
Abstract
Lens transparency relies on proper intercellular communication. Exosomes are crucial mediators of intercellular communication and play a key role in organ homeostasis and development. However, their presence and dynamics in the lens remain unclear. Here, we report endogenous exosomes in the zebrafish lens using cryaa-driven Cd63-AcGFP labeling. Live imaging revealed dynamic exosome movement within lens cells and their potential transfer to adjacent tissues. Additionally, we found that the biogenesis of Cd63+ exosomes in the lens is regulated by the Syntenin-a pathway. And Syntenin-a knockdown delayed lens development by impairing lens cell differentiation, highlighting the potential role of lens cell–derived exosomes. Furthermore, ROR1+ lens progenitor cell-derived extracellular vesicles promoted lentoid differentiation in vitro, with proteomic analysis suggesting underlying mechanisms. Overall, our study addresses the gap in direct observation of endogenous lens exosomes, providing foundational insights into lens pathophysiology and a potential strategy for modulating the lens microenvironment.
Similar content being viewed by others


Introduction
The lens is a transparent organ composed of epithelial and fiber cells1. Since the lens lacks blood vessels, efficient molecule exchange between cells is crucial for its transparency2. Cataracts, characterized by opacification of the lens, are the most common cause of blindness3. Therefore, understanding how the lens interacts with its environment and how molecules are exchanged between lens cells has been a major focus of research. Previous studies have highlighted the importance of gap junctions and the microcirculatory system in lens homeostasis4. However, some studies have shown that gap junctions are rare at the interface between epithelial and fiber cells (EFIs)4. Ultrastructural analyses in both humans5 and zebrafish6 have revealed numerous endocytotic–exocytotic vesicles between lens cells. These findings suggest a role of the vesicles in mediating intercellular communication in the lens.
Exosomes are secreted by a wide range of cell types and are increasingly recognized as crucial mediators of intercellular communication7. Exosomes are a type of extracellular vesicles (EVs), characterized as membrane-bound structures typically ranging from 50 to 200 nm in size, and formed through the multivesicular body pathway7. Exosomes are enriched in various biomolecules that facilitate autocrine, paracrine, and long-distance communication among cells, as well as between cells and their environment8. Exosomes are found in nearly all bodily fluids, including the aqueous humor9. Moreover, owing to their cell-specific cargos and excellent biocompatibility, exosomes show great potential as biomarkers and therapeutic tools for various diseases10. Exosomes are known to play key roles in various ocular diseases11, but their role in lens pathophysiology remains underexplored.
Several indirect studies showed that exosome-mediated intercellular communication may occur in the lens. Exosomes are important components of the aqueous humor12, which directly contact the lens. MiRNA levels in exosomes from the aqueous humor were altered in patients with age-related and diabetic cataracts13. This suggested that lens cells may secrete exosomes into the aqueous humor and these exosomes may play a role in the development of cataracts. Additionally, an ex vivo study provided evidence that the lens can secrete exosomes into the medium, which can cross the lens capsule and be taken up by lens epithelial cells (LECs)14. Various studies have shown that EVs secreted by LECs lines have multiple biological functions, such as inhibiting microglial activation15 and retinal neovascularization16. However, these studies on lens-EVs mainly depend on heterogeneous populations of EVs isolated from cell culture or liquid biopsy samples. The presence and dynamics of endogenous exosomes in the lens in vivo are poorly understood.
Recent advances in imaging techniques and innovative EVs labeling strategies provide opportunities to investigate EVs in vivo17,18. Zebrafish are an ideal model for in vivo EVs research because of their transparent embryos, ease of genetic manipulation, and amenability to live imaging19,20. Fluorescent tags are useful to label exosome-enriched proteins such as CD63 and CD9 to track the biodistribution and behavior of endogenous exosomes17. Among these proteins, CD63 is the most widely used marker and accumulates on exosome membranes by trafficking to late endosomes17,21. Verweij et al. developed a model of CD63-pHluorin-expressing zebrafish embryos for real-time tracking of the production, journey, and fate of exosomes derived from the yolk syncytial layer22. These in vivo zebrafish EVs research techniques pave the way for investigating the roles of endogenous EVs in homeostasis, disease, and cellular processes.
The transparency of the lens and its proximity to the surface of zebrafish make it relatively easier to observe and track exosomes in vivo. The cryaa gene, which encodes the αA-crystallin, is essential for maintaining lens transparency. The expression of the cryaa gene is restricted to the lens in both larval23 and adult24 zebrafish. This specificity enables precise protein expression under the cryaa promoter in transgenic zebrafish25. Moreover, transgenic zebrafish employing cryaa-driven Cre recombinase have been effectively utilized for targeted gene deletion, specifically within the lens25. This allows us to use the cryaa promoter to achieve lens-specific labeling of exosomes.
In this study, we showed that zebrafish embryos expressing Cd63-AcGFP reporter driven by cryaa promoter can be used to track and visualize lens-Exos (exosomes derived from lens) in vivo. Live imaging captured the presence of exosomes in lens cells during development and their movement trajectory under physiological conditions. Exosomes facilitate communication among lens cells and convey signals to adjacent eye tissues. The biogenesis of lens Cd63+ exosomes relies on the Syntenin-a pathway. Interference with Syntenin-a pathway affected lens development, supporting a potential role of these exosomes. ROR1+ lens progenitor cell-derived EVs (ROR1+ LPCs-EVs) promoted lentoid differentiation in vitro. Proteomic analysis revealed insights into the functions of ROR1+ LPCs-EVs. Overall, our research suggests that exosomes mediate intercellular communication within the lens, providing a distinct perspective for studying the lens pathophysiology.
Results
Exosome-like vesicles are present in lens cells and can penetrate through the lens capsule
To determine whether vesicle-mediated intercellular communication occurs in the lens, we first performed TEM on P0 mouse lens and 3.5 dpf zebrafish lens. In the mouse lens, exosome-like EVs were observed in both LECs (Fig. 1A, B) and LFCs (Fig. 1C, D), along with vesicles undergoing phagocytosed or about to be phagocytosed were observed between LECs (Fig. 1E) and between LFCs (Fig. 1F). Interestingly, these exosome-like EVs were also found in the lens capsule, both on the side adjacent to the fiber cells (Fig. 1G) and on the distant side (Fig. 1H). Similarly, in the zebrafish lens, we also observed exosome-like vesicles in LFCs (Fig. 1L) and on the apical side of the LECs (Fig. 1I–K, M, N), with invaginations in the cell membrane near the fiber cells (Fig. 1M2, N2). In our lens samples, no microvilli were observed on either LECs or LFCs, ruling out the possibility that these profiles represent cross-sections of microvilli. These results show that exosomes may mediate communication within the lens and can cross the lens capsule.

A–H TEM images of mouse lens at P0. A Exosome-like vesicles are present on the basal side of LECs. B Magnified image of the white dashed area in (A) shows exosome-like EVs in LECs and an EV in the lens capsule being phagocytosed by a LEC (white arrow). C Exosome-like EVs (white arrows) are present in LFCs: newly phagocytosed (left), inside a cell (right), and within endosomal-like structures (red arrows). D Exosome-like EVs (white arrows) and multivesicular endosomal structures (red arrows) are observed in LFCs. E Vesicle-mediated intercellular communication occurs between LECs. The arrows indicate the process of an exosome-like EV being phagocytosed by another LEC at the cell boundary (white arrow). F Exosome-like vesicles also mediate intercellular communication between LFCs. The white arrows indicate the vesicles to be phagocytosed, and the red arrows indicate the invaginated cell membrane. G, H Exosome-like EVs pass through the lens capsule. G vesicles near the LFCs, and H vesicles distant from the LFCs. I‒N TEM images of zebrafish lens at 3.5 dpf. I–K and M, N Exosome-like EVs are present on the apical side of LECs. J a magnified view of the white dashed box in (I), and (K) further magnifies the white dashed box in (J). N a magnified view of the white dashed box in (M). Cell membrane invaginations are present at the boundary between epithelial and fiber cells. (M2; N2, white arrows) L Exosome-like EVs are present in LFCs.
Labeling, live imaging, and identification of lens cell-derived exosomes in zebrafish embryos
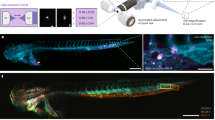
To further investigate the production, transit, and distribution of exosomes in the lens, we used zebrafish for in vivo labeling and tracking. Immunofluorescence confirmed that αA-crystallin was exclusively expressed in the lens at various stages (Supplementary Fig. 1A). For lens-specific EVs labeling, we constructed an expression plasmid for the Cd63-AcGFP fusion protein under the control of the cryaa promoter and used a cryaa-driven AcGFP expression plasmid as a control (Fig. 2A). By injecting these two plasmids into one-cell-stage embryos, we observed at 3.5 dpf that the GFP in the lens of the control group was diffuse, whereas the GFP in the lens of the group expressing cryaa-Cd63-AcGFP was punctate (Fig. 2B). Confocal imaging further confirmed punctate GFP signals across the whole lens and within the central lens section in live zebrafish expressing cryaa-Cd63-AcGFP (Fig. 2C and Supplementary Fig. 1B). Analysis of the labeling efficiency of the cryaa-Cd63-AcGFP revealed a decrease in the GFP signal over time, possibly due to a decrease in the plasmid concentration within the cells. The highest labeling efficiency in the lens was observed between 2.5 dpf and 4.5 dpf, therefore, we performed our subsequent experiments on this period (Supplementary Fig. 1C, D). These findings showed successful labeling of lens exosomes using our strategy.

