
Abstract
Accurate dynamic protein structures are essential for drug design. NMR experiments can detect protein structures and potential dynamics, but the spectrum assignment and structure determination requires expertise and is time-consuming, while deep-learning-based structure predictions may be inconsistent with experimental observations. A symbiosis between experiments and AI methods is therefore essential for solving such problems. Here, we developed a Restraint Assisted Structure Predictor (RASP) model and an iterative Folding Assisted peak ASsignmenT (FAAST) pipeline directly leveraging experimental information to improve the AI-assisted structure prediction and facilitate experimental data analysis in an integrative way. The RASP model improves structure prediction, especially for multi-domain and few-MSA proteins. The FAAST pipeline for NMR NOESY analysis reduces the time consumption to hours and yields high quality structure ensemble. Both methods show high consistency between predicted structures and restraints, provided or iteratively assigned. This strategy can be expanded to other types of sparse experimental information in structure prediction.
Similar content being viewed by others

Introduction
NMR is an experimental technique used to determine structures and detect weak interactions in situ1,2. However, NMR assignment requires both expertise and time. It might take months even years for NMR assignment. Leveraging machine learning and deep learning technologies, researchers have endeavored to automate the NMR assignment protocol. For example, ARTINA3 provides an integrated pipeline which accepts raw NMR spectra, assigns chemical shifts and NOE peaks, and provides structures simultaneously. It utilizes molecular simulations to construct structures, with a focus on achieving automated and accurate chemical shift assignments.
Specifically, the nuclear overhauser effect spectroscopy (NOESY) peak assignment process provides restraints on the distances between hydrogen atoms and is an essential technique in NMR structure analysis. Automated algorithms such as CYANA4, ARIA5,6, CANDID7 have been developed to assist NOE peak assignment, which mostly apply strategies such as molecular dynamics simulation or simulated annealing for structure construction. Rosetta suites or pipelines leveraging sparse NMR restraints from NOE, RDC, and PRE data have also be developed for the data-assisted structure construction8,9,10. These methods rely on molecular simulation techniques and are relatively time consuming.
Recent progress in deep learning provides more efficient and accurate tools to generate protein structure given its sequence. Employing deep learning protein structure prediction models for experimental data analysis has been a problem of interest. In practice, the input data form and distribution of general structure prediction models, such as AlphaFold11, do not necessarily align with the needs of experimental methods. While AlphaFold (AF2) and AlphaFold-multimer12 (AFM) have greatly improved the accuracy of predicting static protein structures, unresolved issues remain, such as generating dynamic structures and predicting structures with extra experimental information provided. Questions remain on how experimental information can facilitate rapid structure prediction and how structure prediction methods can aid in the resolution or acceleration of experimental data analysis. Attempts have been made to provide AF2 structures as templates for X-ray molecular replacement13 or Cryo-EM density map14 templates. However, these approaches rely on use of iterative templates, which includes dense but not necessarily accurate restraints and cannot utilize structural differences directly to improve predictions. Recently, AlphaLink15 fine-tuned AF2 to accept sparse restraints, improving AF2 performance in cross-linking experiments.
To directly integrate experimental or knowledge-based prior information into the AI model and allow the model to feed back to experiments, in this work we propose and train a model named Restraints Assisted Structure Predictor (RASP) and an iterative NMR NOESY peak assignment pipeline called FAAST(iterative Folding Assisted peak ASsignmenT). The architecture of RASP is derived from AF2 Evoformer and structure module, and it accepts abstract or experimental restraints, sparse or dense, to generate structures. This enables RASP to be used in diverse applications, including improving structure predictions for multi-domain proteins and those with few multiple sequence alignments (MSAs). The confidence of RASP can evaluate restraint quality in terms of information efficiency and accuracy. Consequently, by leveraging the model’s ability to accept a flexible number of restraints and evaluate them, together with an NMR assignment protocol adapted from ARIA6, we developed the FAAST pipeline. In addition to structure prediction with prior restraints from experimental or knowledge-based information, this pipeline also in return assists NMR NOESY assignment and simultaneously generates corresponding structures. Using chemical shift and NOE peak lists as input, FAAST assigns NOE peaks iteratively and generates a structure ensemble based on the subsampled restraints, thus accelerating NMR analysis.
Results
RASP takes in restraints directly and helps in structure prediction
To make use of the general experimental information as restraints, we developed the RASP model based on the AF2 architecture. The model takes in sequence and a flexible number of distance restraints, and returns a structure that largely complies with the restraints. Additionally, it measures the consistency between the restraints and the predicted structure. We consider restraints as a form of edge information and input as the edge bias in the Evoformer MSA attention block (defined as MSA bias) as well as invariant point attention block (IPA, defined as IPA bias) (Fig. 1a, b, Supplementary Methods). Moreover, we experimented with the pair representation update in Evoformer (defined as pair bias) and adopted the structure module (defined as structure bias) to ensure that the structure update follows the restraints. We evaluated the impact of the four types of bias information and chose MSA and IPA biases as the baseline RASP model setting for simplicity and stability (Supplementary Fig. 1). Only the first two bias forms were used in the following experiments.

