
Abstract
Glycogen Storage Disease Type III (GSDIII) is a rare genetic disorder caused by mutations in the AGL gene, leading to a deficiency in the glycogen debranching enzyme. This results in the accumulation of abnormal glycogen in various tissues, causing a range of symptoms, including liver enlargement and hypoglycemia. Current animal models do not fully recapitulate the severe phenotypes observed in patients, highlighting the need for improved model systems. To the best of our knowledge, this study presents the first Caenorhabditis elegans model of GSDIII, which successfully exhibits disease-relevant traits, including glycogen accumulation. Using this model, we developed a computational approach based on high-throughput screening methods, enabling the identification of key genetic modulators. Notably, we demonstrate that glycogen accumulation can be rescued by genetic and pharmacological inhibition of CHK1, a gene involved in cell cycle regulation and DNA damage response, in a variant-specific manner. These findings suggest that targeting CHK1 may represent a promising therapeutic strategy for treating GSDIII, particularly when considering specific AGL mutations.
Similar content being viewed by others

Introduction
Glycogen storage disease type III (GSD III, Cori disease, OMIM #232400) is a rare inherited metabolic disorder caused by a deficiency in the glycogen debranching enzyme (GDE), which leads to the accumulation of abnormal glycogen in various tissues. It appears as a multi-systemic syndrome, with a prevalence of 1/100,000 births1,2. Loss-of-function mutations in AGL (locus: 1p21.2), the gene coding for the glycogen debranching enzyme (GDE), have been identified in affected patients through a homozygous recessive transmission3,4. Patients diagnosed with GSDIII disease have significant hyperplasia of the liver, hypoglycemia, mental retardation, and myopathy1,5. The diagnosis is confirmed via GDE dosage, highly ketotic hypoglycemia, or high levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in the blood1,4. The only treatment currently available is cornstarch, which prevents hypoglycemia episodes in patients. Although several GSDIII models (mouse, canine, and cell models) exist, these do not fully replicate the phenotypes observed in patients, especially in the most severe cases, where both glycogen buildup and decreased survival are observed6,7,8,9. Hence, it is essential to continue to generate patient-specific genetic models.
The nematode Caenorhabditis elegans (C. elegans) has proven invaluable in advancing our understanding of the underlying mechanisms of diseases, which is crucial for developing effective treatments10,11. In the case of GSDIII, identifying genetic and pharmacological targets that play a role in the pathogenesis is also an important step in elucidating the disease’s molecular underpinnings.
In this study, we characterized specific mutations in CRISPR/Cas9-edited C. elegans models. We performed high-throughput drug screening and computational analysis, which led us to investigate the unexpected role of the chk-1 gene, a key regulator of cell cycle and DNA damage response, in the context of glycogen storage disease type III. Our computational approach based on a high-throughput pipeline demonstrates that genetic and pharmacological inhibition of CHK1 in C. elegans rescues disease-relevant phenotypes associated with GSDIII, particularly glycogen accumulation. Our results suggest that CHK1 can be considered a novel actionable therapeutic target in the context of GSDIII.
Results
agl-1/AGL mutants show significant glycogen buildup phenotype
The C. elegans genome encodes for one orthologous gene (agl-1) to the human AGL gene, which is well conserved with 43.48% identity and 62.41% similarity and predicted to have both 4-alpha-glucanotransferase activity and amylo-alpha-1,6-glucosidase activity12,13,14. The rare W1099X point mutation is a human pathogenic variant corresponding to a W1044X point mutation in C. elegans15. S1514R is a variant of uncertain significance (VUS) present in population databases aligning with S1444R in C. elegans16 (Fig. 1A, B). As a loss-of-function model, we used a 6955 bp complete deletion of agl-1. Available mutants are referred to as the following: Δagl-1 for agl-1(knu867), W1044X for agl-1(knu860), and S1444R for agl-1(knu864).
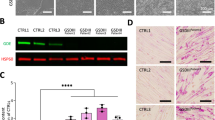
Abnormal glycogen buildup in several tissues is one of the main symptoms of GSDIII17 (Supplementary Fig. 1). Therefore, we sought to perform glycogen quantification in our C. elegans agl-1-mutant strains (Fig. 1C). Glycogen deposits are found in worms’ intestines, hypodermis, and muscles18. Our observations showed significant glycogen buildup in W1044X, S1444R, and Δagl-1 mutant worms compared to wild-type, with no significant difference between W1044X and S1444R mutants (Fig. 1D, E). The Δagl-1 loss-of-function mutant showed the most glycogen accumulation (Fig. 1E, F).
To evaluate the functional impact of agl-1 mutations and confirm the biochemical disruption caused by mutations in agl-1, we performed a glucose intolerance assay. Exposure to low concentrations of glucose significantly increased embryonic lethality in W1044X, S1444R, and Δagl-1 mutant worms compared to wild-type, with no significant difference in brood size between mutants and wild-type (Fig. 1G, H). These data suggest that our patient-specific mutants replicate the abnormal glycogen phenotype of GSDIII.

A Location of the AGL mutations in the glucosidase domain. B Evolutionary conservation of affected residue in C. elegans AGL-1 ortholog (highlighted in yellow). Protein alignment was done using MARVEL. C Imaging protocol for iodine staining from bright field to image processing. From left to right: schematic of 3D printed pad used for staining, an example of an acquired image after staining, and image processing using a macro in Fiji software for quantification. Control and test worms are exposed to iodine simultaneously. D Representative image of glycogen buildup visualized by iodine staining in an agl-1 mutant compared to wild-type. E Iodine staining quantification shows significantly increasing glycogen buildup in agl-1 mutants compared to wild-type at day 1 of adulthood (n ≈ 30). F Glycogen quantification using a calorimetric kit shows significantly increased levels of glycogen in mutants compared to wild-type at day 1 of adulthood (N = 3). G and H Embryonic viability assay is significantly decreased in mutants when exposed to 2% of glucose, with no significant differences in brood size (n ≈ 30). One-way ANOVA was performed for E (DF = 3; F(3,97) = 1.380), F (DF = 3; F(3,41) = 1.058), G (DF = 3; F(3,350) = 18.85) and H (DF = 3; F(3,8) = 1.513). Data are presented as mean ± SEM.