A Schematic diagram of the plasmid construct used for live tracking of Cd63+ exosomes in the lens. cryaa-Cd63-AcGFP pDNA served as the experimental plasmid, whereas cryaa-AcGFP pDNA served as the control. B Expression of Cd63-AcGFP in the lens of zebrafish at 3.5 dpf after the injection of cryaa-Cd63-AcGFP pDNA at the 1-cell stage. C Live confocal images of zebrafish lens at 3.5 dpf showing the expression of Cd63-AcGFP or AcGFP after plasmid injection. The left panel shows the whole lens layer scan overlay, and the right panel shows the central section of the lens. Z-scan images of the whole lens are shown in Fig. S1D. D Still image from a time-lapse video (Video S1) of the lens in a 48 hpf embryo expressing Cd63-AcGFP. Dashed box 1 highlights the area with notable movement of AcGFP-labeled exosomes in the center of the lens, with enlarged images shown in (E). Box 2 shows the AcGFP signal in the retina surrounding the lens, with enlarged images shown in (H2). Box 3 shows the AcGFP signal in the boundary area between the lens and the surrounding retina, with enlarged images shown in (H3). E Enlarged images of box 1 in (D). The dashed areas indicate two examples of regions with notable movement of AcGFP-labeled exosomes, labeled (a and b), with their enlarged images shown in (F and G), respectively. The moving exosomes are marked with white arrows. F, G Magnified views of regions (a and b) in (E), highlighting the moving exosomes indicated by white arrows. The scale bar is 2 μm. H H2 and H3 show magnified images of Box2 and Box3 from (D), respectively. H2 shows the movement of AcGFP signals in the retina surrounding the lens, whereas H3 illustrates the process of AcGFP signals moving from the lens to the surrounding retina. I Immunoelectron microscopy (IEM) image of the lens in the zebrafish expressing cryaa-Cd63-AcGFP at 3.5 dpf, showing exosome-like vesicles positive for GFP-immunogold. J Diagram of zebrafish EVs extraction. K TEM analysis of zebrafish EVs. L Nanoflow cytometry analysis revealed that nearly 72% of the GFP-positive particles were also positive for the EVs membrane dye.
To validate that the Cd63-AcGFP signals represent exosomes, live time-lapse imaging of zebrafish lenses revealed dynamic movement of fluorescence punctate signals within the lens over time (Supplementary Video 1 and Fig. 2D–H). The Cd63-AcGFP signals shifted positions within lens cells over time (Fig. 2D–G), appeared at the lens edge, and extended into the adjacent retina (Fig. 2H2), gradually migrating from the lens to the retina (Fig. 2H3), suggesting the release of lens-derived exosomes and their potential intercellular transfer. This conclusion was supported by the presence of exosome-like EVs in the lens capsule, as shown in Fig. 1.
Given the limitations of optical imaging techniques in their resolution and point spread function when determining the size of endogenous EVs in the context of live imaging, we analyzed these particles ex vivo to evaluate their characteristics. IEM analysis of lens from embryos expressing cryaa-Cd63-AcGFP confirmed the presence of EVs in the exosome size range (~100 nm) (Fig. 2I). Additionally, the representative vesicles (Fig. 2I) and multivesicular structures (Supplementary Fig. 1E) were positive for GFP-immunogold. Next, we extracted EVs from zebrafish expressing cryaa-Cd63-AcGFP at 3.5 dpf (Fig. 2J). TEM images showed that the extracted EVs had typical cup-shaped exosome characteristics (Fig. 2K). Nanoflow cytometry analysis revealed the distribution of the extracted vesicles primarily between 50 and 200 nm (Supplementary Fig. 1F), with 5.3% GFP-positive particles (Supplementary Fig. 1G), which had similar size distributions (Supplementary Fig. 1H). Membrane dye labeling revealed that approximately 80% of the particles were EVs-positive, while 20% were likely contaminants or fragments (Supplementary Fig. 1I). Colocalization analysis revealed that approximately 72% of the GFP-positive particles were also positive for the EVs-membrane dye (Fig. 2L), indicating that most GFP-positive particles represent exosomes. These results showed the effectiveness of our strategy in labeling endogenous exosomes within the zebrafish lens.
Lens-specific exosomes were distributed throughout the lens and surrounding tissues
To analyze the distribution of Cd63-AcGFP-positive exosomes, we evaluated the lens of zebrafish at various developmental stages26 (Fig. 3A). Immunofluorescence detected subcellular Cd63-AcGFP-positive EVs in the lens as early as 36 hpf (Fig. 3B–D). Studies have shown that cryaa expression begins in the lens after 24 hpf27, whereas Cd63-AcGFP-positive exosomes are detected after 36 hpf, compatible with a role in specific stages of lens development. We observed Cd63-AcGFP positive exosomes in both the lens and surrounding tissue cells, indicating that the lens might communicate with neighboring cells through these exosomes during development (Fig. 3D, E).

A Diagram of key stages in zebrafish lens development. B–G Expression of αA-crystallin and distribution of Cd63-AcGFP at different stages of lens development. Scale bar as shown in the image. H Quantification of the mean fluorescence intensity in the central region of the lens at 3.5 dpf, as indicated by the dashed outline in (F). (n = 16 and n = 29, respectively). I At 48 hpf, Cd63-GFP was primarily distributed in lens fiber cells. The scale bar is 10 μm. J Distribution of Cd63-GFP in the lens at 3.5 dpf. The white arrows indicate that Cd63-GFP was primarily localized around the nuclei in differentiating fiber cells, whereas the asterisks indicate the presence of Cd63-GFP in peripheral epithelial cells. The scale bar is 20 μm. K At 3.5 dpf, the distribution of Cd63-GFP in the lens after costaining for F-actin (red) is shown. The dashed box highlights the area that is enlarged in the panels below. The white arrows indicate Cd63-AcGFP in the inner fiber cells. The scale bar is 10 μm. L Magnified views of the dashed box areas in (c and d) from (K). White arrows indicate Cd63-GFP signals outside the lens. The dashed boxes in (b) are shown at higher magnification in (M). The scale bar is 10 μm. M White arrows indicate Cd63-GFP in lens epithelial cells, whereas asterisks indicate Cd63-GFP secreted outside the lens. N Distribution of Cd63-GFP throughout the eye at 4.5 dpf. The dashed boxes are shown at higher magnification on the right. The white arrows in (c) indicate Cd63-GFP signals in the anterior chamber and ocular surface cells. The white arrows in (d) indicate Cd63-GFP signals in surrounding retina cells. The scale bar is 10 μm.
As development progressed, the AcGFP signal in the central lens region gradually diminished in the control group, likely due to translational stop in central LFCs and the natural degradation of GFP (Fig. 3F, G). In contrast, the signal of Cd63-AcGFP-positive exosomes remained in the central region of the lens at 3.5 dpf, indicating that these exosomes originate from surrounding fiber cells, facilitating communication within the LFCs (Fig. 3F, H). F-actin and β-catenin staining were used to delineate the cells and investigate the specific distribution of the exosomes (Fig. 3I–N). The results revealed that at 48 hpf, Cd63-AcGFP-positive exosomes were distributed mainly around the nuclei of fiber cells (Fig. 3I). By 3.5 dpf, in addition to being present within inner LFCs, Cd63-AcGFP-positive EVs were also detected in newly differentiated fiber cells (Fig. 3J, K). Compared with LFCs, LECs contain fewer Cd63-AcGFP-positive exosomes (Fig. 3L, M). We also observed Cd63-AcGFP-positive exosomes around the lens (Fig. 3L, M). Additionally, these exosomes could enter the anterior chamber (Fig. 3N-c1, c2) and reach the retina (Fig. 3N-d1, d2).
