
Abstract
Drug response prediction at the single-cell level provides guidance for drug development and treatment. Existing methods typically rely on integrating data from bulk RNA sequencing and single-cell RNA sequencing, transferring overall cell line drug response labels to individual cells. However, the inherent differences between these data types and the assumption that the cell line response represents each cell’s response can affect prediction accuracy. This study proposes a heterogeneous network transfer learning model, scXDR, for drug response prediction across single-cell datasets. The model uses heterogeneous networks to integrate features and associations among drugs, genes, and cells, performing message passing, feature and structure alignment, structure reconstruction, and drug-cell score prediction. Experimental results show that scXDR outperforms methods transferring from bulk data to single-cell data and methods transferring from single-cell data to single-cell data in multiple scenarios, achieving excellent performance at individual cell and cell group levels. The importance of the model components is also confirmed. Case studies include outcome prediction for cells under drug holiday treatment, melanoma drug screening, multi-tumor drug response analysis, and predictions for approved drugs and potential drug combinations for patient cells. The research strategy and results of scXDR provide references for precision treatment at the cell level.
Similar content being viewed by others

Introduction
Gene expression differences exist between different tumor cells, which are an important factor contributing to tumor heterogeneity and poor treatment outcomes1. Traditional tumor gene expression studies often rely on bulk RNA sequencing (bulk RNA-seq), which tends to overlook heterogeneity between tumor cells. The rise of single-cell technologies, including single-cell RNA sequencing (scRNA-seq), has promoted cell-level tumor research2. Unlike bulk RNA-seq, which provides average gene expression data, scRNA-seq offers detailed expression data for each tumor cell, capturing subtle differences between individual cells in the tumor microenvironment3. This helps researchers better understand tumor mechanisms, improve drug selection, and promote precision medicine4. Aissa et al.5 used scRNA-seq to demonstrate that multiple cancer cell subpopulations exist in cell lines, xenograft tumors, and patient tumors, with these subpopulations exhibiting epigenetic changes and varying treatment sensitivities. França et al6. characterized cell states using scRNA-seq data and discovered that drug resistance develops along trajectories of cell state transitions. Yan et al.7 integrated various data, including scRNA-seq, to analyze the changes in tumor cells before and after neoadjuvant immune checkpoint blockade therapy, revealing significant heterogeneity and dynamic changes in tumor cells’ response to treatment. These studies employed wet lab experiments to investigate single-cell drug research, but the high costs and time investment have led some researchers to explore computational and predictive approaches8. Such methods not only save cost and time but also enable more extensive experimentation.
The inherent differences between bulk RNA-seq and scRNA-seq data, along with other factors, prevent the direct transfer of previous drug response prediction models based on bulk RNA-seq to scRNA-seq data9. Therefore, specific drug response prediction models based on scRNA-seq data need to be designed. Suphavilaid et al10. proposed CaDRReS-Sc, one of the earliest published methods for predicting drug response at the single-cell level. It learns a common pharmacological space by integrating bulk RNA-seq data from cancer cell lines in the Genomics of Drug Sensitivity in Cancer (GDSC) database with scRNA-seq data, thereby transferring bulk-level drug response information to the single-cell level. Beyondcell11, released concurrently with CaDRReS-Sc, and the later method DREEP12 calculate gene markers based on the relationship between bulk RNA-seq data and drug response data. They assume that markers identified at the bulk level are applicable at the single-cell level and predict single-cell drug responses based on marker expression. Later, Chen et al.13 introduced scDEAL, and Zheng et al.14 proposed SCAD, both of which use transfer learning methods that treat bulk-level data as the source domain and single-cell data as the target domain. After bringing the two domains closer, these methods predict drug responses at the single-cell level using models trained on bulk-level data, offering a new approach for applying deep learning, particularly transfer learning, to single-cell drug response prediction.
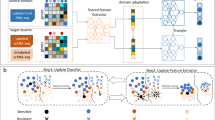
An overview of current single-cell drug response prediction methods reveals that most of these methods rely on expression data and drug response information obtained at the bulk level from cell lines to predict the drug response of each individual cell at the single-cell level. However, bulk expression data and single-cell expression data are produced using different technical platforms and processing methods15, which results in inherent differences between them. This discrepancy might be one of the major reasons why the performance of many single-cell drug response prediction methods is not high. Secondly, the bulk expression and drug response data used in these models come from a few specific databases8,13, while single-cell datasets exhibit multiple variations, including differences in tumor types, individual variability, treatment methods, and microenvironments. Models based on bulk data may not generalize well to the more diverse and variable single-cell datasets. Finally, and perhaps most importantly, bulk drug response is obtained by observing the overall performance of cell lines, whereas single-cell prediction focuses on the drug response of each individual cell16. Even within the same cell group, some cells may be sensitive while others may be resistant. To overcome some of the shortcomings associated with transferring from bulk data to single-cell data, this study proposes scXDR (where “X” denotes cross‑dataset), a drug‑response prediction model across single‑cell datasets. The model first constructs heterogeneous networks from different single‑cell datasets and then applies transfer learning, among other procedures, to predict the drug response of each individual cell. The workflow is shown in Fig. 1.


a The transition between the message passing phase, transfer learning phase, and prediction phase. b The specific operations in the transfer learning phase. c Schematic of the four scenarios. Square node Source domain node, Circular node: Target domain node, Blue Drug; Green: Cell, Orange Target, HGNN Heterogeneous Graph Neural Network, TL Transfer Learning, MLP Multi-Layer Perceptron. Each row of the feature and embedding shapes represents the features or embeddings of each drug, cell, or target.
Due to the wide variety and complex backgrounds of single-cell datasets, and based on differences in tumor and treatment contexts, this study divides 12 datasets into 4 scenarios (see Fig. 1 and Supplementary Table S1). In each scenario, every dataset is assigned prediction tasks both when used as the source domain and when used as the target domain. scXDR consists of three phases. In the message passing phase, the heterogeneous networks of the two domains perform message passing independently using a Heterogeneous Graph Neural Network (HGNN)17. The transfer learning phase includes an autoencoder component, a feature and structure alignment component, and a structure reconstruction component. These phases are executed separately for each type of element present in the heterogeneous networks. The prediction phase treats response prediction as a drug-cell edge prediction task, where the model, once trained on the source domain, is applied to the target domain. These three phases are coherent and integrated in an end-to-end manner. The design, evaluation, and application of scXDR possess the following innovations:
-
The transfer learning across different single-cell datasets is implemented, reducing the differences introduced by technical platforms, sequencing technologies, and other factors when transferring from bulk RNA-seq data to scRNA-seq data. Drug response prediction across single-cell datasets based on transfer learning is feasible.
-
The heterogeneous network fully utilizes the feature and association information of drugs, cells, and genes. The transfer of the heterogeneous network implies the transfer of multiple types of information while maintaining the integrity of the network structure during the process.
-
The experimental setup and training process more closely align with real prediction scenarios, including the source and target single-cell datasets being complete and independent, cells within each domain sharing information rather than existing in isolation, and the drug response labels in the target domain being treated as unknown and not leaked.
-
It outperforms various other methods in multiple scenarios, cell scales, and evaluation metrics. The prediction results in several cases are not only validated by existing evidence but also provide valuable insights for precise drug treatment at the cell level.
Results
Superior performance at the individual cell level compared to various methods
In each scenario, scXDR is compared with two major categories of single-cell drug response prediction methods. The AUC results for each scenario are shown in Tables 1 and 2, while the results for AUPR, ACC, and F1 Score are available in Supplementary Data 1. Visualizations of the four metrics in each scenario or the combined scenario (all values from the four scenarios) are provided in Supplementary Fig. S1 and Fig. 2 as box plots. A comparison of scXDR with seven methods for transferring from bulk to single-cell data shows that in all four scenarios, scXDR outperforms the other methods, exhibiting higher and more concentrated metric values, with values clustering in the high-value region (Supplementary Fig. S1a). The AUC values for the other methods are all lower than those for scXDR. Specifically, in the first, second, and fourth scenarios, the mean AUC for scXDR exceeds 0.82, and the AUPR, ACC, and F1 Score metrics are also higher than those of the other methods (Supplementary Data 1). The combined results from the four scenarios (Fig. 2a) show that scXDR’s AUC and AUPR values concentrate above 0.8, with medians of 0.85 and 0.89, and means of 0.84 and 0.86, respectively. The median and mean ACC are also close to 0.8. In the third scenario (Supplementary Fig. S1a), the median metric values for scXDR approach those of scDEAL (bulk), as scDEAL (bulk) is the advanced model for transferring from bulk to single-cell data. The two datasets in this scenario were used in the original scDEAL study, where the authors provided the optimal parameters. However, the distribution of scDEAL’s metric values in this scenario remains dispersed, with both high and low values, indicating that it does not adapt as well to multiple datasets as scXDR.

a Metric values for scXDR and the 7 models of first category in the combined scenario. b Metric values for scXDR and the 8 models of second category in the combined scenario.
