
Abstract
Membrane proteins constitute ~25–30% of the human proteome, with G protein-coupled receptors (GPCRs) representing the largest family and targets of ~35% of FDA-approved drugs. While cryo-electron microscopy (cryo-EM) has transformed GPCR structural biology, resolving small GPCR complexes, particularly those in the apo and inactive states, remains challenging due to their limited size and conformational flexibility. This limitation is significant, as many therapeutics act by stabilizing inactive GPCR conformations. In this review, we discuss the key obstacles to visualizing apo/inactive GPCRs by cryo-EM, summarize fusion and target-binding strategies that have enabled their structural determination, outline practical considerations for fiducial design, and highlight emerging AI-driven approaches likely to further expand the scope of GPCR structural studies.
Similar content being viewed by others


Introduction
Membrane proteins lie at the heart of virtually all cellular processes, making them prime targets for drug discovery. They facilitate ion and nutrient transport, maintain membrane potential, and coordinate signaling pathways that drive diverse physiological responses1,2,3,4,5,6,7. Among them, G protein-coupled receptors (GPCRs) form the largest and most versatile family, governing functions ranging from vision and olfaction to cardiovascular regulation8,9,10,11,12,13. Their central role in physiology has placed them at the forefront of pharmacology, with nearly 35% of FDA-approved drugs acting on GPCRs1,14.
Notably, advances in structural biology have significantly shaped our understanding of GPCR function at the molecular level. Interestingly, the structural biology of GPCRs has advanced in two distinct phases. In the first phase, X-ray crystallography dominated from 2000 onward, revealing crucial details about inactive and active receptor states and providing the first atomic models of GPCRs15,16,17,18,19,20,21,22,23,24,25,26,27. A second phase in GPCR structural biology began in 2017, when cryo-electron microscopy (cryo-EM) provided the first structure of the Calcitonin receptor (CTR1), a class B GPCR, in complex with heterotrimeric G proteins28. Following the visualization of the Calcitonin receptor (CTR1)28, cryo-EM has successfully resolved active-state structures of GPCRs across all vertebrate classes—A, B, C, and F28,29,30,31,32,33 and has rapidly emerged as the preferred method for studying GPCRs, owing to its ability to capture proteins in near-physiological states without the need for crystallization, while requiring smaller quantities of material compared to X-ray crystallography and NMR. To date, cryo-EM has provided unprecedented insights into receptor complexes with G proteins28,34,35,36,37,38,39,40,41, GPCR kinases (GRKs)42,43, and β-arrestins44,45,46,47,48,49,50,51, illuminating fundamental aspects of receptor activation, effector coupling, and downstream signaling. Collectively, these advances have transformed GPCR biology and have expanded opportunities for structure-based drug discovery52,53,54,55. Yet, despite these advances, apo/inactive-state GPCRs remain strikingly underrepresented. Notably, based on the current GPCRdb release (2025), among the 1716 total GPCR entries, cryo-EM accounts for 1086 active-state structures (~63% of all entries) and only 107 inactive-state structures (~6%). In contrast, X-ray crystallography provides a more substantial representation of inactive-state GPCRs with 358 inactive (~21%) and 130 active (~8%) structures within the same dataset. These crystallographic structures have been instrumental in shaping our current understanding of GPCR activation mechanisms and have significantly contributed to structure-based drug discovery. This disparity highlights that, while inactive-state GPCR architecture is well represented in crystallographic studies, extending such coverage to cryo-EM, particularly for apo and inactive-state, remains a major outstanding challenge. This stark imbalance highlights a critical gap, especially considering that a significant number of currently approved drugs target GPCRs in their inactive state as antagonists or inverse agonizts, underscoring the urgent need for strategies to visualize this pharmacologically important conformational landscape.
In this review, we highlight the specific challenges associated with studying smaller biological samples, particularly apo/inactive-state GPCRs that are not complexed with their transducer elements. We discuss current approaches that are pushing the boundaries of size limits, enabling high-resolution structure determination, outline the practical considerations for designing fiducials, and highlight how complementary strategies integrated with AI can advance the study of sub-100 kDa membrane proteins.
Confronting the challenges in resolving apo and inactive-state GPCR structures with Cryo-EM
Cryo-EM has sparked an exponential growth in the structural elucidation of protein samples, contributing 16,996 coordinate models to the Protein database (PDB) and 28,860 density maps to the Electron Microscopy Data Bank (EMDB) (Accessed 9th July 2025). This transformation has been profound for GPCRs, which are challenging to study due to their inherent flexibility and often low expression56. Yet, cryo-EM structure determination of apo/inactive-state GPCRs with molecular weight <100 kDa57,58 remains disproportionately low (Table 1), despite advances in detectors, grid preparation, sophisticated data acquisition, and data processing software59,60,61,62,63.
Several factors hinder the resolution of apo/inactive-state GPCR structures. Class A GPCRs in their apo/inactive-state are small (35–50 kDa)64, and lack the notably large extracellular domains of Class B and C receptors65, or the extended N-termini of Class F GPCRs65. They are typically solubilized in detergent micelles or lipid mimetics like nanodiscs. While these environments stabilize the receptor, they often exceed the receptor in size and scatter electrons more strongly, reducing signal-to-noise ratio and resulting in diminished contrast66. Moreover, the absence of extramembranous features in apo/inactive-state GPCRs, unlike active-state GPCRs coupled with transducers, makes data averaging during particle alignment difficult56,67, thereby hindering high-resolution reconstruction58. Consequently, much of the structural landscape of apo/inactive-state GPCRs remains unresolved. To address these obstacles, several approaches have been utilized to improve sample68,69,70 and grid preparation71,72, data acquisition73 as well as data processing software (CryoSPARC and RELION)74,75 including strategies that promote thinner ice76, reduce beam-induced motion77, and preserve particle integrity at the air–water interface78, factors that do not increase mass per se but collectively improves signal-to-noise and preserve signal, effectively lowering the size barrier for cryo-EM analysis of small GPCRs. Here, we focus on the fusion-based and target-binding strategies that have been employed to increase the effective molecular weight of apo/inactive-state GPCRs, highlighting successful examples that have enabled their high-resolution structural characterization. By systematically presenting the different strategies in the context of specific GPCRs, the receptor positions targeted, and practical considerations for each approach, this review provides a framework to guide researchers in selecting suitable strategies. The strategies described here for obtaining high-resolution apo/inactive-state GPCRs can be further extended to other low molecular weight membrane proteins beyond GPCRs, as illustrated in Table 2.
Chaperone-assisted strategies for resolving apo/inactive-state GPCRs
To address the challenges associated with the small size of apo/inactive-state GPCRs (without transducers), a common strategy is to increase the protein’s size to make it more suitable for structural determination. One effective approach involves using structural chaperones that bind to the protein of interest and increase its molecular weight. This not only increases the size of the target protein but also introduces polar contacts that facilitate crystal formation in X-ray crystallography16,79,80 or adds distinctive features that aid in single-particle alignment during cryo-EM data processing81,82. Here, we outline the state-of-the art strategies (fusion-binding strategies and target-binding strategies) that have been successfully employed to overcome the obstacle posed by the small size of the apo/inactive-state GPCRs (Fig. 1).

