
Abstract
Innovations in 3D tissue imaging have revolutionized research, but limitations stemming from lengthy protocols and equipment accessibility persist. Widefield microscopy is fast and accessible but often excluded from 3D imaging workflows due to its lack of optical sectioning. Here we combine tissue clearing with a commercial depth-variant deconvolution approach that we optimized for large-volume widefield imaging. By implementing prefiltering with z-brick splitting, we achieve subnuclear axial resolution in tissues to a depth of 500 µm in multi-tile scan images. We illustrate the utility of this method in a model of ileitis and to gain a 3D perspective in thick brain slices from a model of cerebral amyloid angiopathy, where we resolve amyloid deposits along small blood vessels, attaining resolution that compared favorably to confocal microscopy. Finally, we leverage our approach to image hundreds of consecutive z planes for richer evaluation of cleared human kidney biopsies in a simulated, transplant time window and visualized atrophic tubules and winding arterioles associated with glomeruli in 3D. Having achieved subnuclear z-resolution in sections hundreds of microns thick, coupling widefield microscopy of cleared tissue to robust deconvolution now emerges as an accessible and viable method to gain 3D insight in research or clinical evaluations.
Similar content being viewed by others

Introduction
Several methods for tissue preparation and refractive index matching to achieve rapid, even, and thorough tissue transparency have been developed. These include CUBIC1, DISCO2, EZClear3, and ADAPT-3D4. We wondered if these improvements to tissue processing could be combined with cost-effective, fast, and accessible three-dimensional (3D) imaging carried out using widefield epifluorescent microscopy. At first glance, widefield epifluorescent microscopy might seem outdated and the least likely to find utility at the forefront of 3D imaging due to diffuse noise inherent to widefield microscopy that obscures and blurs the signal. Widefield microscopy does not have the advantage of confocal, two-photon, or light-sheet microscopy, where collection of light in a tissue sample is limited to a defined layer of tissue by a pinhole, the temporal compression of photons, or an arrangement of lenses that creates spatial illumination, respectively. However, given that widefield epifluorescent microscopes are often more accessible and user-friendly than other sophisticated systems, we reasoned that improving their utility in deep 3D imaging could have advantages. Moreover, a widefield system with precision z-axis control creates an opportunity to collect x-, y-, and z-plane data. However, much of the emitted light collected will be out of focus as it spreads through the many z-layers of tissue, to the point where widefield microscopy has largely been disregarded for providing meaningful axial information. Correction of this blurring to reconstruct a sharp image that places detected light at its point of origin can be estimated, or deconvolved, with a point spread function (PSF). A PSF is a functional shape that describes the diffraction of light through an optical system5. Both open source (e.g., DeconvolutionLab2 or DeconWolf) and commercial systems (e.g., AutoQuant, Scientific Volume Imaging Huygens) ably estimate the diffraction of light in deconvolution modalities6,7,8. However, advances in 3D deconvolution using widefield microscopy have mainly been demonstrated on samples that are little more than 1–2 cell layers in thickness or are mostly applied to other modalities like light-sheet imaging9.
Application of deconvolution to thicker tissues or cell layers requires PSFs that account for and vary with the depth of the sample. With recent advances in machine learning algorithms and improvements to computer hardware (e.g., graphics processing unit), there have been efforts to scale deconvolution to improve depth-variant analysis. However, depth-variant deconvolution remains a challenge, and it has not yet been demonstrated successfully in large-volume 3D widefield imaging. While tissue thickness is one challenge, widefield epifluorescent microscopy adds the additional challenge of a large diffraction limit that arises from low numerical apertures on objective lenses in combination with large spherical aberration. Spherical aberration is a phenomenon where rays of light do not converge toward a focused point due to a mismatch in refractive indices as light passes through multiple imaging depths10.
To overcome the challenges of unrestrained light collection in widefield microscopy that are accentuated in deep tissue imaging and achieve rapid start-to-finish large-volume 3D tissue imaging with an accessible widefield system, we herein describe how we adapted and combined the commercially available software product Scientific Volume Imaging Huygens, which starts with depth-variant PSFs, with optimized image acquisition using a turn-key commercial microscope. We demonstrate successful depth-variant deconvolution of images acquired during widefield microscopy through a 0.5-mm tissue depth, yielding results approaching the quality of confocal images for many features in a fraction of the time. We go on to further expand the tissue volume that could be analyzed by developing a protocol for x- and y- tiling of thick stacks of depth-variant deconvoluted images to reconstruct significantly larger tissue areas in a volume of approximately 1 cm3. These technical advances allowed us to visualize cerebral amyloid angiopathy (CAA) in 3D volumes, a disease characterized by β-amyloid (Aβ) plaque deposition along the brain vasculature. Finally, we demonstrate the potential of this approach for application in a mock clinical scenario wherein biopsies of human donor kidneys are evaluated for transplant suitability, typically within a constrained time frame11. The speed and quality of our approach allowed for robust 3D information to be collected in a rapid time frame to assess the patency and characteristics of kidney glomeruli.
Results and discussion
Key elements of a turn-key epifluorescent widefield microscope for large-area 3D imaging
To achieve high axial resolution in thick tissue, we configured a system optimized for rapid acquisition of in-focus signal along the axial z-axis. We used a standard base for the microscope with an electronically controlled z-drive fitted with a ×20 immersion objective (NA 1.0, the highest resolution on the market) that had a spherical aberration collar, enabling lossless sampling of focal planes at a Nyquist rate of less than 1-μm apart in axial z. While high-NA lenses typically have limited working distances, our selected objective featured a 6.4-mm working distance, optimal for deep tissue imaging. This configuration permitted the collection of extensive in-focus axial information at greater depths than would be possible in less ideal configurations.
