
Abstract
Osteoporosis is increasingly attracting attention. Studies have indicated a correlation between vitamin K2 levels and bone mass, but the specific mechanisms remain unclear. Therefore, this study aims to further elucidate the specific molecular mechanisms and related target proteins. We found that serum vitamin K2 level in osteoporosis women was significantly lower than that in normal women. Animal experiments confirmed the anti-osteoporotic effects of vitamin K2. The mechanism and target of vitamin K2 therapy for osteoporosis was investigated through metabolomics and transcriptomics sequencing. Molecular docking was employed to identify downstream target proteins of vitamin K2, while EMSA and ChIP-qPCR were used to explore the specific molecular mechanisms. The results indicate that vitamin K2 promotes bone formation. Additionally, vitamin K2 may bind to the downstream target protein Nrf2 and inhibit Keap1-mediated ubiquitination, and Nrf2 has been shown to up-regulate CBR1 to inhibit osteoblast ferroptosis caused by the inflammatory factor PGE2. In summary, our study demonstrates that Vitamin K2 attenuates bone loss by inhibiting osteoblast ferroptosis, highlighting its therapeutic potential for osteoporosis. These findings advance our understanding of Vitamin K2-mediated osteoprotective effects and facilitate the identification of novel drug targets for osteoporosis treatment.
Similar content being viewed by others


Introduction
Osteoporosis, as a common factor leading to fractures in the middle-aged and elderly population, is increasingly garnering attention. Characterized primarily by reduced bone density, osteoporosis results in increased bone fragility and decreased bone strength, leading to fracture1. Under physiological conditions, osteoblasts secrete bone matrix, and osteoclasts are responsible for breaking down and absorbing the bone tissue. A dynamic equilibrium is maintained between the two. However, under pathological conditions, due to the imbalance of bone homeostasis, bone loss occurs2. Various factors can contribute to osteoporosis, such as endocrine levels, alcohol consumption, age, nutritional status, and more3. As a trace nutrient, fat-soluble vitamins participate in various biochemical reactions in the body. Studies have found a close association between fat-soluble vitamins and bone metabolism4 Inadequate intake of vitamins can lead to osteoporosis, and supplementation of vitamins has been shown to improve bone loss5. In recent years, fat-soluble vitamins have been increasingly attracting scholars’ attention in the prevention and treatment of osteoporosis6.
Vitamin K, as a fat-soluble vitamin, is a group of compounds with a quinone ring. Based on the different numbers of side-chain groups, it is divided into Vitamin K1 (mainly found in green leafy vegetables), Vitamin K2 (produced by microbial fermentation), and Vitamin K3 (synthetically manufactured)7,8. Vitamin K, as a clotting factor, plays a crucial role in blood coagulation9. However, recent studies suggest that a deficiency in Vitamin K2 (VK2) increases the risk of osteoporosis, and supplementation with VK2 has been shown to treat and prevent osteoporosis10,11. Abnormalities in VK2 levels have been obsreved among osteoporosis patients. In 1995, M Kaneki and others measured the levels of VK2 in elderly women who experienced osteoporotic fractures and those who did not. The results showed that vitamin K deficiency is one of the risk factors for osteoporotic fractures12.
Ferroptosis is a form of non-apoptotic programmed cell death, primarily characterized by iron-dependent lipid peroxidation and the accumulation of reactive oxygen species (ROS). Since the discovery of ferroptosis, numerous studies have confirmed its involvement in various organ injuries and degenerative diseases13. Investigating the mechanisms of cellular ferroptosis in diseases and its response to drugs is currently a major research focus. Osteoporosis, as a systemic bone metabolism disorder, involves the crucial role of ferroptosis in osteoblasts or osteoclasts during the process of bone loss14,15. Eikan Mishima et al. 2022 demonstrated that vitamin K is an effective inhibitor of ferroptosis16. Our preliminary research found that the up-regulation of ferroptosis in osteoblasts is one of the important causes of bone loss in osteoporosis, and the inhibition of ferroptosis in osteoblasts is a effective method to treat osteoporosis17.
Therefore, we speculate that VK2 may also act as an inhibitor of ferroptosis involved in bone metabolism. However, the exact mechanism remains unclear at present18,19. In this study, we conducted a combined analysis using non-target metabolomics and transcriptomics and found that VK2 inhibits osteoblast ferroptosis mediated by PGE2 by up-regulating CBR1. And methods such as simulated molecular docking, EMSA, Ch-IP, and Co-IP were used to determine the downstream target genes of VK2 and the specific molecular mechanisms regulating osteoblast ferroptosis. This study has explored new potential therapeutic targets for the prevention and treatment of osteoporosis in the future. Meanwhile, it has also provided a new research direction for the mechanism study of VK2 inhibiting cellular ferroptosis.
Results
Differential presence of VK2 in the serum of postmenopausal women and its potential role in improving bone loss in ovariectomized mice
Our previous study found that VK2 was closely related to the occurrence and development of osteoporosis20. We collected venous blood samples from postmenopausal women aged 55-65 (n = 103) and measured their bone density using dual-energy X-ray. Mass spectrometry analysis of the blood revealed differences in the serum levels of VK2 among postmenopausal women with normal bone mass (n = 35), reduced bone mass (n = 35), and osteoporosis (n = 33). Postmenopausal women with osteoporosis exhibited significantly lower levels of VK2 in their serum compared to women with normal bone mass (Supplementary Fig. 1A). We found no significant difference in the level of N-terminal propeptide of type I collagen (TP1NP) in bone turnover markers (Supplementary Fig. 1B). However, the level of serum bone alkaline phosphatase (bsALP) was significantly different between normal bone mass group and osteoporosis group (Supplementary Fig. 1C). Hypothesizing that VK2 might improve bone loss in postmenopausal women, we tested this idea by creating an osteoporosis mouse model (OVX mice) through ovariectomy. The mice were divided into three groups (control, OVX, VK2, n = 6) and fed for 8 weeks before being euthanized for micro-CT scanning of their femurs. Mice fed with VK2 showed significant improvements in bone density, cortical bone density, and BV/TV compared to OVX mice (Fig. 1A–C). Three-dimensional reconstructions of the femurs, both overall and in cross-section, supported these findings (Fig. 1D–F). Subsequent staining of mouse femur sections using the Goldner trichrome method revealed significantly more cortical and trabecular bone in the VK2 group compared to the OVX group (Fig. 1G–I). Calcein staining results (n = 3) showed that the rate of bone formation in OVX mice was significantly lower than that in ctrl group, while VK2 could promote the rate of bone formation in OVX mice (Fig. 1J, K).

A–C Micro-CT scans of mouse femurs (n = 6), showing bone density, cortical bone density, BV/TV, represented by histograms. D–F Three-dimensional reconstructions of mouse femurs in the control group, OVX group, and VK2 group, displaying the overall, cortical, and trabecular structures from left to right. G–I Goldner’s trichrome staining of mouse femur sections, with cortical bone stained in green, cancellous bone in orange-red, and cell nuclei in black (n = 3). Scale, 3000 μm; 1000 μm. J Calcein staining results of mouse femurs (ctrl, OVX, and VK2 groups, n = 3). Scale, 50 μm. K The distance between the two calcein staining (μm), divided by the time between the two calcein injections (10 d), yielded the rate of bone formation in the mouse femur, which is presented using a histogram. L Osteoblasts were treated with VK2 at concentrations of 10−9M, 10−8M, 10−7M, 10−6M, and 10−5M, and cell viability was measured using CCK-8 at 450 nm, represented by Histogram (n = 3). M Protein expression levels of RUNX2 and COL1 in osteoblasts after 7 days of differentiation shown by Western blot. N, O The histograms show the quantitative results of the gray value of WB (n = 3). P Alizarin Red staining to visualize calcium nodules in osteoblasts after 21 days of differentiation. Scale, 100 μm. Q Alkaline phosphatase staining to show the activity of alkaline phosphatase in osteoblasts after 14 days of differentiation. Scale, 100 μm. VK2 vitamin K2, OVX ovariectomy, RUNX2 runt-related transcription factor 2, COL1 collagen I. ns not significant, *P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. Data is presented as mean values ± S.D.