Schematic representation of high-resolution structure elucidation of apo/inactive state GPCRs by using several target binding strategies utilizing Nb6, NabFab, and DARPin and fusion-based strategies utilizing BRIL, PGS, and calcineurin.
Fusion-binding strategies for resolving apo/inactive-state GPCRs by cryo-EM
Beyond traditional crystallography, fusion-based strategies utilizing incorporation of fusion proteins such as BRIL81,83,84,85, PGS81,86, and calcineurin87,88 have been adopted for GPCR structural studies using cryo-EM. These fusion proteins are mostly positioned between the TM5 and TM6 of the GPCRs, one of the most flexible and dynamic regions. By stabilizing this flexible region, these fusion proteins reduce conformational heterogeneity, enhance stabilizing contacts, and facilitate alignment, thereby enabling high-resolution reconstructions. In this section, we specifically focus on the fusion proteins that have enabled structure determination of apo/inactive-state GPCRs by cryo-EM.
BRIL
The apocytochrome b562RIL (BRIL) comprises of a 4-helix bundle structure with its N and C-termini forming a continuous α-helix with TM5 and TM6, respectively81,83,89. Continuous α-helix of the fusion protein with the TM5 and TM6 offers additional structural rigidity, a critical requirement to form a rigid protein assembly and enable structural determination89. This feature has been previously harnessed for structural determination of 117 inactive-state GPCRs for crystallographic studies, according to the current GPCRdb release (2025). Notable examples include the inactive structures of 5-HT1B-BRIL complexed with ergotamine (PDB: 4IAR)90 and A2A adenosine receptor–BRIL structure bound to antagonist ZM241385 (PDB: 4EIY)91,92. However, beyond traditional crystallography, BRIL fusion proteins have also been instrumental in advancing structure determination of apo/inactive-state GPCRs by cryo-EM81,83,84. To date, BRIL has been used as a fusion strategy in 124 cryo-EM GPCR structures, of which 22 (~18%) structures represent inactive-state. Representative examples include the human Frizzled receptor (FZD5)84 and the A2A adenosine receptor81, yielding a global resolution of 3.7 Å and 3.4 Å, respectively. Notably, for A2A, subsequent addition of an anti-BRIL Fab fragment (SRP2070) combined with focused refinement enhanced map quality of the TM domain to 3.2 Å, sufficient for model building81. These results demonstrate that the Fab fragment not only increases particle mass but also enhances image contrast and provides a rigid fiducial for particle alignment, thereby making otherwise small and featureless GPCRs more tractable for high-resolution cryo-EM analysis. Additionally, BRIL’s continuous α-helices have also been utilized to resolve the structure of the prototypical β2-adrenergic receptor (β2AR) bound to the antagonist alprenolol (Fig. 2i-A)83. However, this structure revealed conformational heterogeneity, highlighting the flexibility between BRIL and the GPCR’s transmembrane (TM) domain. To address this issue and improve rigidity, a modified BRIL was engineered by replacing the loop between BRIL helices 2 and 3 with a flexible GS linker, reducing interference and minimizing structural clashes with the receptor83 (Fig. 2i-A). This enhancement stabilized the β2AR-BRIL complex and was later successfully applied to determine the structure of the adhesion receptor ADGRL3 at a global resolution of 3.5 Å83. Interestingly, near-atomic resolution inactive-state GPCR structures by cryo-EM have been achieved when BRIL fusion is combined with additional rigidifying binders. For example, the inactive chemokine receptor CCR6 was resolved at ~3.0 Å resolution using a BRIL fusion together with an anti-BRIL Fab (PDB: 9D3E)93, while the inactive CXCR3 structure reached comparable resolution through the use of BRIL in combination with nanobody 6 (PDB: 8K2W)94. These examples illustrate that augmenting BRIL with auxiliary stabilizers can substantially enhance rigidity and improve achievable resolution.

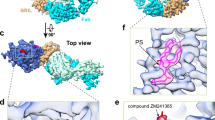
i Structures solved using fusion-based strategies like BRIL, PGS and calcineurin either with antagonist/inverse agonist or in apo state. ii Structures solved using target binding approaches like Nb6, NabFab, and DARPin. iA, iB, iC, iiC Insets highlighting the residues on the receptor to which the fiducial marker has been attached. iiA, iiB Insets highlighting the interacting residues and interfaces between receptor and fiducial marker. *Structures solved by X-ray crystallography.
At the same time, these studies reflect the persistent flexibility between the BRIL-TM interface, underscoring the limitations of BRIL as a stand-alone fusion partner for resolving GPCR structures in the absence of signaling complexes. Importantly, this behavior reflects a broader challenge inherent to fusion-based strategies, where residual flexibility at the fusion–receptor junction can introduce conformational heterogeneity and limit achievable resolution, particularly for inactive-state GPCRs. Recognition of this limitation has directly motivated the incorporation of additional stabilizing elements and the development of multi-point or three-point contact fusion strategies (discussed later in this section), which better restrain inter-domain motion and enhance structural rigidity, enabling higher-resolution cryo-EM analysis of inactive-state GPCRs.
PGS
Similar to BRIL, another widely used fusion protein for facilitating GPCR crystallization is the thermostable glycogen synthase domain (PGS) from Pyrococcus abyssi20,95,96,97. At approximately 20 kDa, PGS is bulkier than BRIL (~12 kDa), but smaller than the BRIL-anti-BRIL Fab fusion81. When grafted onto the intracellular loop three (ICL3) of GPCRs, PGS has enabled the high-resolution determination of several inactive-state GPCR structures by X-ray crystallography20,95,97,98. In Cryo-EM, however, the application of PGS fusion to inactive-state GPCRs remains limited, with only four structures reported to date. Interestingly, the PGS fusion strategy applied to the mouse Smoothened receptor (mSMO) to visualize its apo-state by cryo-EM, explored two distinct constructs with striking observations. In the higher-resolution structure (3.7 Å), the PGS domain (designated PGS2) was inserted into the third intracellular loop (ICL3) and extended approximately one and a half helical turns into transmembrane helix 6 (TM6), adopting a position beneath the receptor (Fig. 2i-B)81. In contrast, the earlier construct (PGS1), in which the PGS domain was inserted between residues 441 (on ICL3) and 445 (on TM6), exhibited a lateral orientation relative to the receptor and yielded a lower-resolution map at 6.0 Å81. While multiple factors, including protein stability, particle number, ice thickness, and data-collection parameters, can influence the final resolution of cryo-EM structures, this comparison highlights how the spatial orientation of the fusion tag can significantly impact particle alignment, ultimately influencing both the resolution and structural quality of cryo-EM reconstructions of GPCRs.