To image cleared samples, we engineered a sample chamber with the objective lens immersed in water and separated from the refractive index-matched sample by a glass coverslip (Fig. 1A). Spherical aberrations from refractive index mismatches in the optical path were mitigated using the lens’ correction collar. A 5-bandpass filter combined with a 7-LED light source enabled rapid, switch-free excitation across channels, a feature that improved upon the inherent speed benefits of camera-based imaging. At the time of this publication, our widefield system cost less than $125,000, required no service contracts, and the one-time cost of the deconvolution software was about $7000.

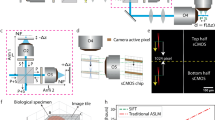
A Standard epifluorescent microscopy build with ×20 immersion lens (Carl Zeiss AG, NA 1.0, working distance of 6.4 mm) (top). Optical path for a refractive index-matched tissue mounted under a glass coverslip in contact with a ×20 lens through a water mount (bottom). B Extended display showing raw image of nuclei in a cleared mouse brain labeled with anti-Histone-H3-ATTO488 (green) acquired on widefield microscope before computational deconvolution. C Nuclei in cleared, transparent brain after deconvolution with depth-variant point spread functions calculated using Huygens Software (Scientific Volume Imaging) and visualized in the axial dimension with Imaris software. D Extended display showing resolution of fine microglial pseudopods labeled by CX3CR1-tdTomato (green) and nuclear staining with anti-Histone-H3-ATTO488 (magenta) captured using epifluorescent widefield microscopy with ×20 immersion lens (NA 1.0); E extended display of microglia as in (D) but captured using line-scanning confocal microscopy at a similar pixel resolution. F Maximum intensity projection of ~200 microns of 5XE4 leptomeninges attached to the brain surface that were stained for Aβ (red) to label CAA plaques along arterioles (smooth-muscle actin: green, CD31-positive vessels: gray), imaged using epifluorescent widefield microscopy with ×5 objective lens (NA 0.16, air), and deconvolved.
Axial nuclear resolution by deconvolution with depth-variant PSFs
Using the widefield microscope build described above, over a 5-minute (min) period, we collected hundreds of z-slices at 0.8-μm step intervals from mouse brain tissue that had been processed using the ADAPT-3D methodology4 and stained with an ATTO 488-conjugated anti-histone H2A–H2B heterodimer nanobody. Whereas tissue processing enabled collection of emitted light to the depth of the entire section of 500 μm, the acquired z-slices were observed as a mixture of blurred emitted light from nuclei and out-of-focus light derived from nuclei in other neighboring z planes (Fig. 1B). To remove out-of-focus light from the image, we used the Huygens deconvolution software package to generate theoretically derived, depth-variant PSFs from the specified acquisition parameters (Fig. 1B, right)8,10. In this approach, we specified three parameters to calculate a custom theoretical PSF: the lens immersion refractive index, the tissue embedding refractive index, and the distance from the coverslip to the start of the tissue. These theoretical PSFs were used across axial depths to deconvolve the image using the Huygens maximum likelihood estimation (MLE) function12. Deconvolution of the acquired image stacks was completed in <5 min on our workstation and successfully removed out-of-focus light to resolve nuclei in the axial dimension (Fig. 1C). Overall, the imaging acquisition and processing of this 0.125-mm3 tissue volume was completed in 10–15 min and achieved nuclear resolution at a z-depth of 500 μm in the mouse brain cortex (Fig. 1C).
To further examine the axial resolution limit of this widefield approach and gauge the extent of time savings, we visualized pseudopod projections of brain macrophages from CX3CR1ERCre-tdTomato reporter mice (Fig. 1D, E). Deconvolution of microglia from imaging volumes acquired using the widefield microscope resolved thin microglial processes in the z dimension, in addition to nuclei (Fig. 1D). Confocal imaging of the same tissue, with acquisition settings adjusted to match the pixel size and z-step size of the widefield system, took 2 h and 50 min compared to the 15-min total acquisition on the widefield system. While confocal microscopy achieved higher resolution of microglial processes (Fig. 1E), widefield imaging was more than 10 times faster and could still resolve major microglial pseudopod projections (Fig. 1D). We conclude that depth-variant deconvolution can be applied to rapidly achieve subnuclear axial resolution with a widefield microscope in optically transparent tissue. Therefore, while confocal microscopy remains the preferred method for applications that demand high resolution of a single 3D tile, our approach has the potential to feasibly image large swaths of tissue at subnuclear resolution in a fraction of the time.
Our widefield approach was also useful in uncleared tissue, when structures of interest were near the tissue surface. We investigated the accumulation of Aβ in the adventitia of leptomeningeal arteries in an experimental mouse model of CAA driven by transgenic expression of mutant amyloid precursor protein and a knock-in of ApoE4 into the mouse ApoE gene locus (5XE4)13. In less than 30 min of acquisition and processing time, widefield images revealed the interspersed and interlocked nature of Aβ deposition between adventitial muscle cells in the leptomeningeal arterial tree (Fig. 1F), although without optical clearing, less than 100 μm of axial information could be resolved.
