
Abstract
Desert ungulates, such as Camelus bactrianus and Hippotraginae antelopes, exhibit extraordinary adaptation to extreme environment. Deciphering these genetic adaptations is critical for understanding evolutionary resilience under climate change. Here, we generate a chromosome-level genome for domestic Bactrian camel and integrate comparative genomics analyses to uncover genomic adaptation in arid-desert ungulates. We find elevated molecular evolution rates with intensified positive selection among desert-adapted lineages. Convergent positively selected genes are mainly involved in energy metabolism, and ion transport and homeostasis. In addition, we identify further evidence reveals numerous parallel amino acid substitution genes associated with lipid/sterol metabolism, particularly cholesterol biosynthesis. Cross-species metabolomics reveal lower steroid-lipid levels in fasting camel serum, suggesting that genetic adaptation promotes metabolic trade-offs for desert survival. INSIG1 involved in cholesterol biosynthesis process emerge as a key candidate. Functional validation reveals that the INSIG1 mutation enhances lipid synthesis in energy-rich hepatocytes and promotes lipolysis during fasting in genome-edited male mice. Altogether, these findings highlight lipid/sterol plasticity as a cornerstone of desert adaptation, providing insights into breeding drought-resistant livestock and advancing therapeutic strategies for human metabolic disorders.
Similar content being viewed by others

Introduction
Deserts, covering approximately one-third of Earth’s terrestrial surface1, are among the most extreme habitats, characterized by limited precipitation, pole-end temperature, and food shortage2. These harsh conditions have driven the evolution of remarkable morphological and physiological adaptations in desert-dwelling species, making deserts natural laboratories for studying convergent evolution3. Amid the pressing threat of rapid global climate change, increasing attention has been given to research on adaptation to desert environments.
Previous studies have cataloged phenotypic adaptations in desert ungulates, including higher stable hematocrit4,5,6, specialized renal morphology7,8,9 and lipid storage and mobilization capacities10. Desert ungulates, such as Bactrian camels (Camelus bactrianus) and Hippotraginae antelopes (e.g., addax (Addax nasomaculatus), scimitar oryx (Oryx dammah), and gemsbok (Oryx gazella)) exemplify “endurers” capable of thriving in hyper-arid conditions3. These lineages diverged from their non-desert relatives (e.g., Laminae and Reduncinae) 15–16 million years ago, yet independently evolved convergent physiological traits, including adaptive heterothermy, enhanced water retention, and lipid-driven metabolic flexibility11,12,13,14. Similarly, desert-inhabiting species, such as fat-tailed sheep15, Merriam’s kangaroo rat (Dipodomys merriami)16, african fat-tailed gecko and fat-tailed gerbil17, exhibit analogous adaptations. Moreover, previous studies have suggested that the Arabian oryx utilizes water derived from fat metabolism, which may account for up to 24% of its total water requirement3. These traits provide a valuable framework for exploring how arid desert adaptations are shaped by evolutionary pressure. However, studies focusing on the genetic basis of traits that facilitate desert adaptation remain relatively scarce.
Genomic studies have revealed that the adaptation of ungulates and rodents to extreme desert environments involves positive selection genes related to energy homeostasis, salt metabolism, and maintenance of water balance18,19,20,21, including AQP422 and GPX323. However, most research has focused on single species or narrow traits, leaving the genetic basis of convergent arid-desert adaptations largely unexplored. Previous studies on convergent evolution have shed light on the genetic foundations of organisms inhabiting similar environments, such as mammalian subterranean24, aquatic adaptation25, and arctic adaptation26. Increased availability high-quality genome data of ungulates11, such as addax12, scimitar oryx27, and gemsbok28, provides resources for investigating the genetic underpinnings of remarkable adaptations to arid-desert environments.
Here, we generated de novo assembled chromosome-level genome for the domestic Bactrian camel using PacBio HiFi and Hi-C sequencing. By using publicly available genomic dataset, we analyzed 22 ungulate genomes to detect convergent evolutionary signatures in desert-adapted of Camelus and Hippotraginae lineages. By integrating genome-wide scan of positively selected genes (CPSG), identification of parallel amino acid substitution (PAAS), and cross-species metabolomics, we searched for genomic signatures of convergence underlying arid-desert adaptation. Specifically, ungulates inhabiting desert environments exhibit parallel substitutions at key regulatory sites of lipid and cholesterol metabolism, suggesting enhanced lipid/sterol metabolic plasticity that may facilitate survival under extreme conditions. To test this hypothesis, we performed functional validation in genome-edited mice and complementary in vitro assays, which provided strong support for the proposed adaptive mechanism. Collectively, these findings provide insights into breeding stress-resilient livestock and understanding human metabolic disorders.
Result
A new domestic Bactrian camel reference genome assembly and annotation
To enhance the power to detect convergent genomic signatures, we firstly assembled a chromosome-level haplotype-resolved genome of the domestic Bactrian camel through a combination of PacBio circular consensus (HiFi) sequencing and high-throughput chromosome conformation capture (Hi-C) sequencing. All 36 autosome pairs and X chromosome are represented by chromosome-level scaffolds (Fig. 1A). Our assembly spanned 2.4 Gb consisting of 715 scaffolds with the contig N50 length of 58.8 Mb and scaffold N50 length of 79.9 Mb, which is better than existing camels’ assemblies (Fig. 1C, Table 1, Supplementary Data 1). The completeness of the new genome assembly that was reflected by the BUSCO value of 96.1%. The GC content was ~41.9%, and the repeats occupied 31.09% of the genome (Fig. 1B). There were more telomeric motifs and centromeric tandem repetitive sequences assembled compared with published reference camel genomes (Supplementary Fig. 1).

A Hi-C heatmaps among all chromosomes of the domestic Bactrian camel generated by Juicebox Assembly Tool91. B Chromosome architecture of the domestic Bactrian camel genome, using 1,000,000 bp windows. From the outer to inner rings: (1) chromosomes; (2) chromosomes length (Mb); (3) the number of genes; (4) the number of repeats; (5) the number of satellites; (6) the number of long interspersed nuclear elements; (7) the number of short interspersed nuclear elements; (8) the number of the long terminal repeats; (9) density of GC content. The domestic Bactrian camel illustration in this figure was entirely created by the authors using original artwork, with no third-party elements. C Comparison of camel genome between de novo assembly and reference in NCBI. Y axis shows scaffold sizes for which x percent of the assembly consisted of scaffolds of at least that size. D State of 18,430 ancestral placental mammal genes in Camelidae. E Syntenic relationships between de novo assembly in our study and published camel genomes.
Subsequently, we employed de novo, transcriptome, and homology-based gene prediction methods to annotate the genomes. In total, 28,877 genes were annotated in the improved genome (Fig. 1B). To further compare assembly quality, we employed TOGA (Tool to Infer Orthologs from Genome Alignments), utilizing 18,430 ancestral placental mammal genes, to determine the highest number of intact genes in de novo genome among other genomes in Camelidae lineages (Fig. 1D; Supplementary Data 2). Our de novo genome assembly showed a highly conserved chromosomal synteny to dromedary and wild Bactrian camel, with chromosomes showing a one-to-one homologous relationship (Fig. 1E). Overall, we assembled a high-quality chromosome-level Bactrian camel genome, providing a robust foundation for advancing our understanding of arid-desert adaptation in ungulates.
Convergent gene-wide patterns of molecular evolution in arid-desert adapted ungulates
To unravel the genetic basis of convergent desert adaptation, we analyzed 22 ungulate genomes, including Camelus and Hippotraginae lineages (Supplementary Data 3). A robust phylogeny was reconstructed (100% bootstrap support; Fig. 2A, B), confirming their divergence ~66 million years ago (Supplementary Fig. 2) and subsequent independent adaptation to arid environments (Fig. 2B). We obtained 12,489 high-confidence orthologous protein-coding genes by multiple genome synteny alignments, providing a foundation for detecting molecular convergence.

A Phylogenetic trees of the 22 species, shaded sections highlight extreme arid-desert-adapted Camelus and Hippotraginae lineages. Different numbers represent different inner and terminal nodes. B Geographical distribution of Camelus, the Bactrian camel (C. bactrianus) and Arabian camel (C. dromedarius), and the Hippotraginae species addax (Addax nasomaculatus), scimitar oryx (Oryx dammah), and gemsbok (O. gazella) in arid and semi-arid regions in the world. Different colors on the map represent different levels of drought. This map is adapted from the Global Aridity Index and Potential Evapotranspiration (Global-AI_PET) Database v3128, available under the CC BY-NC 4.0 license (https://creativecommons.org/licenses/by/4.0/). Photos of different species were adapted from Pexels (pexels.com) and are used under the terms of the Pexels License. The dataset was reclassified based on Aridity Index (AI) thresholds to delineate arid and semi-arid zones. C The boxplot shows the rate of gene-wise molecular evolution (ω, dN/dS) in arid-desert adapted lineages compared to non-arid-desert adapted branches. D The molecular evolutionary patterns of homologous genes in arid-desert adapted lineages. The foreground representing arid-adaptive lineages contrasts sharply with the background of non-arid-adaptive taxa. E Functional GO terms annotated by candidates for convergent positive selection genes.
We initially conducted examination of selective pressures and direction to test the signatures of molecular evolution in two arid-desert adapted lineages. Arid-desert lineages (which defined as foreground) experienced higher gene-wide molecular evolution rates (dN/dS) compared to non-arid-desert lineages (which defined as backgrounds) (p-value < 2.2e−16, Wilcoxon rank-sum test, Fig. 2C), and coupled with a higher proportion of genes under intensification of positive selection (p-value = 0.009, Chisq test, Fig. 2D).