To further demonstrate the impact of VK2 on bone, we used mouse osteoblasts and found that a concentration of 10-5M VK2 optimally enhanced osteoblast activity in a CCK-8 cell viability assay (Fig. 1L). Subsequent cell experiments were conducted using this concentration. Additionally, Western blot results indicated that VK2 could promote the expression of RUNX2 and COL1 in mouse osteoblasts (Fig. 1M–O). Alizarin Red staining results demonstrated that VK2 significantly promoted the formation of calcium nodules in mouse osteoblasts (Fig. 1P). Alkaline phosphatase (ALP) staining results confirmed that VK2 could enhance the activity of ALP in mouse osteoblasts (Fig. 1Q). In summary, the results suggest that VK2 can alleviate bone loss in ovariectomized mice with postmenopausal osteoporosis and promote the differentiation of mouse osteoblasts.
Changes in bone tissue metabolism in OVX Mice and the restorative Effect of VK2
In order to explore the molecular mechanisms through which VK2 improves postmenopausal bone mass, we conducted a non-targeted metabolomics analysis of mouse bone tissues (ctrl group, n = 9; OVX group, n = 8; VK2 group, n = 9). The scatter plot of Partial Least squares discriminant Analysis (PLS-DA) shows the differences in metabolites among the control group, the OVX group, and the VK2 group. The closer R2Y is to 1, the more stable and reliable the model is. Q2Y is used to evaluate the predictive ability of the PLS-DA model. The R2Y in the figure are 0.97 and 0.96, respectively. And R2Y is greater than Q2Y, indicating that the model is well established. (Fig. 2A, B). The Venn diagram results showed 64 different metabolites common to the control and OVX groups, 30 different metabolites common to the OVX and VK2 groups, and 6 different metabolites present in all three groups (Fig. 2E). To identify metabolites with significant changes, we further analyzed the metabolites that exhibited significant upregulation and downregulation between the OVX and VK2 groups, visualizing them with a volcano plot (Fig. 2C, D). Our analysis enriched top 20 KEGG pathways (Fig. 2F). After excluding the pathways not related to bone metabolism, the remaining pathways include: Metabolic pathways, Inflammatory mediator regulation of TRP channels, cAMP signaling pathway, Arachidonic acid metabolism, Endocrine resistance. We found that all these enriched pathways were related to inflammation, and there is evidence suggesting that inflammation plays an important role in the development of osteoporosis21. We further discovered that prostaglandin E2 (PGE2) in the arachidonic acid pathway, as an inflammatory factor, has significantly different levels in bone tissue (n = 3). Previous studies have also shown that PGE2 is closely related to bone metabolism22. Therefore, the differential metabolite PGE2 was selected for the following research. Cluster analysis revealed a significant downregulation of PGE2 levels in OVX mice treated with VK2 (Fig. 2G, H). Using a KEGG pathway bubble chart, we found that PGE2 is produced by arachidonic acid and catabolic by Carbonyl Reductase 1 (CBR1). (Fig. 2I). To verify the results of metabolomic analysis, we measured the PGE2 level in the femur tissue of the mice by ELISA. The results showed that PGE2 level in the bone tissue of OVX mice was significantly higher than that in the ctrl group, and PGE2 level in the femur of OVX mice was reduced by VK2 (Fig. 2J). To further explore the reasons for the decrease in PGE2, we hypothesized that it might be due to an increase in the metabolic enzyme CBR1 for PGE2. Western blotting and immunohistochemistry confirmed this hypothesis (Fig. 2K–P). Furthermore, we used si-CBR1 (si-CBR1) to knockdown the intracellular level of CBR1 in mouse osteoblasts. The results of ELISA showed that the intracellular level of PGE2 was significantly reduced under VK2 treatment, while the level of PGE2 was increased again after si-CBR1 treatment. This suggests that VK2 can reduce the level of intracellular PGE2 through CBR1. (Fig. 2Q). In summary, based on the metabolomics analysis results, we demonstrate that VK2 upregulates the expression of CBR1 to promote the breakdown of PGE2, leading to its reduction.

A, B Score scatter plot of the first principal component (PC1) versus the second principal component (PC2) in PLS-DA analysis. R2Y represents the model’s explanatory rate, and Q2Y is used to evaluate the predictive ability of the PLS-DA model (ctrl group, n = 9; OVX group, n = 8; VK2 group, n = 9). C, D Volcano plot displaying the overall distribution of differential metabolites. The x-axis represents the fold change of metabolites in different groups, and the y-axis represents the significance level. Each point in the volcano plot represents a metabolite, with significantly upregulated metabolites shown in red and significantly downregulated metabolites shown in green. The size of the dots corresponds to the VIP value. E Venn diagram illustrating the shared and unique differential metabolites among different groups based on multiple comparative combinations. F Bubble chart illustrating the enrichment of KEGG pathways. The x/y-axis values represent the degree of enrichment of differential metabolites in the pathway, with larger values indicating higher enrichment. The color of the points represents the P-value from hypergeometric testing, with smaller values indicating greater statistical significance. The size of the points represents the number of differential metabolites in the respective pathway. G Hierarchical clustering analysis depicting the differences in metabolic expression patterns among the three groups. The vertical axis represents sample clustering, and the horizontal axis represents metabolite clustering. H Box plots showing the changes and differences in PGE2 levels in the control group, OVX group, and VK2 group. I Demonstrate the metabolic pathway of PGE2. J Competitive ELISA measuring absorbance at 450 nm to reflect PGE2 concentration, with a regression curve plotted using Origin and presented in a histogram (n = 3). K Western blot showing the expression of CBR1 in osteoblasts treated with VK2(10−5M). L The histogram shows the quantitative results of the gray value of WB (n = 3). M–O Immunohistochemical staining of mouse femur sections (ctrl, OVX and VK2 groups), depicting femoral neck and distal femur. Scale, 300 μm. P The results of IHC staining were quantified using ImageJ and presented as histograms (n = 3). Q Competitive ELISA measuring absorbance at 450 nm to reflect PGE2 concentration, with a regression curve plotted using Origin and presented in a histogram (n = 3). PLS-DA, Partial Least Squares Discrimination Analysis; PGE2, Prostaglandin E2; CBR1, Carbonyl Reductase 1. ns, not significant, *P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. Data is presented as mean values ± S.D.
VK2 inhibits Keap1-induced ubiquitination by binding to the transcription factor Nrf2, which can bind to the CBR1 promoter and promote its transcription
To further investigate how VK2 influences the expression of the CBR1 gene, we hypothesized that VK2 may enhance the activity of a certain transcription factor or directly bind to a transcription factor to promote its expression, thereby activating the upstream promoter of the CBR1 gene and promoting its expression. Nuclear factor erythroid 2-related factor 2 (Nrf2), as a common and essential regulator maintaining oxidative-reductive homeostasis23, may play a role in this process. First, we demonstrated that VK2 could indeed promote the expression of Nrf2 in cells through protein western blotting (Fig. 3A, B), and the results of RT-qPCR indicated that VK2 could promote the expression of Nrf2 by promoting its transcription (Fig. 3C). To further explore the reasons why VK2 promotes the upregulation of Nrf2, we initially explored whether VK2 could bind to Nrf2. As the complete protein structure of Nrf2 is currently unclear, we predicted the protein structure of Nrf2 using Uniprot and simulated the docking of VK2 with Nrf2 using AutoDock software (Fig. 3D), revealing three possible binding modes. To further demonstrate whether VK2 could bind to Nrf2, we retrieved the real protein structures of four partial Nrf2 proteins from Uniprot and displayed the lowest binding mode with VK2 (Fig. 3E). To verify whether VK2’s promotion of CBR1 expression is related to Nrf2, next, we used Electrophoretic Mobility Shift Assay (EMSA) to verify whether Nrf2 could bind to the CBR1 promoter. First, we identified the upstream promoter sequence of CBR1 by consulting the Ensembl database and designed probes based on its sequence (Fig. 3F). In the EMSA assay, we demonstrated that Nrf2 could bind to the promoter fragment of CBR1 in vitro (Fig. 3G). To further confirm this, Chromatin Immunoprecipitation-quantitative Polymerase Chain Reaction (ChIP-qPCR) assays confirmed that Nrf2 could also bind to the CBR1 promoter in osteoblasts (Fig. 3H). Finally, we used siRNA to knockdown the intracellular level of Nrf2, and the results of PCR and WB showed that CBR1 level was also reduced, which was reversed after the addition of VK2 (Fig. 3I–O).