a A scheme of how the pairwise restraints can be taken into the model in the form of MSA, pair, IPA, and structure biases, adopted from the Evoformer and structure module in AlphaFold. b More detailed illustration of the model structure with restraints treated as biases in the Evoformer block (left) and structure block (right). For the PSP validation dataset, RASP with a fixed number of randomly sampled input restraints is compared with AlphaFold on (c) the TM-score between the PDB structure and predicted structure and d on restraint recall.
We implemented RASP using MindSpore16 and trained it on 32 * Ascend910 NPUs. The model was initialized with MEGA-Fold17 weights and fine-tuned on the PSP dataset17 with a true structure: distilled structure ratio of 1:3. We sampled pairwise restraints to tolerate longer restraint distances (refer to Methods). The training converged after 15 k steps (480k samples in total, Supplementary Fig. 2) and demonstrated stable improvement over initial MEGA-Fold.
Although the model supports templates in prediction and could improve performance with template used, for fairness and to avoid data leakage, we chose not to utilize templates in this research. We tested the model’s performance on the PSP validation dataset17 previously constructed along with the PSP training set, which contains 490 samples of CAMEO18,19,20,21 targets and unique proteins between October 2021 and March 2022. This validation set is strictly after the PDB and sequence deposition time of the training set. When restricting the number of restraints to 100, the TM-score22,23 which measures topological similarity between structures greatly improved for the structure prediction in the PSP validation dataset (Fig. 1c). Furthermore, the model followed the randomly sampled short-range restraints much better than those predicted by AF2 or MEGA-fold, as expected (Fig. 1d, Supplementary Fig. 3, Methods). Moreover, the AlphaFold violation loss which measures bond length, bond angle, and atomic clash violation for RASP predictions remained low with a median of 0.0012 (Supplementary Fig. 4, Methods), indicating its capability to predict structures following basic physiochemical principles.
RASP helps structure prediction and evaluation in a broad range of restraints numbers
We discovered that the structure accuracy improves steadily as the number of restraints increases, starting from zero (Fig. 2a). However, restraint recall, defined as the ratio between the number of correctly predicted restraints and the total restraints (see Methods), remains relatively constant, implying that the current model can tolerate different numbers of prior information or restraints without adversely affecting the baseline model performance (Fig. 2b). Additionally, the predicted local-distance difference test score (pLDDT score) of the model serves as an indicator of the model confidence on the restraints. For proteins with varying numbers of restraints, the pLDDT score rises stably though not significantly with an increase in the number of restraints applied. The pLDDT confidence correlates well with the corresponding structure TM-score with an overall correlation of 0.71 (Fig. 2c).

a Violin plot for the TM-score distributions of AlphaFold2, MEGA-Fold, and RASP with increasing number of input restraints from 0 to 200. b Violin plot for the restraint recall of AlphaFold2, MEGA-Fold, and RASP with different numbers of input restraints. The restraint recall of AF2, MEGA-Fold, and RASP with zero restraint are computed with the same restraints as from RASP with 100 restraints. c Scatter plot of the pLDDT of RASP with varied restraints number and the real TM-score of the predicted structure, with an overall correlation coefficient of 0.71. d The distribution of correlation coefficient between the RASP predicted pLDDT confidence and TM-score for the same case with different number of restraints. The distribution is drawn for all validation samples. e Violin plot for the TM-score distribution with decreasing percentage of good restraints, with total input restraints number fixed at 20. f Scatter plot of RASP pLDDT confidence and the TM-scores when loose restraints are present, with an overall correlation coefficient of 0.73. For (a, b, e), the central white dot indicates the median, the bar edges indicate the quantiles, and the whiskers extend to 1.5 times of the interquartile range. For subfigures (a, b, d, e), statistics were performed on 490 samples from the PSP validation dataset with different methods or restraints.
Despite the difference in restraint numbers, due to sampling randomness, some restraints might provide repeating information with MSAs (and potentially templates, although templates are not used here for fairness), and the restraint information provided by experiment may not be free of error. The restraint quality therefore could be considered in two aspects: one is how much additional information it provides aside from that provided by MSA and templates, another is how accurate the restraint information is. To examine the ability of the confidence score to distinguish prediction quality for the same protein with different restraint information, we first examined the confidence-TM-score correlation for each case with restraint numbers from 0 to 200. The averaged correlation score for the 490 cases is 0.353, with 61.4% showing correlation coefficient above 0.3 (Fig. 2d). With MSA kept the same for predictions of the same protein, better structures can therefore be attributed to more effective restraint information. This indicates that the pLDDT score can be used to distinguish better structures and correspondingly more informative restraints. Furthermore, when loose restraints are intentionally used (restraints with a Cβ distance greater than 12 Å, as defined), the TM-score decreases fast along with the increasing loose restraint ratio and fixed number of 20 restraints (Fig. 2e), suggesting that the model is sensitive to inconsistent restraints and can distinguish corresponding bad structures. The TM-scores correlate well with the pLDDT scores with an overall correlation of 0.73 and averaged per-case correlation of 0.89 (Fig. 2f and Supplementary Fig. 5). These findings indicate that the pLDDT score can gauge how well the restraints may assist in structure prediction and the restraint’s quality or self-consistency, both with restraints that may be of little use and with bad restraints present. With this evaluation, the model may find applications in areas such as NMR determination (see section below).