C. elegans agl-1 /AGL mutants display impairments in health phenotypes
Though agl-1 mutants look superficially indistinct from wild-type worms, further characterization showed a significantly decreased lifespan for the point mutation and deletion mutants compared to wild-type (Fig. 2A). To assess global health, we first measured the reproductive capacity through progeny, and we observed significantly decreased offspring in mutants compared to wild-type animals (Fig. 2B). Then, we categorized worms by their larval stages to assess development. W1044X, S1444R, and Δagl-1 mutants exhibited a significant developmental delay compared to wild-type animals (Fig. 2C)
Motility is also an important indicator of health in C. elegans and can be assessed in both liquid and solid media. Using automated phenotyping at Day 1 of adulthood, we detected significant differences in speed, wavelength, and mean amplitude between mutant and wild-type worms (Supplementary Fig. 2). Among the mutants, the S1444R variant exhibited pronounced impairments in liquid motility compared to wild-type controls (Fig. 2D). To investigate whether these defects progressed with age, we assessed paralysis on solid media and observed a significantly higher rate of age-dependent paralysis in S1444R mutants (Fig. 2E). Together, these findings suggest that agl-1 mutations contribute to general health impairments affecting both developmental and aging phenotypes, including motility and lifespan. Since these latter results show impairments in only one mutant, we decided to perform further investigations in S1444R animals.

A agl-1 mutants show significantly decreased survival compared to wild-type (n ≈ 360). B All agl-1 mutants have a significantly reduced number of offspring compared to wild-type animals (n ≈ 30). C agl-1 mutants show significant developmental delay compared to wild-type (n ≈ 300). D S1444R C. elegans mutant displays significantly impaired swimming motility behavior at day 1 of adulthood (n ≈ 360). E S1444R mutants show greater motility defects leading to paralysis on solid media compared to wild-type, W1044X, and deletion mutants (n ≈ 360). Log-rank (Mantel–Cox) tests were used for A (DF = 3) and E (DF = 1). A one-way ANOVA was performed for B (DF = 3, F(3,107) = 0.03231). Two-way ANOVAs were performed for C (DF = 4, F(4, 10) = 137.1) and D (DF = 3, F(3,32) = 39.31). Data are presented as mean ± SEM.
Distinct transcriptomic signature of agl-1 /AGL C. elegans mutants
To learn more about gene expression profiles in C. elegans models of GSDIII, we performed RNA sequencing to understand the transcriptomic profile of our mutants.
To gain deeper insight into the molecular mechanisms underlying the GSDIII phenotype in C. elegans, we performed transcriptome-wide RNA sequencing (RNA-seq) to characterize gene expression changes in agl-1 mutants. This approach allows for an unbiased, high-resolution analysis of differential gene expression, enabling the identification of dysregulated pathways associated with the disease model. Our analysis revealed substantial transcriptomic reprogramming in agl-1 mutants (Δagl-1, W1044X and S1444R), with 20 downregulated genes and 1034 upregulated genes (log₂ fold change (LFC) ≥ 1), compared to wild-type controls (Fig. 3A, Supplementary Fig. 3). Combined with enrichment analysis, these observations showed that several pathways are significantly mobilized in agl-1 mutants (Fig. 3B–F). One interesting result is the disrupted expression of MSP genes (major sperm protein) in agl-1 mutants, consistent with the observed progeny defects (Fig. 3C). MSP genes are integral to the motility apparatus that drives the crawling movement of nematode spermatozoa, a process crucial to successful fertilization19,20. Consistent with the disease phenotype of developmental delay and muscle weakness, larval and muscle development pathways are suppressed in mutants (Fig. 3E, F). In contrast, protein modification and phosphate metabolic pathways seem activated, suggesting a compensatory mechanism (Fig. 3E, F). Despite these broad transcriptomic changes, several top RNA-seq hits could not be validated by qPCR (Supplementary Fig. 4), highlighting the need for additional validation and higher-resolution approaches such as single-cell transcriptomics.

A Fold change analysis shows a higher number of downregulated genes in wild-type compared to mutants (Δagl-1, W1044X, and S1444R); p < 0.05. B Volcano plot shows that MSRB1/2 genes are significantly downregulated, whereas SMPD1 and EPHX1 are significantly up-regulated; p < 0.05. C Heat map shows clustering of the 20 most differentially expressed genes in W1044X, S1444R, and Δagl-1 mutants compared to wild-type; p < 0.05. D Pathway enrichment analysis of combined mutants shows significantly activated and suppressed pathways in GSDIII C. elegans models based on GeneRatio score (Δagl-1, W1044X, and S1444R). E. Interaction analysis of combined mutants suggests interplay between the identified pathways. Data are presented as mean ± SEM.
Pharmacological and genetic rescue of S1444R mutant phenotypes
After two rounds of drug screening from ~ 4000 compounds, we identified 25 compounds significantly improving the swimming motility phenotype in S1444R (Fig. 4A, Supplementary Fig. 5). To validate for an additional relevant phenotype, we counter-screened those hits against glycogen buildup. We identified pimozide and pramoxine HCl as compounds that significantly decreased glycogen accumulation in S1444R (Fig. 4B, Supplementary Fig. 6). Pimozide, an antipsychotic drug, is known to inhibit dopamine receptors21,22. Pramoxine HCl, commonly used as a local anesthetic, blocks sodium channels, which could influence cellular signaling and energy regulation23,24. The discovery of these compounds highlights promising therapeutic avenues by linking their known pharmacological activities to the reduction of glycogen accumulation, a hallmark of GSDIII.