Addressing technical limitations of deconvolution to facilitate large-volume multi-tile imaging
After demonstrating nuclear and fine process resolution in z with our widefield system, we next leveraged its speed to image large tissue volumes in 3D. Prior work imaging millimeters of human mesentery revealed key lymphatic features in Crohn’s disease14, but some acquisitions required up to 72 h or beyond on line-scanning confocal systems, limiting throughput. Prolonged acquisition also led to evaporation of mounting water used for immersion lenses, forcing the use of lower-resolution air objectives. To overcome these barriers, we implemented widefield z-stack stitching to rapidly extend imaging across large x–y areas, enabling scalable 3D tissue analysis.
Applying default depth-variant deconvolution (Huygens) to individual widefield tiles introduced edge artifacts (Fig. 2A), the first challenge of deep imaging not previously observed in samples less than 10-μm thick7. While artifacts could be cropped out in isolated tiles (Fig. 2B, B’), this approach could not be used for large-scale reconstructions including at greater axial depths. Artifacts were most prominent in channels that collected emission of Alexa Fluor 555 or Alexa Fluor 488, both typically prone to higher background, and rare in the channel collecting emission of Alexa Fluor 647, which exhibited low background. Before deconvolution, we subtracted background noise using a small Gaussian kernel and a local minimum filter to correct for low-frequency variations. This eliminated scattered out-of-focus light without generating edge artifacts (Fig. 2C, C’). We observed that the optical noise in a single raw image volume was nonuniform with patches of significantly greater background (Fig. 2D), such that these patches would be classified as real signal during deconvolution of raw images. However, since this signal was produced by tissue autofluorescence and not a real object, the Huygens MLE function most likely failed to solve for the object producing the signal, resulting in the signal being projected onto the tile edge. Our prefiltering approach eliminated the global variance in background (Fig. 2E), thus avoiding this type of artifact during deconvolution. From an x–z perspective, background levels were accentuated by extremely large PSFs in thick sections (Fig. 2F and Supplementary Fig. 1A), but not in sections less than 10 μm in thickness. By using filter kernels smaller than objects or their PSFs, the true background was removed without compromising the object signal (Fig. 2G). We conclude that prefiltering of large volumes removed the varying global background and left behind objects and local PSFs for artifact-free deconvolution.

A 3D display of single tile depicting striping edge artifacts (depicted by arrows) following deconvolution from mouse ileum immunostained with LYVE1 (magenta), smooth muscle actin (SMA, yellow), and S100A9 (cyan). B Extended display of same deconvolved tile as in (A) following cropping of artifacts as indicated with dashed lines and overview 3D display in (B’). C Extended display of the same tile in (A) when local background is subtracted before the deconvolution process without any cropping and a 3D overview (C’). D x–y max intensity projection of 142 microns depth of brain capillaries immunostained with anti-CD31 and anti-podocalyxin before background subtraction and (E) after subtraction of background found with a combined Gaussian and minimum filter. F x–z sum-intensity projection of capillaries of 81 µm of raw image and (G) after background subtraction. H 3D projection of brain cortex following deconvolution of the whole volume as a single brick or (I) after subdividing the volume into seven separate bricks in the Z axis for maximal retention of fine capillaries found in the raw image (D). J x–z sum-intensity projection after deconvolution of the filtered image volume in (G). K x–z sum intensity projection of a vessel event with its corresponding PSFs cropped from the dashed region of the filtered image in (G) or the same region taken from the final deconvolved image (L). M Line profiles along the yellow line in (K–L), normalized to 50,000: raw (white), filtered (purple), deconvolved (green). N Frequency histograms (bin-width of 2000) of all pixel intensities from X–Z sum projections of nine cropped vessel events: filtered (purple) vs deconvolved (green); Kolmogorov–Smirnov test, p < 0.0001, effect size = 0.6790. O Extended display of a stitched 3 × 3 tile of mouse ileum after background subtraction and deconvolution with z bricks immunostained for LYVE1, SMA, and S100A9. P 3D display of the entire overview from the mouse ileum in (O). Q X–Y projection of 50 microns from the ileum that depicts fine resolution of neutrophils in the muscularis layer of the mouse ileum from (O, P).
The second challenge in applying Huygens depth-variant deconvolution to thick samples was the reduced ability of MLE to resolve dim signal across full z-stacks of 500-µm depth, including after prefiltering (Fig. 2H and Supplementary Fig. 1B). Unlike 10-µm sections, which contain a single layer of PSFs, thick stacks required deconvolution across dozens of interdependent layers (Fig. 2G). To address this challenge, we used the bricking function of Huygens, which was originally intended for computer memory-limited systems, to divide the stack into seven thick segments (i.e., “bricks”) in the z-dimension for separate deconvolution and subsequent automatic reassembly. This preserved true signal while removing scattered light (Fig. 2I and Supplementary Fig. 1C). The combination of pre-filtering and bricking deconvolution removed out-of-focus light and improved the resolution of the true signal (Fig. 2J–L and Supplementary Fig. 1D–F). We quantified this by measuring fluorescence along a line through isolated objects in x–z sum-intensity projections before and after each step (Fig. 2K–M and Supplementary Fig. 1E–G). In raw data, line sums were elevated across the profile due to integrated background (Fig. 2M and Supplementary Fig. 1G). However, after background subtraction, intensities distant from the object returned to baseline, leaving a low-amplitude broad peak reflecting the object with PSF (Fig. 2M). Finally, deconvolution sharpened the peak of the object signal, increased the signal-to-noise ratio (SNR), and resolved adjacent peaks (Fig. 2M and Supplementary Fig. 1G). Frequency distributions of all line-profile intensities from the sum x–z projections of nine different objects taken from filtered and deconvolved image volumes were significantly different, indicating greater dynamic range of intensities after deconvolution (Fig. 2N). These differences in distributions replicated across two additional separate tiles (Supplementary Fig. 1H, I) and across three different tiles where standalone objects were analyzed (Supplementary Fig. 1J–L). Pairing background subtraction with deconvolution of z-bricks resulted in effective deconvolution of deep images without edge artifacts.