A comparison with eight methods for transferring from single-cell to single-cell data reveals that in scenarios 2, 3, and 4, the other methods not only have lower metric values than scXDR but also exhibit more scattered distributions, as shown by the standard deviations in Table 2 and the box lengths in Supplementary Fig. S1b. In contrast, scXDR’s metric values are consistently high and stable in all four scenarios and the combined scenario. Table 2 and Supplementary Data 1 indicate that in the first scenario (different batches), scXDR’s performance in AUC and AUPR is inferior to scVI. This could be due to scVI’s advantage in batch effect correction, as the first scenario involves datasets from different batches of the same tumor and drug. However, in the subsequent three scenarios, scVI consistently underperforms compared to scXDR, showing that scVI’s advantage is limited, while scXDR adapts well in different scenarios. Supplementary Data 1 show that scVI loses its advantage in ACC and F1 Score, while scXDR outperforms scVI, achieving the highest values in three scenarios and the second-highest in the remaining one. Supplementary Fig. S1c further illustrates the improvement in scXDR’s metrics compared to the best performing methods for each metric, corresponding to the median lines in the box plots of Fig. 2a, b. It can be observed that scXDR’s AUC improvement is over 50% higher than the first category of methods, with improvements in AUPR and ACC close to 40%, indicating a significant advantage over traditional bulk to single-cell methods. Compared to the second category of methods, the improvements in all four metrics are also generally above 20%, which is still quite high.
The analysis above compares each scenario. Next, ROC and PR curves are plotted for four example datasets (bolded in Supplementary Table S2) from the four scenarios to demonstrate the cell level predictions for each dataset. The results for the classic single-cell dataset GSE11787218 (scenario 3) are shown in Fig. 3. This dataset has been frequently mentioned in previous studies10,13 and is highly representative. The other three datasets, GSE1348395 from scenario 1, GSE10839419 from scenario 2, and GSE147326 patient220 from scenario 4, are shown in Supplementary Figs. S2, S3, and S4, respectively. It can be observed that, compared to both the first and second categories of comparison models, scXDR’s area under the curves is significantly larger, clearly distinguishing it from other models. The specific metric values, including AUC and AUPR, for each experiment of scXDR and the comparison models can be found in Supplementary Data 2. The AUC values for these four datasets all exceed 0.85, with two datasets even surpassing 0.9. Additionally, three datasets have AUPR values greater than 0.9. Together, these results demonstrate the high performance of the model.

a ROC and PR curves of scXDR and the first category of comparison models on the representative dataset. b ROC and PR curves of scXDR and the second category of comparison models on the representative dataset. c Accuracy of scXDR versus the 7 models of the first category at the general cell group level. d Accuracy of scXDR versus the 7 models of the first category at the malignant cell group level. e Accuracy of scXDR versus the 8 models of the second category at the general cell group level. f Accuracy of scXDR versus the 8 models of the second category at the malignant cell group level. “Overall” indicates the combined scenario.
Superior performance at the cell group level compared to various methods
For each dataset, scRNA-seq data are annotated with cell types using conventional tools. At the general cell group level, scXDR is compared with two major categories of methods. The results are shown in Fig. 3c, e. Prediction accuracy of cell groups in each dataset is averaged by scenario, with brighter heatmap colors indicating higher mean accuracy. It is evident that, whether compared with the first category of seven methods or the second category of eight methods, scXDR achieves the highest accuracy at the general cell group level. Then, the dimensionality reduction results are presented again using the four representative datasets mentioned in Section 3.1 as examples. The cell dimensionality reduction plot of GSE117872 is shown in Fig. 4b (first row). The results for the other datasets are shown in the first rows of Supplementary Figs. S6b, S7b, and S8b. A consistent pattern can be observed, where different Louvain clusters correspond to distinct drug response groups and cell type groups, and the predicted labels align well with the true labels.

a The percentage decrease in metrics for each ablation model on the combined scenario, calculated by the median of each metric. b First row: UMAP dimensionality reduction of cells’ true response labels, Louvain clusters, cell types, and predicted response values. Second row: PCA dimensionality reduction of cells’ original features, embeddings after the scXDR message passing phase, embeddings after alignment in the scXDR transfer learning phase, and the scXDR score scatter plot. Third row: PCA dimensionality reduction plots of source and target domains for scDEAL (bulk) and SCAD (bulk), along with score scatter plots. Fourth row: PCA dimensionality reduction plots of source and target domains for scDEAL and SCAD, along with score scatter plots. Red text refers to the Euclidean distance between the mean points of the source and target domains. In the first row, the x and y axes of all plots represent UMAP1 and UMAP2. In rows 2 to 4, the x and y axes of the PCA plots represent Principal Components 1 and 2, and in the score scatter plots, the x-axis corresponds to the cell index and the y-axis corresponds to the score.
A malignant cell annotation tool is further applied for cell grouping. The heatmaps are shown in Fig. 3d, f. Compared with the first category of methods, scXDR outperforms all other methods in three scenarios and in the combined scenario, showing slightly lower performance only in one scenario. When compared with the second category of methods, scXDR achieves the highest accuracy in scenarios 2 and 4 as well as in the combined scenario, and ranks in the upper range in the remaining two scenarios. Therefore, at both the general cell group level and the malignant cell group level, scXDR demonstrates a clear advantage over the two major categories of comparison methods.
Overall, considering the above comparative experiments, scXDR achieves the highest mean AUC, AUPR, ACC, and F1 Score in three of the four scenarios, with very small standard deviations. This corresponds to higher and shorter boxes in the box plots, indicating that scXDR’s performance is both high and stable. In contrast, other models show fluctuating and unstable performance metrics. In the remaining scenario, scXDR also attains the second-highest or near-top results, demonstrating competitive performance. In the combined scenario, scXDR achieves the highest median values for all four metrics and reaches the highest accuracy at both the general and malignant cell group levels. These results indicate that scXDR outperforms other models not only at the individual cell level but also at the cell group level, and it demonstrates a better ability to adapt to different scenarios.
Contribution of model components and architecture to performance
The scXDR framework is built on heterogeneous network transfer learning, which requires the coordinated operation of multiple components. The components in the message passing phase are relatively standard, so the design and arrangement of components in the transfer learning phase are particularly important. To examine the rationality of this phase, ablation experiments are conducted. The complete model and four ablated variants, each missing a specific component, are tested for cross-dataset prediction in each scenario (Supplementary Fig. S5a), and the combined results across all scenarios are shown in Supplementary Fig. S5b. The results show that the complete model achieves the highest values across all four metrics, while the ablation models perform worse and display greater variability, indicating less stable distributions. Figure 4a illustrates the percentage decline in metrics for each ablation model relative to the complete model. Notably, the removal of the transfer learning component leads to the largest impact, with AUC, AUPR, and ACC each decreasing by about 30%, indicating that transfer learning significantly affects prediction outcomes. Based on the degree of decline, the structure alignment component appears less critical than the feature alignment component, however, their removal still weakens all four metrics. The elimination of the structure reconstruction component results in approximately a 20% decline across all four metrics, demonstrating that preserving network structural integrity during alignment is essential.
To further explain the model’s internal mechanism, the original cell features, embeddings after the message passing phase, and embeddings after alignment in the transfer learning phase are visualized using PCA. Additionally, the Euclidean distance between the mean points of the source and target domains is calculated to quantify their proximity. Since scDEAL and SCAD are also transfer learning–based models, comparing their embeddings with those of scXDR is fair. Therefore, their source and target domain embeddings are also reduced in dimension, visualized, and their inter-domain distances are computed. The interpretation is based on the four representative datasets mentioned in Section 3.1, each from one of the four scenarios. The results for the representative dataset GSE117872 are shown in Fig. 4b, while the results for the other datasets are presented in Supplementary Figs. S6, S7, and S8. Overall, these plots share several common patterns. The source and target domain cells are well separated in the original feature space, showing clear distinctions and large distances between them. After message passing, cells within each domain become more compact (as seen from the coordinate scale), and some cells across domains begin to move closer, reducing their distances. After the alignment operation, the source and target domain cells become even closer than in the previous two stages, with inter-domain distances reaching 0.51 or lower. This suggests that message passing encourages intra-domain cells to cluster together, likely those that are functionally similar or interacting within the heterogeneous network, while the alignment step in transfer learning brings cells from different domains closer, indicating that scXDR effectively integrates information across domains. The scXDR score plots also show that sensitive cells have higher scores, whereas resistant cells have lower scores. In contrast, the comparison models show much larger inter-domain distances, particularly SCAD (bulk) and SCAD, where distances are far greater than 1. In these models, the scores of sensitive and resistant cells are mixed and difficult to distinguish, resulting in lower AUC and AUPR values, as shown in Fig. 3. Although the inter-domain distances of scDEAL (bulk) and scDEAL are around 1, closer inspection reveals that these models mainly align the centers of different domains (especially in scenarios 3 and 4), rather than bringing the entire domains closer together as scXDR does. Consequently, their scores are also mixed, and their metric values are lower than those of scXDR.
From the perspective of different scenarios, the relationship between scXDR embeddings and metric values shows a clear pattern. For the four example datasets mentioned above, the ranking of inter-domain distances after alignment in the transfer learning phase is closely related to the ranking of AUC values. In other words, the greater the inter-domain difference, the more difficult the prediction, and the lower the metric values. Even so, the lowest AUC achieved by scXDR is still 0.85, which corresponds to the dataset from scenario 3. This dataset has the most complex background (involving different tumors and different drugs) yet still yields strong predictive performance, outperforming other models (Figs. 3, 4b). This observation indicates that smaller inter-domain differences lead to better prediction performance. Supplementary Table S3 further highlights this relationship by listing the inter-domain distances along with AUC and AUPR values for all datasets. Whether calculated by individual dataset or by scenario, the inter-domain distance after alignment is negatively correlated with both AUC and AUPR. This underscores the importance of the transfer learning components, particularly the alignment process. A well-designed transfer learning mechanism, especially an effective alignment strategy, contributes significantly to improving model performance and provides valuable insights for further enhancement.