The biogenesis of exosomes derived from lens cells relies on a Syntenin-a-dependent pathway
Syntenin-a is essential for exosome formation28, and has been previously reported in zebrafish29. Verweij et al. utilized Syntenin-a knockdown to investigate the function of yolk syncytial layer derived exosomes in vivo22. Similarly, we used this validated Syntenin-a morpholino (SyntA-MO)30, to knockdown Syntenin-a expression and investigate its impact on lens exosome formation, aiming to gain insights into the role of exosomes in lens development (Fig. 4A). The effective knockdown was confirmed by qPCR, showing a reduction in Syntenin-a mRNA levels (Fig. 4B). To minimize potential secondary effects, we specifically selected lower morpholino dose. And the results showed that low-dose of morpholino injections (2 ng/nl) did not significantly disrupt epiboly or axis formation (Fig. 4D, E). We coinjected the morpholino with cryaa-Cd63-AcGFP plasmid into zebrafish embryos and confirmed that the embryos did not display obvious deformities. Compared with control group, the GFP signal in the lens significantly decreased following coinjection of cryaa-cd63-AcGFP and SyntA-MO (Fig. 4C, Supplementary Fig. 2A–C, E, F). Quantitative fluorescence analysis also confirmed this (Fig. 4H), indicating that Syntenin-a affects the biogenesis of Cd63+ exosomes in the lens. Although no significant differences in body length were observed (Fig. 4G), zebrafish treated with SyntA-MO exhibited significantly smaller lenses and eyes (Fig. 4F, I, J), indicating a potential role of Syntenin-a in lens and eye development. Further analysis revealed normal organelle degradation (mitochondria, lysosomes, and nuclei) in the SyntA-MO group, indicating that Syntenin-a does not affect the degradation of lens organelles (Supplementary Fig. 2B–G). And BrdU analysis revealed that Syntenin-a does not significantly affect the proliferation of LECs (Supplementary Fig. 2H, I). These findings indicate that the formation of Cd63+ exosomes in the lens depends on the Syntenin-a pathway. To further clarify the impact of Syntenin-a knockdown on lens development, we performed a rescue experiment. Syntenin-a was knocked down, and simultaneously, a morpholino-insensitive Syntenin-a was overexpressed under the cryaa promoter. We then assessed how effectively this rescued the lens phenotype. We observed that Syntenin-a overexpression markedly increased Cd63-GFP fluorescence (Fig. 4K, L) and partially rescued both lens and overall eye size (Fig. 4M, N). Subsequent qRT-PCR analysis of mature lens-specific protein genes showed that most were significantly downregulated in the SyntA MO knockdown group, whereas Syntenin-a overexpression restored the expression of several of these genes to some extent (Fig. 4O). These results indicate that Syntenin-a knockdown delays lens development by impairing lens cell differentiation, highlighting the potential role of lens cell–derived exosomes in this process.

A Morpholino oligos were designed to silence Syntenin-a expression in zebrafish. B qRT-PCR results showing the knockdown efficiency. ns not significant; ***p < 0.001. C Fluorescence images of zebrafish embryos at 4.5 dpf treated with Ctrl MO or SyntA MO and injected with cryaa-Cd63-AcGFP. The scale bar is 200 μm. D, E Quantification of the morphological features and health status of zebrafish embryos treated with Ctrl MO or SyntA MO from 1 to 3 dpf. Sample size > 100. D Ratio of hatched to non-hatched embryos. E Comparison of health status, categorized as normal morphology (healthy), mild or severe developmental defects, and mortality. F Brightfield images of zebrafish lens and eyes following injection with Ctrl MO or SyntA MO. The scale bar is 200 μm. G Quantification of the size (length) of Ctrl MO- or SyntA MO-treated 3.5 dpf zebrafish embryos (n = 37 and n = 30, respectively). H Quantification of fluorescence intensity in the lenses of 3.5 dpf zebrafish embryos treated with Ctrl MO or SyntA MO (n = 25, n = 55, respectively). I Quantification of the eye area of 3.5 dpf zebrafish embryos treated with Ctrl MO or SyntA MO (n = 30 and n = 14, respectively). J Quantification of the lens area of 3.5 dpf zebrafish embryos treated with Ctrl MO or SyntA MO (n = 30 and n = 14, respectively). K Bright-field and fluorescence images of zebrafish lenses and eyes after rescue experiments: Ctrl MO + OE-Ctrl (cryaa-DsRed), Ctrl MO + OE-Exp (cryaa-Syntenin-a-DsRed), SyntA MO + OE-Ctrl (cryaa-DsRed), and SyntA MO + OE-Exp (cryaa-Syntenin-a-DsRed). L Quantification of fluorescence intensity in the lenses of 3.5 dpf zebrafish embryos treated with SyntA MO + OE-Ctrl or SyntA MO + OE-Exp (n = 41, n = 29, respectively). M, N Quantification of the eye and lens area of 3.5 dpf zebrafish embryos after rescue experiments. (n = 58; 54; 117; 66). O Quantification of relative expression levels of crystallin-related genes by qRT-PCR.
ROR1+ cells sorted from a lentoid induction system exhibit characteristics of lens progenitor cells
Owing to the complex regulation of exosome formation pathways31 and the challenges of designing specific interventions for the lens, it is difficult to precisely study the functions of exosomes in the lens in vivo. Therefore, we aimed to investigate the roles of lens-EVs in vitro. Owing to the small size of the zebrafish lens, it is also challenging to extract enough EVs for in vitro functional experiments. Therefore, we followed Murphy’s approach32, using an hESC-lentoid induction system to isolate ROR1+ cells, which mimic cells in the early stages of lens development.
By optimizing the sorting time and culture conditions (Fig. 5A), we obtained ROR1+ cells and validated the efficiency of MACS (Fig. 5B). Bright-field images of the ROR1+ cells are shown in Fig. 5C. Immunofluorescence showed that the ROR1+ cells expressed markers of lens progenitor cells (LPCs), such as αA-crystallin, Pax6, Six3, Sox2, and BMI1, but did not express the mature lens fiber cell marker γ-crystallin (Fig. 5D–F). Similar to previous findings, ROR1+ cells differentiated into lentoid bodies upon induction (Fig. 5G) and expressed β-crystallin and γ-crystallin (Fig. 5H). RNA-seq was used to further characterize the ROR1+ cells. Consistent with above findings, the RNA-seq results showed that ROR1+ cells expressed genes for LPCs and LECs, but had low or no expression of genes for fiber cells (Fig. 5I). Moreover, ROR1+ cells exhibited low expression levels of endoderm, mesoderm, non-lens ectoderm, and pluripotency-related genes (Supplementary Fig. 3B). Additionally, ROR1+ cells express various genes associated with lens development33, including PITX3, HIF1α, GATA3, OTX2, and MAF (Supplementary Fig. 3C–E). Enrichment analysis showed that feature gene of ROR1+ cells were significantly enriched in lens and eye development, as well as stem cell maintenance and differentiation (Supplementary Fig. 3F, G). Collectively, these findings demonstrated that ROR1+ cells exhibit characteristic features of LPCs. Ultracentrifugation was used to isolate EVs from ROR1+ LPCs. NTA, TEM, and WB confirmed that EVs derived from ROR1+ LPCs exhibited typical characteristics of exosomes (Fig. 5J‒L). Next, these isolated ROR1+ LPCs-EVs were used for subsequent functional experiments.

A Schematic illustration of the induction of lentoid bodies from hESCs and magnetic-activated cell sorting (MASC) of ROR1+ cells. B Flow cytometry analysis of the purity of MACS-sorted ROR1+ cells. C Bright-field image of ROR1+ cells at P0; scale bar, 200 μm. D–F Immunofluorescence staining of ROR1+ cells. These cells expressed the lens progenitor markers αA-crystallin, Pax6, Six3, Sox2, and BMI1 but did not express the mature lens fiber cell marker γ-crystallin. The scale bar is 100 μm. G ROR1+ cells had the ability to differentiate and, upon induction, formed lentoid bodies. H Immunofluorescence staining of lentoid bodies derived from ROR1+ cells revealed the expression of the mature lens fiber cell markers β-crystallin and γ-crystallin. The scale bar is 100 μm. I Heatmap showing lens-related genes identified via RNA-seq analysis of ROR1+ cells. J Nanoparticle tracking analysis of EVs derived from ROR1+ cells. K TEM images of EVs derived from ROR1+ cells. The scale bar is 100 nm. L WB analysis of EVs derived from ROR1+ cells showing the expression of the exosome markers CD63, CD9, and CD81. Uncropped blot images are shown in Fig. S5.
EVs derived from ROR1+ LPCs promote lentoid differentiation
To investigate the role of ROR1+ LPC-derived EVs, we added them to the hESC-lentoid induction system at stage 3. EVs tracking experiments with Dil-labeled EVs showed preferential uptake by lentoid and nearby cells (Fig. 6A, B), consistent with previous studies indicating homotypic targeting of EVs34. At the end of induction, quantitative analysis revealed that exposure to ROR1+ LPCs-EVs from Days 20 to 35 resulted in significantly larger and more abundant lentoid bodies (Fig. 6C, D). Previous studies have shown that the small transparent structures observed via bright-field microscopy are lentoid bodies expressing αA-crystallin and β-crystallin, not cell aggregates35. qRT-PCR analysis further revealed the upregulated expression level of CRYAA, CRYAB, CRYBB2, CRYGC, PAX6, PROX1, MIP, BFSP1, and BFSP2 in the ROR1+ LPCs-EVs treatment group (Fig. 6E). In addition, WB revealed significantly increased expression of α-crystallin, β-crystallin, and γ-crystallin following ROR1+ LPCs-EVs treatment (Fig. 6F–J). These findings suggest that ROR1+ LPCs-EVs promote the differentiation of lentoid bodies.

A Images showing the internalization of ROR1+ LPCs-EVs-Dil by cells in the lentoid body induction system. Dil-labeled EVs are shown in red, cell nuclei in blue, and the lentoid body is indicated by white arrows. The dashed lines outline the lentoid bodies. B Magnified view of the dashed boxes in (A); the asterisks indicate lentoid body cells that have taken up ROR1+ LPCs-EVs, and the arrows point to cells surrounding the lentoid bodies that have taken up ROR1+ LPCs-EVs. C Phase-contrast microscopy was used to observe lentoid bodies in the control and ROR1+ LPCs-EVs-treated groups. D Statistical analyses of the number and area of lentoid bodies were performed on the basis of three independent experiments. E qRT‒PCR analysis of lens-related gene expression (CRYAA, CRYAB, CRYBB2, CRYGC, PAX6, PROX1, MIP, BFSP1, and BFSP2) in lentoid bodies after treatment with ROR1+ LPCs-EVs. *p < 0.05, **p < 0.01, ***p < 0.001. F WB analysis of the expression of proteins (αA-crystallin, β-crystallin, γ-crystallin, and Pax6) in lentoid bodies after treatment with ROR1+ LPCs-EVs. Uncropped blot images are shown in Fig. S5. G–J Quantification of the WB results. *p < 0.05, ***p < 0.001; ns not significant.