To further interrogated evolutionary pressures of orthologous genes across the Camelus and Hippotraginae lineages, HyPhy BUSTED-PH29,30 model was adopted to search for evidence of diversifying positive selection. We identified 538 convergent positive selection genes (CPSGs) after correcting for multiple testing (adjusted p-value (background) >0.05, adjusted p-value (foreground) <0.05, adjusted p-value (difference between background and foreground) <0.05) (Supplementary Data 4), which enriched pathways fall into three main functional categories: energy metabolism (e.g. positive regulation of adipose tissue development, glucose homeostasis, energy homeostasis, positive regulation of cholesterol efflux), ion transport and homeostasis (e.g. cellular calcium ion homeostasis, potassium ion transmembrane transport), development (e.g. metanephros development, vasculogenesis, heart morphogenesis) (Fig. 2E, Supplementary Data 5). Functional enrichments were largely consistent with those identified in previous studies, underscoring the pivotal role of certain biological strategies in shaping the adaptations to extremely harsh desert environment3,31. We obtained novel CPSGs like GPLD1 and ATP2B4, suggesting adaptive shifts in energy utilization32 and blood pressure regulation33. Several genes such as SLC24A3, LAMA5, and CRLS1 demonstrate not only convergent positive selection in desert-dwelling ungulates but also evidence of selection pressure in more distantly related lineages, specifically observed in desert foxes31. Altogether, the adaptive convergent genes and pathways offer significant insights into the complex processes underpinning adaptation to desert ecosystems, highlighting the intricate interplay between genetic evolution and environmental challenge.
Convergent lipid/sterol metabolism adaptive evolution in arid-desert ungulates
To robustly identify convergent evolutionary signatures, we performed pairwise convergence comparisons between each terminal branches and inner nodes across Camelus and Hippotraginae lineages (Supplementary Fig. 3), as adaptive variations can emerge at any evolutionary node or species in response to arid conditions. We then employed multifaceted approach to obtain candidates. Parallel amino acid substitution (PAAS) sites (we collectively refer to “parallel” and “convergent” substitution as PAAS) were primarily detected using PCOC34 (posterior probability [PP] >0.99), considering shifts substitution according the amino acid preference profile. To minimize false positives, we applied the extended Convergence at Conservation Sites (CCS) method35, filtering PAAS to retain those at moderately conserved positions. This stringent approach identified 427 candidate genes with 457 high-confidence PAAS sites. To assess background convergence noise, we performed seven control scenarios by swapping outgroups with target lineages, and yielded 568 genes with 635 substitutions (Supplementary Fig. 3, Supplementary Data 6). Similar to the trend of echolocation in cetaceans and bats36,37,38 and transitions to Viviparity in Cyprinodontiformes39, there was no excessive genome-wide convergence among arid-desert-adapted lineages compared to their outgroups, implying arid adaptation primarily targets specific loci. Subsequently, we further perform analysis to screen candidates with significant convergent signal at the gene level. By comparing observed substitution frequencies to neutral expectations in branch analysis, we confirmed 171 candidate genes exhibit statistical significance in arid-adaptation lineages relative to empirical controls after multiple correction analyses (Conv_cal method, Poisson test, Benjamini–Hochberg, FDR < 0.1)38 (Supplementary Fig. 3, Supplementary Data 6). To further detect the proteins evolved in similar directions among convergent lineages, we applied the CSUBST40 to calculate ωC (ωC = dNC/dSC) by distinguishing the effects of natural selection from genetic drift and phylogenetic errors. This method compares observed-to-expected ratios of nonsynonymous (dNC) and synonymous (dSC) substitutions across various phylogenetic branches. We revealed 157 candidates with ωC > 1 in arid-adapted lineages (Fig. 3A, Supplementary Fig. 3, 4, Supplementary Data 6), exhibiting significantly higher convergent evolutionary rates (ωC) than controls (Wilcoxon Rank Sum Test, p-value = 2.91e−16).

A The number of conservative parallel amino acid substitutions sites (red) and genes (blue) detected in 25 pairwise comparisons among Camelus and Hippotraginae lineages. B Functional GO terms enriched by parallel amino acid substitution genes. C Bar chart showing the percentage of super-class for identified metabolites in fatty acids, sterol lipids, and other metabolites of fasting camel and fasting mouse. Heatmap further showing the differences in identified fatty acid and sterol lipid metabolites super-class between camel and mouse. Red: sterol lipids; Green: fatty acid. n = 4 samples for male mice and camel. Animal silhouettes in (C) were obtained from PhyloPic (http://phylopic.org), which provides free silhouette images of organisms. The silhouettes used here are available for reuse under the Creative Commons Attribution 4.0 International License (CC BY 4.0; https://creativecommons.org/licenses/by/4.0/).
Despite strong signals of convergence, only ~7% of PAAS genes showed evidence of positive selection, likely due to pleiotropic constraints or transient or episodic selection regimes41. Examples include PLEKHA7 act on salt-sensitive hypertension42, LAMA5 associated with kidney disease43, BUD1344 and ECI245 affect lipid/sterol metabolism process, and et al. To resolve this paradox, we employed the Mixed Effects Model of Evolution (MEME)41, which revealed that nearly all PAAS sites (93%) were under episodic positive selection (β + >α, p < 0.05; Supplementary Data 7). These candidates highlight loci under strong selection in arid environments, independent of phylogenetic or stochastic effects.
Functional analysis of PAAS genes revealed significant enrichment in cholesterol biosynthesis (Benjamini–Hochberg; adjust p-value = 0.0293) (Fig. 3B, Supplementary Data 8), a signal absent in control analyses (Supplementary Data 9). To validate these genomic findings, serum metabolomic profiling were conducted on fasting camels and mice, revealing a statistically significant elevation in the proportion of fatty acid-related metabolites, alongside a reduced proportion of steroid lipids in camel serum compared to mice (Fig. 3C, Supplementary Data 10, 11). The results are consist with previous studies that dromedaries have lower plasma cholesterol concentration and higher hepatic cholesterol than sheep and cattle46,47. Collectively, these results underscore lipid/sterol metabolic rewiring as a key adaptive strategy in desert ungulates, balancing energy storage during scarcity and rapid mobilization under fasting conditions, as a critical advantage for survival in extreme aridity.
The parallel amino acid substitution in INSIG1 impact the lipids/sterols metabolism demonstrated by cell experiments
Among candidates in cholesterol biosynthetic process, INSIG1 and NPC1L1 pronounced the strongest convergent signals, with PAAS at highly conserved sites (site-wise posterior probability of convergence >0.95; Supplementary Fig. 5). INSIG1 regulates lipid and sterol synthesis via negative feedback, while NPC1L1 mediates cholesterol transport48,49. PAAS in INSIG1 and NPC1L1 altered the 3D-structure and hydrophobicity (Fig. 4A, B, Supplementary Fig. 6). In addition, INSIG1 and NPC1L1 had lower expression levels in camel liver tissue using available data comparing to cattle, pig and human (Supplementary Fig. 7, Supplementary Data 12). Hence, the parallel substitutions caused the influence on characteristics of proteins, suggesting their involvement in adaptation to severe drought.

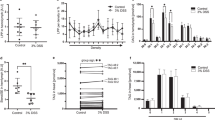
A Parallel substitutions at conserved sites of INSIG1 protein in Camelus and Hippotraginae lineages. B Mutation site in the 3D protein structures of INSIG1 was indicated in green. The red chains represent the normal protein structure, and the blue chains represent protein structure after parallel mutations. The arrows indicate the locations where structural changes occurred before and after the mutation. The hydrophobicity prediction of INSIG1. Amino acid substitution of H150R in INSIG1 reduces the amino acids hydrophobicity from 146 to 154. C Wild-type or mutant INSIG1 Co-IPed with SCAP were detected by western blotting. D Representative immunofluorescence images of Dil-LDL uptake in HepG2 cells from three independent experiments. Confocal microscopic images represent the fluorescence intensity of Dil-LDL (red) and DAPI (blue). Scale bars, 100 μm. E Relative DiI-LDL fluorescence levels were quantified in HepG2 cells transfected with empty vector (Con), INSIG1, or INSIG1(H150R). INSIG1 overexpression enhanced LDL uptake, whereas the H150R mutation significantly reduced LDL uptake compared with wild-type INSIG1. F OA/PA treated HepG2 cells were stained with ORO and lipid accumulation was visualized under a microscope at 100× magnification. Scale bars, 100 μm. G Relative ORO staining levels in the same groups showed increased neutral-lipid accumulation in cells expressing INSIG1(H150R) compared to wild-type INSIG1. Data are presented as mean ± SEM from at least three independent experiments All experiments were repeated at least three times, and representative data are presented. Values and error bars represented means and ±SEM from multiple independent biological replicates (DiI-LDL (n = 4 biologically independent experiments); ORO (n = 5 biologically independent experiments)). Asterisks indicate significant differences. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, NS: not significant by one-way ANOVA with Tukey’s post-hoc test.
Previous research has demonstrated that a single amino acid mutation, D205A, plays a critical role in the function of INSIG1 proteins in regulating cholesterol homeostasis50. Our study found INSIG1 (H150R) substitution under positive selection, located within its functional domain, influenced the stability of the proteins (∆∆G = −0.425) and disrupted binding pockets (Supplementary Fig. 8). INSIG1 can trigger to bind the SREBP cleavage–activating protein (SCAP) to reduce synthesis of cholesterol, fatty acids51. To assess the functional effect of mutations on the binding efficiency of INSIG1 to SCAP, we co-transfected with Flag-SCAP in Human liver cancer cell line (HepG2) cells. The results of CO-IP revealed that mutant INSIG1(H150R) blocked binding of SCAP (Fig. 4C, Supplementary Fig. 9, Supplementary Data 13). To directly assess the impact of mutation on lipid/cholesterol synthesis, we constructed lipid accumulation model using HepG2 cells (hepatoblastoma cell line) treated with the free fatty acid palmitic acid/oleic acid in vitro. Oil Red O (ORO) results showed that the INSIG1(H150R) mutation significantly promoted lipid accumulation in hepatocytes compared with wild-type INSIG1 (Fig. 4F, G; Supplementary Data 14). We then constructed wild-type and mutant INSIG1 plasmids, transfected HA-INSIG1 and HA-INSIG1(H150R) alone, and found that the mutation resulted in lower LDL uptake levels compared with the wild-type in HepG2 cells (Fig. 4D, E; Supplementary Data 15). By decoupling lipids/sterols homeostasis from feedback inhibition, desert ungulates may optimize energy reserves-a vital strategy for surviving unpredictable food scarcity in arid ecosystems.