Prediction of outcomes for cells under different treatment conditions
The first scenario is a cross-dataset prediction for the same tumor and the same drug from different batches. To demonstrate the model’s application capability, a new dataset with the same standard, GSE134841, is selected for prediction. This dataset is originally used to study drug holiday treatment for Erlotinib where one sample receives drug holiday treatment and the other does not5. The two conditions are treated as a whole. scXDR’s prediction distinguishes between the two conditions and produces outcomes for sensitivity and resistance that are consistent with the original literature. In terms of drug response, cells not receiving drug holiday treatment (No Erlotinib) obtain higher scores than cells that received drug holiday treatment (Erlotinib) as shown in Fig. 5a. The proportion of sensitive cells in the No Erlotinib group is also higher than that in the Erlotinib group as seen in Fig. 5c. The determination of sensitive cells is based on whether the score exceeds the optimal threshold for that scenario. The resistance markers provided in the original paper also show significant differential expression between the two conditions as shown in Fig. 5b. These markers are expressed at lower levels in No Erlotinib cells and at higher levels in Erlotinib cells, which is consistent with the original literature. The reason for these results is that cells receiving drug holiday treatment are first deprived of the drug for a period and then continuously treated for several days. As a result, most surviving cells are resistant. They show high expression of resistance markers while their drug scores are not high. In contrast, cells not receiving drug holiday treatment grow in a drug-free environment after drug withdrawal and tend to be sensitive. Therefore, their drug scores are high and the expression of resistance markers is low.

a Drug response scores of cells under two treatment conditions. b Expression of resistance markers. c Cells are classified into sensitive (score is 1) and resistant (score is 0) groups based on a threshold. The vertical axis represents the percentage of sensitive and resistant cells. d UMAP dimensionality reduction plot of the cells. First row: Treatment conditions, cell cycle, positions of No Erlotinib cells, and positions of Erlotinib cells. Second row: Response scores of No Erlotinib cells, response scores of Erlotinib cells, expression of drug resistance markers from the original literature in No Erlotinib cells, and expression in Erlotinib cells. “Markes” refers to the drug resistance markers from the original literature. Third row: Expression of MT-ND6 or TUBA1B in No Erlotinib cells, and expression in Erlotinib cells. MT-ND6 and TUBA1B are the markers newly identified in this study. e, f The correlation between marker expression and drug response scores. g The top 10 enriched pathways.
For visualization at the single-cell level, the first row of Fig. 5d shows the basic status of cells in the dataset. Cells under the two conditions are mixed together and are difficult to distinguish in the dimensionality reduction of gene expression. The cell cycle is arranged from G1 on the left to S in the middle and to G2/M on the right, which is consistent with reality and shows a natural transition5. The second row visualizes the drug scores and the expression of resistance markers from the original literature in cells under the two conditions. The levels of drug scores and resistance marker expression are inversely related as shown in Fig. 5a, b. The drug scores exhibit some ability to distinguish cells under different conditions. The color differences for the original markers in the figure are not pronounced enough. This case identifies two markers, MT-ND6 and TUBA1B, with more significant differences (based on a t-test p value ranking) between the two conditions. Their expression in cells shows opposite trends as seen in the third row of Fig. 5d. In addition to being more distinct than the original markers, the expression of these two markers is highly correlated with drug scores as shown in Fig. 5e, f. Their survival analyses also show significant differences in survival rates between high and low expression groups as shown in Supplementary Figs. S9a and S9b. Specifically, as MT-ND6 expression increases, drug response increases, making cells more susceptible to the drug and resulting in higher patient survival rates. In contrast, as TUBA1B expression increases, drug response decreases, leading to lower susceptibility and lower patient survival rates. Further, KEGG enrichment analysis is performed on the top 100 differentially expressed genes sorted by p-value. The top 10 enriched pathways ranked based on p-value are shown in Fig. 5g, where larger nodes and thicker lines indicate greater enrichment. Pathways closely related to tumors include Transcriptional misregulation in cancer, ECM-receptor interaction, and MicroRNAs in cancer.
Drug screening and drug response markers
The tumor type in the second scenario is melanoma. Comparative experiments show that scXDR performs excellently in predicting the response of melanoma to different drug treatments. Although the samples are from the same tumor type, individual differences may exist, so screening for the optimal drug in a new melanoma dataset is necessary. Drugs from GDSC are processed to ensure they have relevant features and targets and do not hinder constructing the heterogeneous network, resulting in a total of 137 unique drugs. For the melanoma sample, which is of low metastatic potential21, a drug score is calculated for each cell for these drugs. The mean and median of all cell scores are computed, and the drugs are ranked based on the sum of the mean and median values (Supplementary Table S4). Next, databases and literature are searched to validate the drug ranking. Specifically, information from professional databases such as DrugBank, HMDB, and DGIdb regarding drugs used to treat melanoma is regarded as database evidence22. Literature evidence is collected from PubMed using the search query “drug name AND melanoma AND treatment”. Higher-ranked drugs show significantly more literature evidence than lower-ranked ones. Among the top five drugs, up to 80% have literature support (Fig. 6a). For the top half of the ranked drugs (68 in total), 22.1% (15/68) have evidence from professional databases, and 44.1% (30/68) have literature evidence outside of databases (Fig. 6d). Interestingly, drugs with database evidence also tend to have abundant literature evidence. Overall, approximately two-thirds (45/68) of these drugs have supporting literature evidence. Detailed results are in Supplementary Table S5.

a Percentage of drugs with literature evidence among the top-ranked and bottom-ranked drugs. b Density distributions of scores for the top 5 and bottom 5 drugs. c Proportion of cells affected by the top 5 and bottom 5 drugs among all cells. d Percentage of drugs with literature or database evidence among the top half of drugs. e Information on cell cycle, MITF pathway, AXL pathway, and drug scores. f Expression levels of RPL37A in high-scoring and low-scoring cells for the top 5 drugs. g The top 10 enriched pathways. h Survival analysis of RPL37A.
A score distribution plot for the top 5 and bottom 5 drugs is shown in Fig. 6b. According to scenario 2’s optimal threshold, cells are divided into affected (scores above the threshold) and unaffected (scores below the threshold), as seen in Fig. 6c. The scores of the top-ranked drugs are overall higher than those of the lower-ranked drugs, and the percentage of cells affected by the top-ranked drugs is around 80%, far exceeding that of the lower-ranked drugs, which is only about 35%. The top-ranked drug, Fulvestrant, is examined next. The general cell status and score distribution for Fulvestrant are shown in Fig. 6e. Many cells are in the G2/M phase, reflecting high proliferative activity. In addition, high expression of MITF (a marker of proliferation) and low expression of AXL (a marker of invasiveness) indicate strong proliferation but weak invasiveness21, consistent with the low metastatic potential of the sample. Therefore, it is meaningful to focus on targeting the primary tumor for drug screening. Fulvestrant is a selective estrogen receptor down-regulator23, and scXDR predicts that most cells are sensitive to it. Previous studies have already shown that Fulvestrant monotherapy can inhibit melanoma growth by blocking ERα signaling and enhancing tumor immunity24, which supports the drug prediction results.
Moreover, gene expression differential analysis is performed on the high-score cell group (sensitive, scores above the threshold) and the low-score cell group (resistant, scores below the threshold) for the top-ranked drugs. A t-test is used to compute and identify the most significantly differentially expressed genes between the two groups. The comparison reveals that in the high-score and low-score groups for the top 5 drugs, a greater number of genes starting with “RPL” rank high by p-value and exhibit differential expression, accounting for as much as 50% of the top 10 genes (Supplementary Table S6). These genes typically encode ribosomal protein large subunits25, and their expression levels are closely associated with cancer cell proliferation and growth. For example, RPL37A is expressed at low levels in high-score cells and at high levels in low-score cells (Fig. 6f). Survival analysis (Fig. 6h) shows that high expression of RPL37A is associated with lower survival rates, while low expression is linked to higher survival rates. This indicates that cells with low RPL37A expression are more susceptible to drug effects and that patients may experience better survival outcomes, suggesting that RPL37A holds potential as a drug response biomarker. KEGG enrichment analysis is conducted on the top 100 differentially expressed genes, and the top 10 enriched pathways ranked based on p-value are shown in Fig. 6g, with node size and line thickness representing the degree of enrichment. Pathways such as “Mismatch repair,” “DNA replication,” and “Ribosome” are consistent with the cell phase findings, high proliferative activity, and the functions of the RPL gene family26.