RASP improve structure prediction assisted by pseudo and experimental NMR restraints
By incorporating restraints, the model demonstrates improved capability to predict the structures of multidomain or few-MSA proteins. Two cases representative of this improvement in the PSP validation dataset are 6XMV and 7NBV (Fig. 3a, b). 6XMV is a multi-domain protein that exhibits wrongly predicted relative domain positions by both AF2 and MEGA-Fold. However, utilizing randomly sampled restraints corrects the inter-domain positions, improving its TM-score from 0.51 to 0.79. For 7NBV, which is a virus protein and only has three sequences in its multiple sequence alignment, an increase in the number of randomly sampled restraints leads to a stable improvement in structure quality from 0.43 to 0.77, with 50 restraints being used. These outcomes demonstrate the potential for using restraints to aid in the prediction of few-MSA and multi-domain proteins.

a Structures predicted by AF2 (colored blue), MEGA-Fold (colored olive) and RASP (colored salmon) for multi-domain structure 6XMV. The PDB structure of 6XMV is drawn in gray for reference. b Structures predicted by AF2 (colored blue), MEGA-Fold (colored olive) and RASP (colored salmon) for 7NBV which has few MSAs. The PDB structure of 7NBV is drawn in gray for reference. RMSDs for the three predictions are indicated. c Violin plot of overall and long-range (sequence separation\(\ge\)4) restraint recall distributions and d TM-score distributions for AF2, AlphaLink in binary mode and distogram mode, and RASP. The restraint recall of deposited PDB structures are also drawn as reference. For (c, d), each box contains 333 samples, the central white dot indicates the median, the bar edges indicate the quantiles, and the whiskers extend to 1.5 times of the interquartile range.
NMR is a commonly used experimental structure determination method that generates restraints of varied quantity. Despite that in many cases AF2 predictions follow the restraints similar or even better than deposited NMR PDB structures24,25, the AF2 model does not naturally foresee structures compatible with NMR restraints and may produce alternative structures opposed to those deposited in the PDB. Given the continued evolution of NMR data deposit requirements and the diversified time samples may be deposited, some entries in the PDB and BMRB database do not include restraint files. After filtration of NMR samples deposited in the RCSB PDB bank with restraint files (.mr) available and bad AF2 predictions (long-range restraint recall lower than 90%), 333 samples remain available (Supplementary Data 1). The samples exhibit a wide variation in the overall number of NMR restraints, from tens to thousands, with a median restraint quantity of 12.3 per residue.
When leveraging NMR restraints to aid in structure prediction, the predicted structures better adhere to the restraints than AF2 predictions, both for overall restraints and especially for long-range restraints (defined as sequence separation\(\ge \!\)4 in this work), with median restraint recall increasing from 95.2% to 98.8% and 81.0% to 94.4%, respectively (Fig. 3c). In contrast, the deposited PDB structures follow the corresponding deposited restraints with respective restraint recalls of 99.2% and 96.3% for overall and long-range restraints, similar to those of RASP. The structures generated by RASP are also more consistent with the deposited structures (Fig. 3d). Additionally, we evaluated the performance of AlphaLink, another representative restraint-assisted structure prediction model on the experimental NMR dataset, and found that it performs better than AF2 but worse than RASP in both restraint recall (Fig. 3c) and overall structure (evaluated by TM-score, Fig. 3d) with the distogram setup, while worse than AF2 in terms of overall structure with the binary restraint setup (Fig. 3d and Supplementary Data 1).
NMR NOESY assignment pipeline FAAST
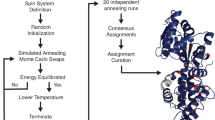
With the ability of the RASP model to take restraints from a wide range of sources and evaluate their quality with pLDDT scores, it has the potential to accelerate NMR NOESY peak assignments. These assignments accumulate over assigning iterations - starting with only a few correctly identified restraints - and lead to refined structure predictions. By combining the RASP model with the ARIA6 assignment protocol (mainly the Calibration - bound Calculation - Violation analysis - Peak assignment pipeline, abbreviated as CCVP), we built an iterative NMR analysis pipeline named FAAST (Fig. 4a). FAAST takes chemical shift and NOE peak lists as input and outputs peak assignment and structure ensembles. Each iteration involves subsampling of the assigned restraints with an increasing ratio from the previous iteration as RASP input and generating an ensemble of 20 structures, which is then used for the subsequent NOE peak assignment (Fig. 4b). As pLDDT scores reflect the restraint quality, if the median pLDDT of the ensemble is lower than 80, we automatically restart the second round of iteration with a lower restraint subsampling ratio to reduce restraints conflict. The protocol allows for a maximum of one restart, resulting in a total ensemble iteration number of 2 or 5.

a The schematic figure of the FAAST workflow and b the current assignment pipeline and parameters applied. In each iteration block, the first parameter is the percentage of input restraints subsampled for RASP structure prediction in order to construct a structure bundle, the second parameter is the cumulative probability to rule out ambiguous restraints in assignment, which is also adjustable in ARIA. c The distribution for number of NOE assignments per residue for the 57 ARTINA samples. The median number is around 15, similar with peak lists from the BMRB database. d The distribution for pairwise mutual RMSD for C-α atoms of the structure bundle. Statistics for (c, d) are performed on all 57 available samples. e The restraint recall of the FAAST structure with the identified restraints compared to the deposited PDB with the deposited NMR restraints. f Scatter plot of backbone RMSDs of FAAST structures and those reported by ARTINA.