Additionally, a recent study employed a PGS-anti-PGS nanobody (Nb) fusion strategy to determine the structure of the Cannabinoid receptor 1 (CB1) in complex with its inverse agonist taranabant, as well as with the peripherally restricted inverse agonizts MRI-1891 and MRI-1867, using cryo-EM86. Initially, the authors attempted to solve the CB1-PGS structure with taranabant using the same receptor construct that had previously yielded high-resolution crystal structures20. However, cryo-EM results showed only a modest ~6 Å resolution86, likely due to flexibility between the receptor transmembrane regions and the PGS fusion. To overcome this flexibility issue, the researchers developed a nanobody (CNB36) that specifically binds to the CB1-PGS fusion, yielding a 3.5 Å structure of CB1-PGS-CNB36 via cryo-EM86, comparable to the previously reported CB1-PGS crystal structure (Fig. 2i-B)20. What distinguishes CNB36 is its dual-contact mechanism86, while CDR2 binds the ICL2 of CB1 (Fig. 2i-B), CDR3 engages the PGS domain, thereby enhancing rigidity and overcoming the flexibility observed in CB1–PGS alone to achieve an improved resolution. Because CNB36 primarily interacts with PGS and only minimally with ICL2, this strategy holds promise for broad application in solving inactive-state GPCR structures by cryo-EM. Although some ICL2 modifications may be necessary, this approach markedly expands the toolkit for structural determination of GPCRs and other small integral membrane proteins.
Calcineurin-based approach
Calcineurin, a heterodimeric protein, is engineered to fuse with GPCRs at three sites—TM5, TM6, and TM7. This provides an additional anchoring point beyond the two-point attachment of BRIL or PGS, which replace only the ICL3 between TM5 and TM6. This three-point fusion creates a more rigid connection, thereby enhancing structural stability and greatly facilitating particle alignment during cryo-EM data processing87,88. To date, this calcineurin-based fusion strategy has enabled the determination of five inactive-state GPCR structures by cryo-EM. This strategy has been successfully applied to β2-adrenergic receptor (β2AR), enabling cryo-EM structures of both the apo and antagonist-bound forms at 3.9 Å and 3.5 Å resolution, respectively and previously unattainable by X-ray crystallography87.
Calcineurin (CN) consists of two subunits: CN-A and CN-B, which are stabilized by four Ca2+ ions and further reinforced through the addition of FK binding protein (FKBP12) and the inhibitor FK506 (Tacrolimus). This provides a rigid fiducial marker that improves particle alignment during data processing87,88. Calcineurin-based strategy has recently enabled structure visualization of ETB both in its apo state and bound to RES-701-3, a selective inhibitor and promising therapeutic candidate. In this approach, CN-B was inserted into ICL3 (between K304 and D313), while CN-A was fused to the C-terminus via a GS linker88. Given its success with β2AR and ETB (Fig. 2i-C), this strategy holds broad promise for inactive-state Family A GPCRs and other small integral membrane proteins, where optimized linker design could further enhance resolution while preserving receptor function. We anticipate that the calcineurin-based approach, harnessed through cryo-EM, will pave the way for resolving an increasing number of GPCR structures in their unliganded or inactive states. This breakthrough holds immense promise for advancing our understanding of GPCR conformational landscapes and their implications for drug discovery.
Target-binding strategies for resolving apo/inactive-state GPCRs by cryo-EM
Target-binding strategies employ high-affinity binders such as Nb682,94,99,100 and NabFab82,101,102, which have been successfully used as fiducial markers to enable structural elucidation of apo- and inactive-state GPCRs by single-particle cryo-EM (Fig. 1). Additionally, engineered binding proteins such as DARPins represent a broader class of target-binding scaffolds that have been widely used in structural biology and cryo-EM scaffolding approaches103,104, and hold potential for future application to GPCR systems. Like fusion-protein approaches, these binders are typically directed toward the flexible ICL3 region of the receptor. However, unlike genetic fusion strategies, target-binding proteins, in some cases, can be introduced as external stabilizers without direct fusion to the receptor. It should be noted, however, that binder-based approaches often still require receptor engineering, such as grafting of intracellular loops (e.g., Nb6 binding to a modified ICL3)82 or modification of receptor termini to create a suitable binding epitope. Exceptions arise when binders are raised or specifically designed against native receptor conformations, as demonstrated for κ-opioid receptor (KOR) and Nb6105. Despite these requirements, target-binding proteins provide a flexible means to enhance complex stability, rigidity, and particle alignment with minimal global perturbation, making them valuable for stabilizing apo- and inactive-state GPCRs for cryo-EM analysis. In the following section, we highlight several such binders that have been successfully applied in cryo-EM studies, illustrating their utility in stabilizing apo- and inactive-state GPCRs for high-resolution structure determination.
Nb-6
In a study carried out by Robertson et al., a single-chain camelid antibody, nanobody 6 (Nb6), originally developed as a BRET-based sensor for the inactive-state κ-opioid receptor (KOR)82,105, has emerged as a valuable tool for solving inactive-state GPCR structures82. The advantage of using a BRET-based biosensor like Nb6 as a fiducial marker lies in its specificity for GPCRs lacking signaling partners such as G proteins or β-arrestins. Interestingly, the structure revealed an off-center binding of Nb6 to KOR, introducing an inherent asymmetry that serves as a reliable orientational guide during alignment, thereby improving cryo-EM map reconstruction82. Notably, unlike the PGS, the off-center binding of Nb6 relative to the receptor core provides a unique positioning which is particularly advantageous during the early stages of low-resolution alignment, as it enables more accurate inference of the receptor’s rotational orientation, thereby enhancing the overall quality of reconstruction82.
Moreover, Nb6 demonstrated a broader applicability for the other four Class A GPCRs (hNT1R, mMOR, hSSTR2, and H2R) that were tested practically and yielded high-resolution cryo-EM structures ranging from 2.4 to 3.1 A° with minimal alterations to their ICL3 regions82. For mMOR, two-point mutations in the ICL3 (M264L and K269R6.24) facilitated Nb6 binding, while hSSTR2 and hNTSR1 required a short swap of residues in their ICL3s82 (Fig. 2ii-A). These results highlight the versatility of Nb6 as a universal nanobody for structural elucidation of inactive-state GPCRs. Furthermore, for hH2R, a modified version of Nb6, termed Nb6M, was developed, where antigen-binding loops of Nb6 were grafted onto an alpaca nanobody scaffold82. This, combined with a nanobody Fab (NabFab)101 directed against Nb6, provided a bulkier and more rigid complex, minimizing flexibility and allowing high-resolution cryo-EM visualization of H2R-Nb6M-NabFab complex, resulting in a well-resolved density map82 (Fig. 2ii-B). This demonstrates that while Nb6 serves as an effective general tool for determining inactive-state GPCR structures, the enhanced Nb6M, combined with NabFab, expands the toolkit to tackle more challenging structural targets82.
Looking ahead, Nb6 is poised to become a widely adopted approach for determining apo/inactive-state structures of GPCRs, offering significant potential for accelerating drug discovery efforts targeting these receptors.