Pan-cancer level drug clusters and tumor clusters
The first two case studies examine the same tumor with the same drug and the same tumor with different drugs. Evaluating the model on more tumors and drugs can enable pan-cancer analyses of similarities and differences among drugs and among tumors, revealing general therapeutic patterns and special cases. Since the Kinker et al. dataset27 includes only solid tumors, after ensuring that scXDR performs well on the solid tumor datasets in scenario 3, it is used to predict the response of 22 cancer types to 137 drugs. For each drug, the mean and median scores of all cells within each cancer type are calculated, and their average, obtained by dividing the sum of the mean and median by two, is taken as the final score for that cancer type with respect to the drug. Then, a heatmap based on the score matrix for all drugs across all cancers is generated, as shown in Fig. 7a, with the positions of drugs and tumors arranged according to Ward hierarchical clustering. It is evident that some drugs exhibit similar response profiles. Drugs on the left generally show lower response scores (darker color), while those on the right mostly display higher scores (brighter color). Regarding differences among drugs, a notable example is Cisplatin, whose color appears much brighter than that of other drugs, with scores mostly around 0.8. This aligns with the fact that Cisplatin is a broad-spectrum anticancer drug that demonstrates good efficacy against various tumors28. Some tumors also show similar response profiles, suggesting that during treatment they can be used as references for each other. The drug clustering tree and tumor clustering tree are pruned at a distance of 1, and the resulting clusters are presented in Fig. 7b, c and Supplementary Tables S7–S8. The drugs are divided into 7 clusters and the tumors into 3 clusters, with each cluster occupying distinct regions and exhibiting similar responses within the cluster. Notably, drug cluster 7 is isolated in the bottom right corner as a single point, precisely the aforementioned Cisplatin, distinguishing it from other drugs and drug clusters. In comparison, the tumor clusters are more dispersed, reflecting the significant heterogeneity among different tumors.

a Scores for each drug in each tumor, where each square represents the sum of the mean and median scores of all cells in that tumor. b PCA dimensionality reduction plot of 7 drug clusters. c PCA dimensionality reduction plot of 3 tumor clusters. d The combined effect of drug clusters on tumor clusters. e Spearman correlation between the p53sen value and drug response scores.
Next, the scores are grouped by tumor clusters and drug clusters and visualized as a network (Fig. 7d), where thicker lines represent higher scores. Both similarities and differences can be observed among drug clusters and tumor clusters. In terms of similarities, for example, drug clusters 7 and 6 both show consistently high responses across all tumor clusters, while tumor clusters 2 and 3 both show consistently low responses to drug cluster 1. Regarding differences, the same drug cluster shows varying responses across different tumor clusters. For instance, drug cluster 2 is most effective for tumor cluster 1, with a score of 0.35 compared to 0.27 and 0.23 for the other two tumor clusters. Crizotinib belongs to drug cluster 2, and neuroblastoma belongs to tumor cluster 1. Neuroblastoma is a common pediatric solid tumor, and clinical studies are exploring the use of Crizotinib for its treatment29. The HER family is often highly expressed in neuroblastoma and is associated with tumor proliferation and poor prognosis, which may explain why drugs in drug cluster 2, such as Afatinib and Lapatinib, show higher response values30. Another difference is that the same tumor cluster can respond differently to various drug clusters. For example, tumor cluster 2 is most sensitive to drug cluster 6 among the six drug clusters (excluding the Cisplatin cluster as a special case), with a score of 0.54, which is higher than the 0.2–0.4 range observed for the other drug clusters. Tamoxifen and Fulvestrant belong to drug cluster 6, and breast cancer is in tumor cluster 2. These two hormone drugs are clinically important agents for treating hormone receptor positive breast cancer31. In addition, Dabrafenib in drug cluster 6 shows high sensitivity to lung cancer in tumor cluster 2, consistent with its standard clinical use for BRAF-mutated lung cancer32. The remaining drug in drug cluster 6, Camptothecin, is therefore inferred to have potential therapeutic value for cancers such as breast and lung cancer, and studies have shown that its derivatives indeed exhibit efficacy against these cancers33,34.
To further understand drug mechanisms and identify potential therapeutic drugs, the Spearman correlation is calculated at the drug cluster and tumor cluster levels between the p53-dependent senescence (p53sen) values provided by the dataset’s original authors27 and the drug scores predicted by this model (Fig. 7e). p53 plays an important role in cell senescence, a protective mechanism that prevents cells from proliferating to mitigate cancer development35. Taking tumor cluster 1 and drug cluster 4, which show the highest correlation, as an example, drug cluster 4 includes the representative drug Nutlin. Nutlin stabilizes and activates p53 by inhibiting MDM2 (a negative regulator of p53), thereby promoting cell senescence36. Therefore, the positive correlation between p53sen values and drug response scores is biologically reasonable and supports the model’s predictions. The drugs within the same cluster share high similarity, suggesting that other drugs in drug cluster 4 may also affect p53. For example, literature shows that Pictilisib is a PI3K inhibitor37. Since the PI3K-AKT-mTOR pathway promotes p53 degradation through MDM2 phosphorylation, Pictilisib inhibits this pathway to reduce MDM2 function, stabilizing and activating p53 in a manner similar to Nutlin, and inducing cell cycle arrest and apoptosis in tumor cells38. This demonstrates the model’s ability to identify potential drugs based on pan-cancer level drug and tumor clustering.
Assessment of approved drugs and potential drug combinations for patient cells
In the fourth case, data come from breast cancer patient cells that are estrogen receptor positive20. Some cells receive drug treatment while others do not. Leo et al39. collect FDA approved drugs for breast cancer dating back to 1949 and divide drugs for biomarker based treatment into three categories: hormone receptor positive, HER2 positive, and gBRCA mutated. These three categories of drugs are applicable for biomarker-based treatment, making them suitable for a fair comparison. In this case, scXDR predicts the sensitivity of cells that have not received drug treatment from two patients to 11 small molecule drugs belonging to those three categories. For each drug, both the mean and median scores of all cells are calculated to generate a heatmap (Fig. 8a). Next, using the optimal threshold from scenario 4 to classify cells as affected or unaffected by drug treatment, the percentage of affected cells is calculated and the drugs are ranked (Fig. 8b). For both patients, the top three drugs belong to the first category. The percentage of affected cells is close to or exceeds 40 percent. The drugs in the other categories rank in the middle and lower positions because they do not target the estrogen receptor positive condition. Therefore, their lower ranking is justified.

a Scores of 3 types of breast cancer drugs on cells from 2 patients. Orange, blue, and green represent first, second, and third class drugs, respectively. b Ranking of the 3 types of drugs based on the percentage of affected cells. c, d For the first drug combination, statistical analysis of the expression of targets of the added drug in cells after the original drug treatment is performed. Ratios are calculated based on the number of resistant cells to sensitive cells expressing these targets and on the expression levels in resistant versus sensitive cells.
Subsequently, scXDR predicts the sensitivity of patient cells that have received drug treatment to all FDA approved breast cancer drugs with the aim of forming an optimal drug combination based on existing drugs. The ideal result is to add a new drug that primarily targets the resistant cells that have already been treated. Only dual drug combinations are considered. In other words, the new drug should affect cells that are not impacted by the original drug. The cells from the two patients are treated with either Carboplatin or Doxorubicin, resulting in four conditions. For all four conditions, responses for a total of 24 drugs are predicted (not limited to those targeting biomarkers). New drugs are ranked by the sum of the mean and median scores for resistant cells and the top ranked drug is paired with the original drug to form the drug combination (Supplementary Table S9). A search of the PubMed and ClinicalTrials databases40 for evidence on these breast cancer drug combinations shows that in all four conditions the predicted combinations have support in the literature and that three combinations receive support from clinical trials.
For example, in the first condition, patient 1’s cells are pretreated with Carboplatin (Drug1) and the resistant cells show the highest sensitivity to Doxorubicin (Drug2). The expression of Drug2 targets in Carboplatin sensitive cells (S) and resistant cells (R) is analyzed statistically using target information from the DrugBank database (Fig. 8c, d). The analysis calculates the number of cells with nonzero expression of each target for Drug2 and then computes the ratio of the number of cells in R to the number in S. Up to 70 percent of Drug2 targets show a ratio greater than 1. In addition, the average expression level for each target in R is divided by that in S and 70 percent of the targets again show a ratio greater than 1. These results indicate that the targets of Drug2 are more strongly expressed in Drug1 resistant cells in terms of both the number of expressing cells and the level of expression. This suggests that Drug2 may preferentially affect Drug1 resistant cells rather than sensitive cells, which supports the rationale behind the drug combination predicted by scXDR. From the perspective of the drug mechanism of action, although Carboplatin and Doxorubicin do not directly share common targeted proteins, they collaborate to disrupt functional pathways related to DNA integrity maintenance. Carboplatin targets DNA itself, causing DNA cross-links, while Doxorubicin primarily targets topoisomerase II, inducing DNA double-strand breaks. Together, they disrupt DNA structure and function through different mechanisms, producing a strong synergistic anti-cancer effect41. Thus, their combination is both mechanistically reasonable and synergistic, reflecting the reliability of the model prediction.
Discussion
Drug response prediction tasks across single-cell datasets are challenging because different single-cell datasets may have biological differences, technical differences, or other types of differences and several differences may coexist42. To facilitate research, various single-cell datasets are divided into different scenarios and scXDR’s predictions are conducted by scenario which reduces differences among datasets to some extent especially for scenarios 1, 2, and 4. It is important to note that from scenario 1 to scenario 3 the represented situations become increasingly complex, ranging from the same tumor with the same drug to the same tumor with different drugs and finally to different tumors with different drugs. Scenario 4 involves the same tumor with different patients including both the same drugs and different drugs. Undoubtedly, scenario 3 is the most complex whereas the datasets in the other three scenarios share some similarity. This is reflected in the prediction results as the evaluation metrics in scenario 3 are always lower than those in the other scenarios whether at the individual cell level or at the cell group level. From the perspective of embedding visualization and inter-domain distance, examining different scenarios and datasets shows that when a dataset within a scenario has a large distance between the source and target domains, both AUC and AUPR are lower. In other words, greater domain discrepancy tends to weaken the model’s predictive performance, which explains the differences in metrics across scenarios. Nevertheless, even in the scenario with the largest domain discrepancy (scenario 3), scXDR still effectively reduces the inter-domain distance (Fig. 4b) and achieves higher metrics than the current advanced models.