We first validated our workflow by imaging a small region of the small intestine from a wild-type (WT) mouse. A custom Python script implemented our prefiltering strategy into Huygens for automated batch processing. Nine 3D tiles were deconvolved and stitched to reconstruct the intestinal images, revealing neutrophils and lymphatics in 3D (Fig. 2H, I). Indeed, pockets of neutrophils were recruited to intestinal villi in WT littermates without overt inflammation (Fig. 2H–J), a spatial pattern not easily appreciated in thin sections or confocal imaging of a singular location. In conclusion, we solved the difficulties of applying Huygens depth-variant deconvolution to very thick image volumes by first filtering out tissue background with high variance and then splitting the image into more manageable sections.
Large-volume widefield imaging enables a comprehensive overview of CAA pathology in 3D
Expanding upon this proof-of-principle, we next imaged a large 3.75 [x] × 2.25 [y] mm area from a 500-µm cleared brain section stained for capillaries and nuclei. Composed of 15 stitched tiles, this image was acquired in under 1 h on a widefield microscope (Fig. 3A), in contrast to the time of nearly 40 h with line-scanning confocal microscopy at a similar pixel resolution. Deconvolved images resolved capillaries and nuclei across the full z-depth, including near dense nuclear regions like the corpus callosum (Fig. 3B and Supplementary Movie 1). This capability enabled the acquisition of volumetric data over large tissue areas rather than isolated cross-sections.

A Representative x–y projection of blood vasculature (CD31 and podocalyxin, red, and Histone-H3-ATTO488, cyan) from a 15-tile acquisition of a sagittal 500-micron section of mouse brain imaged, deconvolved, and stitched. B Extended display of image in (A) illustrating the depth and resolution of fine capillaries and nuclei. C Tilted 3D display of 80-tile image of Aβ plaques (green) in association with vessels (CD31/podocalyxin, blue) or arterioles (SMA, red) across the sagittal section of the brain isolated from the 7-month-old 5XE4 mouse acquired using ×20 immersion lens, deconvolved, and stitched. * indicates bridging vein with deposits of Aβ plaques in SMA-negative vessel (i.e., bridging vein) accompanied by a SMA-positive vessel arteriole along the leptomeninges surface. D Extended display depicting three-dimensional CAA of capillaries zoomed in from (C, dashed box). E Maximum intensity projection after deconvolution of leptomeninges on the surface of an uncleared brain hemisphere from a 9-month-old CAA mouse labeled for SMA (yellow), CD31 (cyan), and Aβ (magenta) and acquired using a ×5 air objective (NA 0.16). The arrow indicates a bridging vein with Aβ deposition.
3D imaging across wide x–y areas better revealed spatial relationships that are lost in two dimensions (2D). CAA is traditionally viewed in limited sections13, where leptomeningeal CAA plaques are observed as a thin line of staining at the edge of a brain section, and CAA along parenchymal vessels is often only observed in small fragments. Using our pipeline, we imaged a 1 cm3 volume of cortex from a 5XE4 mouse brain to a 500-µm depth in 5.5 h (Fig. 3C and Supplementary Movie 2). At sub-capillary z-resolution, we visualized Aβ plaques along fine parenchymal vessels that would otherwise be missed in 2D (Fig. 3D). While the Aβ-laden vessels can be readily viewed in the 150-µm maximum intensity projection, the U shape of the vessel in the x–z view underscores that a standard 20-µm cross-section would only show two lumina that would be challenging to recognize as CAA. We also visualized extensive CAA in the leptomeninges, including along vessels with low smooth muscle actin (SMA) signal (Fig. 3C).
The surface-exposed leptomeninges would be easiest to image without sectioning, but the curved dorsal surface of the brain has a range of at least 1.5 mm and cannot readily be peeled from the brain to prepare a flat mount, as has been done previously to image lymphatics in the dura mater attached to the curved skull15. We imaged the surface of an intact, uncleared brain hemisphere using widefield microscopy in only 20 min (Fig. 3E and Supplementary Movie 3). This approach revealed both expected Aβ deposition along SMA-positive arteries and novel deposition along SMA-negative bridging veins (Fig. 3E). These veins, identifiable by torn ends from dural detachment, are not typically implicated in CAA. However, their perivascular space was recently shown to allow cerebrospinal fluid-derived solutes to enter the dura and access lymphatics16, suggesting a role for bridging veins in Aβ clearance. These findings warrant further study into venous involvement in CAA.
Taken together, our solutions facilitated image processing of tiled image volumes as large as 1 cm3 while still achieving nuclear resolution in the z dimension. These larger 3D fields captured 3D CAA features including previously unreported CAA along bridging veins. We also leveraged widefield imaging to rapidly image the surface of an uncleared brain hemisphere using our image processing pipeline to attain a global view of leptomeningeal CAA. Widefield 3D imaging is a fast and accessible approach to achieve overview-like perspectives while maintaining fine-featured resolution to visualize pathology that is otherwise overlooked in standard 2D sections.