We benchmarked the FAAST pipeline on samples used in ARTINA. Out of the 100 ARTINA samples, 57 had both chemical shift and at least one 3D NOESY peak list deposited on BMRB that can be identified from the nmrstar files (Supplementary Table 1). We validated the NMR pipeline on all of the 57 samples. With a median time of 32 min (minimum and maximum time of 14 and 103 min), we were able to assign a median of 1569 peaks per sample and a median peak number of 14.75 per residue (Fig. 4c). Furthermore, the median pairwise mutual C-α RMSD for the structure ensemble is 0.86 Å (Fig. 4d), indicating consistency between subsampled restraints and the resulted structure ensemble. We note here that pairwise RMSDs from the structure bundle in the initial iteration have a median of 1.99 Å, higher than that from the final structures, indicating that the subsampling strategy is able to generate diversified structures, and that iterative refinement leads to convergence in structure ensemble.
Moreover, the predicted structure by FAAST is consistent with simultaneously assigned restraints as well as the NOE peaks. A median of 99.6% of the identified restraints match the highest confidence structure and the corresponding median is 99.0% for identified long-range restraints. In comparison, the model 1 structure and restraints from the PDB database conform on a median of 98.6% and 98.2% for all restraints and long-range restraints, respectively (Fig. 4e, Supplementary Fig. 6). The RMSD score and correlation score calculated by ANSURR26 and DP score by RPF27 indicate that the structures obtained by FAAST are of comparable or better quality and consistency between predicted structure ensemble and the NOE peak lists, compared to corresponding PDB structures (Supplementary Fig. 7). The predicted structures not only agree with the assigned peaks, but are also consistent with the deposited restraint and structure data. 96.9% of the deposited restraints from PDB database align with the predicted structure ensemble. The median mean structure backbone RMSD against the deposited PDB model 1 is 0.739 Å for structured regions defined by ARTINA. For the median scored structure in the structure ensemble, the backbone RMSD is 0.791 Å against the PDB structure. Both are lower than that reported by ARTINA, in which the median mean structure backbone RMSD is 1.44 Å for all samples and 1.47 Å for the 57 samples with BMRB peak lists (Fig. 4f), the latter utilized raw spectra for both chemical shift and NOE assignments and hence is a more complete pipeline.
Notably, among all the 57 samples, only 7 has AF2 prediction with TM-score lower than 0.7. Despite that FAAST returns relatively similar average TM-scores against the deposited structure (a lower score of 0.627 for FAAST and 0.651 for AF2, Supplementary Data 1), the average TM-score between FAAST and AF2 prediction is 0.75, indicating these two methods could return different structures. Further, for all seven examples, FAAST pipeline returns an average mutual C-α RMSD of 1.53 Å and average assigned restraint recall of 99.7% and 99.5% for overall as well as long-range restraints, indicating good convergence both within structure ensemble and between structure and NOE peak assignment. The predicted structure also aligns well with the deposited assignments with overall and long-range recalls of 97.2% and 96.5%, respectively (Supplementary Data 1). These observations indicate that the FAAST pipeline could provide alternative solutions different from both AF2 structure predictions and the deposited assignment-structure ensemble sets.
Since we adopted the CCVP protocol from ARIA, as an ablation study, we further evaluated the performance of the automated ARIA pipeline with default settings (Supplementary Fig. 8, Supplementary Methods). The internal median restraint recall for overall restraints and long-range restraints are 96.2% and 91.3%, respectively, showing relative consistency between the predicted structures and the assigned restraints. However, the median mutual RMSD for ARIA structure ensemble after eight rounds of iteration is 5.10 Å, with median RMSD to the reference structure of 4.95 Å. These results indicate that the ARIA structure ensemble optimization pipeline under default settings are slow to converge. Again showing the effectiveness of FAAST with the application of the RASP model.
Since only processed NOESY peak lists are available and raw peak lists are absent for samples downloaded directly from BMRB, we validated the pipeline’s performance on the 2MRM case with raw NOESY peak lists. For this YgaP protein, much more restraints can be assigned from the raw peak lists than from the deposited NOE peak lists (14.19 per residue for raw lists compared to 4.93 for deposited ones). The number of assigned long-range restraints is also higher (366 for raw lists and 285 for deposited lists). Despite similar small mutual RMSDs, the predicted structures from raw peak lists and deposited peak lists have similar TM-scores to the deposited structure (0.862 and 0.857, respectively), even though the restraints assigned from the raw peak list are in better consistency than those assigned from deposited lists, with the former exhibiting a restraint recall of 98.2% and the latter of 94.9%. These results indicate that the pipeline doesn’t require strict peak assignment, and we expect the raw peak lists from NOE spectra to provide better assignment in the FAAST pipeline than the deposited peaks.
In summary, we present a fast NMR pipeline that provides accurate structure ensemble and highly consistent NOE peak assignments. Compared to previous methods, this pipeline is fast, and through restraints iteration and subsampling, can provide a structure ensemble plus a full set of NOE peak assignments. We expect this FAAST pipeline to be useful in the NOESY peak assignment and NMR structure determination since it performs well both with raw peak lists and deposited ones, even better with raw peak lists for the example case in this study.