Transfer learning enables cross-domain prediction13, and this study also demonstrates that it is applicable to single-cell datasets with both large and small inter-domain differences. The input of scXDR is a heterogeneous network that includes feature information and association information for drugs, targets and cells. This makes fuller use of available data compared with previous single-cell drug response prediction models that use only gene expression information. Previous studies have also shown that prediction methods based on heterogeneous networks can effectively utilize heterogeneity information to predict drug responses43,44. It is common for existing prediction models to use bulk-level RNA-seq data and drug response information to predict drug response at the single-cell level8,14, but the scXDR proposed in this study achieves cross-dataset drug response prediction at the single-cell level. The basic idea of scXDR is heterogeneous network transfer learning and the core principle is to align the heterogeneous networks of the source domain and target domain in terms of features and structure while preserving the integrity of each network. During alignment both Euclidean distance and cosine similarity are considered. This approach brings the heterogeneous networks of the two domains into the same space with close distances, similar directions, and intact structures. Since the heterogeneous network contains multiple types of nodes and edges, training during the message passing phase and the transfer learning phase is performed for each node type, each edge type, and each meta-path. The source domain and target domain share components such as the HGNN, autoencoder and predictor which means that operations on the heterogeneous networks in the source and target domains are synchronized with no sequence of operations. Training during the prediction phase targets the source domain. After several iterations, prediction and evaluation are performed simultaneously on both the source and target domains. These designs ensure that the drug response labels of the target domain are used only at evaluation time and remain completely hidden during training, while training considers both the source and target domains and achieves high prediction performance in both.
At both the individual cell and cell group levels, the results show that scXDR outperforms not only prediction models that use bulk data to predict single-cell responses but also those that use single-cell data to predict single-cell responses. In the vast majority of prediction tasks across the four scenarios, scXDR achieves the highest values in its four evaluation metrics and the highest cell group accuracy. Because scVI and scDEAL have advantages in specific scenarios or particular datasets, scXDR’s metrics are sometimes slightly inferior to or comparable with theirs. However, no other model maintains high performance in all scenarios as scXDR does, and the metric values of the other models are generally more dispersed, with frequent fluctuations between high and low values, whereas scXDR’s performance consistently remains in the high range. Many models overlook evaluation at the cell group level11,14. Sometimes a model may perform well in certain cell groups while performing poorly in others. In this study, cell groups are more finely divided into general cell groups and malignant cell groups, which represent two different cell group annotation methods. Except for the cell group level in scenario 3, where the accuracy is slightly lower, scXDR achieves the generally highest average accuracy in the other scenarios and overall. In scenario 3, the most complex scenario involving different tumors and different drugs, with tumors including both solid and hematological types, no model achieves very high values, yet scXDR’s accuracies of 75% and 60% remain noteworthy. Ablation experiments and interpretability experiments demonstrate that the components of transfer learning have a significant impact on model performance. In particular, the dimensionality reduction plots for interpretability reveal that message passing and transfer learning each emphasize different functions. Additionally, the comparison of embeddings between other models and scXDR can be quantified using inter-domain distance. The transfer learning component of scXDR clearly has an advantage, as it brings the source and target domains closer together, resulting in a smaller inter-domain distance. These experiments demonstrate that the scADR model design is well structured, achieving high performance in cross-domain prediction.
In this study, case studies are conducted for each of the four scenarios to demonstrate that scXDR can adapt to different situations and handle various prediction tasks. The non-small cell lung cancer PC9 cell line is first cultured in an environment containing Erlotinib for 11 days and then transferred to an environment without Erlotinib. If Erlotinib is added again 6 days later, it is considered treatment with a drug holiday. If it is not added, it is considered treatment without a drug holiday5. In cancer treatment, a “drug holiday” refers to the planned or intermittent cessation of anticancer drugs, which may delay resistance and restore drug sensitivity45. The original paper5 for the first case study’s drug holiday treatment dataset mainly explores drug tolerance. The authors point out that the plasticity of drug tolerance is related to the reversibility of the expression of drug-tolerant markers, which are in fact drug resistance markers. Although drug tolerance and drug resistance are conceptually different, their effects are the same in that the cellular response to drugs is weak. There is no difference in judging drug response scores and sensitivity, so this case is still explained using resistance in place of tolerance. Tumor cells treated after a drug holiday show high expression of drug resistance markers and low drug response scores, while untreated tumor cells show the opposite. The main reason is that the drug holiday allows tumor cells to regain some sensitivity, and subsequent treatment kills the sensitive cells so that most surviving cells are resistant. As a result, the predicted drug response based on Erlotinib scRNA-seq data is low, which is consistent with the results of Aissa et al. The newly predicted markers, MT-ND6 and TUBA1B, exhibit more obvious differential expression between the two treatment conditions, with fold changes of 2.18 and 0.13, respectively, and they also show relatively high Spearman correlation coefficients with the drug response scores. MT-ND6 belongs to mitochondrial DNA, and studies show that alterations in mitochondrial translation affect tumor cell sensitivity to drugs46. Other research indicates that TUBA1B is significantly upregulated in lung cancer and that its expression may strongly influence patients’ treatment responses47. These findings suggest that the two identified markers are related to drug response and are worthy of further investigation.
The drug screening case uses an unlabeled single-cell dataset as the target domain, which means predicting the response of untreated melanoma samples to over 100 drugs. The drugs ranked at the top show higher scores on cells, affect a greater number of cells, and have more supporting literature evidence compared to those ranked lower. Fulvestrant is mainly used for breast cancer at present, but several studies show that Fulvestrant, whether used alone or in combination with other drugs, can inhibit the growth of melanoma cells24,48. This makes its high ranking understandable. Genes beginning with RPL, represented by RPL37A, are significantly differentially expressed between sensitive and resistant cells. When these genes express at low levels, the drug response is higher and the survival probability is greater. Studies show that RPL37A can serve as a marker for pathological complete response and survival in cancer patients25,. Targeting ribosomal proteins is effective in the treatment of melanoma49. These studies prove the potential of RPL genes as markers for drug response. After applying scXDR to a single tumor, the next step is to predict the response of multiple tumors to multiple drugs at a pan-cancer level. Both drug clusters and tumor clusters are divided based on response scores. The responses are similar within each cluster, while the similarity between clusters is low. In the dimensionality reduction plots (Fig. 7b, c), the greater the distance between clusters, the less similar they are. This pattern implies that once an instance is studied, similar drugs or similar tumors can be referenced for each other. For example, Nutlin affects p53, and other drugs in the same cluster may also have some influence on p53. Pictilisib, a PI3K inhibitor in the same cluster, is shown by studies to possibly affect the function or stability of p53 indirectly38. The predicted responses of breast cancer patient cells to FDA approved drugs match the conditions of these samples. Hormone receptor-positive drugs rank at the top among the drugs. Further predicted drug combinations are designed to target resistant cells. In these combinations, cells that are not affected by the first drug are affected by the second drug. Four pairs of drug combinations predicted for two patients are supported by literature or clinical trials. The targets of the second drugs in these combinations are mostly highly expressed in resistant cells. These findings further demonstrate that resistant cells are more likely to be affected and provide additional support for the prediction results.
Conclusion
In summary, by converting single-cell datasets into heterogeneous networks and performing transfer learning, scXDR achieves drug response prediction across single-cell datasets. In each scenario and in the combined scenario, it outperforms advanced methods that predict drug response from bulk data to single-cell data and from single-cell data to single-cell data, whether evaluated at the individual cell level or the cell cluster level. Every phase and component of scXDR is designed to serve heterogeneous networks, and the transfer learning component is the core part. Case studies for each scenario achieve the prediction of cell response outcomes under different treatment conditions, drug screening and identification of related markers, pan-cancer studies of multi-drug and multi-tumor responses, and evaluations of single drugs and drug combinations on patient cells. In conclusion, the methodological innovations and practical applications of scXDR provide guidance for a new model of single-cell drug response prediction and help advance drug treatment at the cell level.
Methods
Data collection and processing
This study focuses on single-cell level datasets. The scRNA-seq data are collected from the Gene Expression Omnibus (GEO) database, and the drug response labels are either provided directly in GEO or determined and provided based on the original literature. Due to differences in disease backgrounds and treatment conditions among various single-cell datasets, the available datasets are categorized into 4 research scenarios (see Supplementary Table S1 and Fig. 1). Specifically, a total of 12 scRNA-seq datasets are divided into 4 scenarios: different batches (same tumor, same drug), different drugs (same tumor, different drugs, including both single drugs and drug combinations), different tumors (including both solid and hematological tumors with different drugs), and different patients (drugs may be the same or different). Overall, this setup encompasses 6 tumor types and 10 drugs, and 20 cross-dataset drug response prediction tasks are conducted.
Each dataset is constructed as a heterogeneous network that includes three types of nodes: drugs, cells, and targets. The features of drugs are represented using Molecular Access System fingerprints50, the features of genes are represented using Composition, Transition, and Distribution fingerprints51, and the features of cells are the expression values of 5000 highly variable genes. The scRNA-seq data are processed using Scanpy, which considers key parameters such as minimum gene count, minimum cell count, total counts, and mitochondrial percentage, along with log transformation and normalization, all based on a previous study’s processing method13. Cell-target interactions consist of each cell and the genes with nonzero expression in that cell. For any two cells, Pearson correlation is computed based on their expression values, and cell-cell interactions are constructed by retaining pairs whose absolute correlation exceeds the median. Drug-target interactions are obtained by querying the Comparative Toxicogenomics Database and PubChem Database, and target-target interactions are filtered from the Human Reference Interactome dataset52. Since 11 of the 12 datasets include only one drug, drug-drug interactions are not considered. Finally, the effect of drugs on cells is treated as the labels.