The Insig1 H132R/H132R mice verified mutation promoted lipid/sterol synthesis and mobilization
To better understand the role of the INSIG1 mutation in vivo, we generated a mice model through point mutation employing CRISPR-Cas9 gene editing. The gene-edited mice were sequenced and confirmed no evidence for off-target effects (Fig. 5A). There was no statistically significant difference between cholesterol and triglyceride concentrations in serum and liver of mutant and wild-type mice after fasting for 14 h in our study (Supplementary Fig. 10A, Supplementary Data 16). Consistent with the Insig1−/− and Insig2−/− mouse phenotype, the levels of plasma cholesterol and free fatty acids showed no change52. We further performed the liver and visceral fat transcriptome sequencing of mutant and wild-type fasting male mice. Sterol regulatory element-binding proteins (SREBPs) as master regulators of cholesterol synthesis, uptake, and lipid metabolism, exhibiting marked downstream effects in the edited mice53,54,55: LDLR, SCD1, and PCSK9 were reduced in liver; ACACB and ACLY were reduced in visceral adipose; and LPIN1 increased in both tissues (Supplementary Fig. 11). The result of differentially expressed genes (DEGs) were significantly enriched pathways related to cholesterol, lipid and glucose metabolism process (Supplementary Fig. 12A, B). Noticeably, the mutant mice markedly enhanced the expression of genes responsible for lipid/sterol degradation, such as LPIN151,56, APOA457,58, PDK459 and et al., as well as CPSG like ADCY560 and CRLS161,62,63, while genes as GOS264,65 and PNPLA366 related to lipid/sterol synthesis were significantly down-regulated (Fig. 5B, C, Supplementary Data 17). We further validated differential expression of PLIN1 and G0S2 between wild-type and mutant mice by RT-qPCR, and the results were concordant with the transcriptomic data (Supplementary Fig. 13, Supplementary Data 18, 19). Consistently, comparing to wild-type mice, the result of gene set enrichment analysis (GSEA)67 of liver and visceral fat revealed significantly upregulated enrichment in “fatty acid oxidation”, “negative regulation of fatty acid biosynthetic process” in the Insig1H132R/H132R mice, while downregulated “response to cholesterol” and “fatty acid derivative biosynthetic process” (Fig. 5D, E). Given previous studies and our own evidences both point out the effect of INSIG1 on metabolism process, we further investigated the downstream influence on the liver metabolite using liquid chromatography–mass spectrometry (LC-MS). We found differential metabolites between Insig1H132R/H132R and wild-type mice associated with lipid/sterol metabolism, including Cholesterol sulfate, 15-KETE, Aminoadipic acid and et al. (Supplementary Fig. 10B, Supplementary Data 20). These results align with transcriptional profiles, suggesting INSIG1 H150R mutation accelerates lipid mobilization during energy scarcity as a key survival strategy for desert ungulates.

A Insig1H132R/H132R gene-edited mice strategies. Human H150R and mouse H132R are orthologous at the same conserved INSIG1/Insig1 site; numbering is species-specific. Animal silhouettes obtained from PhyloPic (http://phylopic.org), which provides free silhouette images of organisms. The silhouettes used here are available for reuse under the Creative Commons Attribution 4.0 International License (CC BY 4.0; https://creativecommons.org/licenses/by/4.0/). Differentially expressed genes in liver (B) and visceral fat (C) of homozygous and wild-type mice. PCA of the liver and visceral fat transcriptomic datasets between mutant and wild-type mice. n = 3 samples of homozygous and wild-type male mice. GSEA analysis showed enrichment of DEGs in liver (D) and visceral fat (E) between wild-type mice and Insig1H132R/H132R mice. NES, normalized enrichment score.
Discussion
Comparative genomic analyses focusing on convergent evolution have emerged as a powerful approach for elucidating the genetic underpinnings of species-specific traits and adaptations to environmental challenges68,69,70. In this study, we de novo assembled a chromosome-level, haplotype-resolved Bactrian camel genome and conducted an analysis of convergent evolution between the Camelus and Hippotraginae, the lineages of ungulates adapted to arid desert environments. Comparative genomics, along with cross-species metabolomic and comparative transcriptomic analyses, confirmed that lipid/sterol metabolism is crucial for arid-desert adaptation. Additionally, experiments and gene-editing of Insig1H132R/H132R mice demonstrated that a single amino acid substitution site putatively underlies the enhanced synthesis and mobilization of lipids/sterols in vivo.
Convergent evolutionary events can be influenced by various biological factors like incomplete lineage sorting (ILS) and introgression71, background noise resulting from theoretical models72 and random convergence36 can all compromise the fidelity of convergent signatures. To enhance the robustness in pinpointing adaptively convergent candidates, we implemented a multi-layered analytical pipeline. Firstly, PCOC34 and CCS approaches73 were combined to identify high-confidence parallel amino acid substitutions (PAAS) sites. Seven non-arid ungulate lineages served as empirical controls to diminish background noise. Finally, gene-level significance was assessed via Conv_cal38 and CSUBST40, ensuring retained candidates reflect arid-specific adaptation rather than random drift. While no method can fully eliminate false positives, this integrative approach significantly enhances confidence in our findings.
Lipid metabolic plasticity is a recurring theme in species adapting to extreme environments. High-altitude yak74, arctic muskox and reindeer26, hibernating species like bears75,76 and cave-dwelling creatures like cavefish77, have developed significant lipid metabolic adaptability. Strong lipid storage and rapid mobilization capabilities are putatively conserved strategies to meet energy demands in extreme environments78,79,80. Similarly, desert animals face dual challenges of energy scarcity and water deprivation15,31,81. Our study revealed that cholesterol biosynthesis exhibits the strongest convergent signal (Supplementary Fig. 5), and highlighted two potential adapted convergent genes INSIG1 and NPC1L1, essential for maintaining cholesterol balance48,49,82. For arid-desert adapted ungulates, boosting lipid metabolism is essential not only for fulfilling energy requirements but also for coping with extreme water scarcity. As the lipid oxidation yields more metabolic water than protein and carbohydrate83. And the cholesterol levels are crucial for maintaining internal water balance in camels under dehydration conditions84.
Furthermore, the convergent substitution of single amino acid in INSIG1 stood out driving lipid/sterol metabolism shift suggested by both in vitro and in vivo experiments. Notably, the mutation of INSIG1 has also been found in other species known for pronounced fat deposition capabilities, in which hibernating bear and polar bears exhibit elevated cholesterol levels (Supplementary Fig. 14)85,86,87. Our results revealed significant upregulation of PDK4 in the visceral fat of Insig1H132R/H132R mice, PDK4 plays a key role in metabolic adaptability during hibernation by facilitating extensive lipid storage and utilization59,80. Our finding provided additional evidence of INSIG1 as a promising therapeutic target for combating diseases related to lipid/sterol dysregulation. This observation is in concordance with previous studies illustrating that single amino acid variations can exert the profound biological impact. For instance, in cavefish, coding mutations in MC4R enhanced appetite and starvation resistance78, P211L mutation in INSRA increased weight and insulin resistance88. The Q247R mutation in RETSAT provided novel insights into the genetic mechanisms of hypoxia adaptation and offers new therapeutic targets for pulmonary arterial hypertension and right ventricular hypertrophy89. Therefore, we propose that PAAS sets may represent loci of significant biological relevance.
Additionally, we detected convergently positively selection genes related to lipid/steroid metabolism, such as GPLD1 and LANCL2. GPLD1 is significantly differentially expressed across species and possesses conserved parallel amino acid substitution sites (pp > 0.99), although it was filtered out in the gene-wise level pipeline (Supplementary Fig. 15). Collectively, our study furnishes additional evidence supporting the indispensable role of lipids/sterols in arid-desert adaptation.
Here, we employed comparative genomics approaches to further elucidate the potential molecular mechanisms underlying the adaptation to extreme desert environments. Future integration of techniques such as ChIP-seq and ATAC-seq with comparative genomics to identify regulatory variations and elements is essential for studying and understanding environmental adaptation. Despite these limitations, by integrating multi-omics approaches, including cross-species comparative genomics, transcriptomics, and metabolomics, our study elucidates the genetic underpinnings of ungulates’ adaptation to arid-desert environments, with a particular focus on the pivotal role in lipid/sterol metabolism. Importantly, we combined multi-omics approaches with cellular assays and gene-edited mouse models, which allowed us to directly validate the functional consequences of key mutations on lipid/sterol metabolism. The in vitro and in vivo experiments provide mechanistic evidence linking specific variants to altered INSIG1-SCAP interactions and downstream metabolic regulation. Moreover, these findings highlight potential gene-editing targets for breeding stress-resistant livestock, inform strategies for developing drought-resistant species under climate change, and deepen our understanding of human metabolic disorders, including obesity, diabetes, and non-alcoholic fatty liver disease (NAFLD).
Method
Samples collection
In this study, the tissue samples used for genomic sequencing were collected from a healthy female domestic Bactrian camel. A healthy adult female domestic Bactrian camel (Camelus bactrianus) was sourced from Inner Mongolia with veterinary certification of good health. The animal was not used in prior experimental procedures and was maintained under standard husbandry before tissue collection.
For terminal sampling, the camel was deeply anesthetized with an intravenous combination of 0.3 g xylazine and 0.9 g ketamine, followed by euthanasia via 59.6 g sodium pentobarbital administered intravenously. Death was confirmed by loss of corneal reflex and cardiac arrest before tissue sampling. Whole blood (~200 mL) was collected via jugular venipuncture into EDTA-coated tubes for genomic DNA extraction. After collection, the blood samples were immediately stored at −80 °C until further processing. Within ~10–15 min post-mortem, liver tissue (more than 500 mg) was dissected, rinsed briefly in ice-cold phosphate-buffered saline (PBS), snap-frozen in liquid nitrogen, and stored at −80 °C until Hi-C library construction. All samples were subsequently shipped on dry ice to Novogene (Beijing, China) for genomic DNA extraction, Hi-C library preparation and sequencing.
For the mouse tissue experiments, male wild-type and genome-edited mice aged 8–12 weeks were bred and housed under specific-pathogen-free conditions at the Animal Core Facility of Northwest A&F University (Shaanxi, China; certificate no. SCXK [SHAAN] 2017-003), with controlled temperature and humidity, a 12 h light/dark cycle, and free access to standard chow and water. For terminal sampling, mice were deeply anesthetized with isoflurane inhalation followed by euthanasia via cervical dislocation. Death was confirmed before dissection. Liver and subcutaneous epididymal white adipose tissue (eWAT) were rapidly collected within 5–10 min post-mortem, rinsed briefly in ice-cold PBS, blotted dry, snap-frozen in liquid nitrogen, and stored at −80 °C until further analyses.