A Western blot showed the expression of Nrf2 in osteoblasts treated with VK2(10−5M). B The histogram shows the quantitative results of the gray value of WB (n = 3). C The ratio of Nrf2 mRNA transcription amount to actin obtained by RT-qPCR and displayed by histogram (n = 3). D Three-dimensional visualization of the predicted molecular structure of Nrf2(green) using Pymol software. E Binding modes of four VK2 molecules(green) with partial structural domains of Nrf2, visualized in three dimensions. F DNA probe sequence used in the EMSA experiment. G EMSA image obtained through chemiluminescence, with lanes 1-5 representing the following: negative control group, cold probe group, labeled probe group, mutated probe group, and super-shift group. H Ct values obtained from ChIP-qPCR for the IgG control group and ChIP group, with enrichment ratios calculated to determine the percentage of chromatin in the two groups relative to total chromatin (%input) and presented in a histogram (n = 3). I The ratio of CBR1 mRNA transcription volume to actin obtained by RT-qPCR and displayed by histogram (n = 3). J, M Western blotting shows the expression of Nrf2 and CBR1 after treatment with siRNA and VK2 (10−5M). K, L, N, O The histograms show the quantitative results of the gray value of WB (n = 3). P Western blot showed the expression of Nrf2 in osteoblasts treated with MG132 (10 μM) and CHX (30 μg/ml) for 2 h, 4 h, and 8 h. Q The histogram shows the quantitative results of the gray value of WB (n = 3). R Immunoprecipitation of ubiquitin and Nrf2 in osteoblasts treated with VK2 (10−5M). S, T Co-immunoprecipitation (Co-ip) of Keap1 and Nrf2 in osteoblasts treated with VK2. U, V Co-localization image of Nrf2 (red) and Keap1 (green) in osteoblasts. The nucleus is stained with DAPI (blue). Scale, 100 μm. Scatter plots resulting from fluorescence quantitative analysis using ImageJ. W The histogram shows the Rcoloc values (Pearson’s for image above thresholds) obtained by quantitative analysis (n = 3). ChIP Chromatin Immunoprecipitation, qPCR quantitative Polymerase Chain Reaction, EMSA Electrophoretic Mobility Shift Assay, CHX cyclohexane. ns not significant, *P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. Data is presented as mean values ± S.D.
The results of simulated molecular docking indicate that VK2 may directly bind to Nrf2, and the key regulation of Nrf2 is mediated by proteasome degradation, which is mainly mediated by the repressor Keap1, resulting in Nrf2 ubiquitination. We speculated whether VK2 could participate in this process by directly binding to Nrf2. Firstly, protein western blotting showed that the degradation of Nrf2 was indeed affected by MG132 (Fig. 3P, Q). Subsequently, the results of immunoprecipitation proved that the addition of VK2 inhibited the ubiquitination level of Nrf2 (Fig. 3R). Next, in order to further explore the reason why VK2 inhibits Nrf2 ubiquitination, we used Co-IP experiment to prove that VK2 inhibits Keap1-mediated Nrf2 ubiquitination (Fig. 3S, T). The immunofluorescence results showed the co-localization changes of Nrf2 and Keap1 in cells after the addition of VK2, and the fluorescence intensity distribution map of pixels drawn by Image J software (Fig. 3U–W). Combined with the above results, we speculated that after VK2 binds to Nrf2, it prevents the binding of the latter to Keap1 and inhibits the ubiquitination degradation of Nrf2. To sum up, we believe that VK2 binds to Nrf2 and inhibits its ubiquitination, and that Nrf2 binds to the CBR1 promoter and promotes its transcription, while VK2 facilitates this process.
VK2 can alter transcriptional levels in mouse osteoblasts
To explore the changes in transcriptional activity induced by VK2 in osteoblasts, we conducted mRNA transcriptome analysis on mouse osteoblasts (control group, VK2-treated group). Principal Component Analysis (PCA) was performed on the two groups, and the results showed dispersion between the sample groups, indicating the reliability of the model (Fig. 4A). The volcano plot visually illustrates the distribution of differentially expressed genes in each comparison group, with significant upregulated genes represented by red dots and downregulated genes by green dots (Fig. 4B). The number of differential genes (both up-regulated and down-regulated) for each comparison combination was shown in a bar chart (Fig. 4C).

A Principal Component Analysis (PCA) of gene expression values (FPKM) for all samples to assess inter-group differences and intra-group sample replicates (n = 5). B Volcano plot illustrating the distribution of differentially expressed genes in each comparison group. Upregulated genes are represented by red dots, while downregulated genes are represented by green dots. C Histogram showing the number of differential differential genes. D Clustering analysis of the union of differentially expressed genes across all comparison groups. Genes with similar expression patterns are grouped together. The heatmap depicts the clustering of genes or samples based on their expression patterns, with colors in each square reflecting the normalized values of the expression data. E Scatter plot displaying the top 20 significantly enriched KEGG pathways from the enrichment results. The size of the points represents the number of genes annotated to the KEGG pathway, and the color gradient from red to purple indicates the significance of enrichment. F Histogram showing the number of genes on the KEGG enrichment pathway. G Immunofluorescence of FPN in mouse femoral sections (ctrl, OVX, and VK2 groups). Scale, 2000 μm. H The histogram shows the quantitative results of immunofluorescence (n = 3). FPN ferroportin, PCA principal component analysis. ns not significant, *P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. Data is presented as mean values ± S.D.
The next figure shows the significance level of the difference in gene expression between the treatment and control groups, with up-regulated genes represented by red dots and down-regulated genes represented by green dots. The differential gene clustering diagram is shown in the figure (Fig. 4D). A total of 358 differentially expressed genes were enriched, among which 102 genes were up-regulated and 256 genes were down-regulated. We employed a KEGG pathway bubble chart to demonstrate the enrichment of differentially expressed genes in KEGG pathways. From the KEGG enrichment results, we selected the top 20 significant KEGG pathways to create a scatter plot (Fig. 4E). For a more intuitive presentation, a bar chart was used to show the number of enriched genes (Fig. 4F). The pathways enriched by KEGG include DNA replication, mismatch repair, cell cycle, Ferroptosis, etc. Recent studies have shown that vitamin K may inhibit cellular ferroptosis16. Therefore, we want to further explore whether the specific mechanism by which VK2 improves bone mass is related to the inhibition of osteoblast ferroptosis. We noted that the cellular ferroptosis pathway was significantly down-regulated after addition of VK2 compared to the control group. Among the potential genes identified, we noticed the upregulation of the Slc40a1 gene (the translation product is ferroportin) by VK2. Ferroportin (FPN), as an iron transport protein, is closely related to ferroptosis and can transport intracellular Fe2+ ions out of the cell, inhibiting cellular ferroptosis24. To demonstrate the changes of FPN in vivo, we performed FPN immunofluorescence in the femur of the mice. The results showed that the expression of FPN in the femur of the OVX group was significantly lower than that of the ctrl group (n = 3), which was reversed by VK2 (Fig. 4G, H). In summary, VK2 upregulates the transcriptional levels of FPN in osteoblasts. VK2 may potentially inhibit osteoblast ferroptosis through this pathway.
VK2 inhibits RSL-3-induced ferroptosis in mouse osteoblasts
Based on the experimental results, we hypothesized that VK2 might inhibit cellular ferroptosis by upregulating FPN. To validate this hypothesis, we used RSL-3, a cellular ferroptosis inducer, to establish a cellular ferroptosis model25 and investigated the relationship between VK2 and ferroptosis in osteoblasts. Protein blotting experiments confirmed that the addition of RSL-3 led to a decrease in intracellular X-CT, Glutathione peroxidase 4 (GPX4), and FPN, while Transferrin receptor (TFRC) increased, indicating an elevated level of cellular ferroptosis. However, the addition of VK2 alleviated these effects (Fig. 5A–E). As ferroptosis involves characteristic changes in mitochondria, such as shrinkage, increased membrane density, and reduced cristae, we directly observed mitochondrial alterations using electron microscopy. The results showed that VK2 mitigated the characteristic changes in mitochondria induced by RSL-3 (Fig. 5F). Next, we used a Fe2+ probe to measure the intracellular Fe2+ content, and the results demonstrated that VK2 reduced the accumulation of intracellular Fe2+ induced by RSL-3 (Fig. 5G). Given that the characteristic of ferroptosis lies in lipid peroxidation, we will next investigate whether VK2 affects the production of malondialdehyde (MDA, the end product of lipid peroxidation) in ferroptosis induced by RSL-3. The results indicated that VK2 inhibited MDA production (Fig. 5H). To further demonstrate the inhibitory effect of VK2 on ferroptosis, we measured the levels of cellular ROS and mitochondrial membrane potential (JC-1). The results were consistent with the observations mentioned above (Fig. 5I, L–N), and flow cytometry results also confirmed this effect (Fig. 5J, K). In conclusion, we demonstrated that VK2 alleviates RSL-3-induced ferroptosis in mouse osteoblasts, consistent with the findings from the transcriptome analysis mentioned earlier.