Model construction
The model is end-to-end, with the input being the heterogeneous networks from the source and target domains, and the output being the scores of drug-cell interactions for both domains. Here, the source and target domains refer to different datasets within each scenario. The model is divided into three phases: the message passing phase, the transfer learning phase, and the prediction phase, as illustrated in Fig. 1.
Message passing phase. In this phase, the heterogeneous networks from the source and target domains execute the same operations. A heterogeneous network is represented as \(G=({{\mathcal{V}}},{{\mathcal{E}}},{{\mathcal{T}}},{{\mathcal{R}}})\) (temporarily without domain identifiers). \({{\mathcal{T}}}\) is the set of node types and consists of 3 types. \({{\mathcal{R}}}\) is the set of edge types, and as mentioned, there are 5 types of edges. Note that drug-cell edges do not participate in the message passing phase and the transfer learning phase, and in the prediction phase the labels for drug-cell edges in the target domain are not disclosed. The remaining 4 types of edges consider reverse edges, which results in a total of 8 edge types that execute message passing and transfer learning. \({{\mathcal{V}}}\) and \({{\mathcal{E}}}\) denote the set of all nodes and all edges respectively. For a node \(v\) (type is \(\tau\), \(\tau\) ∈ \({{\mathcal{T}}}\)), 2 layers of message passing are performed using Eqs. (1) and (2).
For each relation \(r\in {{\mathcal{R}}}\), node \(v\) has a neighborhood set \({{{\mathcal{N}}}}_{r}(v)\), where each node \(u\) in the neighborhood set has the original feature \({x}_{u}^{(\tau )}\). The features of the neighbor nodes (including the node itself) for each relation are aggregated, and the sum of the features across all relations is passed through a nonlinear transformation. In this study, \(\sigma\) represents the activation function, specifically ReLU, as used throughout. The first layer representation \({h}_{v}^{(\tau ,1)}\) is then aggregated to obtain the second layer representation \({h}_{v}^{(\tau ,2)}\).
Transfer learning phase. This phase comprises an autoencoder component, an alignment component, and a structure reconstruction component. Its purpose is to perform cross-domain feature alignment and structure alignment on the heterogeneous network while preserving the integrity of the network structure within each domain.
The autoencoder component includes node encoding, edge encoding, and meta-path encoding. The steps are identical for the two domains, so no distinction is made. For each type of node, encoding is performed followed by decoding. The input is the representation \({h}_{v}^{(\tau ,2)}\) obtained from the message passing phase. The output of the intermediate layer (the encoded embedding) is \({z}_{v}^{(\tau )}\), and the output of the decoding layer is \({\hat{h}}_{v}^{(\tau ,2)}\). Here, \(W\) and \(b\) denote the parameters of the encoding layer, and \(W^{\prime}\) and \(b^{\prime}\) denote the parameters of the decoding layer. Equation (5) is used to compute the mean of the squared L2 norm, that is, the Mean Squared Error (MSE) is used to calculate the loss of the autoencoder. \({{{\mathcal{V}}}}^{(\tau )}\) represents the set of nodes of type \(\tau\), and \(|{{\mathcal{V}}}^{(\tau )}|\) represents the number of nodes of type \(\tau\).
The feature of an edge \({e}_{{uv}}^{(r)}\) is formed by concatenating the features of the two nodes at its ends. Here, \(u\) (of type \(\alpha\)) and \(v\) (of type \(\beta\)) represent the two nodes, and \(\alpha\) and \(\beta\) may be the same. Similar to the previous steps, the concatenated features are first encoded and then decoded. The loss is calculated using MSE. \({{{\mathcal{E}}}}^{(r)}\) represents the set of edges of type \(r\). Note that at this stage, \({{\mathcal{R}}}\) does not include drug-cell edges, and as mentioned earlier, there are a total of 8 edge types.
Meta-paths can be regarded as substructures of the network53. The input to the meta-path encoder consists of numerous two-hop meta-path samples. A meta-path sample’s feature is denoted as \({p}^{(m)}\) and is obtained by concatenating the features of the starting node \(u\) (of type \(\gamma\)), the intermediate node \(v\) (of type \(\delta\)), and the ending node \(w\) (of type \(\epsilon\)). The types \(\gamma\), \(\delta\), and \(\epsilon\) can have at most two identical types. The specific meta-path types are provided in Supplementary Table S10. \({{\mathcal{M}}}\) represents the set of all meta-path types, \({{{\mathcal{P}}}}^{(m)}\) denotes the set of meta-path samples of type \(m\), and \({{|}}{{{\mathcal{P}}}}^{(m)}{{|}}\) indicates the number of meta-path samples of type \(m\).
The alignment component considers both feature alignment and structure alignment. Specifically, the alignment of node encoded features and edge encoded features falls under feature alignment, while the alignment of meta-path encoded features is regarded as structure alignment, or more precisely, substructure alignment. For each type of node, edge, and meta-path, the encoded embedding \(z\) is used to compute a density \(D\) using a kernel density estimation method (Eq. 14)54. High-density points from both domains are then used to calculate the Euclidean distance and cosine similarity to obtain the loss (Eqs.( 15)–(17)). Here, \(K\) denotes the Gaussian kernel, \(N\) is the number of samples, \(h\) is the bandwidth parameter, and \(\left\langle \cdot \right\rangle\) represents the dot product. The symbols \(s\) and \(t\) refer to the source domain and target domain, respectively, while \({N}_{\tau }\), \({N}_{r}\), and \({N}_{m}\) denote the number of high-density samples for specific types of nodes, edges, and meta-paths. Each alignment loss is composed of two parts. The first part is MSE, which represents the Euclidean distance. The second part is the complement of the cosine similarity (1 minus the cosine similarity). The combination of these two components ensures that, after training, the representations between the source and target domains are not only close in distance but also as aligned as possible in direction.
The structure reconstruction component predicts the existence of edges in the heterogeneous network, ensuring that the network structure is maintained during alignment. The operations are identical for both domains and are performed separately for each type of edge. The node-encoded representations are dot-multiplied to predict the edge score (Eq.( 18)). Edges that exist in the network are treated as positive edges with a score \({s}_{p}\), while edges that do not exist are treated as negative edges with a score \({s}_{n}\). A margin loss function (Eq.( 19)) is used to compute the loss, encouraging higher scores for positive edges and lower scores for negative edges. Here, \(r\in {{\mathcal{R}}}\), and drug-cell edges are still not considered.
Prediction phase. In this phase, which is specifically focused on drug-cell edges in the source domain, the drug embeddings and cell embeddings are concatenated and fed into a Multilayer Perceptron (MLP)55 to predict scores. A 5-fold cross-validation is used for testing. The predicted score \({y}_{{uv}}\) and the true label \({Y}_{{uv}}\), where \({Y}_{{uv}}\in \left\{0,1\right\}\), are used to compute the loss via a binary cross-entropy loss function. \({{{\mathcal{E}}}}_{{pred}}^{s}\) represents the set of drug-cell edges in the source domain. Up to this point, the drug-cell information in the target domain remains unknown.
Through the aforementioned steps, a total of 8 losses are established, and the overall loss is the weighted sum of these 8 losses. The first line of Eq. (23) represents the losses of the three autoencoders, which are summed over each node type, each edge type, and each meta-path type. The second line corresponds to the three alignment losses, also computed and summed by type. The third line includes the structure reconstruction loss and the source domain prediction loss. Note that the first 7 losses apply to both domains, whereas the 8th loss is applied only to the source domain. The total loss \(L\) is backpropagated.
After training through the message passing phase, transfer learning phase, and prediction phase, the predictor trained on the source domain is used to predict the scores for drug-cell pairs in the target domain, and these predictions are then compared with the true labels.
Experiment setting
In each scenario, the heterogeneous network of each dataset can serve as either the source domain or the target domain. Therefore, the prediction tasks need to consider both the cases where the network is used as a source domain and as a target domain. For the sake of categorization and targeted expansion, this study does not include cross-scenario predictions. Instead, it focuses on transfer and prediction across datasets within the same scenario. There are a total of 20 single-cell drug response prediction tasks in four scenarios, as shown in Supplementary Table S2. The hyperparameters and other settings are shown in Supplementary Table S11. Evaluation metrics include the Area Under the ROC Curve (AUC), the Area Under the Precision-Recall Curve (AUPR), Accuracy (ACC), and F1 Score. For the calculation of ACC and F1 Score, the optimal threshold is determined using the Youden index56. The scores are then binarized before comparing them with the ground truth labels. To ensure a fair comparison, the optimal thresholds of the comparison models are also determined based on the Youden index before calculating ACC and F1 Score.
To comprehensively evaluate the performance of scXDR, this study compares scXDR with different categories of single-cell drug response prediction methods. The comparisons include methods that transfer from bulk data to single-cell data (first category) and methods that transfer from single-cell data to single-cell data (second category). The evaluation is conducted not only at the individual cell level but also at the cell group level, with the latter including both general cell group level and malignant cell group level.