For metabolomics analysis, additional blood samples were collected from adult male domestic Bactrian camels (n = 4) sourced from Inner Mongolia with veterinary certification of good health and adult male mice (n = 4) from the Animal Core Facility of Northwest A&F University (Shaanxi, China). All animals were fasted for 12 h prior to blood collection (food withdrawn, water available ad libitum). Camel blood was drawn by jugular venipuncture; mouse blood was collected via the retro-orbital plexus under light isoflurane anesthesia using sterile needles. Blood samples were allowed to clot at room temperature for 30 min and then centrifuged at 1500 × g for 10 min at 4 °C to obtain serum. The resulting serum was aliquoted and stored at −80 °C until metabolomic profiling.
We have complied with all relevant ethical regulations for animal use. All procedures were reviewed and approved by the Northwest A&F University Animal Care Committee (Approval No. DK2022049). We took all necessary precautions to ensure animal welfare during tissue collection, and all procedures adhered to ethical guidelines for animal care. To ensure compliance with experimental standards and animal welfare, all operations followed the guidelines of the Animal Ethics Committee, and all sampling processes were carried out under the supervision of trained personnel, ensuring both animal welfare and the reliability of experimental data.
Genome sequencing
Genomic DNA (gDNA) extraction as well as CCS/PacBio long-read library preparation and sequencing, and Hi-C library preparation and sequencing were performed by Novogene (Beijing, China) according to the vendor’s standard operating procedures.
Genomic DNA (gDNA) was extracted from the blood for HiFi reads generation. Genome sequencing was performed using CCS (Circular Consensus Sequencing Mode) on the Pacbio Sequel II platform. Finally, 37.59 Gb, 38.86 Gb, and 40.32 Gb of CCS reads in 3 cells were yielded, respectively. In addition, liver tissue collected from the same camel was used for Hi-C library construction, and 206.173 G raw data were obtained using the Illumina sequence. To ensure the quality of information analysis, raw reads must be filtered by the following criterion: (1) Remove the reads pair with adapter; (2) Remove paired reads that contain more than 10% N content in a single-end read. (3) Remove paired reads that contain low-quality (Q ≤ 5) bases of more than 50% of the read length ratio. We ended up with 204.135 G of clean data, which were used for subsequent analysis.
Genome assembly and annotation
We first extracted fasta from bam files by “samtools fasta”, and then provided merged fasta to HiFiasm90. We used generate_site_positions.py in Juicer91 to pre-calculate the position of the enzyme restriction sites with respect to the draft genome sequence (generate_site_positions.py MboI camel draft.fa). Identifying the precise location of each restriction site within the genome is crucial for accurately mapping the fragments back to the reference genome and ensuring that the observed interactions between genomic regions are based on accurate positional relationships. And a draft assembly was obtained by running Juicer with the raw fastq files from sequencing a Hi-C library (juicer.sh –g draft –s MboI –z draft.fa –y draft_MboI.txt –p assembly). Script run-asm-pipeline.sh in 3D de novo assembly (3D-DNA) pipeline combined with a review step in Juicebox Assembly Tools91 to yield highly accurate assemblies. The candidate 157 chromosome-level scaffoldings were manually reviewed using Juicebox assembly tools until the overall heatmap matched the characteristics of chromosome interaction. Finally, we got three preliminary assemblies including one monoploid assembly and two haploid assemblies, which spanned 2.4 Gb (Haploid), 2.2 Gb (Haploid-1), and 2.3 Gb (Haploid-2). The transposable elements were identified using RepeatMasker (v4.1.1)92 (RepeatMasker -pa 20 -engine ncbi -species Camelidae -xsmall -s -no_is -cutoff 255 -frag 20000 -dir. -gff de novo genome). As a complement, tandem repeats composed of a motif occurring twice or more were predicted using TRF v4.0993. To annotate telomeres, we searched for clusters of (AACCCT)n repeats throughout the genomes using the Telomere Identification toolkit (Tidk, v0.2.0) (https://github.com/tolkit/telomeric-identifier) and RepeatMasker. The completeness of the de novo genome was assessed by Benchmarking Universal Single-Copy Orthologs (BUSCO) with “mammalia_odb10” 94.
To perform gene annotation on the de novo genome, we integrated three complementary approaches: ab initio prediction, transcriptome-guided annotation, and homology-based inference. For ab initio prediction, AUGUSTUS (v3.4.0)95 was trained using an iterative optimization protocol, wherein initial gene models derived from the Bactrian camel reference annotation, wherein initial gene models derived from the Bactrian camel reference annotation20 were randomly partitioned into training (80%) and test (20%) sets, ensuring a minimum of 200 loci for parameter calibration. After five rounds of cross-validation to refine species-specific parameters (e.g., splice site motifs, codon usage), the optimized model was applied to annotate the full genome. We collected publicly available transcriptomic data from 38 samples of six adult Bactrian camel tissues (liver, lung, adipose, renal medulla, muscle, and pancreas) from NCBI for transcriptome annotation. The corresponding SRA accessions are SRP24745396, SRP014573, SRP122491, and SRP14853597 (Supplementary Data 21). The transcriptome data were first aligned to the genome via HISAT2 (v2.2.1), followed by genome-guided assembly with Trinity (v2.15.1)98 using parameters --genome_guided_bam all.sort.bam --max_memory 50G --genome_guided_max_intron 10000 --CPU 6. For homology-based annotation, protein sequences from three camelid species (C. dromedarius, C. ferus, C. bactrianus) were aligned to the genome using miniprot (v0.7) with thresholds of ≥70% sequence identity and coverage98. The three sets of gene models were integrated through EvidenceModeler (EVM v1.1.1)99, prioritizing transcriptome-derived evidence, followed by homology and ab initio predictions. Specifically, we assigned weights of 5, 3, and 2 to the TRANSCRIPT, PROTEIN, and ABINITIO_PREDICTION evidence, respectively, and then performed the integration using the following script: evidence_modeler.pl --genome genome.fa --weights weights.txt --gene_predictions denovo.gff --protein_alignments proteinprediction.gff --transcript_alignments transcripts.fasta.transdecoder.genome.gff3 evm.out. Finally, annotation completeness was validated using TOGA (v1.0)100 against 18,430 conserved mammalian orthologs, achieving >95% intact gene recovery, thereby ensuring high-confidence gene structures for downstream analyses.
One-to-one orthologous genes and genome-scale phylogeny
Orthologous genes between 22 species (Supplementary Data 3) were identified by Multiple genome synteny alignments. Previous evaluations on simulated data have indicated that the differences are minimal in gene region while whole-genome alignments101,102. Over closer evolutionary distances, these alignment tools perform very well across all annotated regions.
The cattle (Bos taurus, UCD1.2) genome as reference, we utilized the LAST (version last1205) for pairwise genomic alignment which means the genome of each species is compared with the reference genome. In this stage, we first tried to find suitable score parameters for aligning the given sequences (lastdb -P0 -uMAM4 -R00; last-train -P0 --revsym --matsym --gapsym -E0.05 -C2 index_name species.fa > species.mat) and align similar sequences (lastal -m 100 -E 0.05 -C2 -P20 -p species.mat index_name species.mat ruminant.species.fa | last-split -m1 > result.maf), after that, multiple alignment files were gained using last-split and maf-swap toolkit. The following step is merging multiple alignment files (MAF) through Multiz (Version 11.2). maf_project was employed to extract MAF blocks that include a given reference sequence or species.
In order to obtain one-to-one orthologous genes, we converted the MAF files into a list format, where each line corresponds to a genomic position and the aligned sequences from each species at that position. Finally, we extracted the CDS regions and related positional information from the cattle’s GFF file. Based on the base correspondence obtained from the previous step, we extracted the gene sequence information for each species at these positions. If multiple transcripts for gene in the same species were recovered, we kept the longest. For each gene, we generated a file containing the FASTA sequences from all 22 species. we adopt in-house Perl scripts to gain 18,719 orthologous genes. After that, we used MACSE v2103 to further align and split each orthologous exon, taking into account the characteristics of protein coding, such as using explicit codon evolution models and considering insertions, deletions, and reading frames. Finally, totally 12489 orthologous protein-coding genes were retained for further analysis after removing the low mass alignment.
We also obtained Fourfold Degenerate Synonymous Site (4DTv) to construct a phylogenetic tree. The following step is that using Gblocks104, and trimal105 methods trim 4DTv to capture the conserved blocks/segments that may be more reliable regions from which to compare evolutionary rates. Finally, the phylogenetic tree was inferred by IQ-TREE106 (iqtree -s 4Dsite.fa-gb -nt 4 -bb 1000 -m TEST) which apply a fast and effective stochastic algorithm by maximum likelihood. Phylogenetic relationships were consistent across different approaches. To evaluate precisely the divergence times in ungulates, our whole genome alignments across 22 species and utilized the MCMCtree program in PAML (v4.9)107 (http://abacus.gene.ucl.ac.uk/software/paml.html) to infer the divergence times. Fossil dates were obtained from the Timetree website (http://www.timetree.org/) and previously published articles11,108.
Molecular evolution analyses
We used coding sequences extracted from the whole-genome alignment to identify evidence for positive selection. Each coding sequence was then realigned using the “refineAlignment” in MACSE v2.06103, which generated both nucleotide and amino acid alignments for each coding region. We further adapted the “exportAlignment” to replace all frameshifts with gaps, convert stop codons at the end of sequences into gaps, and substitute all internal stop codons with “N”.