A Top 25 compounds from the secondary screen that significantly improved mthe edian activity of the swimming motility behavior in S1444R mutants. B Validation of top hits on glycogen buildup phenotype through iodine staining. Pimozide and Pramoxine HCl significantly decreased glycogen buildup in S1444R mutants compared to vehicle controls (n ≈ 30). A one-way ANOVA was used for B (DF = 7, F(7, 304) = 2.454). Data are presented as mean ± SEM.
In parallel to our pharmacological investigation, we developed a computational approach to identify potential disease-relevant targets. Using open databases with previously reported drug targets, we grouped compounds from our drug screening analysis into clusters to highlight shared biological pathways (Fig. 5A, Table S1). We selected and targeted 77 genes through an RNA interference (RNAi) genetic screen (Fig. 5B, Supplementary Figs. 7–9). The initial screen revealed 13 genes that significantly altered glycogen accumulation in S1444R mutants (Fig. 5C). Downregulation of genes such as CYP-13A5, CYP-13A6, CYP-13B2, and ugt-23 significantly increases glycogen accumulation in S1444R mutants. These genes are orthologous to the cytochrome family of enzymes and glycosyltransferase liver enzymes, which is consistent with the clinical involvement of the liver in GSDIII patients25,26,27,28. Knockdown of genes like gpa-15, erm-1, ant-1.1, chk-1, tax-6, pkc-1, let-607, jmjd-1.2, and gnrr-1 significantly decreases glycogen accumulation (Fig. 5C). Interestingly, the literature has previously shown that pimozide decreases CHK1 levels, which suggests a promising avenue for our approach22.

A Symmetrical heatmap representing target overlap between compounds. Compounds were clustered based on the correlation of target overlap scores between rows/columns (drugs). B Description of our workflow based on drug screening data to identify targetable genes. C First round of RNAi targeting shows 13 genes that significantly change glycogen buildup in S1444R mutants (N = 1). One-way ANOVAs with Welch’s correction applied when variances were unequal were performed in D (F(13.00, 35.53) = 27.67)). Data are presented as mean ± SEM.
After multiple rounds of validation (Supplementary Fig. 8), we selected two genes that significantly rescued glycogen buildup in S1444R mutants compared to wild-type worms: chk-1 and pkc-1, which encode checkpoint kinase 1 and protein kinase C, respectively. These genes were prioritized for their high translational potential in clinical contexts. Notably, the reduction in glycogen levels was specific to the S1444R mutant background, suggesting a variant-dependent regulation of glycogen metabolism (Fig. 6A, B, Supplementary Figs. 8, 10). Consistently, RNA sequencing data showed significantly increased chk-1 in agl-1 mutants compared to wild-type, while pkc-1 expression remained unchanged (Fig. 6C, D). To validate our findings in another phenotype, we replicated the RNAi experiment on age-dependent paralysis. We observed that only chk-1 downregulation rescued the dysfunction in S1444R mutants (Fig. 6E, F, Supplementary Fig. 9). To further confirm the specificity of CHK1 inhibition in rescuing impaired phenotypes, we opted for rabusertib compound, a selective CHK1 inhibitor29. Rabusertib treatment significantly rescued glycogen accumulation and age-dependent paralysis phenotypes in S1444R compared to vehicle control (Fig. 6G, H, Supplementary Fig. 10).

A Knockdown of chk-1 in S1444R C. elegans mutant shows significant and specific rescue in glycogen buildup phenotype (n ≈ 30); **p = 0.0071. B Knockdown of pkc-1 in S1444R C. elegans mutant shows significant and specific rescue in glycogen buildup phenotype (n ≈ 30); ****p < 0.0001. C S1444R and Δagl-1 mutants show significantly increased RNA expression of the chk-1 gene (N = 3); **p < 0.01. D W1044X, S1444R, Δagl-1 mutants and wild-type have a similar expression of the pkc-1 gene. E chk-1 downregulation rescues the progressive paralysis on solid media in S1444R mutant (n ≈ 360); ****p < 0.0001. F pkc-1 knockdown does not rescue the progressive paralysis phenotype in S1444R mutants. G Pharmacological inhibition of CHK1 rescues glycogen buildup (n ≈ 30) and H paralysis phenotypes (n ≈ 360); p = 0.0433, ****p < 0.0001. I Knockdown of aak-2 in S1444R mutants shows no change in glycogen buildup phenotype (n ≈ 30) and (J) no changes in chk-1 expression (N = 3). One-way ANOVAs with Welch’s correction applied when variances were unequal were performed for A (DF = 3, F(3, 80) = 1.513), B (DF = 3, F(3, 75) = 0.7404), C (DF = 4, F(3.000, 6.050) = 32.49) and D (DF = 4, F(3.000, 4.360) = 5.601). Log-rank (Mantel–Cox) tests were used for E (DF = 1), F (DF = 1), and H (DF = 4). Unpaired t-tests with Welch correction were performed for G (t = 2.347, DF = 65.20), I (t = 1.480, DF = 43.69), J (t = 1.059, DF = 3.324). Data are presented as mean ± SEM.