Proteomic analysis revealed candidate pathways through which ROR1+ LPCs-EVs regulate lens differentiation
Proteins are one of the key bioactive components loaded in EVs that mediate their functions36. We conducted a label-free proteomic analysis to investigate the cargo proteins within the ROR1+ LPCs-EVs. Our results revealed that most peptide segments contained 7–30 amino acids (Supplementary Fig. 4A). Andromeda analysis showed a high median value, indicating that the data met the quality control standards for LC‒MS/MS. (Supplementary Fig. 4B). In the three ROR1+ LPCs-EV samples, we identified 424, 358, and 359 proteins, respectively (Supplementary Fig. 4C). To minimize inter-batch variation, we focused on the 315 intersection proteins that were present in all samples, which we used for our downstream analysis (Supplementary Fig. 4D). Following the 2018 guidelines for studying EVs37, we investigated three categories of EVs marker proteins: transmembrane or GPI-anchored proteins, secreted or luminal proteins found within EVs, and cytosolic or periplasmic proteins. The results revealed that ROR1+ LPCs-EVs contained all three categories of exosome markers, including CD81, CD9, CD63, HSPA8, TUBA1C, and MFGE8 (Fig. 7A‒C). GO and KEGG analyses revealed significant enrichment in biological processes related to EV biogenesis, secretion, and transport, such as early endosomes, late endosomes, endocytosis, phagocytosis, and focal adhesion, highlighting the core properties of EVs (Fig. 7D). These results confirm the exosomal characteristics of ROR1+ LPCs-EVs.

A–C Gene expression heatmaps of three categories of EV-associated markers in ROR1+ LPCs-EVs: transmembrane or GPI-anchored proteins (A), secreted or luminal proteins recovered in EVs (B), and C cytosolic or periplasmic proteins. D–H Gene Ontology and KEGG analyses of proteins in ROR1+ LPCs-EVs. Enriched terms related to EVs synthesis and secretion (D). Enriched terms related to lens development (E–H).
Furthermore, our proteomic analysis revealed that the proteins found in the ROR1+ LPCs-EVs were significantly enriched in cell proliferation and inhibition of apoptosis (Fig. 7E). The proteins in the ROR1+ LPCs-EVs exhibited RNA-binding activity and were localized to ribosomes, the endoplasmic reticulum, and the Golgi apparatus, suggesting that they play roles in regulating transcription and translation (Fig. 7G). Moreover, these proteins also related to cell motility and actin filament organization, which are essential for fiber cell elongation (Fig. 7E–G). As fiber cells elongate, they establish cell polarity and adhere to surrounding cells, forming organized structures to build transparent lentoid. During this process, proteins related to cell polarity, adhesion, and the Cdc42 signaling pathway play critical roles38. Our analysis revealed that some proteins in ROR1+ LPCs-EVs have either an apical or basal location and are enriched in terms related to cell polarity and adhesion (Fig. 7E–G). Furthermore, the proteins in ROR1+ LPCs-EVs exhibited growth factor-binding activities, such as FGF-binding and PDGF-binding activity (Fig. 7F). KEGG analysis further demonstrated that the proteins in ROR1+ LPCs-EVs were indeed enriched in key signaling pathways critical for lens development, such as the PI3K/AKT, Hippo, and Rap1 signaling pathways (Fig. 7H). GSEA revealed significant enrichment of three processes: cell differentiation and homeostasis, gene expression and protein synthesis, and cellular adhesion and cytoskeletal remodeling (Supplementary Fig. 4E–G). These results suggest that the proteins in ROR1+ LPCs-EVs promote lentoid differentiation by enhancing various biological processes necessary for lens fiber cell maturation.
Discussion
Our study provides direct evidence of endogenous exosome-mediated communication within the lens. We identified three key aspects of lens exosome biology: (1) Ultrastructural and live-imaging analyses confirmed the presence of exosomes in the lens, capable of crossing the lens capsule; (2) Genetic experiments revealed that the biogenesis of Cd63+ exosomes is Syntenin-a-dependent, with Syntenin-a playing a crucial role in lens development by supporting fiber cell differentiation, thus highlighting the potential role of lens-derived exosomes; (3) EVs from ROR1+ lens progenitors promote lentoid differentiation, with proteomic profiling offering insights into potential mechanisms. Together, these findings uncover a previously unrecognized exosome communication network within the lens, providing new perspectives on its microenvironment.
Our labeling strategy employed the cryaa promoter, whose expression in LECs and LFCs is temporally regulated. Specifically, cryaa is first expressed in LFCs at 24 hpf and continues to be expressed in them27. By 55 hpf, central fiber cells start to mature, and as their nuclei and organelles degrade39, cryaa expression stops in these mature cells while continuing in newly differentiating or immature fiber cells. In contrast, cryaa expression in LECs only becomes significant after 6 dpf40. We injected a cryaa-driven Cd63-AcGFP plasmid into zygote, and initially, the plasmid was randomly distributed within the lens placode, later spreading to epithelial and primary fiber cells.
Due to the temporal expression pattern of the cryaa promoter and the limitations of transient plasmid expression, our labeling strategy primarily targeted LFCs and was unable to efficiently label LECs. Since cryaa is primarily expressed in LFCs before 6 dpf, this approach primarily labeled exosomes from fiber cells. The plasmid initially present in LECs becomes diluted as the cells divide, and by the time LECs start expressing cryaa at 6 dpf, the plasmid levels are too low to produce detectable signals. As a result, exosomes from LECs are not effectively labeled. Therefore, the GFP-positive exosomes in LECs observed in our experiment originate from LFCs. Our results showed that a few individual exosomes were present in LECs and surrounding ocular tissue cells, suggesting that LFCs secrete exosomes to convey signals to both LECs and adjacent ocular tissues. Moreover, TEM revealed exosome-like vesicles and multivesicular body structures in the LECs, suggesting that LECs may be capable of exosome secretion. To specifically track exosomes from LECs, it will be necessary to generate a cryaa-Cd63-AcGFP transgenic zebrafish line or use a more specific LEC promoter to drive Cd63-AcGFP expression.
There are several interesting phenomena in exosome labeling and live imaging experiments. First, Cd63-AcGFP fluorescence revealed that exosomes exhibit constitutive secretion during the early stages of lens development. Previous studies have shown that Cd63-GFP labeling can specifically target late endosomes and is incorporated into exosomes before their secretion41. Therefore, in our study, the GFP particles observed in LFCs represented both unreleased exosomes and those taken up from neighboring cells. Whether this constitutive secretion of exosomes is influenced by external stimuli remains to be further explored. Second, GFP driven by the cryaa promoter was detected as early as 25 hpf42, but significant exosome presence was observed at 36 hpf, indicating that exosomes secretion in the lens varies across developmental stages. Third, exosomes derived from lens cells were shown to migrate from the lens to the retina, supported by TEM images demonstrating exosome movement across the lens capsule. Similar findings in corneal research indicate that EVs can cross Descemet’s membrane43, indicating that basement membranes do not block the migration of EVs. However, owing to imaging limitations, we did not capture significant fluorescence accumulation or observe the uptake process in the cells surrounding the lens. Thus, future studies should employ more precise labeling strategies, such as the Cre‒loxP system44, to better identify the recipient cells that take up EVs.
We also observed that at 3.5 dpf, the GFP signal does not completely overlap with cryaa expression. This is also related to the combined effects of cryaa’s expression pattern and plasmid dilution. Primary fiber cells, which begin expressing cryaa at high levels from 24 hpf, maintain a high plasmid copy because they are terminally differentiated, resulting in strong exosome labeling. In contrast, secondary fiber cells, derived from epithelial cells, experience significant plasmid dilution during cell division. At 3.5 dpf, even though cryaa expression was activated in newly differentiated fiber cells, there was not enough plasmid to drive GFP expression to detectable levels. Additionally, in inner fiber cells, cryaa expression decreased with maturation, but the GFP signal persisted due to the long half-life of the Cd63-AcGFP protein. This explains why, at 3.5 dpf, inner fiber cells maintain GFP fluorescence despite low cryaa expression, while newly differentiated outer fiber cells express cryaa but lack detectable GFP signals.