We defined the two arid-adaptive lineages, namely the Camelus and Hippotraginae lineages, as the foreground lineages, while other non-arid-adaptive species were defined as the background. And then, we used RELAX in Hyphy29,109 to test the gene-wide evolved pattern in arid-desert-adapted lineages by inferring a relaxation parameter K. We classified the patterns of selection pressure acting on genes. If a gene meets the criterion dN/dSforeground > dN/dSbackground and K > 1 (Likelihood Ratio Test, LRT, Benjamini–Hochberg, adjust p-value < 0.05), it is considered to be under intensified positive selection. Conversely, if a gene meets dN/dSforeground > dN/dSbackground but K < 1 (Likelihood Ratio Test, LRT, Benjamini–Hochberg, adjust p-value < 0.05), it is under relaxed positive selection. For intensified purifying selection, a gene must meet dN/dSforeground < dN/dSbackground and K > 1 (Likelihood Ratio Test, LRT, Benjamini-Hochberg, adjust p-value < 0.05). Lastly, if a gene satisfies dN/dSforeground <dN/dSbackground and K < 1 (Likelihood Ratio Test, LRT, Benjamini–Hochberg, adjust P-value < 0.05), it is under relaxed purifying selection. Our results showed that intensified positive selection played prominent roles in arid-desert adaptation. Convergent positive selection genes (CPSG) were identified by BUSTED-PH module and correcting for multiple testing (adjusted P-value (background) > 0.05, adjusted P-value (foreground) < 0.05, adjusted P-value (difference between background and foreground) < 0.05), Benjamini–Hochberg. Gene Ontology (GO) enrichment analysis was conducted by KOBAS (Xie et al. 2011).
We also employ the Mixed Effects Model of Evolution (MEME) (Murrell et al. 2012) in hyphy to test whether PAAS is under positive selection pressure. For each site, MEME infers two ω values and the probability of evolving under these ω values for a given branch. To infer ω, MEME calculates α (dS) and two distinct β (dN), namely β− and β+. A site is inferred to be under positive selection pressure when β+ > α and the likelihood ratio test indicates p < 0.05.
Detection of parallel amino acid substitutions
At the beginning of identifying parallel amino acids substitution (PAAS) shared between arid-desert-adapted lineages, we inferred the ancestral sequence through Codeml program PAML (V4.9)107 in the internal nodes of 22 species phylogenetic tree, and then performed paired convergence analysis during Camelinae and Hippotraginae lineages, which includes three species and two ancestral nodes, respectively. In addition, to make a reliable signal of convergent molecular evolution at the amino acid level, four methods were conducted to try to find reliable PAAS. As the first measure, Profile Change with One Change (PCOC) pipeline34, can detect not only the same parallel substitutions of amino acids sites but also convergent shifts with similar biochemical properties that correspond to phenotypically convergent clades. We employed the PCOC to get a relaxed set of convergent genes in 25 pairwise combinations of two arid-desert-adapted linages, and the posterior probabilities (PP) for the PCOC model were set to greater than 0.99. And then, we expand CCS73, a strict method was applied in order to filter out noises as much as possible. In our study, considering that there may be more than one amino acid substitution at a particular site that contributes to the convergent phenotype under some circumstances, so moderate conservative sites follow three characteristics in foreground lineages: from different ancestral state to specific derived state, or from specific ancestral state to specific derived state, or from specific ancestral state to different/any derived state (Supplementary Fig. 3). Seven control groups were analyzed using the same procedure to obtain the corresponding sites and their gene sets. In developing the approach of Convergence Event Counting and Probability Calculation (Conv_cal)38, molecular convergence was considered to include both parallel substitutions which means identical substitutions in the target clades derived from the same ancestral amino acids and convergent substitutions which means the same substitutions changed from different ancestral amino acids. We herein collectively refer to these substitutions in all methods as parallel amino acid substitutions. To further exclude noise resulting from random amino acid substitutions, we put the JTT-fsite model of the Conv_cal method to figure out the observed number of convergence events through protein sequence alignments, and calculate the expected total probability of parallel and convergent events accordingly. Moreover, the Poisson test was employed to eliminate the noise generated by random amino acid substitution, and then applied the Benjamini–Hochberg (BH) method to correct the P-values, setting the false discovery rate (FDR) threshold at 0.1, to eliminate sampling bias between the experimental and control groups. Finally, CSUBST40 were used to detect adaptive substitutions that occur at the same protein site in multiple independent branches by calculating ratio between non-synonymous and synonymous substitution rates. We retained candidates with ωc > 1 in any catalog among convergence: omegaCany2spe and omegaCspe2spe, discordant convergence: omegaCdif2spe, and other profile convergence: omegaCspe2dif and omegaCspe2any. “csubst site” command can produce site-wise convergence/divergence probabilities with bar charts.
Protein 3D structure simulation
We used I-TASSR110,111,112 to generate a homology model of the proteins (INSIG1 and NPC1L1) based on the human protein. Alphafold2113 was used to conduct three-dimensional (3D) structure simulations to examine the possible effects of these mutations. Homology modeling was used the protein sequences of the human in the Alphafold2. Structure visualization and manipulation were done in UCSF Chimera114. ProtScale (https://web.expasy.org/protscale/) were used to predict the hydrophobicity changes of INSIG1 and NPC1L1 between wild-type and mutant amino acids. FoldX115 was used to calculate the impact of point mutations on free energy, thereby assessing the effects on protein stability, folding, and binding.
Metabolome analysis
Animals were fasted for 12 h (food withdrawn, water ad libitum). Housing was adjusted to prevent access to chow during fasting (timed removal, single-housing if necessary). Serum from camels and mice, fasted for twelve hours, was collected for off-target metabolomics analysis. Untargeted metabolomics was performed by Nuomi Metabolomics (Nanjing, China). The company provided the raw mass spectrometry files and the processed quantitative metabolite matrix; both were used for downstream analyses. Through peak detection, peak filtering and peak alignment processing, the material quantity is obtained, and the data is corrected by the area normalization method to eliminate systematic errors. For biochemical identification, Human Metabolome Database (HMDB)116 (http://www.hmdb.ca), massbank117 (http://www.lipidmaps.org), mzclound118 (https://www.mzcloud.org), KEGG119 (https://www.genome.jp/kegg/) and Pano Mick’s self-built standard product database (https://www.panomix.com/) were used. These characteristics include retention time, molecular weight to charge ratio (m/z), and associated chromatographic data (including MS/MS spectra). We ultimately identified 168 and 318 metabolites from the serum metabolomes of camels and mice, respectively, with four biological replicates. Enrichment analysis was conducted in MetaboAnalyst 5.0120 using the compound IDs to map to the 35 super chemical class metabolite sets based on chemcial structures. Differential metabolites between Insig1H132R/H132R edited mice and wild-type mice were screened based on the thresholds of VIP > 1, p-value < 0.05, and abs(log2FoldChange) > 1.
Cell culture and transfection
HepG2 cells (Qingqi, China) were cultured in DMEM medium (Bio-Channel, China) supplemented with 10% fetal bovine serum (TIANHANG, China) and 1% penicillin-streptomycin. HepG2 cells were cultured in a constant-temperature cell culture incubator set at 37 °C with 5% CO2. Before transfection, HepG2 cells were seeded in 6-well plates at a density of 6 × 105 cells/well, and when the cell density reached about 70%, the optimized vector was transfected into HepG2 cells with PEI 40 K Transfection Reagent (Servicebio, China). Cell lysates were collected at 24 h and 48 h post-transfection for subsequent RT-qPCR and co-immunoprecipitation (Co-IP) analyses.
Co-immunoprecipitation
HepG2 cells were cultured in 6-well plates until reaching ~80–90% confluence. After removing the culture medium, cells were gently washed twice with 1× PBS. Each well was then lysed with 200 μL of Co-IP/WB Tissue/Cell Lysis Buffer containing protease inhibitors (Affinibody, AIWB-012) and incubated on ice for 15 min. Lysates were centrifuged at 12,000 × g for 5 min at 4 °C, and the supernatants were collected as total protein extracts.
Protein A/G Magnetic Beads (MedChemExpress, HY-K0202) were thoroughly resuspended prior to use. Fifty microliters of beads were washed three times with 400 μL of binding/wash buffer (PBST, pH 7.4). The washed beads were incubated with HA antibody (Abmart, TT0050; final concentration 50 μg/mL) or isotype control IgG (Abmart, B30011) at 4 °C for 2 h with gentle rotation (10–15 rpm) to form antibody–bead complexes. Following incubation, the beads were separated using a magnetic stand and washed four times with binding/wash buffer.
Prepared cell lysates were then added to the antibody–bead complexes and incubated at 4 °C for an additional 2 h with gentle rotation (10–15 rpm) to capture antigen–antibody complexes. Subsequently, beads were washed four times with binding/wash buffer to remove nonspecific proteins. Bound proteins were eluted with acidic elution buffer (0.15 M glycine, pH 2.8), and the eluates were immediately neutralized with 0.1 M NaOH (1/10 of the total volume). Co-Immunoprecipitation Antibody Information were provided in Supplementary Data 13.
Western blot analysis
Eluted proteins were mixed with 5×SDS-PAGE loading buffer and denatured at 37 °C for 30 min. Equal volumes of samples were separated by SDS-PAGE and transferred onto PVDF membranes (Millipore, USA). The membranes were blocked with a dedicated protein blocking buffer (Servicebio, G2052-500ML) for 30 min at room temperature to minimize nonspecific binding. Subsequently, the membranes were incubated overnight at 4 °C with primary antibodies diluted 1:1000 in TBST containing 5% BSA. The primary antibodies used were anti-HA (Abmart, T62939) and anti-FLAG (Abcepta, AP74805).
After washing three times with TBST, the membranes were incubated with HRP-conjugated goat anti-mouse IgG (H + L) secondary antibody (Proteintech, SA00001-1) at a 1:10,000 dilution for 2 h at room temperature. Protein signals were detected using an enhanced chemiluminescent (ECL) substrate (Affinibody, China) and visualized with a fluorescence/chemiluminescence imaging system (Peiqing, China).
Dil-LDL uptake assay
The LDL-C uptake was determined using fluorescently labeled Dil-LDL (L3482, Thermo Fisher Scientific, MA, USA) in HepG2 cells according to the manufacturer’s protocol. Briefly, cells were cultured in serum-free medium for 24 h and then incubated in serum-free medium containing 5 mg·mL-1 Dil-LDL for 4 h in the dark. Then, cells were stained with DAPI for 20 min and washed 3 times with PBS. The images were obtained with a fluorescence microscope (Spinning Disk Confocal Microscope, Revolution WD, Andor, England). The Dil-LDL-stained cell membrane showed orange-red fluorescence, and the DAPI-stained cell nucleus showed blue fluorescence. For semiquantitative analysis, the staining intensity was measured using ImageJ software. The normalized values have been placed in the Supplementary Data 15.