Rapid optical transparency of human donor kidney in a time-constrained, clinical diagnostic context
Rapid 3D imaging made possible by fast tissue processing and clearing, paired with rapid image acquisition and processing described above, opens a path to clinical application of 3D imaging where time constraints are present. We considered the relevant clinical scenario where donor organs must be evaluated for transplant viability within a finite, defined time. Currently, the evaluation of donor kidney suitability for transplant requires a pathologist’s evaluation of 5-μm frozen sections of a wedge biopsy collected from the putative donor organ. In a short time, while the donor organ is stored, the biopsy is evaluated to determine if it would provide a sufficiently healthy organ to warrant transplant. Considering that frozen sections inherently sample a small area of tissue, even a fraction of the biopsy collected, there may emerge undersampling error, with conclusions drawn that may not be representative of the actual status of the organ. The current standard of care is fraught with interobserver variability and significantly contributes to an overly high rate of discarding suitable tissues11. 3D imaging of the biopsy might be a feasible alternative to current methodologies because it evaluates a larger portion of the organ, which could lead to improved decision-making in the clinic. However, an obstacle to that potential solution is the challenge of carrying out a 3D evaluation within the defined time. To that end, we tested whether ADAPT-3D combined with a widefield imaging evaluation could lead to fast, informative pathological evaluation of human kidneys. Human kidney tissue from a mock wedge biopsy (Fig. 4A) measuring approximately 1 cm × 1 cm × 0.5 mm (x, y, z), equivalent to 150 individual frozen section slides, was fixed, stained with a fluorescent analog of the Periodic Acid-Schiff solution, and cleared with ADAPT-3D refractive index matching solution within 90 min and then imaged and processed using widefield microscopy and deconvolution in another 30 min (Fig. 4B).

A Approximately 0.45-mm wedge biopsy treated with periodic acid, stained with CellBrite Orange, FAM-Hydrazide, and DAPI before refractive index matching (left) and after 30 min of refractive index matching (right). B Timeline overview to obtain optical transparency of wedge biopsy as seen in (A). C–E Five-µm x–y maximum intensity projections of 3 separate glomeruli spaced at 100 µm z-intervals extracted from a single imaging volume of a kidney biopsy labeled with a Periodic Acid Schiff stain (cyan) and nuclei (red). Lower box schematic (yellow) of the image volume from which digital sections were extracted at different depths. F Digital section from 3D image of biopsy constrained to timing shown in (B) depicting intact glomeruli and their neighboring proximal tubules, some of which contain atrophy (arrows). G Three intact glomeruli (*), along with a possible arteriosclerotic vessel (arrowheads) found 100 microns of z-depth below the digital section in (F) and its accompanying nuclear staining with DAPI (H).
To visualize glomeruli and their connecting tubules, a wedge biopsy stained with Periodic Acid Schiff and nuclear staining was imaged across over 300 μm of depth, followed by deconvolution. The resulting image volume captured three whole glomeruli at different depths (Fig. 4C–E), with 3D information allowing for clear distinction of closely situated glomeruli. By preserving overall structure without physical sectioning, we digitally sliced through hundreds of consecutive z-planes, and visualized an afferent arteriole entering its corresponding glomerulus along with its associated proximal tubules at different depths (Supplementary Movie 4). As we established the features of a relatively unremarkable biopsy, we applied this approach in a time-constrained manner to examine potential pathology as outlined in Fig. 4B. We found examples of atrophic tubules with thickened tubular basement membranes (Fig. 4E, F, arrows) and possible arteriosclerotic vessels (Fig. 4C, D, arrowheads). While optimization of this process for clinical utility and standards for evaluation would be needed, along with a collection of comparative studies evaluating 3D images versus frozen sections, these data indicate that tissue processing with a method like ADAPT-3D combined with a rapid method of image acquisition and analysis holds promise for 3D pathology in a time-constrained, clinically applicable setting.
The method is broadly applicable when specimens are effectively cleared and refractive index matched, and when targets are brightly labeled with antibodies or dyes. The ADAPT-3D method we used here has also cleared multiple other tissues—including human intestine and mouse brain, lung, spleen, liver, and intestine. Triglyceride-rich adipose tissue was a notable exception, which requires further optimization, likely requiring an alternative clearing protocol. Pathologies that were not evaluated here (e.g., melanocytic skin lesions, cardiac biopsies) may present additional clearing challenges due to pigment and dense extracellular matrix. In all cases, our widefield-deconvolution approach performs best in optically transparent tissue when labeling achieves high SNRs. High SNRs are preferred in any microscopy approach, but are particularly important in widefield microscopy because camera detection is generally less sensitive than the photomultiplier tubes used in confocal systems. Consequently, although a widefield system with the same numerical aperture and sampling could theoretically approach confocal resolution, very fine, dim features remain more difficult to recover by deconvolution. When clearing and labeling are adequate, widefield enables rapid, large-area volumetric surveys while maintaining nuclear-scale resolution for detailed review, and it can be used in tandem with confocal for targeted, highest-resolution imaging of selected regions.
In conclusion, this study reveals and optimizes how widefield microscopy can be coupled with depth-variant deconvolution and improved rapid tissue processing protocols to provide for much faster and more affordable 3D imaging. The financial aspects of epifluorescent microscope builds and objectives, along with computer and software, reduce costs substantially over confocal and light-sheet systems. Furthermore, whereas the latter typically requires expensive service contracts, a service contract for the widefield system would not be necessary. For work in remote areas, including fieldwork, the widefield microscope build offers greater accessibility. Indeed, as clinical utility is considered, one might envision these types of microscopes on carts for use in operating rooms or clinical offices. For many laboratories, including our own, one might envision the advantages of this system to be one that does not always replace the utility of confocal or light-sheet fluorescence microscopy but does so on a select and even regular basis, such as times when higher throughput is critical, when water-dipping lenses are needed, and evaporation over long imaging time would be an obstacle, and when the quality of the images on a widefield system meets the needs of a project. In busy groups where equipment is desired by several operators, time-saving options can be game-changing. We anticipate that, along with increasing computational power becoming ubiquitous, improvements to this pipeline will continue. Future efforts will focus on the ability to better deconvolve fluorescence signal where signal-to-noise is low and to evolve our protocol to allow multiplex approaches. We also envision stitching in the z-axis to complement stitching in the x- and y-axes.