Designed ankyrin repeat proteins (DARPins)
Designed Ankyrin Repeat Proteins (DARPins) are structural chaperones derived from ankyrin repeat proteins, the most abundant binding proteins in the human genome106,107,108. Structurally, DARPins consist of two antiparallel helices connected by a small beta loop, with each helix containing 3–4 repeat motifs107,109. Each motif comprises 33 amino acids, 27 of which are conserved to maintain structural integrity, while the remaining six are randomized to enable high-affinity protein-protein interactions. DARPins have been engineered as surrogates for antibody fragments110 and have been instrumental in facilitating the structural determination of small proteins103,104,111,112, including GPCRs. The first GPCR that utilized DARPins and enabled the first structural insights into the inactive and apo-state was the neurotensin receptor 1 (NTSR1) bound to non-peptide inverse agonizts (SR48692 and SR142948A) using X-ray crystallography (Fig. 2ii-C)113. In this study, a DARPin (D12) was fused to the C-terminal end of the TM7 of a stabilized mutant form of NTSR1 via a shared helix to facilitate crystal packing rather than to rigidly constrain the receptor. This design enabled extensive crystal contacts while allowing limited conformational accommodation of the receptor in inverse agonist–bound states, which proved advantageous for X-ray crystallography. Later DARPins were also used to resolve the structure of the α-adrenergic receptor (α1BAR) bound to the inverse agonist cyclazosin114 (Fig. 2ii-C), although without invoking the same shared-helix design principles. Although DARPins provide strong crystal contact that proved advantageous for X-ray crystallography, it is important to note that such single-point attachment strategies do not inherently guarantee rigidity and may introduce conformational heterogeneity in cryo-EM datasets. Indeed, DARPins have also been employed as versatile adapters, linking small target proteins to larger symmetric platforms such as homo-tetramers, thereby overcoming the challenge of preferred orientation in cryo-EM104,112. By creating cage-like assemblies, this strategy increases molecular size and symmetry, improving data collection and alignment. This approach has already enabled high-resolution structures of small proteins like KRAS (19 kDa)112 and GFP (28 kDa)104, highlighting the potential of DARPins as modular and adaptable tools to tackle small and otherwise challenging proteins.
However, although DARPins have aided XRD studies of NTSR1113 and α1BAR114, their potential in cryo-EM remains untapped. With their modular design and capacity to enlarge and stabilize targets, DARPins fused to TM7 could provide the extramembranous features needed to overcome alignment challenges, opening new avenues for high-resolution structures of inactive-state GPCRs and other small proteins.
Beyond fusion and target binding: complementary approaches to resolve apo/inactive-state GPCRs
While fusion- and target-binding fiducials address the key challenges of <100 kDa GPCRs by increasing particle mass, introducing extramembranous features, and improving alignment during cryo-EM data processing, additional complementary approaches play an equally critical role in achieving high-resolution reconstructions. Importantly, a well-resolved structure, regardless of its conformational state, begins with a thoughtful construct design and high-quality sample preparation. GPCRs, as dynamic gatekeepers of cellular signaling, often contain flexible regions such as extended terminus and long intracellular or extracellular loops, which hinder expression, purification, and consequently image quality. Introducing thermostabilizing mutations, often guided by previously reported GPCR constructs optimized for stability and crystallization (e.g., β2AR115,116 and NTSR1117,118), can effectively reduce conformational heterogeneity. In fact, community resources such as the GPCRdb “Structure Constructs” tool provide curated information on previously engineered GPCR constructs, offering a practical starting point for rational construct design. In the current AI era, predictive tools such as AlphaFold2 and RoseTTAFold might provide invaluable preemptive insights into construct design119,120. The generated predicted Local Distance Difference Test scores flag structurally uncertain or disordered segments, informing rational strategies like truncation, loop engineering, linker stabilization, and the effect of thermostabilizing mutations, thereby providing a predictive framework for improving protein homogeneity at very early stages, potentially enhancing both sample quality and the resolution of cryo-EM reconstructions.
Volta phase plate (VPP) technology offers a powerful way to enhance optical contrast in cryo-EM, thereby enabling higher-resolution reconstructions of small or low-contrast particles. VPP was instrumental in resolving the first structure of human hemoglobin (~64 kDa, a soluble protein)121 and later the calcitonin receptor (CTR), a Class B GPCR (~150 kDa) in complex with heterotrimeric G proteins28. Since then, VPP has been successfully applied to several other GPCRs29,30,36. However, subsequent quantitative evaluations have shown that VPP generally does not provide resolution benefits for GPCR cryo-EM studies and is therefore rarely used in current workflows122. Nevertheless, its historical contributions to early GPCR structural efforts remain noteworthy. Although, by improving electron beam focus and boosting image contrast, VPP substantially enhances data quality123,124,125,126. However, the method carries practical challenges, including the short operational lifespan of phase plates and the technical difficulty of aligning the small holes with the microscope, which makes it more labor-intensive126,127. While VPP itself does not provide fiducials, when combined with small affinity tags, it could expand opportunities to make otherwise intractable apo- or inactive-state GPCRs more amenable to structural determination. The value of VPP becomes even greater when integrated with advances in AI-driven image analysis. For instance, Topaz, a deep learning-based particle picker, excels at detecting low-contrast particles buried in noisy micrographs, improving data recovery for receptors that are small or sparsely distributed128,129. Similarly, CryoTransformer, trained on large cryo-EM datasets, offers superior accuracy in particle identification and directly contributes to higher-resolution 3D reconstructions158. Beyond particle picking, AI has also transformed the way conformational heterogeneity is handled. cryoDRGN leverages neural networks to reconstruct a continuum of 3D conformations, capturing the structural ensembles of highly dynamic systems like apo-state GPCRs130. In contrast, HetSIREN is optimized for heterogeneous reconstructions in real space, refining particle subsets and improving map densities, particularly useful where GPCRs embedded in detergent micelles or nanodiscs suffer from weak signals131.
Taken together, these innovations spanning rational construct design, contrast enhancement with VPP, and AI-based image analysis represent promising complementary strategies that can significantly improve the resolution of apo/inactive-state GPCRs. Together, these forward-looking strategies, when complemented with fusion- or target-based approaches, can enhance our understanding of receptor regulation, enabling the rational design of state-specific therapeutics.
Designing effective fiducial markers: practical insights from successful reconstructions
-
(i)
Rigid Attachment and beyond: Fiducials must form stable attachments to GPCRs, but stability alone does not guarantee success. For instance, A2AR fused with the BRIL domain (~10 kDa) produced only intermediate 2D averages despite extensive data collection, and the use of a low-pass filtered crystal structure as a ref. 81. This underscores that fiducial effectiveness is receptor-specific and careful selection coupled with additional stabilization is critical.
-
(ii)
Antibody-assisted enhancement: Adding antibodies against fiducials can further increase bulk and rigidity, enhancing resolution. Examples include BRIL–anti-BRIL Fab81,84,93,132, PGS-CNB36 fusion86, and Nb6–NabFab complexes82, all of which improved structural stability and alignment.