In addition to the observed phenomena, several aspects of the exosome labeling strategy need to be discussed. In recent years, CD63-GFP has been widely used to analyze the distribution of exosomes in organs and bodily fluids45. pHluorin, a pH-sensitive form of GFP, is used on the basis that exosomes internalized by recipient cells usually end up in acidic compartments, leading to fluorescence quenching19,22. Thus, the CD63-pHluorin strategy primarily labels extracellular EVs. Given the lens’s simple cellular composition and limited extracellular space, we chose the pH-insensitive AcGFP to label CD63, which allowed the detection of both intercellular and extracellular exosomes in lens cells, as confirmed by our results. However, although Cd63-GFP labels exosomes inside and outside the cell, the high GFP background from exosomes aggregation makes distinguishing individual exosome behaviors difficult. To gain deeper insights into EVs in various states within the lens, combining pHluorin with non-pH-sensitive GFP is needed in future research. Moreover, future studies with cryaa-Cd63-GFP-expressing transgenic lines will be indispensable for improving our understanding of exosomes in the adult lens. Furthermore, CD63-positive EVs represent only a subset of EVs. Other TSPAN markers, such as CD81, CD82, and CD9, may help expand EVs labeling. Additionally, overexpressing CD63-GFP might interfere with the physiological processes of exosomes in vivo. The relatively high percentage (5.3%) of GFP-positive EVs in our study may reflect active EVs secretion during embryonic development. However, we cannot exclude the possibility that Cd63 overexpression may be enhancing EVs production. Although studies have shown that the CD63-GFP labeling strategy has a minimal impact on the exosomes proteome46 and size distribution22, further validation is necessary.
Based on our results and previous studies, we hypothesize that EVs play a key role in intercellular communication in the lens. As secondary fiber cells differentiate and surround the periphery throughout the lifespan, the relative positions of fiber and epithelial cells change47. Researchers have hypothesized that if epithelial and fiber cells rely mainly on gap junctions for intercellular communication, the shifting positions of LECs and LFCs would require constant degradation and resynthesis of these proteins4. From this perspective, EV-mediated communication might be more energy efficient. This hypothesis is also supported by observations of fewer gap junctions and numerous endocytotic–exocytotic vesicles at the EFI4. Moreover, the microcirculation system of the lens, which relies on connexins and aquaporins, mainly facilitates the exchange of ions and small molecules2. In contrast, EVs are likely involved in the transfer of larger molecules, such as growth factors, cholesterol, and lipoproteins48. These large molecules are also essential for lens differentiation and development. Therefore, we propose that vesicle-mediated communication may play a vital role in exchanging large molecules within the lens.
The Syntenin-ALIX-ESCRT-III pathway is an essential pathway for exosome biogenesis28. Our results revealed that after Syntenin-a knockdown, Cd63-GFP fluorescence in the lens significantly decreased, and the sizes of the lens and the eye were notably reduced. Notably, while Syntenin-a knockdown did not affect processes including lens organization and LECs proliferation, it markedly downregulated the mature lens crystallin gene expression profile. This selective regulation, together with the partial phenotypic rescue observed in overexpression experiments, suggests that Syntenin-a likely participates in lens cell differentiation and maturation, potentially through an exosome-dependent regulatory pathway. Research indicated that abnormal lens development can lead to retinal abnormalities42, suggesting that the lens influences surrounding tissues. Our study revealed that exosomes may be a mechanism via which the lens affects the development of surrounding tissues. Furthermore, lens-Exos were found mainly in the cytoplasm of LFCs in our study. During terminal differentiation, LFCs cells undergo denucleation and organelle removal49. Given that exosomes can remove waste components and provide nutrients50, we hypothesize that lens-Exos might also aid in organelle degradation during LF differentiation. However, our analysis of the mitochondria, lysosomes, and nuclei in Syntenin-a-knockdown zebrafish revealed that Syntenin-a does not affect overall organelle degradation. We hypothesize that this might be due to the complex regulatory pathways involved in exosome formation. Knocking down Syntenin-a did not completely block exosome formation, and Cd63-positive exosomes represented only a subset of exosomes, with other types likely involved as well. We acknowledge that morpholinos must be used with caution in zebrafish. Considering the limitations of morpholinos and the role of Syntenin-a in body axis development29, more precise, lens-specific EVs interference methods are needed to clearly define the role of endogenous EVs in lens differentiation.
To gain insight into the function of lens-Exos, we conducted experiments using the hESC-lentoid induction system in vitro. To acquire lens-Exos at the early developmental stages, we sorted ROR1+ cells from lentoid induction system. The lentoid induction system directs hESCs to form lentoids through the LPCs stage51. ROR1 is a membrane surface protein expressed during early lens development52. Murphy et al. sorted ROR1+ cells in a lentoid induction system and demonstrated that further differentiation of ROR1+ cells can lead to large-scale lentoid induction32. Therefore, we optimized the ROR1 sorting method and showed that the sorted ROR1+ cells exhibit LPCs characteristics in terms of proliferation, differentiation, and gene expression profile. This provides an opportunity to explore the potential role of lens cell-derived EVs in lens development.
These EVs were found to enhance lentoid differentiation when added to the lentoid induction system. Proteomic analysis suggested that ROR1+ LPCs-EVs enhance gene expression, promote cytoskeletal remodeling, and facilitate signal transduction, which are key processes in the differentiation of LECs into LFs. This finding aligns with studies in other tissues, where exosomes from stem or progenitor cells were shown to positively regulate tissue cell differentiation or repair53. For example, cardiac progenitor cell exosomes promote cardiomyocyte differentiation54,55, and neural progenitor cell exosomes promote neurogenesis56. Thus, LPCs-EVs might have potential applications in improving the microenvironment after cataract surgery for lens regeneration, as well as in other lens pathologies. Although these experiments have explored the preliminary functions of LPCs-EVs, the difficulty in extracting large quantities of EVs hinders further validation of their precise mechanisms. Therefore, future efforts should focus on optimizing culture conditions to extract more EVs, enabling a more thorough investigation of their specific roles and applications. Additionally, owing to significant differences between in vivo and in vitro environments, the functions of LPCs-EVs can provide only possible insights into the functions of lens-EVs in vivo.
Overall, in our study, we developed methods to label and visualize lens-specific exosomes in vivo and highlighted exosome-mediated intercellular communication both between lens cells and potentially between lens and surrounding tissues. We also optimized the method for enriching LPCs and showed in vitro that LPCs-EVs promote lentoid differentiation. These findings provide new insights into lens pathophysiology, and ROR1+LPCs-EVs may serve as regulators of the lens microenvironment.
Materials and methods
Zebrafish strains
Zebrafish were raised and cared for following standard protocols57. Wild-type AB zebrafish were maintained and bred in the fish facility at the Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, adhering to local animal welfare guidelines. The fish were kept at a controlled temperature of 28.5 °C and fed a diet of live and pellet food. Zebrafish embryos were collected through natural spawning and staged according to hours post-fertilization (hpf) or days post-fertilization (dpf). All experimental procedures were approved by the local ethical review committee of the Zhongshan School of Medicine, Sun Yat-sen University.
Construction of pCS2(+)-Cd63-AcGFP
To construct the pCS2(+)-cryaa promoter, the CMV IE94 promoter in the pCS2+ plasmid (#CZP1, CZRC, Hubei, China) was replaced with the cryaa promoter25. Then, Cd63-AcGFP (cd63: National Center for Biotechnology Information, NM_199543) and AcGFP were synthesized and cloned into the pCS2(+)-cryaa promoter, respectively. The cryaa promoter, Cd63-AcGFP, and AcGFP were synthesized by GENEWIZ (Jiangsu, China). To induce lens-specific expression of Cd63-AcGFP, embryos were injected with the plasmid at the one-cell stage. The following day, the embryos were examined under a stereomicroscope. Embryos that were dead or had developmental abnormalities were removed, and only those with a normal appearance were retained for further analysis.
Morpholino injection
Morpholinos (denoted by “MO” below) were purchased from Gene Tools (Philomath, USA). Syntenin-a-MO was adopted from a previously published study22. The sequence is: 5′-TACAACGACATCCTTTCTGCTTTCA-3′. The sequence of standard control morpholino is 5′- CCTCTTACCTCAGTTACAATTTATA-3′. Different doses of MO, including 2, 4, and 8 ng/nl, were injected for testing. To minimize the potential off-target effects, 2 ng/nl of ctrl MO or 2 ng/nl of Syntenin-a-MO was used. Real-time PCR was used to verify the knockdown efficiency. The primers used are listed in Supplementary information: Table S1.
Construction of pCS2(+)-Syntenin-a-DsRed
For the rescue experiments, morpholino-insensitive Syntenin-a–DsRed and DsRed were synthesized by GENEWIZ (Jiangsu, China) and individually cloned downstream of the cryaa promoter in the pCS2(+) vector. Zebrafish embryos at the one-cell stage were co-injected with pCS2(+)-Cd63-AcGFP, either Ctrl MO or SyntA MO, and either cryaa–Syntenin-a–DsRed or cryaa–DsRed, yielding four groups: Ctrl MO + OE-Ctrl (cryaa–DsRed), Ctrl MO + OE-Exp (cryaa–Syntenin-a–DsRed), SyntA MO + OE-Ctrl (cryaa–DsRed), and SyntA MO + OE-Exp (cryaa–Syntenin-a–DsRed). The following steps are as described above.
Live fluorescence imaging
At 24 hpf, PTU (200 µM) was added to the embryo-rearing water to inhibit melanin production. At 48 hpf, the zebrafish were anesthetized with tricaine (0.016%) and embedded in 1.5% low-melting-point agarose for positioning. After positioning at the lens, 3D images with a z-stack and time series videos were captured via a ZEISS LSM980 confocal microscope, and the images were reconstructed and analyzed using ZEN software (3.1).