Methods
Mice
Studies in mice were approved by the Washington University Institutional Animal Care and Use Committee, protocol #22-0433 to GJR. We have complied with all relevant ethical regulations for animal use. All mice, including WT C57BL/6 mice, were bred in-house in specific pathogen-free facilities. Other mouse strains were also maintained on the C57/BL6 background. 5xFAD (line 7031) bearing one or two human apoE4 alleles and zero or one mouse apoE allele that develop Aβ plaques characteristic of CAA and Alzheimer’s disease were obtained from David Holtzman (Washington University) with a Materials Transfer Agreement between the Cure Alzheimer’s Fund in place for the humanized apoE4 line with GJR. CX3CR1ERCre-tdTomato reporter mice were generated by crossing previously described CX3CR1ERCre mice17 with TdTomfl/fl mice18. TNFΔARE/+ mice19 were obtained from the Cleveland Clinic in 2016 and bred in-house.
Human kidney biopsies and tissue processing
Human kidney samples were acquired from deceased donors whose kidneys were rejected for transplant through an agreement with Mid-America Transplant Foundation, with a waiver of consent and associated IRB approval to J.P.G. All ethical regulations relevant to human research participants were followed. For time-constrained processing of human kidneys, kidneys were biopsied into wedges first fixed in 4% paraformaldehyde (PFA) and stored until the trialed experiments. They were pre-treated in 0.5% periodic acid for 28 min, washed with 1× phosphate-buffered saline (PBS) containing 10 U/mL heparin, incubated in blocking buffer containing CellBrite Orange (Biotium, 1:200), FAM-Hydrazide (Lumiprobe, 15170, 1:100), and DAPI (Sigma-Aldrich, D9542, 1:200) for 25 min, washed with 1× PBS containing 10 U/mL heparin, and incubated in ADAPT-3D refractive index matching solution for 25 min before mounting the sample for imaging. The kidney tissue in Fig. 4 without evident pathology was incubated in ADAPT-3D Decolorization/Delipidation Buffer with 0.2× PBS for 24 h, pretreated with periodic acid, stained with the same dyes for 1.5 h, and then incubated in refractive index matching solution for 1.5 h.
Preparation and staining of tissues
The last two cm of the mouse ileum was collected after flushing the mouse small intestine with PBS and then was fixed in 4% (w/v) PFA. For preparation of the sagittal brain sections, mouse brains were fixed in 4% w/v PFA at 4 °C for 24 h, washed in PBS containing 10 U/mL heparin overnight, and sectioned on a LeicaVT1200 vibratome at 500-µm intervals. All subsequent steps were performed at room temperature. Sections were incubated in Decolorization/Delipidation Buffer with 0.2× PBS for 3–10 h, washed 3× 30 min with PBS containing 10 U/mL heparin, and then incubated in partial delipidation buffer for 15–48 h. Samples were then washed 3× 30 min in PBS, with the first wash performed using 0.1× PBS and then blocked in ADAPT-3D blocking buffer for 3 h. Samples were stained for 15–36 h in blocking buffer containing antibodies against histone H2A-H2B heterodimers (Histone-Label Atto488, Chromotek, tba488, 1:200), CD31 (R&D Systems, AF3628, 1:200), Podocalyxin (R&D Systems, AF1556, 1:200), Aβ (Clone D54D2, Cell Signaling Antibodies, 8243L, 1:200) conjugated to CF647 (Biotium Mix-n-Stain, 92259), and αSMA-AF488 (Clone 1A4, Sigma-Aldrich, 1:200). After 3× 1 h washes in PBS with 0.2% tween 20 and 10 U/mL heparin, thick brain sections were stained 1:200 with secondary antibodies prepared at 1.5 mg/mL (Cy3 F(ab’)2 Donkey Anti-Sheep IgG, Jackson ImmunoResearch, 713-166-147), (Cy3 Donkey Anti-Rat IgG, Jackson ImmunoResearch, 712-165-153). Endogenous tdTomato signal in CX3CR1-tdTomato brain sections was boosted by staining with CF568-conjugated rabbit anti-RFP in blocking buffer (1:200, Biotium, 20477). Intestinal tissues were only processed with the Decolorization/Delipidation Buffer before proceeding to the blocking step. Mouse ileum was stained with antibodies against alpha SMA-Cy3 (clone 1A4, 1:200, Sigma-Aldrich, C6198) along with LYVE1 (Abcam, ab14917, 1:200) and S100A9 (Bio-techne R&D, AF2065, 1:200) that were conjugated to CF647 and CF488, respectively, using Mix-n-Stain™ Antibody Labeling Kit (Biotium, #92259, #92253) in ADAPT-3D blocking buffer. Antibody-labeled tissues were incubated in ADAPT-3D refractive index matching solution diluted 1:1 with PBS for 30 min and then in full-strength refractive index matching solution for 3 h directly prior to image acquisition.