A Western blot showing the expression of GPX4, TFRC, FPN, and XCT in osteoblasts under treatment with RSL3 and VK2. B–E The histograms show the quantitative results of the gray value of WB (n = 3). F Electron transmission microscopy images illustrating changes in mitochondrial morphology in osteoblasts under treatment with RSL3(1 μM) and VK2(10-5M). The green arrows point to normal mitochondria, and the red arrows point to mitochondria showing ferroptosis. Scale, 2 μm, 500 nm. G–I Fluorescence microscopy images displaying the distribution of intracellular Fe2 + , MDA, and ROS in osteoblasts under treatment with RSL3 and VK2. Scale, 100 μm. J, K Osteoblasts treated with RSL3 and VK2 were analyzed using flow cytometry to detect changes in intracellular ROS levels and mitochondrial membrane potential. L–N Fluorescence microscopy images showing changes in mitochondrial membrane potential in osteoblasts under different wavelength illuminations. Images from left to right correspond to osteoblasts at 520 nm, 590 nm, and the merged image obtained from both wavelengths. Scale, 100 μm. GPX4 glutathione peroxidase 4, TFRC transferrin receptor, xCT Solute Carrier Family 7 Member 11, MDA Malondialdehyde, ROS reactive oxygen species. ns not significant, *P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. Data is presented as mean values ± S.D.
VK2 inhibits osteoblast ferroptosis by upregulating CBR1/FPN
As mentioned above, we hypothesized that VK2 inhibits cell ferroptosis by upregulating FPN transcription, which was confirmed by western blotting (Fig. 6A, B). To further prove this point, we used small interfering RNA (siRNA) to transfect cells and knock down the expression of FPN. q-PCR results demonstrated that VK2 upregulates FPN transcription, and this promotion effect disappears when VK2 is added to cells after FPN knockdown (Fig. 6C). Protein blotting experiments showed that VK2 inhibits cellular ferroptosis by upregulating FPN, and this inhibitory effect is diminished after FPN knockdown (Fig. 6D–H). To explore the mechanism of VK2-induced FPN upregulation, we combined the results of previous metabolomic analysis with the above results. We hypothesized that PGE2, as an intracellular inflammatory molecule, may be involved in the process of cellular ferroptosis. To further prove this point, we measured cell viability with different concentrations of PGE2 using a CCK-8 assay. The results showed a decrease in cell viability with PGE2 treatment at concentrations of 0.1 μM, 1 μM, 10 μM, 0.1 mM, and 1 mM. Due to the significant impact of high concentrations of PGE2 on cell viability, we selected a concentration of 10 μM for subsequent experiments (Fig. 6I). and used ELISA to confirm that, under RSL-3 induction, the levels of intracellular PGE2 increase, and VK2 can inhibit this effect (Fig. 6J). Therefore, we believe that CBR1, as a metabolic enzyme for PGE2, may also play a role in inhibiting cellular ferroptosis. We first used CBR1 siRNA to transfect cells. q-PCR confirmed that the transcription of FPN was affected by CBR1, and knocked down the mRNA level of CBR1 in cells to inhibit the transcription of FPN. (Fig. 6K). Western blotting experiments indicated that, under PGE2 treatment, osteoblast ferroptosis was promoted, and VK2 inhibited this effect (Fig. 6L–P). Western blotting and Fe2+ ion probe experiments confirmed that after CBR1 knockdown, the accumulation of PGE2 would increase the level of ferroptosis in osteoblasts (Fig. 6Q–W). In summary, we have demonstrated that VK2 inhibits osteoblast ferroptosis by upregulating the expression of CBR1, promoting the breakdown of PGE2.

A Western blot showing the expression of FPN under VK2 treatment(10−5M). B The histograms show the quantitative results of the gray value of WB (n = 3). C Histogram showing the transcriptional levels of intracellular FPN in osteoblasts transfected with FPN-siRNA, as detected by qPCR (n = 3). D Western blot depicting the expression of GPX4, TFRC, FPN, and XCT in osteoblasts 48 h after FPN-siRNA transfection. E–H The histograms show the quantitative results of the gray value of WB (n = 3). I Osteoblasts treated with different concentrations of PGE2 (0.1 μM, 1 μM, 10 μM, 0.1 mM, 1 mM) were analyzed using CCK-8 to measure absorbance at 450 nm, represented by a histogram (n = 3). J Competitive ELISA measuring absorbance at 450 nm to reflect PGE2 concentration in osteoblasts treated with VK2, with a regression curve plotted using Origin and presented in a histogram (n = 3). K Histogram displaying the transcriptional levels of intracellular FPN in osteoblasts transfected with CBR1-siRNA, as detected by qPCR (n = 3). L Western blot showing the expression of GPX4, TFRC, FPN, and XCT in osteoblasts 48 h after PGE2 treatment. M–P The histograms show the quantitative results of the gray value of WB (n = 3). Q Western blot depicting the expression of CBR1, GPX4, TFRC, FPN, and XCT in osteoblasts 48 h after CBR1-siRNA transfection. R–V The histograms show the quantitative results of the gray value of WB (n = 3). W Fluorescence microscopy images showing the distribution of intracellular Fe2+ in osteoblasts transfected with CBR1-siRNA under treatment with PGE2 and VK2. Scale, 100 μm. FPN ferroportin, PGE2 prostaglandin E2, CBR1 carbonyl reductase 1, GPX4 glutathione peroxidase 4, TFRC transferrin receptor, XCT Solute Carrier Family 7 Member 11. ns not significant, *P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. Data is presented as mean values ± S.D.
Discussion
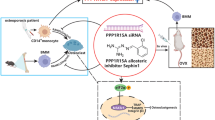
VK2, as a fat-soluble vitamin, has long been recognized as a coagulation factor in the past26. However, recent research has found a close relationship between vitamin K and bone metabolism27. In this study, we evaluated the anti-osteoporotic effects mechanisms and key targets of VK2 through a combined analysis of metabolomics and transcriptomics. Additionally, through molecular docking simulations and related experiments, we identified the target protein Nrf2 within osteoblasts for VK2. It was also demonstrated that VK2 can inhibit Nrf2 ubiquitination through the Nrf2/Keap1 signaling pathway. In summary, our results show that VK2 activates the transcription and expression of downstream gene CBR1 by binding Nrf2 and inhibiting Keap1-mediated ubiquitination, and then promotes the metabolism of inflammatory factor PGE2, thereby inhibiting ferroptosis in osteoblasts. (Fig. 7).

Upon entering the cell, VK2 can bind to the transcription factor Nrf2, competitively inhibit the binding of Keap1 to Nrf2, and reduce the ubiquitination of Nrf2 caused by Keap1. Nrf2 can bind to the upstream promoter of CBR1 to promote its transcription. As a metabolic enzyme for PGE2, CBR1 can break down PGE2 into PGF2α. PGE2, acting as an inflammatory factor, can promote cell iron death by downregulating the expression of GPX4, FPN, Slc7A11, and upregulating TFRC. Additionally, VK2 can upregulate the expression of ALP, COL1, and RUNX2 in osteoblasts to promote osteoblast differentiation and alleviate osteoporosis.
Multiple studies have indicated that patients with osteoporosis often experience disruptions in iron metabolism, which promote the occurrence of ferroptosis28,29. With the discovery of ferroptosis30, inhibiting ferroptosis in osteoblasts has gradually become a new target for the treatment of osteoporosis31. Recent studies suggest an increasingly close relationship between VK2 and ferroptosis32,33. It wasn’t until 2022 that research discovered vitamin K as an effective inhibitor of ferroptosis16. Subsequent studies demonstrated that the vitamin K epoxide reductase complex subunit 1-like 1 (VKORC1L1) is a potent ferroptosis suppressor in the vitamin K cycle. VKORC1L1 protects cells from ferroptosis by producing a reduced form of vitamin K34. Notably, our mRNA transcriptome analysis also supports this view. We found VK2 promotes the transcription of the intracellular slc40a1 gene, whose translation product is the iron transporter FPN, which is the only known intracellular iron effector protein and plays a crucial role in the regulation of cellular ferroptosis17. We also verified the mRNA transcriptome analysis results through experiments. Therefore, we believe that VK2 can inhibit the ferroptosis of osteoblasts by promoting the expression of FPN, these results suggest that VK2 may have new targets and mechanisms for inhibiting ferroptosis in cells. Our studies have shown that VK2 may be closely related to ferroptosis, suggesting that VK2 may be related to inhibiting ferroptosis in addition to promoting bone loss.