-
(iii)
Orientation matters: The placement of fiducials relative to the receptor strongly influences resolution. This is best illustrated in mouse smoothened receptor (mSMO) studies where PGS positioned at the receptor base (mSMO-PGS2) yielded higher resolution (3.7 Å) compared to a side placement (mSMO-PGS1)81, demonstrating the importance of strategic orientation.
-
(iv)
Distinctive features for alignment: Smaller fiducials should provide unique features to aid orientation. In the KOR–Nb6 complex, Nb6 bound at the base of TM5–TM6 created distinct structural cues that facilitated early particle alignment82.
-
(v)
Optimizing marker size: Both small and medium-sized fiducials: BRIL (~10 kDa)81,83,84,132,133, PGS (~20 kDa)81, and Nb6 (~12 kDa)82,99,100 have driven successful reconstructions, proving that even modestly sized markers can aid alignment. However, generating synthetic antibodies for each GPCR remains challenging, and a single antibody fragment may not always provide sufficient stabilization.
Taken together, these fiducial markers play a pivotal role in cryo-EM studies of GPCRs by adding extra-membranous features that stabilize receptors, reduce dynamics, and enhance particle alignment, key factors required for high-resolution reconstructions. However, it is crucial to recognize that even with optimized fiducial markers, the ultimate resolution depends heavily on the biochemical quality and behavior of the biological sample. With advancing methodologies, fiducial markers are positioned to unlock apo/inactive-state GPCR structures and reveal mechanisms of receptor inactivation, offering powerful new opportunities for drug discovery.
Conclusion
Apo- and inactive-state GPCRs remain among the most challenging targets for cryo-EM, primarily due to their small molecular size and inherent conformational flexibility. In this review, we focus on fusion-based and target-binding strategies that have been successfully employed to overcome these limitations, highlighting representative examples where these approaches enabled high-resolution GPCR structures. We also outline key practical considerations in the rational design of such fiducial markers. However, at the same time, it is important to recognize that neither fusion-based nor target-binding strategies are universally applicable. Fusion partners can adversely affect receptor folding, expression, stability, or function and often require empirical screening to identify optimal insertion sites, while target-binding proteins may be limited by binding affinity, epitope accessibility, or state selectivity. Accordingly, no single fiducial strategy can be expected to work across all receptors or functional states. In this context, this review is intended as a decision-making guide to help researchers choose suitable approaches for apo and inactive-state GPCRs. In this context, the review is intended as a decision-making guide to help researchers choose suitable strategies for apo and inactive-state GPCRs. Finally, when combined with careful sample preparation, advances in optical imaging, and emerging AI-driven tools, these fusion and target-binding strategies continue to expand the scope of cryo-EM, enabling structural insights into GPCR states that were previously unattainable.
Future perspective
Cryo-EM has transformed the landscape of GPCR structural biology. Fusion-based and target-binding strategies have been instrumental in overcoming challenges of instability and conformational flexibility, yielding a diverse repertoire of structures across receptor families. These advances have laid a strong foundation for linking GPCR structure to function and pharmacology. A major frontier now lies in resolving small (<100 kDa) GPCRs in their inactive/apo states, where technical limitations persist. Progress in detector technology, AI-driven image processing, and novel scaffolds such as megabodies, fiducial markers promise to push the boundaries of size and resolution. Intriguingly, non-neuronal RGS proteins, which naturally interact with certain GPCRs, could also serve as inhibitory scaffolds to stabilize receptors in their apo states. While this should be viewed as a conceptual strategy rather than a broadly generalizable tool, exploring this possibility offers an exciting avenue for future research, where structural and systems biology approaches may uncover new layers of GPCR regulation extending well beyond the CNS. We anticipate that these synergistic developments will empower a deeper and more comprehensive understanding of GPCR biology, paving the way for structure-guided therapeutics.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
References
Sriram, K. & Insel, P. A. G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol. 93, 251–258 (2018).
Arinaminpathy, Y., Khurana, E., Engelman, D. M. & Gerstein, M. B. Computational analysis of membrane proteins: the largest class of drug targets. Drug Discov. Today 14, 1130–1135 (2009).
Santos, R. et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017).
Overington, J. P., Al-Lazikani, B. & Hopkins, A. L. How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996 (2006).
Rask-Andersen, M., Masuram, S. & Schiöth, H. B. The druggable genome: evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu. Rev. Pharmacol. Toxicol. 54, 9–26 (2014).
Huang, Y. et al. Membrane transporters and channels: role of the transportome in cancer chemosensitivity and chemoresistance. Cancer Res. 64, 4294–4301 (2004).
Jelokhani-Niaraki, M. Membrane proteins: structure, function and motion. Int. J. Mol. Sci. 24, 468 (2023).
Schöneberg, T. Modulating vertebrate physiology by genomic fine-tuning of GPCR functions. Physiol. Rev. 105, 383–439 (2025).
Heng, B. C., Aubel, D. & Fussenegger, M. An overview of the diverse roles of G-protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol. Adv. 31, 1676–1694 (2013).
Rosenbaum, D. M., Rasmussen, S. G. & Kobilka, B. K. The structure and function of G-protein-coupled receptors. Nature 459, 356–363 (2009).
Gulati, S. & Palczewski, K. Structural view of G protein-coupled receptor signaling in the retinal rod outer segment. Trends Biochem. Sci. 48, 172–186 (2023).
Spehr, M. & Munger, S. D. Olfactory receptors: G protein-coupled receptors and beyond. J. Neurochem. 109, 1570–1583 (2009).
Li, Y., Li, B., Chen, W.-D. & Wang, Y.-D. Role of G-protein coupled receptors in cardiovascular diseases. Front. Cardiovasc. Med. 10, 1130312 (2023).
Lorente, J. S. et al. GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 24, 458–479 (2025).
Palczewski, K. et al. Crystal structure of rhodopsin: AG protein-coupled receptor. Science 289, 739–745 (2000).
Cherezov, V. et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein–coupled receptor. Science 318, 1258–1265 (2007).
Rasmussen, S. G. et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 469, 175–180 (2011).
Manglik, A. et al. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326 (2012).
Rasmussen, S. G. et al. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 450, 383–387 (2007).
Shao, Z. et al. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 540, 602–606 (2016).
Claff, T. et al. Structural insights into partial activation of the prototypic G protein-coupled adenosine A2A receptor. ACS Pharmacol. Transl. Sci. 7, 1415–1425 (2024).
Asada, H. et al. Molecular basis for anti-insomnia drug design from structure of lemborexant-bound orexin 2 receptor. Structure 30, 1582–1589.e1584 (2022).
Qin, J. et al. Molecular mechanism of agonism and inverse agonism in ghrelin receptor. Nat. Commun. 13, 300 (2022).
Im, D. et al. Structure of the dopamine D2 receptor in complex with the antipsychotic drug spiperone. Nat. Commun. 11, 6442 (2020).
Rasmussen, S. G. et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature 477, 549–555 (2011).
Xu, F. et al. Structure of an agonist-bound human A2A adenosine receptor. Science 332, 322–327 (2011).