Discussion
The question of how experimental results and AI methods can mutually benefit each other has been a topic of discussion, particularly with the emergence of advanced biochemical deep learning models. Here, we present the RASP model and the FAAST pipeline, wherein the former utilizes prior knowledge or restraints to improve in silico structure predictions, while the latter employs the former’s flexible number restraint-taking capability and evaluation of restraint-structure quality to accelerate NOESY peak assignment. This model and pipeline underscore the self-consistency of the two questions as an AI method capable of being assisted by external knowledge has the potential to facilitate the acquisition and/or validation of that external knowledge in return.
Despite the application of the RASP model on NMR restraints, due to its improvement of structure prediction with abstract randomly sampled restraints, flexibility in restraint number, and ability to evaluate restraints, we expect it to be useful for broad knowledge types, such as cross-linking or covalent labeling data as in AlphaLink, even abstract prior knowledge such as closeness of two residues regardless of the knowledge source, and in this way may help the generation of dynamic structures or states guided by restraints.
While we have currently applied our standard automated pipeline in FAAST for benchmark, parameters for RASP and CCVP steps can be flexibly adjusted by users to accommodate their particular peak quality and expectations on peak-structure convergence. When benchmarking the pipeline, we did not employ parallel computation considering the possibly limited computational resources for users. However, both the RASP prediction and relaxation can be executed parallelly, which is expected to accelerate the process up to 20 times, which is the ensemble size, depending on the hardware available. Since the chemical shift and NOE peak assignment could be iteratively improved, merging the chemical shift and peak assignment pipelines is also expected to produce more comprehensive and accurate NMR protein assignment pipelines.
Moreover, in this study we only used restraints generated from 3D NOESY spectra. The current pipeline could be readily expanded to other NMR data types such as 4D NOESY spectra, as long as the experimental data could be formatted as pairwise restraints. More diversified forms of experimental data also exist that might provide information for different molecule types, such as NMR for protein-small molecule interactions. In addition to the conventional paired restraints, we also expect to incorporate additional information forms (e.g., torsion angle in NMR) into our structure prediction. These restrained structure prediction models hold the potential to introduce an alternative approach to restrained design.
Methods
Structure of RASP
To incorporate restraint information, we developed the RASP model derived from the AlphaFold Evoformer and structure module. Four additional biases were added, which draw on restraint information: pair bias, MSA bias, IPA bias, and structure bias. To handle inter-residue restraints as edge information, the first 3 biases are introduced as edge biases to the Evoformer and IPA modules. This inter-residue information can be naturally converted into features of shape (\({N}_{{res}},{N}_{{res}}\),\({C}_{{channels}}\)), similar to the pair activation in the Evoformer module of the original model. Following the strategy of merging pair activation and MSA activation in the Evoformer module, an extra contact bias is added to the row-wise attention and the outer-product mean module by pair bias. The merging of inter-residue information and per-residue information also occurs in the Invariant Point Attention. The contact information is added to the IPA attention weight matrix as IPA bias in the same way as the MSA bias. In addition to the biases in the attention, an additional bias is introduced in the structure generation process. When generating the 3D structure, near-residue pairs identified by restraint information are moved into close distances, whereas the rest of the residue pairs connected to the pairs are then moved accordingly by optimizing the inter-residue distance in the violation loss of AlphaFold2. For simplicity and stability, only the RASP model with MSA bias and IPA bias are used for result analysis.
Restraint loss and tasks
All AlphaFold2 losses are retained, including the auxiliary loss from the Structure Module (a combination of averaged FAPE and torsion losses on the intermediate structures), averaged cross-entropy losses for distogram and masked MSA predictions, model confidence loss, experimentally resolved loss, and violation loss.
We introduce restraint loss into the model training to reinforce the input restraint information in the final prediction. This loss comprises three components, each corresponding to a restraint-related task. The first task is a 0/1 classification task with a loss called contact classification loss. In this task, residue-wise distogram prediction of input restraints is computed, and reorganized into 2 classes (whether or not the contact exists) with cross entropy calculated using the ground truth label. The second task is to minimize the distance RMSD difference of input restraints using a loss called dRMSD contact loss. The last task is to make local structures similar to the ground truth structures, and takes a reduced version of backbone FAPE loss called contact FAPE loss, in which the errors of all atom positions are calculated in the local backbone frames of all residues in the restraints. The contact FAPE loss and dRMSD contact loss are weighted equally at 0.5 so that the three losses are of the same order of magnitude at the beginning of training. We clip the sum of the last two losses by 1.5 to avoid training clashes in abnormal training examples.
Sampling strategy
The model was trained using the PSP dataset17, which was previously constructed by us. The PSP dataset is a compilation of true and distilled protein structures, and it includes sequence, structure, template, and MSA data for each protein sequence. Training data for RASP are sampled with replacement from both the true structure and distillation datasets and mixed in a ratio of 1:3.
To simulate the restraints observed in real experiments, the residue-wise distance map of the protein structure is computed using the pseudo-Cβ atom position of the residue, where the pseudo-Cβ atom is the C-\({{{\rm{\alpha }}}}\) atom position for glycine and Cβ for other amino acids. The restraints are sampled based on a probability distribution that decreases with residue-wise distance. When the distance is <7 Å, the probability is equal, and it decays exponentially from 7 Å to 10 Å. With this distribution, 90% of the sampled restraints are at a distance less than or equal to 8 Å, and 10% of the restraints are at a distance greater than or equal to 8 Å. This setting provides the model with a tolerance to longer restraints. The number of restraints is also randomly sampled from a distribution with equal probability for 16–128 and an exponential decay from 128 to 2048. The expected value of this distribution is 115.