One of the characteristic features of ferroptosis is an increase in intracellular oxidative stress levels. VK2, as an antioxidant vitamin, can inhibit intracellular oxidative stress levels35. Nrf2 is a transcription factor that is sensitive to oxidation and can regulate the expression of various intracellular antioxidant genes. The main regulation of Nrf2 is mainly mediated by the inhibitory protein Kelch-like ECH associated protein 1 (Keap1), which has a structure that contains the cysteine residues of the E3 ubiquitin ligase ligand domain. In normal physiological conditions, Nrf2 can be ubiquitinated by the E3 ubiquitin ligase linked to Keap136, while in oxidative stress conditions, the ability of Keap1 to regulate the ubiquitination of Nrf2 is inhibited, resulting in the accumulation of active Nrf2, which promotes the expression of antioxidant stress genes in the cell37. Therefore, the Nrf2/Keap1 signaling pathway plays an important regulatory role in cellular oxidative stress, and the ferroptosis process is also accompanied by an increase in oxidative stress levels and the Nrf2/Keap1 pathway may also play an important role in it38. By simulating molecular docking, we found that the target of VK2 in osteoblasts may be Nrf2, which is a typical antioxidant-related transcription factor that plays a crucial role in cellular defense against oxidative stress by inducing the transcription of antioxidant genes39. Increased oxidative stress can inhibit the differentiation and proliferation of osteoblasts and even ferroptosis40,41. Studies indicate that Nrf2, as an antioxidant transcription factor, plays a significant role in inhibiting the process of cell ferroptosis42. Therefore, improving the accumulation of Nrf2 in cells to protect cells from the effects of ferroptosis may be one of the future directions to explore ferroptosis. Keap1, as a negative regulator of Nrf2, can cause Nrf2 ubiquitination. Our study proved that VK2 could directly bind to Nrf2. According to the above experimental results, we speculated that after VK2 binds to Nrf2, it competitively prevents the binding of Nrf2 to Keap1, reduces the ubiquitination of Nrf2, and thus increases Nrf2.
In order to further explore the specific mechanism of VK2 up-regulating Nrf2 level and inhibiting ferroptosis of osteoblasts, we validated through ChIP-qPCR and EMSA that Nrf2 can bind to the upstream promoter of CBR1 and directly regulate its transcription, confirming CBR1 as a downstream gene of VK2/Nrf2 involved in the inhibition of ferroptosis by VK2. CBR1 is a metabolic enzyme for PGE2, converting the pro-inflammatory factor PGE2 into PGF2α, thereby inhibiting intracellular inflammation43. PGE2, generated through the metabolism of arachidonic acid, is produced by cyclooxygenase (COX). PGE2 primarily acts by binding to four different G protein-coupled receptors (GPCRs): EP1, EP2, EP3, and EP444. With the gradual deepening of research on bone metabolism, chronic systemic inflammation is one of the factors that lead to osteoporosis and decreased bone mass45. Consistent with our research findings, through non-target metabolomics analysis of mouse bone tissue, we found that a large amount of PGE2 was accumulated in OVX mouse bone tissue. At present, it has been proved that the excessive accumulation of PGE2 is one of the inducements of ferroptosis in cells. Excessive accumulation of PGE2 increases intracellular oxidative stress levels, downregulating GPX4 levels and promoting cellular ferroptosis46. In vitro experiments proved that high concentration of PGE2 promoted ferroptosis of osteoblasts. However, VK2 reduces the levels of PGE2 in osteoblasts by upregulating Nrf2/CBR1, thereby inhibiting ferroptosis in osteoblasts.
In summary, our study demonstrated that PGE2 accumulation in OVX mouse bone tissue is one of the important reasons leading to bone loss. It was further determined that VK2 inhibits Nrf2 ubiquitination through the Nrf2/Keap1 pathway, and its Nrf2 downstream target gene CBR1. Our study elucidates a potential therapeutic mechanism of VK2 in postmenopausal osteoporosis, demonstrating its ability to alleviate bone loss through suppression of PGE2-mediated ferroptosis in osteoblasts. Our research opens up new potential therapeutic targets for the treatment of osteoporosis in the future, and simultaneously, it offers a fresh direction for further investigations into the mechanisms through which VK2 inhibits ferroptosis. However, our study also has shortcomings. First, because the complete structure of Nrf2 is not known at present, the results obtained by simulating molecular docking may have limitations. Secondly, we are not yet clear about the specific binding sites where VK2 and Keap1 compete to bind Nrf2. Our subsequent experiments will further determine their binding sites by constructing truncation of Nrf2.
Materials and methods
Reagents
Reagents used in the study include α-MEM purchased from Viva Cell (C04001-050×10), fetal bovine serum from Biological Industries (C04001-050×10), VK2 (MK-4) from MedChemExpress (HY-B2156), hematoxylin staining reagent kit from Beyotime (C0138), alkaline phosphatase (ALP) staining kit from Solarbio (G1481), RSL-3 from Solarbio (IR1120), JC-1 mitochondrial membrane potential assay kit from Beyotime (C2003S), reactive oxygen species (ROS) detection kit from Beyotime (S0033S), and PGE2 from MedChemExpress (HY-B2156). Antibodies used for western blotting are detailed in Table 1.
Patient recruitment and data collection
This study was approved by the Ethics Committee of the First Hospital of China Medical University ([2020]-248). All ethical regulations relevant to human research participants were followed. From March to September 2021, postmenopausal women aged 55-65 were recruited from the community. They underwent eligibility screening, with exclusion criteria as follows: (1) history of cardiovascular diseases; (2) patients with malignant tumors; (3) those who had undergone radiotherapy; (4) past or current use of medications affecting bone density. All participants were fully informed about the procedures and potential risks associated with this study, and they signed informed consent forms before participation.
Bone density measurement
All participants underwent dual-energy X-ray absorptiometry (DXA) to measure the bone density of the left femoral neck and lumbar spine (L1-L4). According to the World Health Organization (WHO) standards, T scores were interpreted as follows: T score ≥ −1 indicates normal bone mass, T score greater than −2.5 and less than −1 indicates low bone mass, and T score ≤ −2.5 indicates osteoporosis.
Postmenopausal women: collection of venous blood and determination of VK2 levels
All participants were in a fasting state (≥10 h), and venous blood was collected by a nurse into heparinized tubes. The blood samples were placed in ice packs on the same day and transported to the laboratory by study participants. Serum was extracted using a centrifuge (4000 × rpm, 10 min) within 4 h of blood collection, and the samples were stored in a −80 °C freezer until further use. Blood was drawn via venipuncture at 7 AM. For the determination of serum fat-soluble vitamin levels, liquid chromatography-tandem mass spectrometry (LC-MS/MS, SCIEX 6500 QTRAP) was employed to analyze serum samples. Standard curves were generated according to the manufacturer’s instructions, and the corresponding concentrations were calculated.
Animal experiment
The animal experiments designed in this study have been approved by the Animal Welfare Ethics Committee of China Medical University, with the approval number: CMU20242040. We have complied with all relevant ethical regulations for animal use. Eight-week-old female mice (C57BL6/J) were purchased from Beijing HFFK Biotechnology Company. Mice with abnormal physical development, trauma stimulation and poor health were excluded. The laboratory temperature is maintained between 22 °C and 26 °C, with noise levels below 60 dB. Light is provided alternately for 12 hours every day, based on sufficient drinking water and food for the mice. After one week of acclimatization in the animal experimental center, the mice were randomly divided into three groups by using the random number table method: ctrl group (n = 24), OVX group (ovariectomy, n = 24), and VK2 group (n = 24). After general anesthesia of the mice, the back fur was removed, and the back skin was incised longitudinally after routine disinfection. Blunt dissection was performed to expose the left ovary behind the peritoneum. After ligating and removing the left ovary, the peritoneum was sutured. The same procedure was repeated on the right side, and after removing both ovaries, the skin was sutured and disinfected. The control group underwent similar procedures, except for the ovary removal. One week post-surgery, mice in the VK2 group were orally administered VK2 dissolved in corn oil (20 mg/kg/d) via gavage. The remaining groups received an equivalent amount of corn oil. After 8 weeks of feeding, the mice were euthanized, and the bilateral femurs and tibias were stored in 4% paraformaldehyde or embedded in paraffin.