Transmission electron microscopy (TEM)
Lens from 3.5 dpf zebrafish and postnatal (P) 0 mice were collected and transferred into TEM fixative (G1102, Servicebio, Hubei, China) for 2 h at room temperature (RT) and another 24 h at 4 °C. The samples were subsequently dehydrated at RT, penetrated with resin, embedded, and polymerized. After positioning, ultrathin sections were cut and stained with 2% uranium acetate and 2.6% lead citrate. Finally, the sections were examined and imaged via a TEM (JEM-1400Flash, Rigaku, Japan).
Immunoelectron microscopy (IEM)
Zebrafish embryos injected with the cryaa-Cd63-AcGFP plasmid at 3.5 dpf were fixed in IEM fixative (G1124, Servicebio). The samples were washed 3 times with prechilled 0.1 M PBS (pH 7.4) in an ice box. After dehydration in a gradient of prechilled alcohol, resin infiltration, embedding, polymerization, and ultrathin sectioning, the sections were subjected to immunogold labeling. In brief, after being blocked with 1% BSA at RT for 30 min, the sections were incubated with an anti-GFP antibody (A-11122, Thermo Fisher, Massachusetts, USA) at 4 °C overnight. GFP antibody was subjected to immunogold labeling with PAG 10 (G7402, Sigma, Missouri, USA) the next day. After uranium counterstaining, the sections were observed and imaged via a TEM.
EVs isolation from zebrafish embryos (ZF-EVs)
One hundred zebrafish embryos injected with the cryaa-Cd63-AcGFP plasmid at 3.5 dpf were anesthetized according to welfare standards, the rearing water was removed, and the embryos were washed 3 times with Ringer’s solution (containing 116 mM NaCl, 2.9 mM KCl, and 5.0 mM HEPES). During this process, the sample was intermittently blown with a sterile 200 µl tip to completely remove the yolk syncytial layer. The embryos were subsequently placed in a 35 mm petri dish with 2 ml of 0.25% trypsin/ 1 mM EDTA at 37 °C for digestion. During this period, the embryos were blown with a sterile 200 µl tip every 10 min until the digestion was complete, which ensured that no large cell clumps or tissue blocks were visible. The entire process took approximately 30–45 min. CaCl2 (1 mM) and 10% fetal bovine serum (FBS, Biowest, Nord, France) in PBS were used to stop trypsin activity. The final volume was transferred to centrifuge tubes. Differential centrifugation at 300 × g (2 × 10 min), 1200 × g (2 × 10 min), and 10,000 × g (1 × 60 min) was performed to eliminate cellular debris. The supernatant was filtered through a 0.22 μm filter and then centrifuged at 100,000 × g (2 × 90 min). The pellet was resuspended in PBS and stored at −80 °C until use.
The morphology of the ZF-EVs was evaluated via TEM. Fifty (50) micrograms of ZF-EVs were diluted (1:40) with PBS, and then the diluted EVs suspension (20 μL) was dropped onto a 300-mesh cell strainer and incubated for 10 min. Excess liquid was absorbed with filter paper, and the samples were counterstained with 1% phosphotungstic acid for 30 s and observed under a TEM. Nanoparticle tracking analysis (NTA, NanoSight NS300, Malvern, United Kingdom) was used to determine the particle number and size distribution of the ZF-EVs.
Nanoflow cytometry
EVs Membrane Red Stain dye (NEPU-638, NanoFCM, Fujian, China) was diluted with PBS to prepare a 100× working solution. One μL of the diluted 100× working solution was added to 99 μL of EVs sample at a concentration of 2 ~ 6 × 108 particles/ml. The mixture was mixed thoroughly and incubated at 37 °C for 15–20 min in the dark. Detection and analysis were subsequently performed via nanoflow cytometry using a Flow Nano Analyzer (U30E, NanoFCM, Fujian, China) equipped with three lasers (405, 488, and 640 nm wavelengths).
Human embryonic stem cells (hESCs) culture
The H1 hESCs line was kindly provided by Professor Shuyi Chen at the Zhongshan Ophthalmic Center. Mycoplasma contamination was routinely checked using a PCR-based assay to ensure the cell lines were free of contamination. The cells were cultured on plates coated with growth factor-reduced Matrigel matrix (354277, Corning, New York, USA) in mTeSR1 medium (# 85850, STEMCELL Technologies, Vancouver, Canada) without feeder cells. For maintenance, the cells were plated in 6-well plates at 37 °C, 5% CO2, and the culture medium was changed daily. The cells were passaged with Gentle Cell Dissociation Reagent (# 100-0485, STEMCELL Technologies, Vancouver, Canada).
Lentoid body induction
When the density of the H1 cells reached approximately 80–90%, the culture medium was replaced with lentoid body induction culture medium according to the protocol51. The basal medium used for lentoid body induction was Dulbecco’s modified Eagle’s medium/F-12 (C11330500BT, Gibco, Waltham, MA, USA) supplemented with 1% minimum essential medium nonessential amino acid solution (11140050, Gibco), 2 mM GlutaMAX (35050061, Gibco), 0.05% BSA (199897, MP Biomedicals, Ohio, USA), N-2 supplement (17502048, Thermo Fisher Scientific, Waltham, MA, USA), and B-27 supplement (17504044, Thermo Fisher Scientific). Growth factors were then added sequentially to the medium as follows: 100 ng/mL Noggin (120-10C-20, PeproTech, New Jersey, USA) from Days 0 to 6 (stage 1), 100 ng/mL FGF2 (100-18B‒100, PeproTech) and 20 ng/mL BMP4 (120-05‒5, PeproTech)/BMP7 (120-03 P‒10, PeproTech) from Days 7 to 18 (stage 2), and 100 ng/mL FGF2 and 20 ng/mL Wnt3a (315-20‒10, PeproTech) from Days 19 to 35 (stage 3). The culture medium was replaced every other day. For EVs treatment, 50 μl of 0.5 μg/μl ROR1-EVs was added to the medium from days 20 to 35. The lentoid bodies were photographed via a Zeiss Axio Observer Z1 microscope (Zeiss, San Diego, CA, USA) on Day 35 and were collected for further analysis.
Magnetic-activated cell sorting
A magnetic-activated cell sorting (MACS) method was used to purify ROR1+ cells32. First, we prepared single-cell suspensions using TrypLE (Thermo Fisher Scientific) on Day 30 of lentoid induction. Next, an Anti-ROR1 MicroBead Kit (130-103-929, Miltenyi Biotec, Bergisch Gladbach, Germany) was used to label the cells, which were sorted with a MACS cell separator (130-042-201, Miltenyi Biotec). The resulting ROR1+ cells were plated on Matrigel-coated culture plates in M199 medium (Thermo Fisher Scientific) containing 10 ng/ml FGF2 (100-18B-100, Peprotech), 10 ng/ml IGF1 (100-11-100, PeproTech, New Jersey, USA), 10 μg/ml insulin (HY-P0035, MedChemExpress, New Jersey, USA) and 0.05% BSA (199897, MP Biomedicals, Ohio, USA). The culture medium was replaced every other day. To analyze the purity of the ROR1+ cells, we performed flow cytometry using an ROR1 antibody (357806, Biolegend, California, USA). ROR1+ cells from passages 0 to 3 were used for the experiments. To induce the differentiation of ROR1+ cells, the maintenance medium was switched to lentoid induction medium as mentioned above, supplemented with 100 ng/μL FGF and 20 ng/μL Wnt3a. After one-week, lentoid formation was observed, followed by examination of lens differentiation markers via immunofluorescence staining.
RNA-seq
Total RNA was extracted from ROR1+ cells using TRIzol reagent (15596-018; Thermo Fisher, USA). The analysis was performed with 3 biological replicates. The RNA was quantified via the K5500 system (Beijing Kaiao, China), and RNA integrity was assessed via the Agilent 2200 TapeStation (Agilent Technologies, USA). The sequencing libraries were generated via the NEBNext® Ultra™ II RNA Library Prep Kit for Illumina (#7775, NEB, USA) following the manufacturer’s instructions. The libraries were sequenced on the Illumina NovaSeq platforms (Illumina, USA) with 150 bp paired-end reads at RiboBio Co., Ltd. (Ribobio, Guangdong, China). After filtering, the clean reads were aligned to the human reference genome (GRCh38) via HISAT258. Then, feature counts were used to determine the number of reads mapped to each gene, and the transcripts per kilobase of exon model per million mapped reads (TPM) value was calculated for each gene. GO and KEGG functional enrichment analyses were performed via the DAVID59,60 (https://david.ncifcrf.gov/tools.jsp), followed by visualization using an online platform (https://www.bioinformatics.com.cn). Gene expression heatmap were performed using ComplexHeatmap packages (version 2.13.1).