Comparison experiments at the individual cell level
Methods that transfer bulk data to single-cell data are used for comparison:
-
scDEAL (bulk)13 represents published transfer learning methods. In this approach, the source domain consists of cell line data from GDSC and CCLE57 while the target domain is single-cell data.
-
SCAD (bulk)14 represents another class of published transfer learning methods. The data and model architecture are similar to scDEAL, but in this approach, the common genes between the source domain and the target domain are used as features, and the evaluation is conducted on the split target domain.
-
CaDRReS-Sc (bulk)10 represents published machine learning methods. The training set consists of cell line data from GDSC and the test set is single-cell data.
-
DREEP (bulk)12 represents published marker-based methods. The cell line data come from GDSC, PRISM26, and CTRP258 and the method predicts drug response at the single-cell level.
-
MLP (bulk) represents conventional deep learning methods. The training set consists of cell line data from GDSC and the test set is single-cell data. Features only consider common gene expression (the same applies below) and do not take into account association information.
-
SVM (bulk) represents conventional machine learning methods. The training and test sets are set up in the same way as for MLP, with support vector machines used for training and prediction.
-
Pearson (bulk) represents conventional statistical methods. The method pairs cells from single-cell data with cell lines from bulk data based on the highest Pearson correlation in gene expression and assigns drug response labels accordingly.
Methods that transfer single-cell data to single-cell data are used for comparison:
Currently, there is no specific method for drug response prediction across single-cell datasets, so other available methods need to be modified for comparison. The methods listed below are not originally designed for drug response prediction across single-cell datasets, but with slight modifications, they can be used, where only the data input is changed without altering the model architecture.
-
scDEAL represents transfer learning methods where both the source and target domains are single-cell data.
-
SCAD represents another class of transfer learning methods. The data is the same as scDEAL, but the use of features and the evaluation of the target domain are the same as SCAD (bulk).
-
CaDRReS-Sc represents machine learning methods where both the training and test sets consist of single-cell data.
-
scVI59 represents deep learning methods for batch effect correction. After correcting for batch effects in different single-cell datasets, they are combined into a single dataset. The data is then divided into a training set and a test set, and an MLP is employed to predict drug scores for the cells.
-
ComBat60 represents non-deep learning methods for batch effect correction. After correction, the training and prediction steps are carried out in the same way as for scVI.
-
MLP represents conventional deep learning methods with both the training and test sets consisting of single-cell data.
-
SVM represents conventional machine learning methods with the same training and test set setup as MLP.
-
Pearson represents conventional statistical methods. Cells from different single-cell datasets compute Pearson correlation and labels are transferred between cells with the highest correlation.
Comparison experiments at the cell group level
General cell group level:
Evaluation at the cell group or cell type level can better confirm whether the model performs well at a higher level than individual cells. In this study, MACA61 is used for clustering and cell type annotation. Since the obtained Louvain clusters mostly correspond to cell types and drug response groups (sensitive or resistant), the percentage of correctly predicted cells within each Louvain cluster is calculated, and then the results from all Louvain clusters are averaged. This average result is considered as the cell group level prediction accuracy. After performing the above operations for scXDR and the two major categories of single-cell drug response prediction methods, the results are compared.
Malignant cell group level:
Most conventional cell type annotation methods can primarily identify normal cell types, while a significant proportion of tumor cells may be malignant. Evaluating the prediction accuracy of drug responses in malignant cell groups is an aspect that previous models have rarely focused on. Using Cancer-Finder62 for malignant cell annotation, the percentage of correctly predicted cells within the malignant cell group is calculated, and this percentage is considered as the accuracy at the malignant cell group level. After performing the above steps for scXDR and the two major categories of methods, the accuracy results are compared.
Ablation experiment
The core components of scXDR are primarily located in the transfer learning phase, which include feature alignment and structure alignment. In addition, structure reconstruction preserves network integrity during alignment. To evaluate the importance of these components and the advantage of the complete model, experiments on 20 prediction tasks are conducted after ablating these components from the full scXDR model.
-
Feature ablation. Ablate the feature alignment component while retaining structure alignment and structure reconstruction. The message passing phase and prediction phase are retained by default.
-
Structure ablation. Ablate the structure alignment component while retaining feature alignment and structure reconstruction. The message passing phase and prediction phase are retained.
-
Reconstruction ablation. Ablate the structure reconstruction component while retaining feature alignment, structure alignment, and other components.
-
Transfer ablation. Ablate all content in the transfer learning phase, retaining only the message passing phase and prediction phase.
Interpretability experiment
The roles of some phases and components in scXDR are studied by observing the changes in features during its different phases, thereby explaining the model’s internal operating mechanism. Specifically, the original features of cells from the source and target domains, the embeddings after the message passing phase, and the aligned embeddings from the transfer learning phase are reduced in dimensionality using Principal Component Analysis (PCA) and visualized. The original features are gene expression, while the other two embeddings are taken from the last iteration, prior to the prediction phase. Each point represents a cell, with different colors indicating the source and target domains. In addition to visualizing the scXDR embeddings, the cell embeddings from the source and target domains of the models scDEAL and SCAD are also reduced and colored using PCA. These embeddings are also taken from the last iteration, and the original authors used them to compute domain discrepancy loss, ensuring the two domains are closely aligned. The comparison with scXDR embeddings is therefore fair.
Case study
This study conducts a case study for each scenario, with the order of the cases corresponding to the order of the scenarios, and the first three cases use additional new datasets. In each scenario, the original dataset serves as the source domain and the new dataset as the target domain, ensuring that after the model performs well on the original datasets in that scenario, it is applied for prediction on the new dataset. The best threshold for each scenario is set as the average of the optimal thresholds obtained from all benchmark single-cell datasets within that scenario.
In the first case, the background involves different samples with the same drug and tumor, consistent with the first scenario in terms of drug and tumor. The GSE134841 dataset5 contains scRNA-seq data from the non-small cell lung cancer PC9 cell line treated with Erlotinib. Samples can be distinguished by whether they received a drug holiday treatment. If the cells are treated with the drug after a period of withdrawal, they are considered to have received drug holiday treatment5 and are labeled “Erlotinib”; if the cells are not treated with the drug after the withdrawal period, they are considered not to have received drug holiday treatment and are labeled “No Erlotinib”. In this case, cells under different treatment conditions are treated as a whole to construct a heterogeneous network, and scXDR is used to predict the sensitivity and resistance of cells that receive or do not receive drug holiday treatment, thereby inferring the outcome of cells under different treatment approaches. The prediction results are validated using the outcomes reported in the original literature. Although drug tolerance differs from resistance in the original literature, they are similar in cellular drug response and are therefore treated equivalently here.
The background of the second case is the same tumor with different drugs, leading to a drug screening study, and the tumor type is identical to that in the second scenario. Based on the poorly metastatic melanoma cell line from the GSE156750 dataset21, drug screening is conducted on anticancer drugs in the GDSC to identify those that produce the highest response in this cell line. The results are analyzed and validated using enrichment analysis, survival analysis, differential analysis, database mining, and literature mining, and drug response markers are also identified.
The background of the third case is different tumors, so a pan-cancer study is conducted. Using scRNA-seq data from 22 cancer types provided by Kinker et al.27, response predictions are performed for 137 anticancer drugs in the GDSC. Based on the response scores, clustering analysis is conducted separately on drugs and tumors, and then the resulting drug clusters and tumor clusters are investigated.
In the fourth case, the background centers on different patients. Based on the dataset from the fourth scenario (breast cancer) and drugs approved by the US Food and Drug Administration (FDA) for treating breast cancer, scXDR predicts the optimal drug or drug combination for patient cells, some of which have received drug treatment and some of which have not. Literature and clinical trial databases40 are searched to validate the results.
Statistics and reproducibility
All the data used in this study are publicly available. Detailed information on the datasets, including sample sizes, data processing, construction methods, and testing procedures, can be found in Supplementary Table 1 and in the Methods section. To facilitate reproducibility, we provide the code, the environment information required for running the experiments, and the server configuration details in our GitHub repository.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data used in this study are publicly available. The single-cell level data are obtained from GEO (GSE134839, GSE149214, GSE108394, GSE164614, GSE230538, GSE117872, GSE127298, GSE140440, GSE147326, GSE134841, GSE156750, and GSE157220) and from the Broad Institute’s Single Cell Portal (https://singlecell.broadinstitute.org/single_cell/study/SCP542/pan-cancer-cell-line-heterogeneity). The bulk level data are obtained from GDSC (https://www.cancerrxgene.org/downloads/bulk_download), HuRI (http://www.interactome-atlas.org/download), and CTD.
Code availability
The code is available on GitHub (https://github.com/QiGuan1920/scXDR2025) and Zenodo (https://doi.org/10.5281/zenodo.17857697)63.
References
Gambardella, G. et al. A single-cell analysis of breast cancer cell lines to study tumour heterogeneity and drug response. Nat. Commun. 13, 1714 (2022).
Grün, D. & van Oudenaarden, A. Design and analysis of single-cell sequencing experiments. Cell 163, 799–810 (2015).
Mohammadi, S., Davila-Velderrain, J. & Kellis, M. A multiresolution framework to characterize single-cell state landscapes. Nat. Commun. 11, 5399 (2020).
Lawson, D. A. et al. Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 20, 1349–1360 (2018).
Aissa, A. F. et al. Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nat. Commun. 12, 1628 (2021).
França, G. S. et al. Cellular adaptation to cancer therapy along a resistance continuum. Nature 631, 876–883 (2024).
