1102) following the manufacturer’s instructions. Begin by centrifuging cells at 300 g for 5 min at 4 °C. After removing supernatant, wash cells twice with PBS using identical centrifugation conditions. Proceed to resuspend cells in 500 μL Binding Buffer. Then add 5 μL Annexin V-FITC, mix gently, followed by 5 μL Propidium Iodide with additional mixing. Protect the stained cells from light and incubate at room temperature for 5–15 min. Finally, analyze cell apoptosis by flow cytometry (BD FACSCalibur). The apoptosis data were analyzed using FlowJo_v10.8.1. Q2 represents the proportion of cells in late apoptosis, while Q3 represents the proportion of cells in early apoptosis. The sum of these two proportions indicates the overall rate of apoptosis. Cell apoptosis gating was performed as described in Supplementary Fig. 2.
Cell cycle analysis
The cell cycle was assessed using the commercially Detection Kit (Keygen Bio TECH, KGA9101) in accordance with the manufacturer’s instructions. The cells were subsequently digested, washed, and centrifuged with a 70% ethanol solution to obtain a single-cell suspension. Next, 500 µL of the PI/RNase A staining solution, prepared in advance, was added, and the samples were protected from light at room temperature for 30–60 min. After staining, the cells were analyzed using a flow cytometer (BD FACSCalibur). The data were processed and analyzed using FlowJo_v10.8.1 software to fit the cell cycle curves and accurately quantify the percentage of cells in each phase of the cell cycle. Cell cycle gating was performed as described in Supplementary Fig. 2.
Immunofluorescence assay (IF)
GCs were grown on coverslips and fixed with 4% paraformaldehyde when the cell density reached 60% to 70%. The cells were then incubated with primary antibodies: rabbit anti-FSHR (1:100, Proteintech, 22665-1-AP), anti-AMH (1:50, Proteintech, 14461-1-AP), anti-AMHR2 (1:50, CUSABIO, CSB-PA614985ESR1HU), and BMPR1B (1:50, Abclonal, A2005). Following this, the cells were incubated with secondary goat anti-rabbit IgG (1:500, Proteintech, Wuhan, China). The nuclei were stained with Hoechst 33342. Finally, the slides were observed using a fluorescence microscope (Zeiss LSM710 META, Jena, Germany).
ELISA assay
Cell supernatant was collected 24 h after the transfection of the interference sequence and overexpression vector into Granulosa cells. The expression levels of hormones were then determined using the E2 (AiFang Biological, AF3907-A) ELISA kit, following the manufacturer’s protocol.
RNA isolation, library construction, and sequencing
GCs were cultured in 6-well plates and transfected with plasmids or siRNAs. After 24 h of transfection, the cells were collected for RNA extraction (n = 3) and subsequently sent to Majorbio company for identification, cDNA library preparation, and Illumina sequencing. Transcriptome sequencing of 12 samples was completed, yielding a total of 84.25 Gb of clean data. The clean reads of each sample were aligned with the designated reference genome (Oar_aries v2.0) for sequence alignment. Gene expression levels were calculated as the normalized number of Transcripts Per Million (TPM). DESeq2 was used to identify differentially expressed genes (DEGs) between the SI-AMH and NC groups, as well as between the OE-AMH and OE groups, using a fold change> |1.5| and a P-value < 0.05 as the screening criteria.
Statistics and reproducibility
All data are presented as the mean ± SEM. Each experiment was conducted a minimum of three times (n = 3 biologically independent samples/animals/independent experiments). Statistical analysis was performed using SPSS software (version 24.0), utilizing either a two-tailed Student’s t-test or a one-way analysis of variance (ANOVA), followed by the Tukey post hoc test. A P-value of less than 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All other data are available from the corresponding author on reasonable request. The RNA-seq data were deposited in the China National Center for Bioinformation database under PRJNA1253545. All plasmids generated from this study have been deposited in Addgene. All data supporting the findings of this study are available within the paper and its Supplementary Information. Numerical source data for all figures was provided in Supplementary Data of Supplementary Information. Uncropped and unedited western blot images for the Figs. 1, 2, 3, 5, 6 are presented in Supplementary Fig. 1 of Supplementary Information. Gating strategy for the Figs. 2I, 5E are presented in Supplementary Fig. 2 of Supplementary Information.