Oil Red O staining
For Oil Red O staining, cells were first washed three times with PBS, then fixed with 4% paraformaldehyde for 20–30 min, and washed again three times with PBS. After fixation, cells were incubated with freshly prepared Oil Red O working solution for 10–20 min. The dye was then removed, and cells were washed three times with PBS. Finally, PBS was added to uniformly cover the cells, and images were acquired under a light microscope. For semiquantitative analysis, the staining intensity was measured using ImageJ software. The normalized values have been placed in the Supplementary Data 14.
Plasmid constructs
The expression plasmids used in this study were generated based on the pcDNA3.1(+) vector (Invitrogen, USA) using standard molecular cloning techniques. The coding sequences of human INSIG1 (GenBank accession No. NM_005542) and SCAP (GenBank accession No. NM_012235) were amplified and inserted into the pcDNA3.1(+) backbone. For INSIG1, a C-terminal HA tag was introduced to generate the HA-INSIG1(WT) construct, while the H150R point mutation (A449G substitution, (CAC → CGC)) was introduced by site-directed mutagenesis to produce HA-INSIG1(H150R). The SCAP construct was designed with a C-terminal FLAG tag to obtain FLAG-SCAP. All recombinant plasmids were confirmed by Sanger sequencing to ensure the accuracy of the open reading frame and the presence of the desired mutation. The plasmid information is summarized in Supplementary Data 24.
cDNA preparation
Total RNA was reverse-transcribed into complementary DNA (cDNA) using the SmArt RTMaster Premix (5×) (Cat. No. DY10502; Deeyee, China) according to the manufacturer’s instructions. Each 20 μL reaction contained 4 μL of 5× SmArt RTMaster Premix, an appropriate amount of total RNA (10 ng–2 μg), and RNase-free water to reach a final volume of 20 μL. The premix included SmArt RTase (a thermostable M-MLV reverse transcriptase lacking RNase H activity), RNase inhibitor, random primers, oligo(dT) primers, dNTP mixture, and an optimized reaction buffer containing Mg2+. The reverse transcription reaction was performed under the following thermal conditions: 25 °C for 5 min, 50 °C for 15 min for reverse transcription, and 85 °C for 5 s to inactivate the enzyme, followed by holding at 4 °C. The resulting cDNA was used as the template for subsequent real-time quantitative PCR (RT-qPCR).
Real-time quantitative PCR
Real-time quantitative PCR (qPCR) was performed using the CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA) with SYBR Green chemistry121. Each 20 μL reaction contained 10 μL of 2× Universal Blue SYBR Green qPCR Master Mix (G3326-05, Servicebio), 0.4 μL of forward primer (10 μM), 0.4 μL of reverse primer (10 μM; final concentration 0.2 μM each), 1 μL of cDNA template diluted 1:10 from reverse transcription, and nuclease-free water to volume. The thermal cycling program was as follows: initial denaturation at 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s, during which fluorescence signals were collected. Melting curve analysis was performed using a stepwise protocol: 95 °C for 15 s, 60 °C for 1 min, and then an incremental increase from 60 °C to 95 °C at 0.3 °C per step with a 15 s hold at each step for signal acquisition. Amplification specificity was confirmed by the presence of a single melting peak. All reactions were conducted with at least three technical replicates per sample. two-tailed Student’s t test. The sequence of primers used for qPCR is listed in Supplementary Data 19.
Generation of Insig1 H132R/H132R mutation mice
For in vitro assays in HepG2 (human), we used the human INSIG1(H150R) construct following human residue numbering; for the knock-in mouse line we edited the orthologous mouse residue Insig1(H132R), as determined by human–mouse INSIG1/Insig1 sequence alignment.
CRISPR-Cas9-mediated genome editing was employed to generate mice harboring the Insig1(H132R) point mutation at Cyagen (Suzhou) Biotechnology Co. Ltd. (Jiangsu, P. R. China). A CRISPR/Cas9 knock-in strategy was used to introduce the Insig1 p.H132R missense substitution (CAC → CGC). Cas9, a single guide RNA (sgRNA, gRNA-A1: 5′-TCCCTGTATTGACAGTCACCTGG-3′, matching the forward strand of Insig1), and a single-stranded donor oligonucleotide (ssODN) were co-injected into fertilized mouse zygotes. The ssODN sequence was: 5′-TCCTGTTTTGTTTTTCTTTAAACAGCTGTTGTCGGTTTACTGTATCCCTGTATTGACAGTCGCCTGGGAGAACCACACAAGTTCAAGAGAGAATGGGCCAGCGTTATGCGCTGTATTGCCGTG-3′ (where the wild-type codon CAC was replaced by CGC to generate the p.H132R substitution). Founder (F0) animals were identified by PCR amplification of the target locus followed by Sanger sequencing. A total of four positive F0 mice were obtained; among these, the F1 animals delivered to the client colony all derived from a single positive male founder (F0-6).
Genotyping and sequence validation
PCR genotyping was performed with primers F1 (forward, 5′-TGCCGAGGAAAATAAGTGGTTTGG-3′) and R1 (reverse, 5′-AGCCACAGTAAACCTCTGCTTCTA-3′), using LongAmp Taq DNA polymerase (NEB M0323L) for 33 cycles at an annealing temperature of 60 °C, which yielded a 403-bp amplicon. A no-template control (NTC; water) and a wild-type genomic DNA control were included in each PCR run to monitor contamination and verify assay performance. PCR products were verified by Sanger sequencing using F1 as the sequencing primer. Representative allele sequences at the edited motif were: WT …TATTGACAGTCACCTGGGAGA… and mutant …TATTGACAGTCGCCTGGGAGA…. Tail DNA was prepared either with a silica-column kit (TaKaRa MiniBEST, code 9765) or a proteinase-K lysis buffer (50 mM KCl, 10 mM Tris-HCl pH 9.0, 0.1% Triton X-100, 0.4 mg mL⁻¹ proteinase K).
Colony establishment
Positive founder F0-6 (♂) was bred to WT C57BL/6J to assess germline transmission and establish F1 animals. The cross yielded six F1 offspring (3 males, 3 females; DOB 2023-01-15), each carrying the p.H132R allele by PCR and Sanger sequencing. Heterozygous F1 mice were intercrossed to generate F2 litters; genotyping and sequencing (as above) were used to assign Insig1H132R/H132R, Insig1+/H132R and Insig1+/+ genotypes.
Experimental cohorts and background strain
Experiments used male Insig1H132R/H132R mice and WT littermates aged ≥3 months. We did not take off-target effects into account. The line was maintained on a C57BL/6JCya background. For reference, C57BL/6JCya identifiers are MGI: 7786639.
Randomization
Edited and wild-type mice were housed at the Animal Core Facility of Northwest A&F University under identical husbandry conditions. Cage assignments and processing order were determined according to a randomization schedule, with allocation performed by a researcher who did not take part in outcome assessment. To minimise potential confounding, measurements and sample processing were performed in balanced batches, and the order of animals within each batch was randomized.
Breeding, husbandry and ethics
All experimental mice were bred and reared under standard conditions in the investigator’s colony at the Animal Core Facility of Northwest A&F University (Shannxi, P. R. China, certificate no. SCXK [SHAAN] 2017-003). Approval for all experimental procedures was obtained from Northwest A&F University.
Transcriptomic analysis
For cross-species transcriptomic analysis, we downloaded publicly available transcriptome data comprising eight samples each of camels, cattle, and humans. Data underwent quality control using Trimmomatic122. Subsequently, clean fastq were mapped to their respective NCBI reference genomes, namely Ca_bactrianus_MBC_1.0, ARS-UCD1.2, and GRCh38.p14, using HISAT2123. Gene expression levels were quantified using StringTie124. We have provided detailed information in the attached Supplementary Data 12 regarding the publicly available data downloaded from NCBI, including Accession (PRJNA857334, PRJEB35350, PRJNA844027, PRJNA665193, PRJNA522422, PRJNA518006), Run, Sample, Species, Tissue, Years, Sex, and Condition. The last two columns in Supplementary Data 12 represent the FPKM values for gene-INSIG1 and gene-NPC1L1.
Liver and epididymal white adipose tissue (eWAT) from three genome-edited and three wild-type mice were submitted to Novogene (Beijing, China) for library preparation and sequencing125. We received the raw FASTQ files. Reads were quality-filtered with Trimmomatic, aligned to GRCm39 using HISAT2, and gene expression was quantified with StringTie. The reference genome version for mice was GRCm39. And then we calculated differentially expressed genes (DEGs) using DESeq2126 with threshold of adjust p-value < 0.05 and abs(log2FoldChange) > 1. Gene Set Enrichment Analysis (GSEA)67 were used to interpreted gene expression data between gene-edited and wild-type mice liver and visceral fat. KEGG pathways and GO terms were enriched by KOBAS127 with threshold of adjust p-value 0.05 based DEGs from gene-edited and wild-type mice liver and visceral fat.
Statistics and reproducibility
All experiments were repeated at least three times, and representative data are presented. For Dil-LDL uptake assay, we conducted 4 biologically independent experiments. For Oil Red O staining, 5 biologically independent experiments were conducted. One-way ANOVA with Tukey’s post-hoc test were used.
For metabolomics, blood samples from both camels (n = 4 male) and mice (n = 4 male) were collected using non-invasive methods. Differential metabolites between Insig1H132R/H132R edited mice and wild-type mice were screened based on the thresholds of VIP > 1, p-value < 0.05, and abs(log2FoldChange) > 1.
For transcriptomics, liver and epididymal white adipose tissue (eWAT) from 3 genome-edited and 3 wild-type male mice. DEGs identified by threshold of adjust p-value < 0.05 and abs(log2FoldChange) > 1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The genome data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number: BioProject ID: PRJNA1158569; BioSample ID: SAMN43543267. Submitted GenBank assembly GCA_048773025.1 in this study has now been listed on the NCBI website as NCBI RefSeq assembly GCF_048773025.1 Addgene ID: 250155, 250158, 250159 The transcriptome data of mice has also been uploaded to NCBI with accession number: SRP620610. Detailed information can be found in the Supplementary Data 23. Numerical source data for graphs and charts can be found in Supplementary Data 4–23. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code availability
The in-house Perl scripts have been uploaded to github (https://github.com/1221li/comparative-genome.git).