To better understand the potential mechanisms by which chk-1 inhibition rescues glycogen buildup in the S1444R mutant, we focused on AMP-activated protein kinase (AMPK, aak-2), a key cellular energy sensor and regulator of the cell cycle. AMPK modulates gene expression and maintains energy homeostasis by promoting catabolic pathways and sensing glycogen availability30,31,32. In C. elegans, AMPK activity has been linked to glycogen depletion and lifespan extension under high-glucose conditions33. However, genetic knockdown of aak-2 did not alter glycogen levels or chk-1 expression in S1444R mutants (Fig. 6I, J, Supplementary Fig. 10). These findings suggest that CHK1 regulates glycogen metabolism in a variant-specific and AMPK-independent manner, highlighting a distinct mechanism underlying the disease-relevant phenotype in GSDIII.
Discussion
To enhance our comprehension of disease mechanisms and identify potential therapeutic targets for GSDIII, we investigated disease-associated characteristics in our C. elegans mutants. Among the variants analyzed, only the S1444R mutant exhibited swimming defects. While muscle weakness has been reported in mouse models and in some patients, it is not a universal feature across all GSDIII forms. The literature distinguishes several subtypes (GSDIIIa, GSDIIIb, and GSDIIIc), in which glycogen accumulation and clinical manifestations may occur in the liver, muscle, or both3. This raises the possibility that the mutations we studied correspond to different GSDIII subtypes, with motility impairment being specific to only one of them.
Our computational analysis led to the identification of chk-1 as a novel potential genetic modulator of glycogen. Our findings suggest that downregulation of chk-1 might improve disease phenotypes in the C. elegans S1444R mutant. Our computational, genetic, and pharmacological validations have shown their potential to influence pathways relevant to GSD III. The advent of shared databases has facilitated the execution of computational analysis that can be validated at the biological level. The overlap between the RNA-seq data and the drug screen GO analysis is depicted in the pathway enrichment analysis, which highlights that phosphorylation-related pathways are activated, consistent with the upregulation of proteins such as PKC-1 and CHK-134,35. In contrast, pathways associated with larval development appear to be suppressed, as reflected by differential expression of genes including gpa-15, erm-1, tax-6, jmjd-1.2, ugt-23, and members of the solute carrier family such as ant-1.136,37,38,39,40,41. These findings suggest that the transcriptional changes observed in the S1444R mutant align with functionally relevant pathways, linking altered gene expression to processes that may also be targeted in the computational drug screen. These approaches can potentially conserve a considerable amount of time and facilitate extensive investigations.
While CHK1 is primarily recognized for its role in the DNA damage response, it is becoming evident that it also has DNA damage response (DDR)-independent functions. In C. elegans, the CHK1 ortholog has been primarily characterized for its importance in germline cells, with its function in somatic cells remaining less understood42. Our study provides evidence for a potential role of CHK1 in somatic cells in the context of GSDIII, warranting further investigation into its functions beyond the germline in C. elegans.
It is worth noting that ATR/CHK1 inhibitors are primarily used in cancer therapy, exploiting the concept of synthetic lethality, where the simultaneous mutation of two genes, such as CDC25A and WEE1, leads to cell death43,44,45,46. The non-specific CHK1 inhibitor drug UCN-01 was previously used in C. elegans, demonstrating extended lifespan, increased hsp-4 expression, and enhanced thermal stress resistance47. Furthermore, studies on medulloblastoma cells showed that CHK1 inhibition modulates GSK-3 activity by changing its phosphorylation state, also known to regulate glycogen metabolism48. These findings are consistent with our results, showing that genetic and pharmacological inhibition of CHK1 improves health phenotypes in our GSDIII C. elegans model. CHK1 inhibition could have therapeutic benefits in the context of GSDIII, potentially by modulating glycogen metabolism and improving cellular stress responses47.
Previous studies have shown that CHK1 activity is regulated by USP1, which is also involved in autophagy and proteasome activity22,49. Furthermore, research on AGL mutants has revealed increased ubiquitination of the protein. Specifically, proteasomal inhibition has been shown to significantly increase mutant AGL levels, leading to an increase in glycogen-like structures (polyglucosans) due to the expected compensatory activity of glycogen synthase in response to AGL loss50,51. These findings highlight the role of ubiquitination as a regulatory mechanism for AGL50,51. These observations, combined with our findings, suggest a potential link between CHK1, ubiquitination, and AGL regulation in the context of GSD III.
One interesting result is pimozide, which came out in several drug screens in our previous work for neurodegenerative diseases52. This molecule, known as a USP1 inhibitor, has also been shown to accelerate CHK1 degradation22,46,53. USP1 is a ubiquitin-specific protease that contributes to the deubiquitination of proteins. It participates in several mechanisms, such as cell cycle regulation, DNA repair, apoptosis, gene expression, and cell motility54.
Finally, recent research has shown that CHK1 inhibition leads to PARP-1 cleavage. Our previous work showing that PARP inhibition reduces neurodegeneration is consistent with the overall health improvements observed in our GSDIII disease worm model55. These findings suggest a potential interplay between CHK1, USP1, and AGL, with ubiquitination as a critical link. Further investigation into this relationship could provide valuable insights into the molecular mechanisms underlying GSD III and identify potential therapeutic targets.
Top candidate genes identified through RNA-seq could not be validated by qPCR, underscoring the limitations of bulk transcriptomic approaches and the need for higher-resolution methods such as single-cell RNA sequencing. As this study was conducted exclusively in C. elegans models of GSDIII, the conclusions are exclusive to the S1444R variant of agl-1 in this organism. Further investigations in mammalian models and patient-derived cell lines need to be performed to confirm our findings in phylogenetically higher species.
Methods
Amino acid alignment
Amino acid alignments were performed using BLAST sequences for the AGL gene from Homo Sapiens (AGL; Gene ID: 178) and Caenorhabditis elegans (agl-1; Gene ID: 175091) NCBI sequences and MARVELL software.