Image acquisition: epifluorescent and confocal microscopy
Zeiss Examiner Z1 was used as a base microscope and was fitted with a motorized x–y–z stage for fine control. The Colibri 7 LED light source was used to generate 7 different excitation lines, and a multiband pass filter set (Zeiss 112 HE) was used to collect emitted light without switching filter cubes. Images were acquired using either a ×20 immersion lens (spherical aberration collar, NA 1.0) or ×5 air lens (NA 0.16) with a Prime BSI express camera. All cleared tissues were imaged with the ×20 lens immersed in water separated from the sample by a coverslip while uncleared tissues were submersed in PBS and imaged directly with either the ×20 immersion or ×5 air lens. Images were acquired at 0.8–1.2 µm z intervals with the ×20 immersion lens and at 15 µm intervals with the ×5 air lens in accordance with the Nyquist rate. For comparison images in Fig. 1, images were taken on a Leica SP8 confocal microscope equipped with a ×20 oil immersion lens (NA 0.75) with the same pixel size as the ×20 immersion lens.
Image processing: deconvolution
Raw images were input into Huygens Professional software (Scientific Volume Imaging, Version 21.1.1) on a workstation with (Operating System: Windows 11 Pro, CPU: AMD Ryzen Threadripper PRO 5975WX 32-Core 3.6 GHz, GPU: NVIDIA GeForce RTX4090, RAM: 512 GB). For images acquired with the Zeiss epifluorescent microscope, .czi files were loaded into Huygens. Within the software, the image microscopic parameters were adjusted to the following: the lens embedding medium was set to a refractive index of 1.33; the sample embedding medium was adjusted to a refractive index of 1.50. The coverslip distance was adjusted by selecting the beginning of the real signal in the coverslip distance editor with the imaging direction set to downward. Theoretical PSFs computed by the Huygens software were used for all deconvolutions. After parameterization, the background was found using a small Gaussian filter followed by a minimum filter and then subtracted from the original image in accordance with our custom code20. Single tiles were deconvolved using the deconvolution wizard to create a deconvolution template. All images were deconvolved using the Quick Maximum Likelihood Estimation (QMLE) algorithm except for images of the human kidney, which were deconvolved using the classic MLE algorithm. The SNR of unfiltered images was often 40 and above, while the SNR of filtered images was lowered to 20 and below. Background was calculated on auto settings for unfiltered images but was manually set to zero for filtered images. Before starting the deconvolution, the acuity was either set to zero or increased to run more QMLE iterations, such as on the images with microglial processes in Fig. 1. Bricking was set to “few bricks,” and GPU processing was enabled. For tiled images, each image was individually filtered and saved by running a custom Python script in the Huygens Python shell, and all images were deconvolved in the batch processor using parameters and deconvolution templates. The Huygens stitcher module was used to select a folder containing deconvolved images numbered by their order of acquisition, specify the image overlap and acquisition pattern, and the alignment between all tiles was calculated for all channels in x, y, and z dimensions before stitching. For tiled images, image stacks were either corrected for vignetting using a flatfield reference or originally acquired with a cropped camera chip to reduce vignetting. All deconvolved images were saved as unsigned integer ICS2 file types. As a note, for the 80-tile brain scan, the required memory was roughly four times that of the raw image file to both simultaneously load all 32-bit float-type tiles and allocate space for the entire newly stitched image. As a workaround solution, virtual memory was allocated on the SSD C:/ drive. See Supplementary Protocol for step-by-step instructions.
Measurement of sum intensities before and after deconvolution
Three tiles from the frontal, middle, and posterior portions of the cerebral cortex in a 500-micron-thick sagittal section of the mouse brain that was antibody labeled for Aβ directly conjugated to CF647 and CD31 + podocalyxin-Cy3 were selected for intensity analysis before and after filtering and deconvolution. Each tile was converted to a 16-bit unsigned integer, and intensities were preserved without scaling. For a given raw tile, five plaque events distributed throughout an individual tile were cropped and extracted as a volume (80 µm, 69 µm, 116 µm in x, y, z). Nine vessel events were selected from each raw tile by reducing the image to three 25 µm-thick x–z planes evenly distributed across the y axis and then selecting vessel events from the bottom, middle, and top third of these planes along the z axis each with a crop dimension of 46 µm in x, 25 µm in y, and 73 µm in z. Matched cropped regions were then selected using identical coordinates in the filtered and deconvolved versions of the images. Each cropped image was resliced in Fiji (ImageJ v2.16.0) to a y–z view for plaque and x–z view for vessels by sampling at the width of the xy voxel (0.36 µm). x–z and y–z stacks were then summed into a projection. To compare sum intensities relative to their positions, a line was created as a region of interest and applied to all images in the comparison, and intensities were measured along the length of the line using the plot profile feature. For comparison of all sum intensities from the whole x–z projection of each cropped event, the x–z projection was saved as a text image, and all intensity values were pooled with the intensities of other event replicates from the same tile. A separate frequency distribution was prepared using GraphPad Prism (v10.5.0) for each tile, and the distributions from within the same tile were compared using a non-parametric Kolmogorov–Smirnov test.
Image visualization
3D image volumes were visualized on Imaris v10.1.1 software (Bitplane Inc.).
Statistics and reproducibility
All imaging analyses were repeated across multiple independent regions. Sum intensity x–z and y–z projections were generated from raw, filtered, or deconvolved volumes, and normalized intensity distributions were compared between conditions. For vessel imaging comparisons, histograms were constructed from nine cropped vessel events across three independent volumes. For plaque imaging comparisons, histograms were constructed from cropped plaque events across three independent volumes. Intensity distributions of filtered versus deconvolved datasets or raw versus deconvolved datasets were compared using the non-parametric Kolmogorov–Smirnov test where p value ≤.0.05 was set to be statistically significant. Representative examples of raw versus deconvolved images are shown, as each example was confirmed in at least three independent imaging volumes.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Primary data can be made available upon request to G.J.R., B.H.Z., or D.D.L. The source intensity data for all plots are included in Supplementary Data 1.