References
Greenville, A. The Biology of Deserts David Ward, 2nd edition. Oxford University Press, Oxford, 2016. xv + 370 pp. Price AUD $66 (paperback, also available as hardback and Ebook). Austral Ecology 43, e20-e20 (2018). https://doi.org/10.1111/aec.12523.
Newby, J. et al. Desert Antelopes on the Brink. In Antelope Conservation (eds Bro-Jørgensen J. & Mallon, D. P.) https://doi.org/10.1002/9781118409572.ch13 (2016).
Rocha, J. L., Godinho, R., Brito, J. C. & Nielsen, R. Life in deserts: the genetic basis of mammalian desert adaptation. Trends Ecol. Evol. 36, 637–650 (2021).
Faye, B., Saint-Martin, G., Bonnet, P., Bengoumi, M. & Dia, M. L. Guide de l'élevage du dromadaire (Sanofi, 1997).
Ostrowski, S., Williams, J. B., Bedin, E. & Ismail, K. Water influx and food consumption of free-living oryxes (oryx leucoryx) in the arabian desert in summer. J. Mammal. 83, 665–673 (2002).
Rymer, T. L., Pillay, N. & Schradin, C. Resilience to droughts in mammals: a conceptual framework for estimating vulnerability of a single species. Q. Rev. Biol. 91, 133–176 (2016).
Bornstein, S. The ship of the desert. The dromedary camel (camelus dromedarius), a domesti- cated animal species well adapted to extreme conditions of aridness and heat. Rangifer 3, 231–236 (1990).
Boyers, M., Parrini, F., Owen-Smith, N., Erasmus, B. F. N. & Hetem, R. S. Contrasting capabilities of two ungulate species to cope with extremes of aridity. Sci. Rep. 11, 4216 (2021).
Ostrowski, S., Williams, J. B., Mésochina, P. & Sauerwein, H. Physiological acclimation of a desert antelope, arabian oryx (oryx leucoryx), to long-term food and water restriction. J. Comp. Physiol. B. 176, 191–201 (2006).
Ouajd, S. & Kamel, B. Physiological particularities of dromedary (camelus dromedarius) and experimental implications. Scand. J. Lab. Anim. Sci. 36, 21511 (2009).
Chen, L. et al. Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science 364, eaav6202 (2019).
Hempel, E. et al. Diversity and paleodemography of the addax (addax nasomaculatus), a saharan antelope on the verge of extinction. Genes 12, 1236 (2021).
Robinson, T. J. & Ropiquet, A. Examination of hemiplasy, homoplasy and phylogenetic discordance in chromosomal evolution of the bovidae. Syst. Biol. 60, 439–450 (2011).
Toljagić, O., Voje, K. L., Matschiner, M., Liow, L. H. & Hansen, T. F. Millions of years behind: slow adaptation of ruminants to grasslands. Syst. Biol. 67, 145–157 (2018).
Xu, Y. et al. Whole-body adipose tissue multi-omic analyses in sheep reveal molecular mechanisms underlying local adaptation to extreme environments. Commun. Biol. 6, 159 (2023).
Tracy, R. L. & Walsberg, G. E. Kangaroo rats revisited: re-evaluating a classic case of desert survival. Oecologia 133, 449–457 (2002).
Wharton, D. A. Life at the Limits: Organisms in Extreme Environments (Cambridge University Press, 2007).
Ababaikeri, B. et al. Whole-genome sequencing of tarim red deer (cervus elaphus yarkandensis) reveals demographic history and adaptations to an arid-desert environment. Front. Zool. 17, 31 (2020).
Colella, J. P. et al. Limited evidence for parallel evolution among desert-adapted peromyscus deer mice. J. Hered. 112, 286–302 (2021).
Consortium, T. B. C. G. Genome sequences of wild and domestic bactrian camels. Nat. Commun. 3, 1202 (2012).
Jirimutu et al. Correction: corrigendum: genome sequences of wild and domestic bactrian camels. Nat. Commun. 4, 2089 (2013).
Wu, H. et al. Camelid genomes reveal evolution and adaptation to desert environments. Nat. Commun. 5, 5188 (2014).
Yang, J. et al. Whole-genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Mol. Biol. Evol. 33, 2576–2592 (2016).
Partha, R. et al. Subterranean mammals show convergent regression in ocular genes and enhancers, along with adaptation to tunneling. Elife 6, e25884 (2017).
Foote, A. D. et al. Convergent evolution of the genomes of marine mammals. Nat. Genet. 47, 272–275 (2015).
Li, M. et al. Convergent molecular evolution of thermogenesis and circadian rhythm in arctic ruminants. Proc. Biol. Sci. 290, 20230538 (2023).
Humble, E. et al. Chromosomal-level genome assembly of the scimitar-horned oryx: insights into diversity and demography of a species extinct in the wild. Mol. Ecol. Resour. 20, 1668–1681 (2020).
Marta Farr é, Q. L. Y. Z., G. Chemnick, J. K. O. A. & Denis M. Larkin, A. H. A. L. A near-chromosome-scale genome assembly of the gemsbok (Oryx gazella): an iconic antelope of the Kalahari desert. Gigascience 8, giy162 (2018).
Kosakovsky Pond, S. L. et al. HyPhy 2.5—a customizable platform for evolutionary hypothesis testing using phylogenies. Mol. Biol. Evol. 37, 295–299 (2020).
Murrell, B. et al. Gene-wide identification of episodic selection. Mol. Biol. Evol. 32, 1365–1371 (2015).
L Rocha, J. et al. North african fox genomes show signatures of repeated introgression and adaptation to life in deserts. Nat. Ecol. Evol. 7, 1267–1286 (2023).
Cao, J., Zhou, A., Zhou, Z., Liu, H. & Jia, S. The role of GPLD1 in chronic diseases. J. Cell. Physiol. 238, 1407–1415 (2023).
Kobayashi, Y. et al. Mice lacking hypertension candidate gene ATP2b1 in vascular smooth muscle cells show significant blood pressure elevation. Hypertension 59, 854–860 (2012).
Rey, C., Guéguen, L., Sémon, M. & Boussau, B. Accurate detection of convergent amino-acid evolution with PCOC. Mol. Biol. Evol. 35, 2296–2306 (2018).
He, Z., Xu, S. & Shi, S. Adaptive convergence at the genomic level—prevalent, uncommon or very rare? Natl. Sci. Rev. 7, 947–951 (2020).
Marcovitz, A. et al. A functional enrichment test for molecular convergent evolution finds a clear protein-coding signal in echolocating bats and whales. Proc. Natl. Acad. Sci. USA 116, 21094–21103 (2019).
Thomas, G. W. C. & Hahn, M. W. Determining the null model for detecting adaptive convergence from genomic data: a case study using echolocating mammals. Mol. Biol. Evol. 32, 1232–1236 (2015).
Zou, Z. & Zhang, J. Are convergent and parallel amino acid substitutions in protein evolution more prevalent than neutral expectations? Mol. Biol. Evol. 32, 2085–2096 (2015).
Yusuf, L. H., Saldívar Lemus, Y., Thorpe, P., Macías Garcia, C. & Ritchie, M. G. Genomic signatures associated with transitions to viviparity in cyprinodontiformes. Mol. Biol. Evol. 40, msad208 (2023).
Fukushima, K. & Pollock, D. D. Detecting macroevolutionary genotype–phenotype associations using error-corrected rates of protein convergence. Nat. Ecol. Evol. 7, 155–170 (2023).
Murrell, B. et al. Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8, e1002764 (2012).
Endres, B. T. et al. Mutation of plekha7 attenuates salt-sensitive hypertension in the rat. Proc. Natl. Acad. Sci. 111, 12817–12822 (2014).
Savige, J. & Harraka, P. Pathogenic LAMA5 variants and kidney disease. Kidney360 2, 1876–1879 (2021).
Fu, Q. et al. Effects of polymorphisms in APOA4-APOA5-ZNF259-BUD13 gene cluster on plasma levels of triglycerides and risk of coronary heart disease in a chinese han population. PLoS One 10, e0138652 (2015).
Van Weeghel, M. et al. Functional redundancy of mitochondrial enoyl-CoA isomerases in the oxidation of unsaturated fatty acids. FASEB. J. 26, 4316–4326 (2012).
Bengoumi, M. Biochimie clinique du dromadaire et mécanismes de son adaptation à la déshydratation. Thèse de Doctorat ès Sciences (IAV Hassan II, 1992).
Faye, B. & Bengoumi, M. Camel Clinical Biochemistry and Hematology (Springer International Publishing AG, 2018).
Luo, J., Wang, J. K. & Song, B. L. Lowering low-density lipoprotein cholesterol: from mechanisms to therapies. Life Metabol. 1, 25–38 (2022).
Carobbio, S. et al. Adaptive changes of the insig1/SREBP1/SCD1 set point help adipose tissue to cope with increased storage demands of obesity. Diabetes 62, 3697–3708 (2013).
Gong, Y., Lee, J. N., Brown, M. S., Goldstein, J. L. & Ye, J. Juxtamembranous aspartic acid in insig-1 and insig-2 is required for cholesterol homeostasis. In Proc. Natl. Acad. Sci. USA 103, 6154–6159 (2006).
Ishimoto, K. et al. Sterol-mediated regulation of human lipin 1 gene expression in hepatoblastoma cells. J. Biol. Chem. 284, 22195–22205 (2009).
Engelking, L. J. et al. Schoenheimer effect explained-feedback regulation of cholesterol synthesis in mice mediated by insig proteins. J. Clin. Investig. 115, 2489–2498 (2005).
Bertolio, R. et al. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat. Commun. 10, 1326 (2019).
Su, F. & Koeberle, A. Regulation and targeting of SREBP-1 in hepatocellular carcinoma. Cancer Metastasis Rev. 43, 673–708 (2024).
Ruiz, R. et al. Sterol regulatory element-binding protein-1 (SREBP-1) is required to regulate glycogen synthesis and gluconeogenic gene expression in mouse liver. J. Biol. Chem. 289, 5510–5517 (2014).
Peterson, T. R. et al. MTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420 (2011).
Cheng, C. et al. Apolipoprotein a4 restricts diet-induced hepatic steatosis via SREBF1-mediated lipogenesis and enhances IRS-PI3k-akt signaling. Mol. Nutr. Food Res. 66, 2101034 (2022).