NMR NOE assignment pipeline
The assignment pipeline used in this study was based on ARIA 2.36 (Ambiguous Restraints for Iterative Assignment), which was developed with Python 2 by Institut Pasteur. The FAAST assignment pipeline refered to part of the ARIA method, mainly the Calibration – Bound Calculation – Violation Analysis – Peak Assignment (CCVP for simplicity) functions, these functions perform assignment of peaks by comparing distance of restraints atom pairs in reference structure and theoretical distance calculated from intensity volume of the peaks. The original Python 2 code is first simplified and translated to Python 3 to cooperate with other parts of FAAST. Also, as the protein structure predicted by RASP does not distinguish equivalent hydrogens in amino acids, we collected equivalent groups of 20 common amino acids and redesigned the CCVP assignment algorithm based on distances between equivalent atomic groups in the group list.
The initial assignment is performed by comparing the chemical shift and NOE lists. Most of the restraints generated by initial assignments are ambiguous restraints (ARs), that is, a single peak is assigned with more than one possibility. While some peaks are naturally unambiguous (URs), without prior structure template information, a random/noise NOE peak could be assigned to hydrogen pairs with very similar chemical shift but with no correspondence in structure. The quality of the initial URs could therefore be very low, with more than half exceeding a distance of 6.0 Å. Thus, for the initial assignment, we filtered out initial URs with distances larger than 12 Å in the reference prediction without restraints. For each iteration, the current URs are fed into RASP to generate 20 structures with the UR subsampling rate of 5%, 10%, or 20%, depending on the iteration step (Fig. 4b, first parameters in each iteration block), and the structures are relaxed by OpenMM28. The structure bundle is then used to assign NOE peaks by CCVP.
In the standard pipeline, the hyper-parameters used for restraint subsampling and CCVP are iteratively tightened, with a restraint subsampling rate of 10% and partial assignment cumulative acceptance of 0.9 for the first iteration and 20% and 0.8 for the second iteration. ARs are transformed into URs iteratively. If after the second iteration, the median pLDDT score is lower than 80, a second round of iteration is initiated with restraint subsampling rates and CCVP parameters for partial assignment cumulative acceptance of (5%, 0.9), (5%, 0.8), and (10%, 0.8). The entire process takes 2–5 structure generation iterations, and the number of iterations, as well as the iterating parameters, can be flexibly adjusted.
Benchmarking data
The benchmarking data for our method consist of three parts:
PSP validation dataset: This dataset is the validation set of the PSP dataset17 and is used to evaluate the performance of the RASP model. The restraints in this dataset are sampled in the same way as during model training.
MR dataset: For most NMR structure in RCSB PDB database29, restraint .mr files are also deposited. We selected all the NMR .mr files from the RCSB PDB database in which a) the restraint numbering followed the PDB numbering, and b) the restraint recall for long-range restraints of the structure predicted by AF2 is less than 90%. This resulted in 333 samples.
NMR dataset: The NMR dataset was obtained to evaluate the NMR FAAST protocol. We obtained the .star file (including the chemical shift and NOE list), .mr file (submitted restraints), and .pdb file (structure) for 100 sequences in the ARTINA dataset by crawling the BMRB30 and RCSB PDB databases. After filtering out the .star files with missing chemical shift or NOE lists, 57 sequences were available for testing our protocol.
Additionally, as the NOE list in .star files from the BMRB dataset before submission could be filtered, we used the raw NOE peak list for pdb id 2MRM to evaluate the peak quality.
Statistics and reproducibility
In this study, sample sizes for the PSP validation dataset, MR dataset, and NMR dataset are 490, 333, and 57, respectively. For each sample in the PSP validation set, 0, 10, 20, 50, 100, and 200 restraints were sampled to generate cases of different restraint numbers for RASP evaluation. Sampling of 20 restraints with 10%, 20%, and 50% loose ratio were additionally used to generate cases of different restraint quality. All the sampled restraints are provided in the data depository for reproducibility. General statistics including average, median, and quantiles were computed using python numpy package. Violin plots were drawn using matplotlib.pyplot.violinplot.
Evaluation methods
We evaluated the structures and their consistency with restraints mainly with TM-score, restraint recall, and root mean square deviation (RMSD).
TM-score22,23 is a metric for assessing the topological similarity of protein structures. This score falls between 0 and 1, and higher TM-score indicates higher similarity between the two compared proteins. We used the TM-align31 package downloaded from Zhang lab for calculation of TM-scores. Specifically, in FAAST evaluation, since this pipeline returns an ensemble of structures, we calculate the TM-score of the most confident structure (structure with the highest predicted confidence) as the TM-score for each case. Since ARIA does not return predicted confidence for its structure ensemble, we use the highest TM-score of all 20 structures for each case as the TM-score for evaluation of ARIA performance.