Nematode strains construction and maintenance
C. elegans were maintained under standard conditions, meaning 15 °C for maintenance and 20 °C for assays56. NGM agar plates with E. coli OP50 culture were used as a living environment. Strains used for this study were as follows: wild-type; N2, COP2004; agl-1(knu860), COP2008; agl-1(knu864), and COP2012; agl-1(knu867) strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
To confirm genotypes, PCR was performed for deletions. High-resolution melting (HRM) and genome sequencing were performed for point mutations. HRM MeltDoctor reagents (Applied Biosystems) and HRM software (Applied Biosystems) were used before verification by Sanger sequencing (Genome Quebec). Designed primers for PCR and sequencing were: agl-1(knu860)-F: CGA CACTTGGCCATCTCAACTC;agl-1(knu860)-R:ATTCTGCGCACTGGATCCTC; agl-1(knu864)-F:G GTAAGTTAGCTAGAAATGCAACAAGG;agl-1(knu864)-R:GGAATGAAGACCCTTGATCCAAC AG, agl-1(knu867)-F:AAGAAAGTCACGTTGCGCTGC and agl-1(knu867)-R:TCTGAATATGAGG GCATTATGACCC.
Liquid culture motility assay
Worms synchronized at day 1 and day 5 of adulthood were collected, rinsed with M9 buffer, and placed into standard 96-well plates, each well containing approximately 50–70 worms in 100 µL of M9 buffer. Their motility was recorded over 10 h using the WMicrotracker ONE device (Phylum Tech).
Lifespan assay
A total of 40 synchronized L4-stage worms were placed on nematode growth media NGM agar plates inoculated with E. coli OP50. They were transferred to fresh plates every two days and monitored for their status: alive, dead, or missing. Worms are considered dead when they no longer react to external stimuli. This experiment was replicated three times.
Progeny assay
Thirty synchronized L4-stage worms were placed individually on NGM agar plates containing E. coli OP50 (one worm per plate). Viable progenies were considered as the offspring hatched by day 1 of adulthood. Each condition was tested in triplicate, and the entire experiment was repeated three times.
Development assay
Synchronized L1-stage C. elegans were placed on NGM agar plates seeded with E. coli OP50. Worms were counted daily based on their developmental stage until the N2 wild-type worms reached adulthood (day 1). Each condition was tested in triplicate with ∼100 worms per experiment, and the experiment was repeated three times.
WormLab analysis
MBF Bioscience’s WormLab software (2020 version) was used to quantify movement parameters such as speed, wavelength, and mean amplitude. A 30 s video of the worm’s movement was recorded, and movement parameters were analyzed by manually selecting individual worms. Data were exported and statistically analyzed with GraphPad Prism 9 software. The experiment was repeated three times, with ∼ 50 worms per experiment.
Paralysis assay on solid media
Thirty-five to 40 wild-type and mutant worms were placed on NGM plates starting from day 1 of adulthood and observed daily over 12 days at 20 °C. Worms were considered paralyzed if they did not respond to gentle prodding with a worm pick. They were scored dead if they showed no head movement upon nose prodding and had no pharyngeal pumping activity. Each condition was tested in triplicate with ∼120 worms per experiment, and the experiment was repeated three times.
Glycogen quantification
Iodine staining
To observe glycogen buildup, we exposed worms to iodine crystals between 40 and 60 s before image acquisition with a Leica S6E microscope. Iodine was purchased from Sigma Aldrich (326143). Quantification was performed using Fiji software by manually selecting worm images processed using the following macro: (run(“Invert”); run(“RGB Stack”); run(“8-bit”); run(“Stack to Images”); run(“Subtract Background”… “,” OK”);). The integrated density was used to calculate the percentage of staining. Test conditions for each replicate were normalized to their respective controls. Each condition was tested in triplicate with ∼25 worms per experiment, and the experiment was repeated three times.
Calorimetric kit
Glycogen Assay Kit II (Calorimetric) was provided by Abcam, Cambridge, UK (ab169558). The analysis was based on the colorimetric method and was performed according to the manufacturer’s instructions. Briefly, samples were prepared by homogenization in ddH2O and quantified at ∼10 mg. The main steps are reagent preparation, standard curve preparation, sample preparation, positive control, reaction mix, and measurement and calculation. Raw data was provided from the plate reader (OD450). The final concentration of glycogen was expressed in micrograms (µg). The experiment was repeated three times.
Embryonic viability and brood size assays
Embryonic viability and brood size assays were performed based on standard protocols57. Ten individual L4-stage hermaphrodites were transferred into 0.2% glucose NGM plates and were allowed to self-fertilize and lay progeny over three consecutive 24-h periods at 20 °C. Each worm was transferred to a new plate daily (Days 1–3), and progeny from each timepoint were scored two days later (Days 3–5) for live larvae and unhatched embryos. Unhatched embryos were considered nonviable, while unfertilized oocytes were excluded from analysis. The experiment was repeated three times.
Drug screening in C. elegans
Small-molecule libraries from the Prestwick Chemical Library, Sigma Aldrich LOPAC Library, Microsource Drug Library, and the BML Natural Products Library from Enzo Life Sciences (totaling 3942 molecules) were used for primary screening. S1444R mutants were grown on compound NGM plates until day 1 of adulthood, and the efficacy of the drugs on motility was monitored using WMicrotracker ONE instruments (Phylum Tech). Each compound was tested in triplicate (1 drug/well, [20 μM]). Molecules were considered hits if they could increase the motility of mutants.
The secondary screen was performed in quadruplicates on the top 150 hits from the primary screen. Robust Z scores were defined as (ActivitySample−Median(ActivityAll within-plate samples))/Median absolute deviation(ActivityAll within-plate samples).