Park, J. H., Scheerer, P., Hofmann, K. P., Choe, H.-W. & Ernst, O. P. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454, 183–187 (2008).
Liang, Y.-L. et al. Phase-plate cryo-EM structure of a class B GPCR–G-protein complex. Nature 546, 118–123 (2017).
Draper-Joyce, C. J. et al. Structure of the adenosine-bound human adenosine A1 receptor–Gi complex. Nature 558, 559–563 (2018).
Liang, Y.-L. et al. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor–Gs complex. Nature 555, 121–125 (2018).
Kang, Y. et al. Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558, 553–558 (2018).
Shaye, H. et al. Structural basis of the activation of a metabotropic GABA receptor. Nature 584, 298–303 (2020).
Qi, X. et al. Cryo-EM structure of oxysterol-bound human smoothened coupled to a heterotrimeric Gi. Nature 571, 279–283 (2019).
Yin, J. et al. Structure of a D2 dopamine receptor–G-protein complex in a lipid membrane. Nature 584, 125–129 (2020).
Kumari, P., Inoue, A., Chapman, K., Lian, P. & Rosenbaum, D. M. Molecular mechanism of fatty acid activation of FFAR1. Proc. Natl. Acad. Sci. USA 120, e2219569120 (2023).
Liang, Y.-L. et al. Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561, 492–497 (2018).
García-Nafría, J., Nehmé, R., Edwards, P. C. & Tate, C. G. Cryo-EM structure of the serotonin 5-HT1B receptor coupled to heterotrimeric Go. Nature 558, 620–623 (2018).
Kato, H. E. et al. Conformational transitions of a neurotensin receptor 1–Gi1 complex. Nature 572, 80–85 (2019).
Maeda, S., Qu, Q., Robertson, M. J., Skiniotis, G. & Kobilka, B. K. Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 364, 552–557 (2019).
Xu, W. et al. Structural basis for strychnine activation of human bitter taste receptor TAS2R46. Science 377, 1298–1304 (2022).
de March, C. A. et al. Engineered odorant receptors illuminate the basis of odour discrimination. Nature 635, 499–508 (2024).
Duan, J. et al. GPCR activation and GRK2 assembly by a biased intracellular agonist. Nature 620, 676–681 (2023).
Chen, Q. et al. Structures of rhodopsin in complex with G-protein-coupled receptor kinase 1. Nature 595, 600–605 (2021).
Bous, J. et al. Structure of the vasopressin hormone–V2 receptor–β-arrestin1 ternary complex. Sci. Adv. 8, eabo7761 (2022).
Wang, Y. et al. Cryo-EM structure of cannabinoid receptor CB1-β-arrestin complex. Protein Cell 15, 230–234 (2024).
Yin, W. et al. A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res. 29, 971–983 (2019).
Staus, D. P. et al. Structure of the M2 muscarinic receptor–β-arrestin complex in a lipid nanodisc. Nature 579, 297–302 (2020).
Lee, Y. et al. Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature 583, 862–866 (2020).
Chen, K. et al. Tail engagement of arrestin at the glucagon receptor. Nature 620, 904–910 (2023).
Huang, W. et al. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 579, 303–308 (2020).
Maharana, J. et al. Structural snapshots uncover a key phosphorylation motif in GPCRs driving β-arrestin activation. Mol. Cell 83, 2091–2107.e2097 (2023).
Scapin, G., Potter, C. S. & Carragher, B. Cryo-EM for small molecules discovery, design, understanding, and application. Cell Chem. Biol. 25, 1318–1325 (2018).
Robertson, M. J., Meyerowitz, J. G. & Skiniotis, G. Drug discovery in the era of cryo-electron microscopy. Trends Biochem. Sci. 47, 124–135 (2022).
Duan, J., He, X.-H., Li, S.-J. & Xu, H. E. Cryo-electron microscopy for GPCR research and drug discovery in endocrinology and metabolism. Nat. Rev. Endocrinol. 20, 349–365 (2024).
Zhang, X. et al. Evolving cryo-EM structural approaches for GPCR drug discovery. Structure 29, 963–974.e966 (2021).
Nygaard, R., Kim, J. & Mancia, F. Cryo-electron microscopy analysis of small membrane proteins. Curr. Opin. Struct. Biol. 64, 26–33 (2020).
Harrison, P. J., Vecerkova, T., Clare, D. K. & Quigley, A. A review of the approaches used to solve sub-100 kDa membrane proteins by cryo-electron microscopy. J. Struct. Biol. 215, 107959 (2023).
Wentinck, K., Gogou, C. & Meijer, D. H. Putting on molecular weight: enabling cryo-EM structure determination of sub-100-kDa proteins. Curr. Res. Struct. Biol. 4, 332–337 (2022).
Bai, X.-C., McMullan, G. & Scheres, S. H. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 40, 49–57 (2015).
Cheng, Y. Single-particle cryo-EM—How did it get here and where will it go. Science 361, 876–880 (2018).
McMullan, G., Faruqi, A., Clare, D. & Henderson, R. Comparison of optimal performance at 300 keV of three direct electron detectors for use in low dose electron microscopy. Ultramicroscopy 147, 156–163 (2014).
Lyumkis, D. Challenges and opportunities in cryo-EM single-particle analysis. J. Biol. Chem. 294, 5181–5197 (2019).
Kühlbrandt, W. The resolution revolution. Science 343, 1443–1444 (2014).
García-Nafría, J. & Tate, C. G. Structure determination of GPCRs: cryo-EM compared with X-ray crystallography. Biochem. Soc. Trans. 49, 2345–2355 (2021).
Venkatakrishnan, A. et al. Structured and disordered facets of the GPCR fold. Curr. Opin. Struct. Biol. 27, 129–137 (2014).
Thonghin, N., Kargas, V., Clews, J. & Ford, R. C. Cryo-electron microscopy of membrane proteins. Methods 147, 176–186 (2018).
Lander, G. C. & Glaeser, R. M. Conquer by cryo-EM without physically dividing. Biochem. Soc. Trans. 49, 2287–2298 (2021).
Passmore, L. A. & Russo, C. J. Specimen preparation for high-resolution cryo-EM. Methods Enzymol. 579, 51–86 (2016).
Noble, A. J. et al. Reducing effects of particle adsorption to the air–water interface in cryo-EM. Nat. Methods 15, 793–795 (2018).
D’Imprima, E. et al. Protein denaturation at the air-water interface and how to prevent it. Elife 8, e42747 (2019).
Palovcak, E. et al. A simple and robust procedure for preparing graphene-oxide cryo-EM grids. J. Struct. Biol. 204, 80–84 (2018).
Russo, C. J. & Passmore, L. A. Progress towards an optimal specimen support for electron cryomicroscopy. Curr. Opin. Struct. Biol. 37, 81–89 (2016).
Cheng, A. et al. High resolution single particle cryo-electron microscopy using beam-image shift. J. Struct. Biol. 204, 270–275 (2018).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018).