Isolation of EVs from ROR1+ cells cultured in vitro
To isolate ROR1+ LPCs-EVs, the medium was collected after the cells were cultured for 48 h, and EVs were extracted via differential centrifugation. The supernatant was centrifuged sequentially at 300 × g for 10 min and 2000 × g for 30 min to remove cell debris. The resulting mixture was ultracentrifuged at 10,000 × g for 60 min and then filtered through a 0.22 μm filter. Next, we extracted EVs using ultracentrifugation (Optima XE 100 centrifuge) at 100,000 × g for 90 min, as described above. The final pellet was resuspended in PBS and stored at −80 °C until use. TEM, NTA, and WB were used to detect the morphology, particle number, and size distribution, and surface markers of the ROR1+ LPCs-EVs, respectively. The protein concentrations of the ROR1+ LPCs-EVs were measured using a BCA protein assay kit (A55864, Thermo Scientific, USA). The doses used in all subsequent experiments were based on the measured protein concentrations.
Western blot analysis (WB)
Lentoid bodies collected on Day 35 were lysed with RIPA buffer. The protein concentration was quantified via a BCA protein assay kit (A55864, Thermo Scientific, USA). The proteins were mixed with 5× SDS sample buffer, and equal amounts of protein were separated via sodium dodecyl-polyacrylamide gel electrophoresis (SDS‒PAGE). The proteins were then transferred onto polyvinylidene fluoride (PVDF) membranes via electroblotting. The membranes were blocked with 5% nonfat milk and incubated overnight at 4 °C with specific primary antibodies. After being washed with TBS containing 0.1% Tween 20 (TBST), the membranes were incubated with HRP-conjugated secondary antibodies. The target protein bands were detected using an enhanced chemiluminescence reagent (NcmECL Ultra, NCM Biotech, Suzhou, China) and a chemiluminescence detection system (ChemiDoc™ Touch, Bio-Rad Laboratories, California, USA). The antibodies used: PAX6 (1:1000, ab195045, Abcam, Cambridge, UK), αA-crystallin (1:500, sc-28306, Santa Cruz, Texas, USA), β-crystallin (1:200, sc-48335, Santa Cruz), γ-crystallin (1:200, sc-22746, Santa Cruz), and β-actin (1:1000, 4970, Cell Signaling Technology, Massachusetts, USA).
RNA isolation and quantification
Total RNA was extracted from cells using TRIzol reagent (15596-018; Thermo Fisher). Reverse transcription was performed using a PrimeScript RT Master Mix kit (TaKaRa, Kyoto, Japan), and quantitative PCR was performed using a SYBR Premix Ex Taq kit (TaKaRa, Japan) on a LightCycler 480 (Roche, Basel, Switzerland). GAPDH was used as an internal control. Relative gene expression was determined by the 2−ΔΔCt method. The primers used are listed in Supplementary information: Table S1.
Immunofluorescence staining
Zebrafish cryosections or cultured cells were fixed with 4% paraformaldehyde for 10 min at RT. After washing with PBST, the cryosections or cells were permeabilized with 0.5% Triton ×-100 for 10 min and blocked with 5% normal donkey serum for 30 min. Afterward, the sections or cells were incubated with different primary antibodies at 4 °C overnight in a humidified chamber. The next day, Alexa Fluor 488-conjugated or Alexa Fluor 568-conjugated secondary antibodies were applied, and the samples were incubated for 1 h at RT. After washing with PBST, the samples were counterstained with DAPI for nuclear staining. The samples were observed using an LSM980 confocal microscope or an inverted fluorescence microscope. The antibodies used: αA-crystallin (1:100, sc-28306, Santa Cruz), β-crystallin (1:100, sc-48335, Santa Cruz, Texas, USA), γ-crystallin (1:200, sc-22746, Santa Cruz), F-actin (1:200, C2201S, Beyotime, Shanghai, China), β-catenin (1:200, sc-7963, Santa Cruz), GFP (1:200, A-11122, Thermo Fisher, Massachusetts, USA), TOM20 (1:200, 11802-1-AP; Proteintech Group, Illinois, USA), LAMP1 (1:200, ab24170; Abcam, Cambridge, UK), PAX6 (1:300, ab195045, Abcam), Six3 (1:200, sc-398797, Santa Cruz), Sox2 (1:400, 23064, Cell Signaling Technology), and BMI1 (1:100, 5856S, Cell Signaling Technology).
Bromodeoxyuridine (BrdU) labeling and immunostaining
At 3.5 dpf, zebrafish embryos were positioned laterally in a drop of embryo medium for microinjection. A pulled glass capillary loaded with a 10 mM BrdU solution (Sigma-Aldrich) was used to deliver approximately 0.5 nL into the cardiac region adjacent to the caudal yolk sac. After a 2-h period in fresh water, embryos were fixed, permeabilized, and processed for immunofluorescence. Finally, incorporated BrdU was detected by incubating the specimens with a mouse anti-BrdU monoclonal antibody (1:300, RPN20, GE Healthcare) following standard immunofluorescence protocols.
Extracellular vesicle uptake assay
To monitor the internalization of ROR1+ LPCs-EVs, the isolated ROR1+ LPCs-EVs were labeled with DiI Cell-Labeling Solution (V22885, Thermo Fisher, USA). In brief, 100 μl of EVs were diluted to 1 ml with PBS, 10 μl of Dil was added to the suspension, and the mixture was incubated at 37 °C for 10 min. Subsequently, the mixed suspension was centrifuged at 100,000 × g for 60 min. After the supernatant was discarded, the pellet was resuspended in PBS to obtain Dil-labeled ROR1+ LPCs-EVs. Then, the labeled ROR1+ LPCs-EVs were added to a lentoid induction system, which was subsequently fixed and stained for immunofluorescence after 48 h of incubation. A ZEISS LSM980 confocal microscope was used to acquire fluorescence images.
Label-free mass spectrometry analysis
For label-free proteomic analysis, three biological replicates were prepared. The proteins in the ROR1+ LPCs-EVs samples were prepared and digested with trypsin. The resulting peptides were analyzed via liquid chromatography‒tandem mass spectrometry (LC‒MS/MS) using an UltiMate 3000 RSLCnano system coupled with a Q Exactive HF mass spectrometer through a Nanospray Flex ion source (Thermo). The MS data were analyzed using MaxQuant software (version 1.6.6) and searched against the human protein sequence database downloaded from UniProt. Proteins were identified and quantified, and the results were filtered with a 1% false discovery rate (FDR) at both the peptide and protein levels. Protein levels were normalized to the iBAQ values [%] for expression analysis. The total protein list for the ROR1+ LPCs-EVs was generated by identifying proteins that were simultaneously present in three samples (315 identified proteins). We utilized the DAVID bioinformatics resources to conduct GO and KEGG enrichment analyses. GSEA analysis was performed using gseGO and gseKEGG function of clusterprofiler R packages (version 4.4.4). The enrichment analysis results were visualized via https://www.bioinformatics.com.cn.
Statistical analysis
We performed statistical analysis using GraphPad Prism version 8.0 (GraphPad software). Student’s two-tailed t test was used for two-group comparisons. All quantitative data are shown as the mean ± standard deviation (SD). P value less than 0.05 was considered to indicate statistical significance.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data from this study can be obtained from the corresponding author upon reasonable request. RNA-sequencing data have been deposited in GEO under accession codes GSE298454. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX partner repository (accession: PXD064544). Source data underlying graphs can be obtained from Supplementary data 1.
References
Liu, Z. et al. The lens epithelium as a major determinant in the development, maintenance, and regeneration of the crystalline lens. Prog. Retin. Eye Res. 92, 101112 (2023).
Donaldson, P. J., Grey, A. C., Maceo Heilman, B., Lim, J. C. & Vaghefi, E. The physiological optics of the lens. Prog. Retin Eye Res. 56, e1–e24 (2017).
Blindness, G. B. D. Vision impairment C, Vision loss expert group of the Global Burden of Disease S. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: the Right to Sight: an analysis for the Global Burden of Disease Study. Lancet Glob. Health 9, e144–e160 (2021).
Dahm, R., van Marle, J., Quinlan, R. A., Prescott, A. R. & Vrensen, G. F. Homeostasis in the vertebrate lens: mechanisms of solute exchange. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 1265–1277 (2011).
Yang, Q. et al. Capsule-epithelium-fibre unit ultrastructure in the human lens. Int. J. Med. Sci. 20, 542–550 (2023).
Dahm, R., Schonthaler, H. B., Soehn, A. S., van Marle, J. & Vrensen, G. F. Development and adult morphology of the eye lens in the zebrafish. Exp. Eye Res. 85, 74–89 (2007).
van Niel, G., D’Angelo, G. & Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19, 213–228 (2018).
van Niel, G. et al. Challenges and directions in studying cell-cell communication by extracellular vesicles. Nat. Rev. Mol. Cell Biol. 23, 369–382 (2022).
Verta, R. et al. Phenotypic and functional characterization of aqueous humor derived extracellular vesicles. Exp. Eye Res. 228, 109393 (2023).
Kalluri, R. & LeBleu, V. S. The biology, function, and biomedical applications of exosomes. Science 367, eaau6977 (2020).
Liu, J. et al. Roles of exosomes in ocular diseases. Int. J. Nanomed. 15, 10519–10538 (2020).
Dismuke, W. M., Challa, P., Navarro, I., Stamer, W. D. & Liu, Y. Human aqueous humor exosomes. Exp. Eye Res. 132, 73–77 (2015).
Gao, C. et al. Exosomal miRNA analysis of aqueous humour of diabetes and cataract patients. Curr. Eye Res. 46, 324–332 (2021).