Rabusertib (LY2603618) compound was purchased from Selleckchem (#S2626-5). Worms were treated with 20 µM in solid media.
Computational analysis and target selection
We began with a list of compounds associated with the effects of a disease of interest. This list was obtained through the Connectivity Map tool from the Broad Institute (https://clue.io/command?q=/conn%20AGL). This database provides pharmacological and genetic perturbagens about a specific gene. This association could be based on gene expression experiments or other experimental results. We then predicted targets for each compound in this list of disease-associated drugs. An online machine learning application called. Similarity ensemble approach or SEA was used to perform target prediction. We filtered out compounds that do not have >4 targets predicted by SEA.
We then determined the overlap between predicted target sets for all compound pairs. This overlap was calculated as the log-transformed quotient of the number of shared targets between each pair of target sets divided by the square root of the size of each set.
These scores were used to calculate a symmetrical heatmap representing target overlap between all disease-associated compounds. The compounds were then clustered based on the correlation of target overlap scores between rows/columns (drugs). Specific clusters of compounds with high target overlap were then selected for further analysis to reveal common biological pathways.
For each analyzed cluster, a list of top targets (proteins) was identified. Because some proteins were targeted much more often than others, we first needed to calculate the baseline frequency for each target across the predicted target sets for all compounds in our library of 3449 drugs. Two methods were used to calculate target enrichment using this baseline. The first method (delta enrichment) uses the difference between a target’s baseline frequency and its frequency in a cluster. In contrast, the second method (ratio enrichment) is calculated as the quotient of these same terms. Target rankings based on delta enrichment scores appeared biased towards high-frequency targets, while the opposite bias was observed when using ratio enrichment scores, so these rankings were combined to produce a composite target enrichment score used to rank top targets for each cluster.
These lists of top enriched targets were then used to perform Gene Ontology enrichment analyses (GOEA) for each analyzed cluster. The protein IDs for each cluster’s top 30 human targets were converted to Entrez gene IDs using Uniprot’s online mapping function (https://www.uniprot.org/uploadlists). The Python library GOATools was then used to determine which annotated biological processes were statistically enriched in each set of top targets/genes relative to the expected frequency of these annotations among all human protein-coding genes. The GOEA for each cluster produces confidence scores for each enriched annotation, allowing us to produce a ranked list of which biological processes are most significantly enriched in each cluster of interest.
RNA interference experiments
All C. elegans RNAi experiments were carried out using the feeding method based on standard protocols58. Ampicillin-resistant clones were in the pL4440-DEST vector from the Ahringer RNAi library. The E. coli host strain is HT115 (DE3). For all assays, synchronized L1 worms were placed on RNAi plates seeded with either empty vector (EV) bacteria or bacterial clones expressing double-stranded RNA targeting the gene of interest. Worms were raised on RNAi plates, and the second generation was used for the subsequent assays. All RNAi clones were sequence-verified.
RNA sequencing
Sample preparation and RNA extraction
Nematodes were homogenized in 1 ml of QIAzol buffer with 0.5 mm glass beads (Qiagen) on a Precellys 24 (Bertin) at 6400 rpm for 2 × 10 s. Total RNA was extracted using the miRNeasy micro kit on-column DNase (Qiagen) treatment following the manufacturer’s instructions. The quantity of total RNA was measured using a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies). Prior to library preparation, total RNA quality was assayed on an Agilent TapeStation 4150 using High Sensitivity RNA ScreenTape (Agilent Technologies). All samples had a RIN > 9.3.
Library preparation and sequencing
The NEBNext Ultra II directional RNA library prep kit for Illumina (New England Biolabs Inc., Ipswich, MA, USA) was used to prepare mRNA sequencing libraries according to the manufacturer’s instructions. Briefly, poly(A)+ mRNA was isolated from 900 ng of total RNA for 12 samples using the NEBNext poly(A) (New England Biolabs Inc., Ipswich, MA, USA) and was used as a template for cDNA synthesis by reverse transcriptase with random primers. The quantification was done using the QuBit 3.0 fluorometer (ThermoFisher Scientific, Canada). mRNA-seq libraries were sequenced for paired-end 100 pb sequencing on NovaSeq 6000 at the Next-Generation Sequencing Platform, Genomics Center, CHU de Québec-Université Laval Research Center, Québec City, Canada. The expected mean coverage/sample will be 16 M paired-end reads.
Quality control
Raw sequencing reads were subjected to quality control using the FastQC tool (Galaxy Version 0.74) on the Galaxy platform (Galaxy version 24.1.2.dev0). After adapter trimming, the sequencing reads were of high quality (99.8% of reads > Q15, 100% of reads > Q12).
Mapping
High-quality reads were aligned to the C. elegans reference genome (WBcel235/ce11) using Spliced Transcripts Alignment to a Reference (STAR) aligner (v2.7.11a) on the Galaxy platform. The following parameters were used for STAR mapping: --runThreadN 10, genomeLoad, NoSharedMemory –genome Dir /data/db/data_managers/rnastar/2.7.4a/ce11/ce11/dataset_6c12215f-bb36-4fb5-82cc-
With a BAM file output. The resulting BAM files were used for gene quantification.
Quantification
Gene expression quantification was performed using featureCounts (v2.0.3) with the following parameters: -T 8 -p -t exon -g gene_id -a C. elegans annotation file (WBcel235.112.gtf) -Gene-ID “It” read-count (MultiQC/DESeq2/edgeR/limma-voom compatible).
Differential expression analysis
Differential expression analysis was conducted using DESeq2 (v1.42.1) in R (v4.3.3). Raw counts were normalized using the DESeq2 normalization method, and differential expression was assessed by fitting a negative binomial generalized linear model. Genes with <10 counts were excluded, and those with an adjusted p-value < 0.05 and a log2-fold change > 1 were considered significantly differentially expressed.