Kim, L. Y. et al. Benchmarking cryo-EM single particle analysis workflow. Front. Mol. Biosci. 5, 50 (2018).
Sgro, G. G. & Costa, T. R. Cryo-EM grid preparation of membrane protein samples for single particle analysis. Front. Mol. Biosci. 5, 74 (2018).
Scheres, S. H. Beam-induced motion correction for sub-megadalton cryo-EM particles. Elife 3, e03665 (2014).
Abe, K. M., Li, G., He, Q., Grant, T. & Lim, C. J. Small LEA proteins mitigate air-water interface damage to fragile cryo-EM samples during plunge freezing. Nat. Commun. 15, 7705 (2024).
Steyaert, J. & Kobilka, B. K. Nanobody stabilization of G protein-coupled receptor conformational states. Curr. Opin. Struct. Biol. 21, 567–572 (2011).
Koide, S. Engineering of recombinant crystallization chaperones. Curr. Opin. Struct. Biol. 19, 449–457 (2009).
Zhang, K., Wu, H., Hoppe, N., Manglik, A. & Cheng, Y. Fusion protein strategies for cryo-EM study of G protein-coupled receptors. Nat. Commun. 13, 4366 (2022).
Robertson, M. J. et al. Structure determination of inactive-state GPCRs with a universal nanobody. Nat. Struct. Mol. Biol. 29, 1188–1195 (2022).
Guo, Q. et al. A method for structure determination of GPCRs in various states. Nat. Chem. Biol. 20, 74–82 (2024).
Tsutsumi, N. et al. Structure of human Frizzled5 by fiducial-assisted cryo-EM supports a heterodimeric mechanism of canonical Wnt signaling. Elife 9, e58464 (2020).
Wang, D. et al. Molecular mechanism of antihistamines recognition and regulation of the histamine H1 receptor. Nat. Commun. 15, 84 (2024).
Kumari, P. et al. Structural mechanism of CB1R binding to peripheral and biased inverse agonists. Nat. Commun. 15, 10694 (2024).
Xu, J. et al. Calcineurin-fusion facilitates cryo-EM structure determination of a Family A GPCR. Proc. Natl. Acad. Sci. USA 121, e2414544121 (2024).
Shihoya, W. et al. Structure of a lasso peptide bound ETB receptor provides insights into the mechanism of GPCR inverse agonism. Nat. Commun. 16, 3446 (2025).
Chun, E. et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976 (2012).
Wang, C. et al. Structural basis for molecular recognition at serotonin receptors. Science 340, 610–614 (2013).
Liu, W. et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236 (2012).
Asada, H. et al. Crystal structure of the human angiotensin II type 2 receptor bound to an angiotensin II analog. Nat. Struct. Mol. Biol. 25, 570–576 (2018).
Wasilko, D. J. et al. Structural basis for CCR6 modulation by allosteric antagonists. Nat. Commun. 15, 7574 (2024).
Jiao, H. et al. Structure basis for the modulation of CXC chemokine receptor 3 by antagonist AMG487. Cell Discov. 9, 119 (2023).
Yin, J., Mobarec, J. C., Kolb, P. & Rosenbaum, D. M. Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant. Nature 519, 247–250 (2015).
Liu, K. et al. Structural basis of CXC chemokine receptor 2 activation and signalling. Nature 585, 135–140 (2020).
Yin, J. et al. Structure and ligand-binding mechanism of the human OX1 and OX2 orexin receptors. Nat. Struct. Mol. Biol. 23, 293–299 (2016).
Zhang, S. et al. Inactive and active state structures template selective tools for the human 5-HT5A receptor. Nat. Struct. Mol. Biol. 29, 677–687 (2022).
Toyoda, Y. et al. Structural basis of α1A-adrenergic receptor activation and recognition by an extracellular nanobody. Nat. Commun. 14, 3655 (2023).
Jiao, H. et al. Structural insights into the activation and inhibition of CXC chemokine receptor 3. Nat. Struct. Mol. Biol. 31, 610–620 (2024).
Bloch, J. S. et al. Development of a universal nanobody-binding Fab module for fiducial-assisted cryo-EM studies of membrane proteins. Proc. Natl. Acad. Sci. USA 118, e2115435118 (2021).
Yu, J. et al. Structural basis of μ-opioid receptor targeting by a nanobody antagonist. Nat. Commun. 15, 8687 (2024).
Lu, X. et al. A large, general and modular DARPin–apoferritin scaffold enables the visualization of small proteins by cryo-EM. IUCrJ 12, 393–402 (2025).
Yao, Q., Weaver, S. J., Mock, J.-Y. & Jensen, G. J. Fusion of DARPin to aldolase enables visualization of small protein by cryo-EM. Structure 27, 1148–1155.e1143 (2019).
Che, T. et al. Nanobody-enabled monitoring of kappa opioid receptor states. Nat. Commun. 11, 1145 (2020).
Stumpp, M. T., Binz, H. K. & Amstutz, P. DARPins: a new generation of protein therapeutics. Drug Discov. Today 13, 695–701 (2008).
Sennhauser, G. & Grütter, M. G. Chaperone-assisted crystallography with DARPins. Structure 16, 1443–1453 (2008).
Batyuk, A., Wu, Y., Honegger, A., Heberling, M. M. & Plückthun, A. DARPin-based crystallization chaperones exploit molecular geometry as a screening dimension in protein crystallography. J. Mol. Biol. 428, 1574–1588 (2016).
Milovnik, P., Ferrari, D., Sarkar, C. A. & Plückthun, A. Selection and characterization of DARPins specific for the neurotensin receptor 1. Protein Eng. Des. Sel. 22, 357–366 (2009).
Schilling, J. et al. Thermostable designed ankyrin repeat proteins (DARPins) as building blocks for innovative drugs. J. Biol. Chem. 298, 101403 (2022).
Liu, Y. & Huynh, D. T. & Yeates, T. O. A 3.8 Å resolution cryo-EM structure of a small protein bound to an imaging scaffold. Nat. Commun. 10, 1864 (2019).
Castells-Graells, R. et al. Cryo-EM structure determination of small therapeutic protein targets at 3 Å-resolution using a rigid imaging scaffold. Proc. Natl. Acad. Sci. USA 120, e2305494120 (2023).
Deluigi, M. et al. Complexes of the neurotensin receptor 1 with small-molecule ligands reveal structural determinants of full, partial, and inverse agonism. Sci. Adv. 7, eabe5504 (2021).
Deluigi, M. et al. Crystal structure of the α1B-adrenergic receptor reveals molecular determinants of selective ligand recognition. Nat. Commun. 13, 382 (2022).
Heydenreich, F. M., Vuckovic, Z., Matkovic, M. & Veprintsev, D. B. Stabilization of G protein-coupled receptors by point mutations. Front. Pharmacol. 6, 82 (2015).
Rosenbaum, D. M. et al. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science 318, 1266–1273 (2007).
Shibata, Y. et al. Thermostabilization of the neurotensin receptor NTS1. J. Mol. Biol. 390, 262–277 (2009).