Code availability
The code20 accompanied with step-by-step instructions are available on https://github.com/wowdanlee/3D-Widefield-with-ADAPT-3D-and-Huygens or https://doi.org/10.5281/zenodo.17156203.
References
Matsumoto, K. et al. Advanced CUBIC tissue clearing for whole-organ cell profiling. Nat. Protoc. 14, 3506–3537 (2019).
Erturk, A. et al. Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nat. Protoc. 7, 1983–1995 (2012).
Hsu, C. W. et al. EZ Clear for simple, rapid, and robust mouse whole organ clearing. Elife https://doi.org/10.7554/eLife.77419 (2022).
Lee, D. D. et al. ADAPT-3D: accelerated deep adaptable processing of tissue for 3-dimensional fluorescence tissue imaging for research and clinical settings. Sci. Rep. 15, 31841 (2025).
Shaw, P. J. in Handbook of Biological Confocal Microscopy (ed. Pawley, J. B.) 453–467 (SpringerScience+Business Media, 2006).
Sage, D. et al. DeconvolutionLab2: an open-source software for deconvolution microscopy. Methods 115, 28–41 (2017).
Wernersson, E. et al. Deconwolf enables high-performance deconvolution of widefield fluorescence microscopy images. Nat. Methods 21, 1245–1256 (2024).
Model, M. A., Fang, J., Yuvaraj, P., Chen, Y. & Zhang Newby, B. M. 3D deconvolution of spherically aberrated images using commercial software. J. Microsc. 241, 94–100 (2011).
Guo, M. et al. Rapid image deconvolution and multiview fusion for optical microscopy. Nat. Biotechnol. 38, 1337–1346 (2020).
Gibson, S. F. & Lanni, F. Experimental test of an analytical model of aberration in an oil-immersion objective lens used in three-dimensional light microscopy. J. Opt. Soc. Am. A 9, 154–166 (1992).
Moeckli, B. et al. Evaluation of donor kidneys prior to transplantation: an update of current and emerging methods. Transpl. Int 32, 459–469 (2019).
Holinirina Dina Miora, R. et al. Experimental validation of numerical point spread function calculation including aberration estimation. Opt. Express 32, 21887–21908 (2024).
Liao, F. et al. Murine versus human apolipoprotein E4: differential facilitation of and co-localization in cerebral amyloid angiopathy and amyloid plaques in APP transgenic mouse models. Acta Neuropathol. Commun. 3, 70 (2015).
Randolph, G. J. et al. Lymphoid aggregates remodel lymphatic collecting vessels that serve mesenteric lymph nodes in Crohn disease. Am. J. Pathol. 186, 3066–3073 (2016).
Louveau, A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015).
Smyth, L. C. D. et al. Identification of direct connections between the dura and the brain. Nature 627, 165–173 (2024).
Yona, S. et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 (2013).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010).
Kontoyiannis, D., Pasparakis, M., Pizarro, T. T., Cominelli, F. & Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 10, 387–398 (1999).
Lee, D. D. & Telfer, K. A. 3D widefield with ADAPT-3D and Huygens. Zenodo https://doi.org/10.5281/zenodo.17156203 (2025).
Acknowledgements
This work was supported by NIH U01 AI163064, NIH R01AI168044, and DP1DK130660 to G.J.R., the Imaging Core Component of NIH PO1AG078106 (GJR Core PI; Jonathan Kipnis, overall PO1 PI), and the Washington University Digestive Diseases Research Core Center (P30DK052574). D.D.L. was funded in part by NIH T32HL007081 and by a SPARK grant from the Department of Pathology & Immunology at Washington University. H.S.R. was funded by NIH T32DK007130. We thank David Holtzman for 5XE4 mice and are grateful to the Cure Alzheimer’s Fund for the provision of the humanized apoE4 line. TNFΔARE/+ mice were obtained through the Cleveland Digestive Disease Research Core Center (NIH P30 DK097948).
Author information
Authors and Affiliations
Contributions
D.D.L. and B.H.Z. conceptualized and initiated the study. D.D.L., K.A.T., H.K., H.L., and H.S.R. designed and conducted experiments. D.D.L. and B.H.Z. selected the microscope build. Y.K.L. and M.A.J.K. provided key advice for deploying the depth-variant point spread function. D.D.L., K.A.T., K.W.N., B.T.S., and M.A.J.K. analyzed data and provided troubleshooting to overcome obstacles and generate large-volume tiling. J.P.G. provided key advice for clinical application. G.J.R. and J.P.G. obtained regulatory compliance and funding. B.H.Z. and G.J.R. provided supervision. D.D.L., K.A.T., and G.J.R. wrote the manuscript draft with editing input from all authors.
Corresponding authors
Ethics declarations
Competing interests
Some of the authors have filed a provisional patent for the ADAPT-3D clearing method used in this manuscript.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary handling editors: Marco Fritzsche and Kaliya Georgieva.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, D.D., Telfer, K.A., Koenis, M.A.J. et al. Depth-variant deconvolution applied to widefield microscopy for rapid large-volume tissue imaging. Commun Biol 8, 1599 (2025). https://doi.org/10.1038/s42003-025-08992-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08992-y