Restraint recall is used to measure the consistency between a structure and a set of restraints. It is defined as the ratio between the number of rightly followed restraints by the structure and the total number of ground truth restraints, similar to the definition of recall in the machine learning field. In RASP evaluation, since the restraints are at residue level, we define a pairwise restraint to be followed by the structure as the distance between pseudo-Cβ atoms (see sampling strategy in method) in the residue pair is closer than 8 Å. In FAAST pipeline evaluation, since the NMR restraints are at atomic level, we define a pairwise restraint to be followed only when the closest hydrogen atomic distance in the two equivalent groups from the structure is lower than 6 Å.
In FAAST evaluation, two types of RMSD calculations are used. For measurement of mutual similarity within a structure ensemble, we calculate the pairwise C-\({{{\rm{\alpha }}}}\) RMSD between all pairs of different structures within the bundle using TM-align, and average them to obtain the pairwise mutual RMSD. For measurement of structure similarity between the deposited PDBs and processed structure ensemble, we follow the ARTINA3 evaluation and calculated the mean structure backbone atom RMSD using PyMOL for structured regions defined by ARTINA.
We further evaluated the goodness-of-fit of our predicted structures by FAAST to the experimental data using correlation score, RMSD score, and DP score. ANSURR26 (v2.0.55) (https://github.com/nickjf/ANSURR2) was used to calculate the correlation score and RMSD score. ANSURR accesses the accuracy of query structures by comparing their local rigidity with the random coil index (RCI). Both correlation score and RMSD score fall between 0 and 100, with higher scores indicating higher accuracy of structures in the aspects of secondary structure and overall rigidity, respectively. We re-referenced chemical shifts before calculating RCI by specifying “-r” as recommended and ran with “ansurr -p xxxx.pdb -s xxxx.str -r” for each structure.
The discrimination power (DP) score is the final output of the NMR structure quality assessment web-server tool RPF27 (https://montelionelab.chem.rpi.edu/rpf/), implying the correctness of the overall fold of query structure. We ran RPFs in batch using “dpsimple” from ASDP (v2.3) (https://github.rpi.edu/RPIBioinformatics/ASDP_public).
The AlphaFold violation loss is defined as
Following the definition given by AlphaFold11, where
It is used as a rough evaluation of structure violation in RASP benchmark.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The training set and PSP validation dataset are from our previous work17 and have been publicly available at http://ftp.cbi.pku.edu.cn/psp/. The PDB ID of the 333 samples used for restraints analysis in this work are available in Supplementary Data 1 and the PDB and restraint .mr files can be downloaded at RCSB PDB database(https://www.rcsb.org/). The information of the 57 samples used for FAAST pipeline benchmark are provided in Supplementary Table 1, and the structure .pdb files, restraint .mr files, and NMRSTAR .str files are available at RCSB PDB(https://www.rcsb.org/) and BMRB databases, according to their PDB and BMRB entry IDs. All processed data including the restraint files, predicted AF2 structures, AlphaLink structures, RASP structures, as well as FAAST structures are released at osf.io/wkspr32.
Code availability
The RASP and FAAST code are available at our gitee repository (https://gitee.com/mindspore/mindscience/tree/r0.6/MindSPONGE/applications/research/FAAST) and github repository33 (https://github.com/mindspore-ai/mindscience/tree/v0.7.0/MindSPONGE/applications/research/FAAST) under Apache 2.0 license. We additionally provide a colab notebook (https://colab.research.google.com/drive/1uaki0Ui1Y_gqVW7KSo838aOhXHSM3PTe?usp=sharing) for ease of use.
References
Kay, L. E. NMR studies of protein structure and dynamics. J. Magn. Reson. 213, 477–491 (2011).
Wüthrich, K. Protein structure determination in solution by NMR spectroscopy. J. Biol. Chem. 265, 22059–22062 (1990).
Klukowski, P., Riek, R. & Güntert, P. Rapid protein assignments and structures from raw NMR spectra with the deep learning technique ARTINA. Nat. Commun. 13, 6151 (2022).
Güntert, P. & Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 62, 453–471 (2015).
Nilges, M., Macias, M. J., O’Donoghue, S. I. & Oschkinat, H. Automated NOESY interpretation with ambiguous distance restraints: the refined NMR solution structure of the pleckstrin homology domain from beta-spectrin. J. Mol. Biol. 269, 408–422 (1997).
Rieping, W. et al. ARIA2: automated NOE assignment and data integration in NMR structure calculation. Bioinformatics 23, 381–382 (2007).
Herrmann, T., Güntert, P. & Wüthrich, K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 (2002).
Kuenze, G. & Meiler, J. Protein structure prediction using sparse NOE and RDC restraints with Rosetta in CASP13. Proteins 87, 1341–1350 (2019).
Kuenze, G., Bonneau, R., Leman, J. K. & Meiler, J. Integrative protein modeling in RosettaNMR from sparse paramagnetic restraints. Structure 27, 1721–1734 (2019).
Ovchinnikov, S. et al. Structure prediction using sparse simulated NOE restraints with Rosetta in CASP11. Proteins 84, 181–188 (2016).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. BioRxiv, biorxiv:2021.10.04.463034 (2021).
Terwilliger, T. C. et al. Improved AlphaFold modeling with implicit experimental information. Nat. Methods 19, 1376–1382 (2022).
Terwilliger, T. C. et al. Accelerating crystal structure determination with iterative AlphaFold prediction. Acta Crystallogr. Sect. D: Struct. Biol. 79, 234–244 (2023).