Lee, S., Bhattacharya, S., Tate, C. G., Grisshammer, R. & Vaidehi, N. Structural dynamics and thermostabilization of neurotensin receptor 1. J. Phys. Chem. B 119, 4917–4928 (2015).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Khoshouei, M., Radjainia, M., Baumeister, W. & Danev, R. Cryo-EM structure of haemoglobin at 3.2 Å determined with the Volta phase plate. Nat. Commun. 8, 16099 (2017).
Danev, R. et al. Routine sub-2.5 Å cryo-EM structure determination of GPCRs. Nat. Commun. 12, 4333 (2021).
Buijsse, B., Trompenaars, P., Altin, V., Danev, R. & Glaeser, R. M. Spectral DQE of the Volta phase plate. Ultramicroscopy 218, 113079 (2020).
Danev, R., Tegunov, D. & Baumeister, W. Using the Volta phase plate with defocus for cryo-EM single particle analysis. Elife 6, e23006 (2017).
Danev, R. & Baumeister, W. Expanding the boundaries of cryo-EM with phase plates. Curr. Opin. Struct. Biol. 46, 87–94 (2017).
Danev, R. & Baumeister, W. Cryo-EM single particle analysis with the Volta phase plate. Elife 5, e13046 (2016).
Danev, R., Buijsse, B., Khoshouei, M., Plitzko, J. M. & Baumeister, W. Volta potential phase plate for in-focus phase contrast transmission electron microscopy. Proc. Natl. Acad. Sci. USA 111, 15635–15640 (2014).
Bepler, T., Kelley, K., Noble, A. J. & Berger, B. Topaz-Denoise: general deep denoising models for cryoEM and cryoET. Nat. Commun. 11, 5208 (2020).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Zhong, E. D., Bepler, T., Berger, B. & Davis, J. H. CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 18, 176–185 (2021).
Herreros, D. et al. Real-space heterogeneous reconstruction, refinement, and disentanglement of CryoEM conformational states with HetSIREN. Nat. Commun. 16, 3751 (2025).
Skiba, M. A. et al. Antibodies expand the scope of angiotensin receptor pharmacology. Nat. Chem. Biol. 20, 1577–1585 (2024).
Choi, C. et al. Understanding the molecular mechanisms of odorant binding and activation of the human OR52 family. Nat. Commun. 14, 8105 (2023).
Mao, C. et al. Conformational transitions and activation of the adhesion receptor CD97. Mol. Cell 84, 570–583.e577 (2024).
Yan, P. et al. The binding mechanism of an anti-multiple myeloma antibody to the human GPRC5D homodimer. Nat. Commun. 15, 5255 (2024).
Hou, J. et al. Structural basis of antagonist selectivity in endothelin receptors. Cell Discov. 10, 79 (2024).
Ye, X. et al. Structural insights into physiological activation and antagonism of melanin-concentrating hormone receptor MCHR1. Cell Discov. 10, 124 (2024).
O’Brien, E. S. et al. A µ-opioid receptor modulator that works cooperatively with naloxone. Nature 631, 686–693 (2024).
Sun, D. et al. Structural basis of antibody inhibition and chemokine activation of the human CC chemokine receptor 8. Nat. Commun. 14, 7940 (2023).
Deneka, D. et al. Allosteric modulation of LRRC8 channels by targeting their cytoplasmic domains. Nat. Commun. 12, 5435 (2021).
Shang, G., Zhang, C., Chen, Z. J., Bai, X. -c & Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature 567, 389–393 (2019).
Coleman, J. A. et al. Serotonin transporter–ibogaine complexes illuminate mechanisms of inhibition and transport. Nature 569, 141–145 (2019).
Niu, Y. et al. Structural basis of inhibition of the human SGLT2–MAP17 glucose transporter. Nature 601, 280–284 (2022).
Bloch, J. S. et al. Structure and mechanism of the ER-based glucosyltransferase ALG6. Nature 579, 443–447 (2020).
Coupland, C. E. et al. Structure, mechanism, and inhibition of Hedgehog acyltransferase. Mol. Cell 81, 5025–5038.e5010 (2021).
Li, F. et al. Ion transport and regulation in a synaptic vesicle glutamate transporter. Science 368, 893–897 (2020).
Yan, R., Zhao, X., Lei, J. & Zhou, Q. Structure of the human LAT1–4F2hc heteromeric amino acid transporter complex. Nature 568, 127–130 (2019).
Mishra, A. K. et al. CryoEM structure of a therapeutic antibody (favezelimab) bound to human LAG3 determined using a bivalent Fab as fiducial marker. Structure 31, 1149–1157.e1143 (2023).
Parker, J. L. et al. Structural basis of antifolate recognition and transport by PCFT. Nature 595, 130–134 (2021).
Kim, J. et al. Structure and drug resistance of the Plasmodium falciparum transporter PfCRT. Nature 576, 315–320 (2019).
Trinco, G. et al. Kinetic mechanism of Na+-coupled aspartate transport catalyzed by GltTk. Commun. Biol. 4, 751 (2021).
Straub, M. S., Alvadia, C., Sawicka, M. & Dutzler, R. Cryo-EM structures of the caspase-activated protein XKR9 involved in apoptotic lipid scrambling. Elife 10, e69800 (2021).
Xie, P. et al. A fiducial-assisted strategy compatible with resolving small MFS transporter structures in multiple conformations using cryo-EM. Nat. Commun. 16, 7 (2025).
Goutam, K., Ielasi, F. S., Pardon, E., Steyaert, J. & Reyes, N. Structural basis of sodium-dependent bile salt uptake into the liver. Nature 606, 1015–1020 (2022).
Bärland, N. et al. Mechanistic basis of choline import involved in teichoic acids and lipopolysaccharide modification. Sci. Adv. 8, eabm1122 (2022).
Wu, X. & Rapoport, T. A. Cryo-EM structure determination of small proteins by nanobody-binding scaffolds (Legobodies). Proc. Natl. Acad. Sci. USA 118, e2115001118 (2021).
Acknowledgements
P.K.’s lab is supported by IISER Bhopal Start-up Grant (IISERB/R&D/2024-25/66) and ANRF (ANRF/ECRG/2024/000398/LS). M.B.’s lab is supported by the Early Career Fellowship from the India Alliance Wellcome Trust (IA/E/20/1/505691), Institute Seed Grant- IIT Jammu (SGT-100081), SERB-SRG Grant (SRG/2023/000424) and ICMR Grant (IIRP-2023-2121).
Author information
Authors and Affiliations
Contributions
All authors contributed to the development of this manuscript. S.K.S. and M.A. contributed to drafting and figure design. A.P. provided support in reviewing the draft and figures. P.K. and M.B. conceptualized and structured the overall content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary handling editors: Laura Rodriguez Perez.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Singh, S.K., Agrawal, M., Pattanayak, A. et al. Strategic advances for cryo-EM structural studies of small (<100 kDa) GPCRs. Commun Biol 9, 237 (2026). https://doi.org/10.1038/s42003-026-09516-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-026-09516-y