References
Chang, H. M., Klausen, C. & Leung, P. C. Antimüllerian hormone inhibits follicle-stimulating hormone-induced adenylyl cyclase activation, aromatase expression, and estradiol production in human granulosa-lutein cells. Fertil. Steril. 100, 585–592.e581 (2013).
Estienne, A. et al. Anti-Müllerian hormone regulation by the bone morphogenetic proteins in the sheep ovary: deciphering a direct regulatory pathway. Endocrinology 156, 301–313 (2015).
Jost, A. The age factor in the castration of male rabbit fetuses. Proc. Soc. Exp. Biol. Med. 66, 302 (1947).
Zhang, H., Adhikari, D., Zheng, W. & Liu, K. Combating ovarian aging depends on the use of existing ovarian follicles, not on putative oogonial stem cells. Reproduction 146, R229–R233 (2013).
Russell, N., Gilmore, A. & Roudebush, W. E. Clinical utilities of anti-müllerian hormone. J. Clin. Med. 11 https://doi.org/10.3390/jcm11237209 (2022).
Iwase, A. et al. Anti-Müllerian hormone beyond an ovarian reserve marker: the relationship with the physiology and pathology in the life-long follicle development. Front. Endocrinol.14, 1273966 (2023).
Vanorny, D. A., Prasasya, R. D., Chalpe, A. J., Kilen, S. M. & Mayo, K. E. Notch signaling regulates ovarian follicle formation and coordinates follicular growth. Mol. Endocrinol. 28, 499–511 (2014).
Carlsson, I. B. et al. Anti-Müllerian hormone inhibits initiation of growth of human primordial ovarian follicles in vitro. Hum. Reprod. 21, 2223–2227 (2006).
Durlinger, A. L. et al. Anti-Müllerian hormone inhibits initiation of primordial follicle growth in the mouse ovary. Endocrinology 143, 1076–1084 (2002).
Gigli, I., Cushman, R. A., Wahl, C. M. & Fortune, J. E. Evidence for a role for anti-Mullerian hormone in the suppression of follicle activation in mouse ovaries and bovine ovarian cortex grafted beneath the chick chorioallantoic membrane. Mol. Reprod. Dev. 71, 480–488 (2005).
Chen, X., Liu, S., Peng, X. & Zong, X. The roles of anti-Müllerian hormone in breast cancer. Endocr. Relat. Cancer 30 https://doi.org/10.1530/erc-23-0060 (2023).
Delamuta, L. C. et al. Antimüllerian hormone levels and IVF outcomes in polycystic ovary syndrome women: a scoping review. JBRA Assist Reprod. 28, 299–305 (2024).
Yu, X. et al. Synergistic regulatory effect of inhibin and anti-Müllerian hormone on fertility of mice. Front. Vet. Sci. 8, 747619 (2021).
Francoeur, L., Scoville, D. M. & Johnson, P. A. Investigations of the function of AMH in granulosa cells in hens. Gen. Comp. Endocrinol. 349, 114454 (2024).
Poole, D. H., Ocón-Grove, O. M. & Johnson, A. L. Anti-Müllerian hormone (AMH) receptor type II expression and AMH activity in bovine granulosa cells. Theriogenology 86, 1353–1360 (2016).
Turgut, A. O. & Koca, D. Anti-Müllerian hormone as a promising novel biomarker for litter size in Romanov sheep. Reprod. Domest. Anim. 59, e14692 (2024).
Cetin, N. & Koca, D. Anti-Müllerian hormone concentrations can be reliably determined by a single measurement, irrespective of cycle, in synchronised ewes during non-breeding season. Reprod. Domest. Anim. 60, e70010 (2025).