Statistics
The data analysis was conducted using GraphPad Prism 9 software. To evaluate statistical significance among multiple groups, we employed one-way or two-way ANOVA followed by post hoc tests, specifically the Bonferroni or Tukey tests. For comparisons between two groups, a two-tailed Student’s t-test was used. To analyze paralysis and lifespan data, we generated survival curves and assessed them using the Log-rank (Mantel–Cox) test for survival curve comparison. Data are presented as mean ± SEM.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available as “supplementary data 1–3” files. RNA-Seq data were deposited into the NIH Sequence Read Archive (SRA) under accession number PRJNA1378342 and are available at the following URL: https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA1378342.
Code availability
The code that supports the findings of this study is available upon request.
References
Dagli, A. et al. GeneReviews is a Registered Trademark of the University of Washington, Seattle. All Rights Reserved (University of Washington, 1993).
Gremse, D. A., Bucuvalas, J. C. & Balistreri, W. F. Efficacy of cornstarch therapy in type III glycogen-storage disease. Am. J. Clin. Nutr. 52, 671–674 (1990).
Schreuder, A. B. et al. GeneReviews is a Registered Trademark of the University of Washington, Seattle. All Rights Reserved (University of Washington, 1993).
Goldstein, J. L. et al. Molecular analysis of the AGL gene: identification of 25 novel mutations and evidence of genetic heterogeneity in patients with Glycogen Storage Disease Type III. Genet. Med. 12, 424–430 (2010).
Kishnani, P. S. et al. Glycogen storage disease type III diagnosis and management guidelines. Genet. Med. 12, 446–463 (2010).
Liu, K. M., Wu, J. Y. & Chen, Y. T. Mouse model of glycogen storage disease type III. Mol. Genet. Metab. 111, 467–476 (2014).
Pagliarani, S. et al. Glycogen storage disease type III: A novel Agl knockout mouse model. Biochim Biophys. Acta 1842, 2318–2328 (2014).
Yi, H. et al. Characterization of a canine model of glycogen storage disease type IIIa. Dis. Model Mech. 5, 804–811 (2012).
Yi, H. et al. Correction of glycogen storage disease type III with rapamycin in a canine model. J. Mol. Med. (Berlin) 92, 641–650 (2014).
Moreno-Arriola, E. et al. Caenorhabditis elegans: a useful model for studying metabolic disorders in which oxidative stress is a contributing factor. Oxid. Med. Cell. Longev. 2014, 705253 (2014).
Kraft, F. et al. Brain malformations and seizures by impaired chaperonin function of TRiC. Science 386, 516–525 (2024).
Wormbase. agl-1 (gene)—WormBase: Nematode Information Resource https://wormbase.org/species/c_elegans/gene/WBGene00011050#0-9fe-10 (2024).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Altschul, S. F. et al. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 272, 5101–5109 (2005).
NCBI. VCV000188819.11—ClinVar—NCBI https://www.ncbi.nlm.nih.gov/clinvar/variation/188819/ (2024).
NCBI. VCV001470163.6—ClinVar—NCBI https://www.ncbi.nlm.nih.gov/clinvar/variation/1470163/ (2024).
Sentner, C. P. et al. Glycogen storage disease type III: diagnosis, genotype, management, clinical course and outcome. J. Inherit. Metab. Dis. 39, 697–704 (2016).
Rollins, J. A., Howard, A. C., Dobbins, S. K., Washburn, E. H. & Rogers, A. N. Assessing health span in Caenorhabditis elegans: lessons from short-lived mutants. J. Gerontol. A Biol. Sci. Med. Sci. 72, 473–480 (2017).
Roberts, T. M. Major sperm protein. Curr. Biol. 15, R153 (2005).
Smith, H. Sperm motility and MSP. WormBook 1–8 https://doi.org/10.1895/wormbook.1.68.1 (2006).
Elmaci, I. & Altinoz, M. A. Targeting the cellular schizophrenia. Likely employment of the antipsychotic agent pimozide in treatment of refractory cancers and glioblastoma. Crit. Rev. Oncol. Hematol. 128, 96–109 (2018).
Lee, J. K. et al. USP1 targeting impedes GBM growth by inhibiting stem cell maintenance and radioresistance. Neuro Oncol. 18, 37–47 (2016).
Pagnoni, A. et al. Lack of burning and stinging from a novel first-aid formulation applied to experimental wounds. J. Cosmet. Sci. 55, 157–162 (2004).
(MD), B. PubChem Compound Summary for CID 73957, Pramoxine Hydrochloride (National Library of Medicine (US), National Center for Biotechnology Information, PubChem, 2004).
Meech, R. et al. The UDP-glycosyltransferase (UGT) superfamily: new members, new functions, and novel paradigms. Physiol. Rev. 99, 1153–1222 (2019).
Chiew, A. L., Gluud, C., Brok, J. & Buckley, N. A. Interventions for paracetamol (acetaminophen) overdose. Cochrane Database Syst. Rev. 2, Cd003328 (2018).
Andrade, R. J. et al. Drug-induced liver injury. Nat. Rev. Dis. Prim. 5, 58 (2019).
Zhou, Z. et al. Diverse functions of cytochrome c in cell death and disease. Cell Death Differ. 31, 387–404 (2024).
Nieto-Jimenez, C. et al. Checkpoint kinase 1 pharmacological inhibition synergizes with DNA-damaging agents and overcomes platinum resistance in basal-like breast cancer. Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21239034 (2020).
Carling, D. Branching out on AMPK regulation. Cell Metab. 9, 7–8 (2009).