Wang, Z. et al. Apolipoprotein a-IV involves in glucose and lipid metabolism of rat. Nutr. Metab. 16, 41 (2019).
Buck, M. J., Squire, T. L. & Andrews, M. T. Coordinate expression of the PDK4 gene: a means of regulating fuel selection in a hibernating mammal. Physiol. Genomics 8, 5–13 (2002).
Hodson, D. J. et al. ADCY5 couples glucose to insulin secretion in human islets. Diabetes 63, 3009–3021 (2014).
Han, Y. et al. Tianhuang formula ameliorates non-alcoholic fatty liver diseases in type 2 diabetic mice through CRLS1-ATF3/ChREBP pathway. J. Holist. Integr. Pharm. 4, 147–156 (2023).
Sustarsic, E. G. et al. Cardiolipin synthesis in brown and beige fat mitochondria is essential for systemic energy homeostasis. Cell Metab. 28, 159–174 (2018).
Tu, C. et al. Cardiolipin synthase 1 ameliorates NASH through activating transcription factor 3 transcriptional inactivation. Hepatology 72, 1949–1967 (2020).
Heckmann, B. L., Zhang, X., Xie, X. & Liu, J. The g0/g1 switch gene 2 (g0s2): regulating metabolism and beyond. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1831, 276–281 (2013).
Zhang, X., Heckmann, B. L., Campbell, L. E. & Liu, J. G0s2: a small giant controller of lipolysis and adipose-liver fatty acid flux. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 1146–1154 (2017).
Bruschi, F. V., Tardelli, M., Claudel, T. & Trauner, M. PNPLA3 expression and its impact on the liver: current perspectives. HEPATIC Med. Evid. Res. 9, 55–66 (2017).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 102, 15545–15550 (2005).
Cole, T. L. et al. Genomic insights into the secondary aquatic transition of penguins. Nat. Commun. 13, 3912 (2022).
Fu, T. et al. The highest-elevation frog provides insights into mechanisms and evolution of defenses against high UV radiation. Proc. Natl. Acad. Sci. USA 119, e2212406119 (2022).
Ovchinnikov, V. et al. Caecilian genomes reveal the molecular basis of adaptation and convergent evolution of limblessness in snakes and caecilians. Mol. Biol. Evol. 40, msad102 (2023).
Mendes, F. K., Hahn, Y. & Hahn, M. W. Gene tree discordance can generate patterns of diminishing convergence over time. Mol. Biol. Evol. 33, 3299–3307 (2016).
He, Z. et al. Convergent adaptation of the genomes of woody plants at the land–sea interface. Natl. Sci. Rev. 7, 978–993 (2020).
Xu, S. et al. Genome-wide convergence during evolution of mangroves from woody plants. Mol. Biol. Evol. 34, 1008–1015 (2017).
Qiu, Q. et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 44, 946–949 (2012).
Arinell, K. et al. Brown bears (ursus arctos) seem resistant to atherosclerosis despite highly elevated plasma lipids during hibernation and active state. CTS-Clin. Transl. Sci. 5, 269–272 (2012).
Harlow, H. J., Lohuis, T., Grogan, R. G. & Beck, T. D. I. Body mass and lipid changes by hibernating reproductive and nonreproductive black bears (ursus americanus). J. Mammal. 83, 1020–1025 (2002).
Lam, S. M. et al. Quantitative lipidomics and spatial MS-imaging uncovered neurological and systemic lipid metabolic pathways underlying troglomorphic adaptations in cave-dwelling fish. Mol. Biol. Evol. 39, msac050 (2022).
Aspiras, A. C., Rohner, N., Martineau, B., Borowsky, R. L. & Tabin, C. J. Melanocortin 4 receptor mutations contribute to the adaptation of cavefish to nutrient-poor conditions. Proc. Natl. Acad. Sci. USA 112, 9668–9673 (2015).
Krishnan, J. et al. Genome-wide analysis of cis-regulatory changes underlying metabolic adaptation of cavefish. Nat. Genet. 54, 684–693 (2022).
Olsen, L., Thum, E. & Rohner, N. Lipid metabolism in adaptation to extreme nutritional challenges. Dev. Cell 56, 1417–1429 (2021).
Blank, D. & Li, Y. Antelope adaptations to counteract overheating and water deficit in arid environments. J. Arid Land 14, 1069–1085 (2022).
von Toerne, C. et al. MASP1, THBS1, GPLD1 and ApoA-IV are novel biomarkers associated with prediabetes: the KORA f4 study. Diabetologia 59, 1882–1892 (2016).
Candlish, J. Metabolic water and the camel’s hump — a textbook survey. Biochem. Educ. 9, 96–97 (1981).
Alvira-Iraizoz, F. et al. Multiomic analysis of the arabian camel (camelus dromedarius) kidney reveals a role for cholesterol in water conservation. Commun. Biol. 4, 779 (2021).
Giroud, S. et al. Hibernating brown bears are protected against atherogenic dyslipidemia. Sci. Rep. 11, 18723 (2021).
Hurt-Camejo, E. & Pedrelli, M. Why are brown bears protected against atherosclerosis even though their plasma cholesterol levels are twice that of humans? Clín. Investig. Arterioscler. 34, 322–325 (2022).
Liu, S. et al. Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 157, 785–794 (2014).
Riddle, M. R. et al. Insulin resistance in cavefish as an adaptation to a nutrient-limited environment. Nature 555, 647–651 (2018).
Xu, D. et al. A single mutation underlying phenotypic convergence for hypoxia adaptation on the qinghai-tibetan plateau. Cell Res. 31, 1032–1035 (2021).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Durand, N. C. et al. Juicebox provides a visualization system for hi-c contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinforma. 4, 4–10 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: assessing genomic data quality and beyond. Curr. Protoc. 1, e323 (2021).
Hoff, K. J. & Stanke, M. WebAUGUSTUS—a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res. 41, W123–W128 (2013).
Zhang, D., Pan, J., Cao, J., Cao, Y. & Zhou, H. Screening of drought-resistance related genes and analysis of promising regulatory pathway in camel renal medulla. Genomics 112, 2633–2639 (2020).
Lado, S. et al. Nucleotide diversity of functionally different groups of immune response genes in old world camels based on newly annotated and reference-guided assemblies. BMC Genomics 21, 606 (2020).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Kirilenko, B. M. et al. Integrating gene annotation with orthology inference at scale. Science 380, eabn3107 (2023).
Armstrong, J. et al. Progressive cactus is a multiple-genome aligner for the thousand-genome era. Nature 587, 246–251 (2020).
Earl, D. et al. Alignathon: a competitive assessment of whole-genome alignment methods. Genome Res. 24, 2077–2089 (2014).
Ranwez, V., Douzery, E. J. P., Cambon, C., Chantret, N. & Delsuc, F. MACSE v2: toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 35, 2582–2584 (2018).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577 (2007).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. Trimal: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Nguyen, L., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Wang, Y. et al. Genetic basis of ruminant headgear and rapid antler regeneration. Science 364, eaav6335 (2019).
Wertheim, J. O., Murrell, B., Smith, M. D., Kosakovsky Pond, S. L. & Scheffler, K. RELAX: detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 32, 820–832 (2015).
Yang, J. & Zhang, Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181 (2015).
Zheng, W. et al. Folding non-homologous proteins by coupling deep-learning contact maps with i-TASSER assembly simulations. Cell Rep. Methods 1, 100014 (2021).
Zhou, X. et al. I-TASSER-MTD: a deep-learning-based platform for multi-domain protein structure and function prediction. Nat. Protoc. 17, 2326–2353 (2022).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Pettersen, E. F. et al. UCSF chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Schymkowitz, J. et al. The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388 (2005).
Wishart, D. S. et al. HMDB: the human metabolome database. Nucleic Acids Res. 35, D521–D526 (2007).
Horai, H. et al. MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 45, 703–714 (2010).
Abdelrazig, S. et al. Metabolic characterisation of magnetospirillum gryphiswaldense MSR-1 using LC-MS-based metabolite profiling. RSC Adv. 10, 32548–32560 (2020).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Pang, Z. et al. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49, W388–W396 (2021).
Xia, B. et al. Urolithin a exerts antiobesity effects through enhancing adipose tissue thermogenesis in mice. PLoS Biol. 18, e3000688 (2020).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Wang, W. et al. Integrating genome- and transcriptome-wide association studies to uncover the host–microbiome interactions in bovine rumen methanogenesis. iMeta 3, e234 (2024).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Xie, C. et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322 (2011).
Zomer, R. J., Xu, J. & Trabucco, A. Version 3 of the global aridity index and potential evapotranspiration database. Sci. Data 9, 409 (2022).
Acknowledgements
We thank the High-Performance Computing Center (HPC) of Northwest A&F University (NWAFU) for providing computing resources. We thank Mallory Eckstut, PhD for editing the English text of a draft of this manuscript. This work was supported by grants from National Key Research and Development Program of China (2021YFF1001000), and Postdoctoral Innovative Talents Support Program of China (BX20200282), and National Natural Science Foundation of China (Grant No. 32570633).
Author information
Authors and Affiliations
Contributions
Conceptualization: Yu Wang, Bo Xia, Chao Tong Formal Analysis: Xinmei Li Investigation: Xinmei Li, Ziyi He, Anguo Liu, Fanxin Meng, Xiao Zhang, Huan Liu, Nana Li, Yuyi Lu, Zhipei Wu, Huimei Fan, Xixi Yan, Nange Ma, Zhenyu Wei, Wei Wang, Xixi He, Kunyu Ma, Yu Jiang Experimental Verification: Bo Xia, Ziyi He, Xiao Zhang, Huan Liu, Nana Li Funding Acquisition: Yu Wang Supervision: Yu Wang Visualization: Xinmei Li, Ziyi He, Anguo Liu, Fanxin Meng, Xiao Zhang, Huan Liu, Nana Li Writing – Original Draft Preparation: Xinmei Li Writing – Review & Editing: Yu Wang, Bo Xia, Chao Tong.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: John Mulley & Rosie Bunton-Stasyshyn.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., He, Z., Liu, A. et al. INSIG1 parallel substitution drives lipid/sterol metabolic plasticity mediating desert adaptation in ungulates. Commun Biol 9, 245 (2026). https://doi.org/10.1038/s42003-026-09523-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-026-09523-z