Micro-CT
Micro-computed tomography (micro-CT, SkyScan1276, Bruker, Germany) was employed to assess the microstructure of mouse femurs. The technical parameters of Micro-CT are as follows: Voltage 80 kV, current 90uA, scanning time 4 min. The spacing of the reconstructed sections was 7.5 μm, and the number of layers was 150. The cortical bone and trabeculae were automatically identified by the software. Bone morphometric parameters, including bone mineral density (BMD), cortical bone mineral density (Cortex Mean BMD), and bone volume fraction (Bone volume/tissue volume, BV/TV), were measured using micro-CT. Three-dimensional reconstruction was performed to visualize the morphology of mouse femurs and the number of cross-sectional bone trabeculae.
Bone tissue sectioning and Goldner’s trichrome staining
Mouse bone tissue was fixed in 4% paraformaldehyde for 48 h, preserved in 70% ethanol, and then embedded in paraffin using an embedding machine (Junjie, JB-P5). Sections of 4 μm thickness were cut using a pathological microtome (Leica Shanghai, RM2016). Bone tissue sections were stored at room temperature in light-protected conditions on glass slides. Goldner’s trichrome staining was performed to observe the mineralization of paraffin-embedded bone tissue sections. The bone sections were stained according to the manufacturer’s instructions (Servicebio, G1064). In brief, equal amounts of Solution A and Solution B were mixed as required. After dewaxing and rehydration, the sections were stained in the mixture for 20 min, followed by rinsing with water. Subsequently, a rapid differentiation in 1% hydrochloric acid alcohol for 2 s, rinsing with water, and further washing with distilled water were performed. The sections were then immersed in Goldner’s Solution C, D, C, and E for 5 min each, with intermittent rinsing in 0.2% acetic acid. The glass slides were dehydrated in ethanol for 5 min, cleared in xylene for 5 min, and fixed in neutral resin.
Calcein staining
Calcein (Servicebio, G1278) 20 mg/Kg was injected intraperitoneally 14 days before and 4 days before the mice were sacrificed. The femur of mice was taken under dark condition, and the bone tissue was fixed with 4% paraformaldehyde. After fixation, the bone tissue was decalcified, embedded and sectioned. Bone formation was observed under a fluorescence microscope.
Cell culture, induction of differentiation, and cell viability assay
MC3T3-E1 subclone 14 mouse pre-osteoblast cells were purchased from Procell Life Science & Technology. The cells were cultured in 10% fetal bovine serum/α-MEM, and the cell culture flasks were placed in a cell incubator (37 °C, 5% CO2). Osteoblast differentiation was induced using osteogenic differentiation medium (100 nM dexamethasone, 50 μM ascorbic acid, 10 mM β-glycerophosphate) for 7–21 days.
Cell viability was assessed using the CCK-8 cell viability assay kit (K1018, APExBIO). Mouse osteoblasts were seeded in a 96-well plate at a density of 5 × 10^3 cells per well. After 24 h of drug treatment, the cells were incubated in the dark at 37 °C, 5% CO2, and 10 μl of CCK-8 reagent was added to each well and incubated for 1 h. Absorbance at 450 nm was measured to assess cell viability.
Alizarin red staining and alkaline phosphatase (ALP) staining
Mouse osteoblasts were seeded in a 6-well plate at a density of 2 × 10^4 cells per well. When the cell density reached 70–80%, the culture medium was replaced with osteogenic induction medium containing VK2. The medium was changed every 48 h, and the induction of differentiation was carried out for 21 days. On day 21, the culture medium was removed, and the cells were fixed with 4% paraformaldehyde at room temperature for 20 min. After washing with PBS, Alizarin Red staining solution was added, and after 30 minutes of staining, the cells were observed under a microscope following three washes with PBS.
For ALP staining, mouse osteoblasts were seeded in a 6-well plate using the same density as mentioned above. The cells were induced to differentiate for 14 days following the same method. After removing the culture medium, PBS was used to wash the cells three times. Then, according to the manufacturer’s instructions, reagent A was added and incubated for 60 min, followed by the addition of reagent B and a 5 min incubation at 37 °C. After washing with PBS, reagent C was added and incubated for 2 minutes. After washing with PBS for 10 min, the cells were observed under a microscope.
Western blotting
Mouse osteoblasts were seeded in 100 mm culture dishes. When the cell density reached 70–80%, different drug treatments were applied, with the optimal concentration of VK2 being 1 × 10−5 M. After cell lysis with RIPA buffer at 4 °C for 15 min, sonication for 10 s was performed, and the lysates were centrifuged at 14,000 × rpm for 30 min. Protein concentration was quantified using the BCA Protein Assay Kit (Beyotime P0010). Equal amounts of protein (20 μg) were loaded into wells of 7.5–12.5% SDS-PAGE gels, separated by electrophoresis at 200 V, and then transferred onto PVDF membranes. After blocking, the membranes were incubated overnight at 4 °C with primary antibodies targeting specific proteins. Subsequently, membranes were incubated at 4 °C for 2 h with secondary antibodies against the species of the primary antibodies. Finally, chemiluminescence detection was carried out using the ECL method for exposure and observation.
Non-targeted metabolomics sequencing
Mouse bone tissue samples, ground after liquid nitrogen freezing, were collected and separated using a chromatography column (Hypesil Gold column 100 × 2.1 mm, 1.9 μm Thermo Fisher USA). Mass spectrometry analysis was performed using a mass spectrometer (Q Exactive™ HF-X Thermo Fisher Germany). The post-sequencing data files were imported into the CD 3.1 search software for processing. Each metabolite was subjected to simple screening, followed by the prediction of molecular formulas. The data were then compared with the mzCloud (https://www.mzcloud.org/), mzVault, and Masslist databases. The original quantitative results were standardized, resulting in the identification and relative quantification of metabolites.
Transcriptomics sequencing
mRNA was extracted from each sample using RNAiso Plus (Takara Japan), and the integrity and quantity of RNA were accurately assessed on the Agilent 2100 bioanalyzer. The cDNA was filtered with AMPure XP beads, subjected to PCR amplification, and then purified again using AMPure XP beads to obtain the library. After library construction, initial quantification was performed using the Qubit 2.0 Fluorometer. qRT-PCR was employed for accurate quantification of the effective concentration of the library to ensure its quality. Once the library passed quality control, Illumina sequencing was conducted for different libraries based on their effective concentrations, ultimately obtaining the sequence information of the target fragments. For differential gene expression analysis, edgeR software package (version 3.22.5) was utilized. The statistical enrichment of differentially expressed genes in KEGG pathways was analyzed using the clusterProfiler software (version 3.8.1). Additionally, GO enrichment analysis of differentially expressed genes was carried out. The GSEA analysis tool (http://www.broadinstitute.org/gsea/index.jsp) was employed for gene set enrichment analysis of GO and KEGG datasets for the species in question.
Immunohistochemistry (IHC), immunofluorescence (IF)
Bone tissue was fixed in 4% paraformaldehyde for 48 h. The fixed tissue was preserved in 70% ethanol and then embedded in paraffin using an embedding machine (Junjie, Wuhan, JB-P5) and sectioned at 4 μm using a pathology microtome (Leica Shanghai, RM2016). The prepared bone tissue sections were used for immunohistochemical staining of CBR1 and immunofluorescence staining of FPN. The excitation wavelength of immunofluorescence was 465–495 nm, and the emission wavelength was 515–555 nm.
ELISA
According to the manufacturer’s instructions, we utilized the PGE2 ELISA kit (Elabscience E-EL-0034) to measure the levels of PGE2 in cell and bone tissue of mice samples. In brief, after collecting samples, we used sonication to break them, followed by centrifugation at 1500 × g for 10 min, and the supernatant was collected for analysis. In each well of the microplate, 50 μL of biotinylated antibody working solution was added and incubated at 37 °C for 45 minutes. After washing, the HRP enzyme conjugate working solution was added, followed by a 30 min incubation at 37 °C. Substrate solution was then added, and after a 15 min incubation at 37 °C, the reaction was terminated by adding stop solution. The absorbance of each well was immediately measured at a wavelength of 450 nm using a microplate reader.
Molecular docking simulation
Initially, we retrieved five known structures of the transcription factor Nrf2 (Uniprot ID: Q60795) from the Uniprot database (https://www.uniprot.org/). Four of these structures were obtained from the Protein Data Bank (PDB) (https://www.rcsb.org/) based on X-ray crystallography data, while the remaining structure was predicted using the AlphaFold database (https://alphafold.ebi.ac.uk/) with MMseq2 and Foldseek algorithms. It is important to note that the structures provided by PDB do not represent the complete form of the Nrf2 protein but rather its structural domains in complex with other proteins. Consequently, we used Pymol software (version 2.3.4, https://pymol.org/2/) to extract and separately save the relevant portions of Nrf2.