McBride, A., Ghilagaber, S., Nikolaev, A. & Hardie, D. G. The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 9, 23–34 (2009).
Ma, Y. et al. SCFβ-TrCP ubiquitinates CHK1 in an AMPK-dependent manner in response to glucose deprivation. Mol. Oncol. 13, 307–321 (2019).
Gusarov, I. et al. Glycogen controls Caenorhabditis elegans lifespan and resistance to oxidative stress. Nat. Commun. 8, 15868 (2017).
Freeley, M., Kelleher, D. & Long, A. Regulation of protein kinase C function by phosphorylation on conserved and non-conserved sites. Cell Signal. 23, 753–762 (2011).
Wang, J., Han, X. & Zhang, Y. Autoregulatory mechanisms of phosphorylation of checkpoint kinase 1. Cancer Res. 72, 3786–3794 (2012).
Wormbase. gpa-15 (gene)vWormBase: Nematode Information Resource (Wormbase, 2024).
Wormbase. erm-1 (gene)—WormBase: Nematode Information Resource https://wormbase.org/species/c_elegans/gene/WBGene00011050#0-9fe6-10 (Wormbase, 2024).
Wormbase. tax-6 (gene)—WormBase: Nematode Information Resource (Wormbase, 2024).
Wormbase. jmjd-1.2 (gene)—WormBase: Nematode Information Resource (Wormbase, 2024).
Wormbase. ugt-23 (gene)—WormBase: Nematode Information Resource (Wormbase, 2024).
Wormbase. ant-1.1 (gene)—WormBase: Nematode Information Resource (Wormbase, 2024).
Kalogeropoulos, N., Christoforou, C., Green, A. J., Gill, S. & Ashcroft, N. R. chk-1 is an essential gene and is required for an S-M checkpoint during early embryogenesis. Cell Cycle 3, 1196–1200 (2004).
Scagliotti, G. et al. Phase II evaluation of LY2603618, a first-generation CHK1 inhibitor, in combination with pemetrexed in patients with advanced or metastatic non-small cell lung cancer. Investig. N. Drugs 34, 625–635 (2016).
Smith, J., Tho, L. M., Xu, N. & Gillespie, D. A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108, 73–112 (2010).
Zannini, L., Delia, D. & Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 6, 442–457 (2014).
Li, X. Y. et al. Inhibition of USP1 reverses the chemotherapy resistance through destabilization of MAX in the relapsed/refractory B-cell lymphoma. Leukemia 37, 164–177 (2023).
Olsen, A., Vantipalli, M. C. & Lithgow, G. J. Checkpoint proteins control survival of the postmitotic cells in Caenorhabditis elegans. Science 312, 1381–1385 (2006).
Krüger, K. et al. Multiple DNA damage-dependent and DNA damage-independent stress responses define the outcome of ATR/Chk1 targeting in medulloblastoma cells. Cancer Lett. 430, 34–46 (2018).
Raimondi, M. et al. USP1 (ubiquitin specific peptidase 1) targets ULK1 and regulates its cellular compartmentalization and autophagy. Autophagy 15, 613–630 (2019).
Cheng, A. et al. A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori’s disease. Genes Dev. 21, 2399–2409 (2007).
Cheng, A., Zhang, M., Okubo, M., Omichi, K. & Saltiel, A. R. Distinct mutations in the glycogen debranching enzyme found in glycogen storage disease type III lead to impairment in diverse cellular functions. Hum. Mol. Genet. 18, 2045–2052 (2009).
Patten, S. A. et al. Neuroleptics as therapeutic compounds stabilizing neuromuscular transmission in amyotrophic lateral sclerosis. JCI Insight 2, https://doi.org/10.1172/jci.insight.97152 (2017).
Mistry, H. et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol. Cancer Ther. 12, 2651–2662 (2013).
García-Santisteban, I., Peters, G. J., Giovannetti, E. & Rodríguez, J. A. USP1 deubiquitinase: cellular functions, regulatory mechanisms and emerging potential as target in cancer therapy. Mol. Cancer 12, 91 (2013).
Tossing, G. et al. Genetic and pharmacological PARP inhibition reduces axonal degeneration in C. elegans models of ALS. Hum. Mol. Genet. 31, 3313–3324 (2022).
Stiernagle, T. Maintenance of C. elegans. WormBook 1–11 https://doi.org/10.1895/wormbook.1.101.1 (2006).
Kwah, J. K. & Jaramillo-Lambert, A. Measuring embryonic viability and brood size in Caenorhabditis elegans. J. Vis. Exp. https://doi.org/10.3791/65064 (2023).
Conte, D. Jr., MacNeil, L. T., Walhout, A. J. M. & Mello, C. C. RNA interference in Caenorhabditis elegans. Curr. Protoc. Mol. Biol. 109, 26.23.21–26.23.30 (2015).
Acknowledgements
This study was funded by The Rare Diseases: Models & Mechanisms Network. We gratefully acknowledge Claudia Maios for the liquid culture experiments efforts and Audrey Labarre for technical and academic assistance.
Author information
Authors and Affiliations
Contributions
E.S., J.D., and H.D. conceived the study. H.D. and E.S. designed the experiments and analyses. C.M., H.D., and J.D. performed the experiments. H.D. and B.P. analyzed the data and generated the figures. H.D. wrote and prepared the manuscript. E.P. generated the strains. J.A.P. supervised the study. All authors have reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Kaliya Georgieva.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Daghar, H., Pyman, B., Maios, C. et al. CHK1 inhibition rescues abnormal glycogen buildup in a Caenorhabditis elegans model for glycogen storage disease III. Commun Biol 9, 257 (2026). https://doi.org/10.1038/s42003-026-09535-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-026-09535-9