Stahl, K. et al. Protein structure prediction with in-cell photo-crosslinking mass spectrometry and deep learning. Nat. Biotechnol. 41, 1810–1819 (2023).
Liu, S. et al. PSP: million-level protein sequence dataset for protein structure prediction. arXiv, arXiv:2206.12240 (2022).
Robin, X. et al. Continuous Automated Model EvaluatiOn (CAMEO)—Perspectives on the future of fully automated evaluation of structure prediction methods. Proteins Struct. Funct. Bioinform. 89, 1977–1986 (2021).
Haas, J. et al. Introducing “best single template” models as reference baseline for the Continuous Automated Model Evaluation (CAMEO). Proteins Struct. Funct. Bioinform. 87, 1378–1387 (2019).
Haas, J. et al. Continuous Automated Model EvaluatiOn (CAMEO) complementing the critical assessment of structure prediction in CASP12. Proteins Struct., Funct. Bioinform. 86, 387–398 (2018).
Haas, J. et al. The Protein Model Portal—a comprehensive resource for protein structure and model information. Database 2013, bat031 (2013).
Zhang, Y. & Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710 (2004).
Xu, J. & Zhang, Z. How significant is a protein structure similarity with TM-score=0.5?. Bioinformatics 26, 889–895 (2010).
Tejero, R., Huang, Y. J., Ramelot, T. A. & Montelione, G. T. AlphaFold models of small proteins rival the accuracy of solution NMR structures. Front. Mol. Biosci. 9, 877000 (2022).
Li, E. H. et al. Blind assessment of monomeric AlphaFold2 protein structure models with experimental NMR Data. J. Magn. Reson. 352, 107481 (2023).
Fowler, N. J., Sljoka, A. & Williamson, M. P. A method for validating the accuracy of NMR protein structures. Nat. Commun. 11, 6321 (2020).
Huang, Y. J., Mao, B., Xu, F. & Montelione, G. T. Guiding automated NMR structure determination using a global optimization metric, the NMR DP score. J. Biomol. NMR 62, 439–451 (2015).
Eastman, P. et al. Openmm 7: Rapid development of high performance algorithms for molecular dynamics. PLOS Comput. Biol. 13, 1–17 (2017).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000).
Hoch, J. C. et al. Biological magnetic resonance data bank. Nucleic Acids Res. 51, D368–D376 (2023).
Zhang, Y. & Skolnick, J. TM-align: a protein structure alignment algorithm based on TM-score. Nucleic Acids Res. 33, 2302–2309 (2005).
Liu, S. et al. Dataset for RASP. OSF https://doi.org/10.17605/OSF.IO/WKSPR (2025).
i-robot et al. mindspore-ai/mindscience: v0.7.0 (v0.7.0). Zenodo. https://doi.org/10.5281/zenodo.15704096 (2025).
Acknowledgements
The authors thank Yupeng Huang for helpful discussions on data processing, and would like to extend our gratitude to Yuanpeng Janet Huang, the author of RPF, for his patience and guidance on how to use dpsimple. This work was supported by the National Science and Technology Major Project (2022ZD0115001 to S.L., Z.W., and Y.Q.G.), the National Natural Science Foundation of China (No. 92353304, and No. T2495221 to Y.Q.G., No. 22274050 to S.W. and 21825703 to C.T.), New Cornerstone Science Foundation (NCI202305 to Y.Q.G.), the Shanghai Science and Technology Commission (contract number: 23J21900300 and 24HC2810700 to S.W.), the Fundamental Research Funds for the Central Universities (to S.W.), the Strategic Priority Research Program of Chinese Academy of Sciences (XDB37000000 to C.T.), and Collaborative Innovation Program of Hefei Science Center, CAS (2022HSC-CIP011 to F.W.). We thank the staff members of the NMR Spectroscopy System (https://cstr.cn/31125.02.SHMFF.SM3.NMR) at the Steady High Magnetic Field Facility, CAS (https://cstr.cn/31125.02.SHMFF), for providing technical support and assistance in data collection and analysis.
Author information
Authors and Affiliations
Contributions
S.L., Z.W., and Y.Q.G. developed overall concepts in the paper and supervised the project. S.L., H.C., and Y.X. wrote the initial draft of manuscript. S.L., H.C., F.M., J.W., N.N., C.W., J.W., J.Z., M.C., J.L., and F.Y. developed and validated model and pipeline. S.L., H.C., Y.X., F.W., F.M., J.W., H.F., S.W., and C.T. carried out the data processing and analyses. Specifically, F.W. and C.T. provided the raw YgaP NOESY peak data. All authors contributed ideas to the work and assisted in editing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare the following competing interests: Changping Laboratory and Huawei Technologies Co., Ltd. are in the process of applying for a patent (202310400042.3) covering the FAAST and RASP methods, that lists S.L., H.C., N.N., Y.Q.G., Z.W., J.W., Y.X., F.M., J.L., and C.W. as inventors. All other authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Lucien Krapp and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Aylin Bircan, Laura Rodriguez Perez.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, S., Chu, H., Xie, Y. et al. Assisting and accelerating NMR assignment with restrained structure prediction. Commun Biol 8, 1067 (2025). https://doi.org/10.1038/s42003-025-08466-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08466-1