Subsequently, we obtained the chemical structure information of VK2 from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). Using the Open Babel GUI tool version 3.1.1 (http://openbabel.org/wiki/Main_Page), we converted the obtained file format into a format suitable for molecular docking. With Autodock software (version 4.2.6, http://autodock.scripps.edu/), we conducted virtual molecular docking between VK2 and the structural domains of Nrf2. Finally, the docking results were subjected to three-dimensional visualization using Pymol software to assess the interaction between VK2 and Nrf2. The results, including binding energies and inhibition constants, are summarized in Table 2.
qRT-PCR
Total RNA was extracted from cells using the RNA extraction reagent (TaKaRa, 9767). Subsequently, the RNA was reverse transcribed into cDNA using the Prime Script RT reagent kit (TaKaRa, RR047A). Finally, qRT-PCR was performed using Trans Start Green qPCR SuperMix (TransGen, AQ101-01) and the LightCycler480 system. The qRT-PCR conditions included an initial denaturation step at 94 °C for 30 s, followed by denaturation at 94 °C for 5 s, annealing at 55 °C for 15 s, and a total of 40 cycles. The obtained results were used to analyze the gene transcription levels. The primer sequences are provided in Table 3.
Immunoprecipitation (IP)
According to the manufacturer’s instructions, we used the Immunoprecipitation Kit (Proteintech, PK10008) to perform the IP experiment. First, after collecting the cells, we added an appropriate amount of IP lysis buffer and incubated at 4 °C for 30 min. Then, we sonicated the cells and centrifuged to collect the supernatant. We took an appropriate amount of the supernatant as the Input, and then added Incubation buffer, antibody, and control IgG to the supernatant and incubated overnight. The next day, we added an appropriate amount of rProtein A/G beads slurry to the mixture and incubated for 3 h. Then, we washed the immunoprecipitated complex with Washing buffer four times, and finally, we washed it with Elution buffer twice and collected the elution liquid. We added an appropriate amount of Sample buffer and Alkali neutralization buffer, boiled it in a water bath at 100 °C for 5 min, and finally, we used the method of Western blotting to detect the IP results.
Cell immunofluorescence (IF)
First, we planted the cells on coverslips, fixed them with 4% formaldehyde, and washed them with PBS three times. We performed fluorescent homologous dual-labeling of the cells on the coverslips with Nrf2 and Keap1 using fluorescent dyes. We incubated the coverslips with anti-Nrf2 monoclonal antibody and goat anti-rabbit IgG (HRP) (1:500, Servicebio, GB23301), and then treated them with FITC-tyrosine (Servicebio, G1222). After protein recovery, we incubated the coverslips with anti-Keap1 monoclonal antibody and goat anti-rabbit IgG (CY3) (1:300, Servicebio, GB21303). For each fluorescent group, we obtained images using specific excitation and emission wavelengths. DAPI Excitation Wavelength: 330–380 nm, Emission Wavelength: 420 nm. FITC Excitation Wavelength: 492 nm, Emission Wavelength: 520 nm. CY3 Excitation Wavelength: 510–560 nm, Emission Wavelength: 590 nm.
Electrophoretic mobility shift assay (EMSA)
According to the manufacturer’s instructions, we conducted the EMSA experiment using the EMSA chemiluminescent kit (Beyotime GS009). In essence, cell nuclear proteins, probes, antibodies, and loading buffer were mixed in a system of 20 μL and left to stand at room temperature for 30 min. The resulting mixture was then loaded into a 4% polyacrylamide gel for electrophoresis. The proteins, probes, and the complexes of proteins and probes were transferred to a nylon membrane. UV cross-linking was performed using a UV cross-linker with a wavelength of 254 nm, 120 mJ/cm², and cross-linking for 45–60 s.
The nylon membrane was then blocked with a sealing solution containing Streptavidin-HRP Conjugate for 20 min. After washing with a wash buffer three times, the reaction balance solution was added. The nylon membrane was placed in the prepared BeyoECL Moon working solution, and the final exposure was observed.
ChIP-qPCR
Following the manufacturer’s instructions, we used the ChIP-qPCR assay kit (Beyotime P2083S) for ChIP-qPCR experiments. In summary, cells were initially fixed with formaldehyde. After cell collection, BufferA, BufferB, and ChIP Buffer were sequentially added. After sonication, DNA fragments of 100–200 bp were obtained. A portion of the supernatant was taken as an input control, and the remaining portions were incubated with antibodies against IgG and Nrf2, respectively, overnight on a rocking platform at 4 °C. Next, 30 µl of pre-mixed Protein A/G Magnetic Beads/Salmon Sperm DNA was added, and the mixture was shaken for 60 min at 4 °C. During the magnetic separation process, Low Salt Immune Complex Wash Buffer, High Salt Immune Complex Wash Buffer, LiCl Immune Complex Wash Buffer, and TE Buffer were used for washing. After adding 0.5 M NaCl, the mixture was heated at 65 °C for 2 h in a metal bath. Following this, EDTA, Tris, and Proteinase K were added to separate the DNA-protein complexes. After adding Tris-equilibrated phenol, the solution was centrifuged at 12,000 × g for 5 min to collect the supernatant. Then, an equal volume of chloroform was added, and the mixture was centrifuged at 12,000 × g for 5 min. The supernatant was transferred to another centrifuge tube, and 20 µg of glycogen, 1/10 volume of 3 M NaAc, and 2.5 times the volume of anhydrous ethanol were added. After thorough mixing, the mixture was precipitated at −70 °C for 1 h to obtain DNA precipitate. The subsequent DNA amplification was carried out using the aforementioned qRT-PCR method.
Fluorescent inverted microscope, flow cytometry, and transmission electron microscope
JC-1, DCFH-DA, Fe2+ probe, and malondialdehyde (MDA) were examined using the fluorescent inverted microscope (Nikon Eclipse Ti, Nikon Corporation) and FACScan flow cytometer (Beckman Coulter, Inc.). Regarding the gating strategy, first, circle the target cell population in the FSC-A vs SSC-A plot, and exclude debris and cell aggregates. Then, gate the positive cells according to the instructions provided in the reagent kit manual. The transmission electron microscope was employed to observe the mitochondrial morphology within osteoblasts.
Detection of mitochondrial membrane potential and ROS
Following the manufacturer’s instructions, cells were thoroughly mixed with prepared JC-1 working solution and DCFH-DA, each at a final concentration of 1 μM. After light-protected incubation, observations of mitochondrial membrane potential and ROS in cells were conducted using an inverted fluorescent microscope and flow cytometer. When observed with the inverted microscope, red fluorescence was observed at 590 nm, and green fluorescence was observed at 520 nm. During flow cytometry analysis, red and green fluorescence were respectively detected by PE and FITC channels.
Detection of intracellular Fe2+ and lipid peroxidation (MDA)
After seeding cells in a 6-well plate, cells were treated with C11-BODIPY 581/591 (Invitrogen) and FerroOrange (Cell Signaling Technology, 36104S) according to the manufacturer’s instructions, each at a final concentration of 1 μM, to respectively detect cellular lipid peroxidation and iron. Following incubation in a culture chamber, images were obtained using an inverted fluorescent microscope.
siRNA transfection
The siRNA was purchased from Msbio-tech, and three different siRNAs were simultaneously transfected. Cells were seeded in a 6-well plate or culture dish, and 24 hours prior to transfection, the culture medium was replaced with antibiotic-free medium. Using antibiotic-free Opti-MEM reduced serum medium (Gibco), a mixture of 50 nM siRNA and Lipofectamine™ 2000 reagent (Thermo Fisher Scientific) was prepared. The two liquids were combined and left at room temperature for 20 min. The resulting mixture was then added to the cell culture medium, and after 8 h of transfection, it was replaced with regular culture medium. The siRNA sequences are provided in Table 4.
Schematic diagram
The Schematic diagram (Fig. 7) in this article was created by Hongyang Li. The elements in the picture were referenced from the open website (https://www.figdraw.com/static/index.html#/), and we drew and made appropriate modifications in Adobe Illustrator software.
Statistics and reproducibility
SPSS 26.0 software was used for statistical analysis of each value. The data involved in this study were expressed as mean ± standard deviation (SD). GraphPad Prism 9.0 was used for statistical analysis of the data and graphics production. Kolmogorov-Smirnov test was used to assess the normality of the data, and Student’s t-test or one-way ANOVA was used for comparison. In this study, all experiments were repeated three times. Quantification of Western blot and other Image results. Ratios were obtained by quantifying images using Image J software. All sample replicates are labeled in the figure legends. In this study, all experiments were performed in triplicate. ns, not significant, *P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. Data is presented as mean values ± S.D.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data available on request from the authors. Transcriptomic data can be accessed with the BioProject number PRJNA1284009 at the NCBI BioProject database. Source data underlying graphs in the study can be found in supplementary data.