Suarez-Henriques, P. et al. Exploring AMH levels, homeostasis parameters, and ovarian primordial follicle activation in pubertal infected sheep on a high-protein diet. Res. Vet. Sci. 169, 105158 (2024).
Lahoz, B. et al. Anti-Müllerian hormone concentration in sheep and its dependence of age and independence of BMP15 genotype: an endocrine predictor to select the best donors for embryo biotechnologies. Theriogenology 81, 347–357 (2014).
Brochado, C. et al. Reduced embryo yield obtained from superstimulated ewes with low circulating AMH concentration is improved by lengthening the FSH treatment. Theriogenology 218, 79–88 (2024).
Xu, Q. et al. Correlation analysis of serum reproductive hormones and metabolites during multiple ovulation in sheep. BMC Vet. Res. 18, 290 (2022).
Alward, K. J. & Bohlen, J. F. Overview of Anti-Müllerian hormone (AMH) and association with fertility in female cattle. Reprod. Domest. Anim. 55, 3–10 (2020).
Humeres, C., Venugopal, H. & Frangogiannis, N. G. Smad-dependent pathways in the infarcted and failing heart. Curr. Opin. Pharm. 64, 102207 (2022).
di Clemente, N., Racine, C., Pierre, A. & Taieb, J. Anti-Müllerian hormone in female reproduction. Endocr. Rev. 42, 753–782 (2021).
Hayes, E. et al. Intra-cellular mechanism of Anti-Müllerian hormone (AMH) in regulation of follicular development. Mol. Cell Endocrinol. 433, 56–65 (2016).
Cimino, I. et al. Novel role for anti-Müllerian hormone in the regulation of GnRH neuron excitability and hormone secretion. Nat. Commun. 7, 10055 (2016).
Trbovich, A. M. et al. Müllerian inhibiting substance lowers testosterone in luteinizing hormone-stimulated rodents. Proc. Natl. Acad. Sci. USA 98, 3393–3397 (2001).
Abbara, A. et al. Anti-Müllerian hormone (AMH) in the diagnosis of menstrual disturbance due to polycystic ovarian syndrome. Front. Endocrinol.10, 656 (2019).
Pellatt, L. et al. Granulosa cell production of anti-Müllerian hormone is increased in polycystic ovaries. J. Clin. Endocrinol. Metab. 92, 240–245 (2007).
Silva, M. S. B. & Giacobini, P. New insights into anti-Müllerian hormone role in the hypothalamic-pituitary-gonadal axis and neuroendocrine development. Cell Mol. Life Sci. 78, 1–16 (2021).
Torres-Rovira, L. et al. Predictive value of antral follicle count and anti-Müllerian hormone for follicle and oocyte developmental competence during the early prepubertal period in a sheep model. Reprod. Fertil. Dev. 26, 1094–1106 (2014).
Cushman, R. A. et al. An extreme model of fertility in sheep demonstrates the basis of controversies surrounding antral follicle count and circulating concentrations of anti-Müllerian hormone as predictors of fertility in ruminants. Anim. Reprod. Sci. 259, 107364 (2023).
Campbell, B. K., Clinton, M. & Webb, R. The role of anti-Müllerian hormone (AMH) during follicle development in a monovulatory species (sheep). Endocrinology 153, 4533–4543 (2012).
Nilsson, E. E., Schindler, R., Savenkova, M. I. & Skinner, M. K. Inhibitory actions of Anti-Müllerian Hormone (AMH) on ovarian primordial follicle assembly. PLoS One 6, e20087 (2011).
Yang, M. Y., Cushman, R. A. & Fortune, J. E. Anti-Müllerian hormone inhibits activation and growth of bovine ovarian follicles in vitro and is localized to growing follicles. Mol. Hum. Reprod. 23, 282–291 (2017).
Meinsohn, M. C. et al. Single-cell sequencing reveals suppressive transcriptional programs regulated by MIS/AMH in neonatal ovaries. Proc. Natl. Acad. Sci. USA 118 https://doi.org/10.1073/pnas.2100920118 (2021).