Yan, Y. et al. Multi-omic profiling highlights factors associated with resistance to immuno-chemotherapy in non-small-cell lung cancer. Nat. Genet 57, 126–139 (2025).
Wu, Z. et al. Single-cell techniques and deep learning in predicting drug response. Trends Pharm. Sci. 41, 1050–1065 (2020).
Zhang, P. et al. A deep learning framework for in silico screening of anticancer drugs at the single-cell level. Natl. Sci. Rev. 12, nwae451 (2025).
Suphavilai, C. et al. Predicting heterogeneity in clone-specific therapeutic vulnerabilities using single-cell transcriptomic signatures. Genome Med 13, 189 (2021).
Fustero-Torre, C. et al. Beyondcell: Targeting cancer therapeutic heterogeneity in single-cell RNA-seq data. Genome Med 13, 187 (2021).
Pellecchia, S. et al. Predicting drug response from single-cell expression profiles of tumours. BMC Med 21, 476 (2023).
Chen, J. et al. Deep transfer learning of cancer drug responses by integrating bulk and single-cell RNA-seq data. Nat. Commun. 13, 6494 (2022).
Zheng, Z. et al. Enabling single-cell drug response annotations from bulk RNA-seq using SCAD. Adv. Sci. 10, e2204113 (2023).
Ziegenhain, C. et al. Comparative analysis of single-cell RNA sequencing methods. Mol. Cell 65, 631–643.e634 (2017).
Shin, D. et al. Multiplexed single-cell RNA-seq via transient barcoding for simultaneous expression profiling of various drug perturbations. Sci. Adv. 5, eaav2249 (2019).
Li, J. et al. Higher-order attribute-enhancing heterogeneous graph neural networks. Ieee Trans. Knowl. Data Eng. 35, 560–574 (2023).
Sharma, A. et al. Longitudinal single-cell RNA sequencing of patient-derived primary cells reveals drug-induced infidelity in stem cell hierarchy. Nat. Commun. 9, 4931 (2018).
Ho, Y. J. et al. Single-cell RNA-seq analysis identifies markers of resistance to targeted BRAF inhibitors in melanoma cell populations. Genome Res 28, 1353–1363 (2018).
Chi, F. et al. A ‘one-two punch’ therapy strategy to target chemoresistance in estrogen receptor positive breast cancer. Transl. Oncol. 14, 100946 (2021).
Kim, Y. S. et al. Single-cell RNA sequencing reveals the existence of pro-metastatic subpopulation within a parental b16 murine melanoma cell line. Biochem Biophys. Res Commun. 613, 120–126 (2022).
Rappaport, N. et al. Malacards: An amalgamated human disease compendium with diverse clinical and genetic annotation and structured search. Nucleic Acids Res 45, D877–D887 (2017).
Slamon, D. J. et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: Monaleesa-3. J. Clin. Oncol. 36, 2465–2472 (2018).
Rae J. M., Lippman M. E. The role of estrogen receptor signaling in suppressing the immune response to cancer comment. J. Clin. Investig. 131, e155476 (2021).
Carrara, G. F. A. et al. Analysis of RPL37A, MTSS1, and HTRA1 expression as potential markers for pathologic complete response and survival. Breast Cancer 28, 307–320 (2021).
Hoof, T., Fislage, R. & Tummler, B. Primary sequence of the human ribosomal-protein L37a. Nucleic Acids Res 20, 5475–5475 (1992).
Kinker, G. S. et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat. Genet 52, 1208–1220 (2020).
Galanski, M. Recent developments in the field of anticancer platinum complexes. Recent Pat. Anti-Cancer Drug Discov. 1, 285–295 (2006).
Mossé, Y. P. et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A children’s oncology group phase 1 consortium study. Lancet Oncol. 14, 472–480 (2013).
Wee P. & Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 9, 52 (2017).
Davies, C. et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 378, 771–784 (2011).
Planchard, D. et al. Dabrafenib plus Trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 18, 1307–1316 (2017).
Cheng, S. et al. Chemotherapy for relapsed small cell lung cancer: A systematic review and practice guideline. J. Thorac. Oncol. 2, 348–354 (2007).
Tesauro, C. et al. Topoisomerase i activity and sensitivity to camptothecin in breast cancer-derived cells: A comparative study. BMC Cancer 19, 1158 (2019).
Weitzman, J. B. et al. Jund protects cells from p53-dependent senescence and apoptosis. Mol. Cell 6, 1109–1119 (2000).
Polanski, R. et al. Senescence induction in renal carcinoma cells by nutlin-3: A potential therapeutic strategy based on mdm2 antagonism. Cancer Lett. 353, 211–219 (2014).
Zeng, S. X. et al. The phosphatidylinositol 3-kinase pathway as a potential therapeutic target in bladder cancer. Clin. Cancer Res 23, 6580–6591 (2017).
Abraham, A. G. & O’Neill, E. Pi3k/akt-mediated regulation of p53 in cancer. Biochem Soc. Trans. 42, 798–803 (2014).
Leo, C. P., Leo, C. & Szucs, T. D. Breast cancer drug approvals by the us fda from 1949 to 2018. Nat. Rev. Drug Discov. 19, 11 (2020).
Asiimwe, I. G. & Rumona, D. Publication proportions for registered breast cancer trials: Before and following the introduction of the clinicaltrials.Gov results database. Res. Integr. peer Rev. 1, 10–20 (2016).
Pujade-Lauraine, E. et al. Pegylated liposomal doxorubicin and carboplatin compared with paclitaxel and carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J. Clin. Oncol. 28, 3323–3329 (2010).
Hoa Thi Nhu T. et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 21, 12 (2020).
Zhang, F. et al. A novel heterogeneous network-based method for drug response prediction in cancer cell lines. Sci. Rep. 8, 3355 (2018).
Güvenç Paltun, B., Mamitsuka, H. & Kaski, S. Improving drug response prediction by integrating multiple data sources: Matrix factorization, kernel and network-based approaches. Brief. Bioinform 22, 346–359 (2021).
Sun, C. et al. Reversible and adaptive resistance to BRAF(v600e) inhibition in melanoma. Nature 508, 118–128 (2014).
Ma, J. et al. Rna binding protein: Coordinated expression between the nuclear and mitochondrial genomes in tumors. J. Transl. Med 21, 512 (2023).
Qi, J. et al. TUBA1B as a novel prognostic biomarker correlated with immunosuppressive tumor microenvironment and immunotherapy response. Front Pharm. 16, 1517887 (2025).
Chakraborty B. et al. Inhibition of estrogen signaling in myeloid cells increases tumor immunity in melanoma. J. Clin. Invest. 131, e151347 (2021).
Catez, F. et al. Ribosome biogenesis: An emerging druggable pathway for cancer therapeutics. Biochem Pharm. 159, 74–81 (2019).
Kuwahara, H. & Gao, X. Analysis of the effects of related fingerprints on molecular similarity using an eigenvalue entropy approach. J. Cheminform 13, 27 (2021).
Govindan G., Nair A. S. Composition, transition and distribution (CTD) - a dynamic feature for predictions based on hierarchical structure of cellular sorting. In: Annual IEEE India Conference - Engineering Sustainable Solutions) (2011).
Luck, K. et al. A reference map of the human binary protein interactome. Nature 580, 402–408 (2020).
Hao Z. et al. A selection-pattern-aware recommendation model with colored-motif attention network. Neurocomputing 538, 756–767 (2023).
Wang, Y., Ding, Y. & Shahrampour, S. Takde: Temporal adaptive kernel density estimator for real-time dynamic density estimation. IEEE Trans. Pattern Anal. Mach. Intell. 45, 13831–13843 (2023).
Tang, J., Deng, C. & Huang, G.-B. Extreme learning machine for multilayer perceptron. IEEE Trans. Neural Netw. Learn. Syst. 27, 809–821 (2016).
Bantis, L. E., Nakas, C. T. & Reiser, B. Construction of confidence regions in the roc space after the estimation of the optimal youden index-based cut-off point. Biometrics 70, 212–223 (2014).
Barretina, J. et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012).
Tsherniak, A. et al. Defining a cancer dependency map. Cell 170, 564–576 (2017).
Lopez, R. et al. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1068 (2018).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical bayes methods. Biostatistics 8, 118–127 (2007).
Xu, Y. et al. Maca: Marker-based automatic cell-type annotation for single-cell expression data. Bioinformatics 38, 1756–1760 (2022).
Zhong Z. et al. Domain generalization enables general cancer cell annotation in single-cell and spatial transcriptomics. Nat Commun 15, 1929 (2024).
Qi, G. scXDR. https://doi.org/10.5281/zenodo.17857697 (2025).
Acknowledgements
This study is supported by the National Natural Science Foundation of China (No. 12271216, 92370131) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX24_2645).
Author information
Authors and Affiliations
Contributions
G.Q. designed the model, developed the analysis pipelines, performed the modeling, and conducted the experiments. L.W. contributed to the design and analysis. M.N., Y.F., Q.R., Y.Z., Z.S., Z.S., and J.L. contributed to the data processing, model testing, and optimization. J.G. supervised the project, offered guidance, and critically reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Michiaki Hamada, Aylin Bircan and Tobias Goris.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qi, G., Wang, L., Nan, M. et al. scXDR: drug response prediction across single-cell datasets via heterogeneous network transfer learning. Commun Biol 9, 139 (2026). https://doi.org/10.1038/s42003-025-09418-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09418-5