References
Bullamore, J. et al. Effect of age on calcium absorption. Lancet 2, 535–537 (1970).
Zhivodernikov, I. V et al. Molecular and cellular mechanisms of osteoporosis. Int. J. Mol. Sci. 24, 15772 (2023).
Khiyali, Z. et al. Smoking, alcohol consumption, drug abuse, and osteoporosis among older adults: a cross-sectional study on PERSIAN cohort study in Fasa. BMC Geriatr. 24, 80 (2024).
Levy, C. & Lindor, K. D. Management of osteoporosis, fat-soluble vitamin deficiencies, and hyperlipidemia in primary biliary cirrhosis. Clin. Liver Dis. 7, 901–910 (2003).
Ensrud, K. E. & Crandall, C. J. Osteoporosis. Ann. Intern. Med. 177, Itc1–itc16 (2024).
Liu, Y. et al. The anti-osteoporosis effects of vitamin K in postmenopausal women. Curr. Stem Cell Res Ther. 17, 186–192 (2022).
Wang, H., Ma, Y., The potential of vitamin K as a regulatory factor of bone metabolism-a review. Nutrients. 15, 4935 (2023).
Fusaro, M. M. et al. Vitamin K and bone. Clin. Cases Min. Bone Metab. 14, 200–206 (2017).
Aaseth, J. O. et al. Vitamin K and calcium chelation in vascular health. Biomedicines. 11, 3154 (2023).
Kaneki, M. [Vitamin K2 as a protector of bone health and beyond]. Clin. Calcium 15, 605–610 (2005).
Huang, Z. B. et al. Does vitamin K2 play a role in the prevention and treatment of osteoporosis for postmenopausal women: a meta-analysis of randomized controlled trials. Osteoporos. Int 26, 1175–1186 (2015).
Kaneki, M. et al. [Serum concentration of vitamin K in elderly women with involutional osteoporosis]. Nihon Ronen Igakkai Zasshi 32, 195–200 (1995).
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282 (2021).
Yang, Y. et al. Prevention and treatment of osteoporosis with natural products: Regulatory mechanism based on cell ferroptosis. J. Orthop. Surg. Res. 18, 951 (2023).
Hu, Y. et al. Identification of ferroptosis-associated biomarkers for the potential diagnosis and treatment of postmenopausal osteoporosis. Front Endocrinol.13, 986384 (2022).
Mishima, E. et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 608, 778–783 (2022).
Tao, L. et al. Exerkine FNDC5/irisin-enriched exosomes promote proliferation and inhibit ferroptosis of osteoblasts through interaction with Caveolin-1. Aging Cell 23, e14181 (2024).
Jin, C. et al. A Novel Anti-Osteoporosis Mechanism of VK2: Interfering with Ferroptosis via AMPK/SIRT1 Pathway in Type 2 Diabetic Osteoporosis. J. Agric Food Chem. 71, 2745–2761 (2023).
Jiang, Z. et al. Ferroptosis in osteocytes as a target for protection against postmenopausal osteoporosis. Adv. Sci. 11, e2307388 (2024).
Wang, J. et al. Development and validation of a novel prediction model for osteoporosis: from serotonin to fat-soluble vitamins. Bone Jt. Res 14, 111–123 (2025).
Li, C. et al. Concrete-inspired bionic bone glue repairs osteoporotic bone defects by gluing and remodeling aging macrophages. Adv. Sci.11, e2408044 (2024).
Zhao, D. et al. Connexin hemichannels with prostaglandin release in anabolic function of bone to mechanical loading. Elife. 11 (2022).
Saha, S., Rebouh, N. Y., Anti-osteoarthritis mechanism of the Nrf2 signaling pathway. Biomedicines. 11 (2023).
Kajarabille, N., Latunde-Dada, G. O., Programmed cell-death by ferroptosis: antioxidants as mitigators. Int. J. Mol. Sci. 20 (2019).
Qi, J. et al. Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation-mediated cell death in mice. Am. J. Pathol. 190, 68–81 (2020).
van den Besselaar, A. et al. Defining a metrologically traceable and sustainable calibration hierarchy of international normalized ratio for monitoring of vitamin K antagonist treatment in accordance with International Organization for Standardization (ISO) 17511:2020 standard: communication from the International Federation of Clinical Chemistry and Laboratory Medicine-SSC/ISTH working group on prothrombin time/international normalized ratio standardization. J. Thromb. Haemost. (2023).
Wang, H. et al. Vitamin K2 improves osteogenic differentiation by inhibiting STAT1 via the Bcl-6 and IL-6/JAK in C3H10 T1/2 clone 8 cells. Nutrients. 14, 2934 (2022).
Liu, P. et al. Ferroptosis: a new regulatory mechanism in osteoporosis. Oxid. Med. Cell Longev. 2022, 2634431 (2022).
Ni, S. et al. Hypoxia inhibits RANKL-induced ferritinophagy and protects osteoclasts from ferroptosis. Free Radic. Biol. Med. 169, 271–282 (2021).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Gao, Z. et al. Ferroptosis—a new target of osteoporosis. Exp. Gerontol. 165, 111836 (2022).
Hirschhorn, T. & Stockwell, B. R. Vitamin K: a new guardian against ferroptosis. Mol. Cell 82, 3760–3762 (2022).
Wang, Z. Vitamin K inhibits ferroptosis via VKORC1L1. Nat. Cell Biol. 25, 1240 (2023).
Henning, L. et al. Reactive microglia are the major source of tumor necrosis factor alpha and contribute to astrocyte dysfunction and acute seizures in experimental temporal lobe epilepsy. Glia 71, 168–186 (2023).
Muszyńska, M. et al. Protective effects of vitamin K compounds on the proteomic profile of osteoblasts under oxidative stress conditions. Molecules. 25, 1990 (2020).
Dinkova-Kostova, A. T. et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 99, 11908–11913 (2002).
Yang, G. et al. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal 18, 1906–1919 (2013).
Fan, Z. et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6, e371 (2017).
Zhang, J. et al. Nrf2 and mitochondria form a mutually regulating circuit in the prevention and treatment of metabolic syndrome. Antioxid Redox Signal. 41, 744-768 (2024).
Pan, J. X. et al. APP promotes osteoblast survival and bone formation by regulating mitochondrial function and preventing oxidative stress. Cell Death Dis. 9, 1077 (2018).
Almeida, M. et al. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J. Biol. Chem. 282, 27298–27305 (2007).
Ma, H. et al. Melatonin suppresses ferroptosis induced by high glucose via activation of the Nrf2/HO-1 signaling pathway in type 2 diabetic osteoporosis. Oxid. Med. Cell Longev. 2020, 9067610 (2020).
Mao, L. et al. Carbonyl reductase 1 attenuates ischemic brain injury by reducing oxidative stress and neuroinflammation. Transl. Stroke Res. 12, 711–724 (2021).
Kulesza, A., Paczek, L., Burdzinska, A. The role of COX-2 and PGE2 in the regulation of immunomodulation and other functions of mesenchymal stromal cells. Biomedicines. 11 (2023).
Di, D. et al. Frailty phenotype as mediator between systemic inflammation and osteoporosis and fracture risks: a prospective study. J. Cachexia Sarcopenia Muscle 15, 897–906 (2024).
Liu, Y. et al. PGE2 pathway mediates oxidative stress-induced ferroptosis in renal tubular epithelial cells. Febs J. 290, 533–549 (2023).
Acknowledgements
The authors particularly thank the participants and their contribution to this study. This work was supported by the National Natural Sciences Foundation of China [32200943 and 82072387].
Author information
Authors and Affiliations
Contributions
L.T. and H.L. contributed Conceptualization, Methodology, Validation, Formal analysis, Resources, Writing—Original Draft, and Visualization; J.W. contributed Methodology, Validation; Q.L. contributed Resources, Data Curation; W.C. contributed Validation, Data Curation; Y.Z. contributed Conceptualization, Writing—Review and Editing, Supervision, Project administration, and Funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Aline Bozec and Joao Valente.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tao, L., Li, H., Wang, J. et al. Vitamin K2 inhibits PGE2-mediated osteoblast ferroptosis by upregulation of CBR1 via the Nrf2/Keap1 pathway. Commun Biol 8, 1116 (2025). https://doi.org/10.1038/s42003-025-08564-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08564-0