Zhang, J. et al. An ex vivo study on release, uptake, and miRNA profile of exosomes in rat lens. J. Ophthalmol. 2022, 6706172 (2022).
Wu, Y. et al. Human lens epithelial-secreted exosomes attenuate ocular angiogenesis via inhibiting microglial activation. Exp. Eye Res. 241, 109837 (2024).
Pan, T. et al. Lens epithelial cell-derived exosome inhibits angiogenesis in ocular pathological neovascularization through its delivery of miR-146a-5p. FASEB J. 37, e23192 (2023).
Verweij, F. J. et al. The power of imaging to understand extracellular vesicle biology in vivo. Nat. Methods 18, 1013–1026 (2021).
Levy, D. et al. Genetic labeling of extracellular vesicles for studying biogenesis and uptake in living mammalian cells. Methods Enzymol. 645, 1–14 (2020).
Verweij, F. J., Hyenne, V., Van Niel, G. & Goetz, J. G. Extracellular vesicles: catching the light in zebrafish. Trends Cell Biol. 29, 770–776 (2019).
Lieschke, G. J. & Currie, P. D. Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 8, 353–367 (2007).
Li, Y. J., Wu, J. Y., Wang, J. M., Hu, X. B. & Xiang, D. X. Emerging strategies for labeling and tracking of extracellular vesicles. J. Control Release 328, 141–159 (2020).
Verweij, F. J. et al. Live tracking of inter-organ communication by endogenous exosomes in vivo. Dev. Cell 48, 573–589 e574 (2019).
Farnsworth, D. R., Posner, M. & Miller, A. C. Single cell transcriptomics of the developing zebrafish lens and identification of putative controllers of lens development. Exp. Eye Res. 206, 108535 (2021).
Runkle, S., Hill, J., Kantorow, M., Horwitz, J. & Posner, M. Sequence and spatial expression of zebrafish (Danio rerio) alphaA-crystallin. Mol. Vis. 8, 45–50 (2002).
Posner, M. et al. The zebrafish as a model system for analyzing mammalian and native alpha-crystallin promoter function. PeerJ 5, e4093 (2017).
Greiling, T. M. & Clark, J. I. New insights into the mechanism of lens development using zebra fish. Int. Rev. Cell Mol. Biol. 296, 1–61 (2012).
Marvin, M. et al. Developmental expression patterns of the zebrafish small heat shock proteins. Dev. Dyn. 237, 454–463 (2008).
Baietti, M. F. et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 14, 677–685 (2012).
Lambaerts, K. et al. Syntenin, a syndecan adaptor and an Arf6 phosphatidylinositol 4,5-bisphosphate effector, is essential for epiboly and gastrulation cell movements in zebrafish. J. Cell Sci. 125, 1129–1140 (2012).
Stainier, D. Y. R. et al. Guidelines for morpholino use in zebrafish. PLoS Genet. 13, e1007000 (2017).
Krylova, S. V. & Feng, D. The machinery of exosomes: biogenesis, release, and uptake. Int. J. Mol. Sci. 24, 1337 (2023).
Murphy, P. et al. Light-focusing human micro-lenses generated from pluripotent stem cells model lens development and drug-induced cataract in vitro. Development 145, dev155838 (2018).
Cvekl, A. & Zhang, X. Signaling and gene regulatory networks in mammalian lens development. Trends Genet. 33, 677–702 (2017).
Zhu, S. et al. Augmented cellular uptake and homologous targeting of exosome-based drug loaded IOL for posterior capsular opacification prevention and biosafety improvement. Bioact. Mater. 15, 469–481 (2022).
Han, C. et al. Wnt5a contributes to the differentiation of human embryonic stem cells into lentoid bodies through the noncanonical Wnt/JNK signaling pathway. Invest Ophthalmol. Vis. Sci. 59, 3449–3460 (2018).
Wang, X. et al. The updated role of exosomal proteins in the diagnosis, prognosis, and treatment of cancer. Exp. Mol. Med. 54, 1390–1400 (2022).
Thery, C. et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 7, 1535750 (2018).
Chen, Y., Stump, R. J., Lovicu, F. J. & McAvoy, J. W. A role for Wnt/planar cell polarity signaling during lens fiber cell differentiation?. Semin. Cell Dev. Biol. 17, 712–725 (2006).
Morishita, H. et al. Organelle degradation in the lens by PLAAT phospholipases. Nature 592, 634–638 (2021).
Posner, M. et al. Loss of alphaBa-crystallin, but not alphaA-crystallin, increases age-related cataract in the zebrafish lens. Exp. Eye Res. 244, 109918 (2024).
Verweij, F. J. et al. Quantifying exosome secretion from single cells reveals a modulatory role for GPCR signaling. J. Cell Biol. 217, 1129–1142 (2018).
Kurita, R. et al. Suppression of lens growth by alphaA-crystallin promoter-driven expression of diphtheria toxin results in disruption of retinal cell organization in zebrafish. Dev. Biol. 255, 113–127 (2003).
Zieske, J. D., Hutcheon, A. E. K. & Guo, X. Extracellular vesicles and cell-cell communication in the cornea. Anat. Rec. (Hoboken) 303, 1727–1734 (2020).
Zomer, A. et al. In vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell 161, 1046–1057 (2015).
Yoshimura, A. et al. Generation of a novel transgenic rat model for tracing extracellular vesicles in body fluids. Sci. Rep. 6, 31172 (2016).
Corso, G. et al. Systematic characterization of extracellular vesicle sorting domains and quantification at the single molecule - single vesicle level by fluorescence correlation spectroscopy and single particle imaging. J. Extracell. Vesicles 8, 1663043 (2019).
Cvekl, A. & Ashery-Padan, R. The cellular and molecular mechanisms of vertebrate lens development. Development 141, 4432–4447 (2014).
Dixson, A. C., Dawson, T. R., Di Vizio, D. & Weaver, A. M. Context-specific regulation of extracellular vesicle biogenesis and cargo selection. Nat. Rev. Mol. Cell Biol. 24, 454–476 (2023).
Wride, M. A. Lens fibre cell differentiation and organelle loss: many paths lead to clarity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 1219–1233 (2011).
Russell, A. E. et al. Biological membranes in EV biogenesis, stability, uptake, and cargo transfer: an ISEV position paper arising from the ISEV membranes and EVs workshop. J. Extracell. Vesicles 8, 1684862 (2019).
Yang, C. et al. Efficient generation of lens progenitor cells and lentoid bodies from human embryonic stem cells in chemically defined conditions. FASEB J. 24, 3274–3283 (2010).
Oishi, I. et al. Spatio-temporally regulated expression of receptor tyrosine kinases, mRor1, mRor2, during mouse development: implications in development and function of the nervous system. Genes Cells 4, 41–56 (1999).
Riazifar, M., Pone, E. J., Lotvall, J. & Zhao, W. Stem cell extracellular vesicles: extended messages of regeneration. Annu. Rev. Pharm. Toxicol. 57, 125–154 (2017).
Ibrahim, A. G., Cheng, K. & Marban, E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep. 2, 606–619 (2014).
Barile, L., Moccetti, T., Marban, E. & Vassalli, G. Roles of exosomes in cardioprotection. Eur. Heart J. 38, 1372–1379 (2017).
Ma, Y. et al. Exosomes released from neural progenitor cells and induced neural progenitor cells regulate neurogenesis through miR-21a. Cell Commun. Signal 17, 96 (2019).
Westerfield, M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio) (Institute of Neuroscience, 2000).
Lopez-Fernandez, H., Blanco-Miguez, A., Fdez-Riverola, F., Sanchez, B. & Lourenco, A. DEWE: a novel tool for executing differential expression RNA-Seq workflows in biomedical research. Comput. Biol. Med. 107, 197–205 (2019).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Acknowledgements
The authors greatly acknowledge the staff of Core Facilities at State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center for technical support. This study was supported by the National Natural Science Foundation of China (No. 82070940 and No. 32070817); the Fundamental Research Funds of the State Key Laboratory of Ophthalmology (No. 83000-32030003); the Guangzhou Basic Research Program, City & University (Institute) Joint Funding Project (No. 2024A03J0245).
Author information
Authors and Affiliations
Contributions
Conceptualization, L.L. and J.M.; Methodology, J.M., Q.S., Y.C., Jinyan Li, S.O. and H.L.; Validation, J.M., Q.S., Y.C., Jinyan Li, S.O., S.K., Jiani Li, H.L.; Formal Analysis, J.M., Q.S., Y.C., Jinyan Li; Investigation, J.M., Q.S., Y.C., Jinyan Li, S.O., S.K., Jiani Li, H.L.; Writing–Original Draft, J.M.; Writing–Review & Editing, J.M., Q.S., Y.C., Jinyan Li, S.O., S.C. and L.L.; Visualization S.O., S.K., Jiani Li; Supervision L.L. and S.C.; Funding Acquisition, L.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Zongbo Wei, Frederick Verweij, and Bárbara Adem Ribeiro for their contribution to the peer review of this work. Primary Handling Editor: Ophelia Bu.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, J., Sun, Q., Chen, Y. et al. In vivo imaging reveals exosome-mediated intercellular communication in lens development. Commun Biol 8, 1774 (2025). https://doi.org/10.1038/s42003-025-09159-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09159-5