Wang, L. et al. BMP6 regulates AMH expression via SMAD1/5/8 in goat ovarian granulosa cells. Theriogenology 197, 167–176 (2023).
Hoshiya, Y. et al. Mullerian inhibiting substance promotes interferon gamma-induced gene expression and apoptosis in breast cancer cells. J. Biol. Chem. 278, 51703–51712 (2003).
Zhao, Z. et al. BMP15 regulates AMH expression via the p38 MAPK pathway in granulosa cells from goat. Theriogenology 118, 72–79 (2018).
Taieb, J. et al. FSH and its second messenger cAMP stimulate the transcription of human anti-Müllerian hormone in cultured granulosa cells. Mol. Endocrinol. 25, 645–655 (2011).
Devillers, M. M. et al. FSH inhibits AMH to support ovarian estradiol synthesis in infantile mice. J. Endocrinol. 240, 215–228 (2019).
Roy, S. et al. Oocyte-derived factors (GDF9 and BMP15) and FSH regulate AMH expression via modulation of H3K27AC in granulosa cells. Endocrinology 159, 3433–3445 (2018).
Fuentes, N. & Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 116, 135–170 (2019).
Pierre, A. et al. Dysregulation of the anti-Müllerian hormone system by steroids in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 102, 3970–3978 (2017).
Grynberg, M. et al. Differential regulation of ovarian anti-müllerian hormone (AMH) by estradiol through α- and β-estrogen receptors. J. Clin. Endocrinol. Metab. 97, E1649–E1657 (2012).
Jakimiuk, A. J., Weitsman, S. R., Yen, H. W., Bogusiewicz, M. & Magoffin, D. A. Estrogen receptor alpha and beta expression in theca and granulosa cells from women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 87, 5532–5538 (2002).
Xu, J., Bishop, C. V., Lawson, M. S., Park, B. S. & Xu, F. Anti-Müllerian hormone promotes pre-antral follicle growth, but inhibits antral follicle maturation and dominant follicle selection in primates. Hum. Reprod. 31, 1522–1530 (2016).
Lecot-Connan, T. et al. Anti-Müllerian hormone induces autophagy to preserve the primordial follicle pool in mice. Faseb J. 38, e23506 (2024).
Acknowledgements
Supported by the high-performance computing platform of Bioinformatics Center, Nanjing Agricultural University. This work was supported by the National Natural Science Foundation of China (No. 32272879), Project of Seed Industry Revitalization in Jiangsu Province (JBGS[2021]113), and the Fundamental Research Funds for the Central Universities (KYT2024002).
Author information
Authors and Affiliations
Contributions
Hui Xu, Yu Cai: Writing-original draft, Writing-review and editing, Methodology, Investigation, Formal analysis. Hua Yang: Supervision, Writing-review and editing. Chong Zhang: Methodology. Wanru Liu: Methodology. Binru Zhao: Methodology. Feng Wang: Resources. Yanli Zhang: Validation, Supervision, Investigation, Project administration, Writing-review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
We have complied with all relevant ethical regulations for animal use. All experimental procedures were conducted in accordance with the guidelines set forth by the Institutional Animal Care and Use Committee of Nanjing Agricultural University, China (SYXK 2022–0031).
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Rosie Bunton-Stasyshyn & Kaliya Georgieva. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, H., Cai, Y., Yang, H. et al. AMH regulates granulosa cell function via ESR2/ p38-MAPK signaling pathway in sheep. Commun Biol 8, 824 (2025). https://doi.org/10.1038/s42003-025-08259-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08259-6
This article is cited by
-
GraphBAN-based identification of active ingredients and key gene targets in compound Chinese herbal medicines for polycystic ovary syndrome
Naunyn-Schmiedeberg's Archives of Pharmacology (2026)
-
Integrated transcriptomic and co-expression network analysis identifies immune-metabolic biomarkers of polycystic ovary syndrome in granulosa cells
Journal of Ovarian Research (2025)
