
Abstract
Liver fibrosis is a major global health burden with limited treatment options. Transforming growth factor-beta-induced protein (TGFBI) is crucial in fibrotic diseases and tumors, however, its precise mechanism in liver fibrosis remains unclear. Here we show that TGFBI promotes liver fibrosis in male C57BL/6 mice. TGFBI is upregulated in fibrotic livers and derived from non-parenchymal cells. Genetic TGFBI deficiency alleviates liver fibrosis in both CCl4 (carbon tetrachloride) injection and bile duct ligation (BDL) models. Mechanistically, PDGFRβ is identified via RNA sequencing as a key downstream molecule upregulated by TGFBI in hepatic stellate cells (HSCs) via the integrin αvβ3-FAK-STAT3 pathway, promoting HSC proliferation and activation. Meanwhile, TGFBI increases PDGF-B expression in macrophages through the integrin αvβ3-AKT-ERK pathway, driving their proliferation, migration and differentiation into the profibrotic TREM2+CD9+ subpopulation. Elevated PDGF-B reversely stimulates TGFBI production in macrophages, which creates a positive feedback loop. This TGFBI-mediated interaction between HSCs and macrophages remodels the profibrotic microenvironment to promote liver fibrosis, identifying a potential therapeutic target.

Similar content being viewed by others


Introduction
Hepatic fibrosis is a severe response to a variety of chronic forms of liver hepatitis and may ultimately progress to cirrhosis and even hepatocellular carcinoma, which is severely harmful to human health1,2. However, effective clinical treatments are still lacking. Thus, clarifying the mechanism of the development of liver fibrosis is crucial for exploring effective therapeutic approaches.
During the development of liver fibrosis, hepatic stellate cells (HSCs) are the primary source of activated myofibroblasts1, which produce abundant extracellular matrix proteins and subsequently lead to extensive deposition, which is regarded as the key event resulting in liver fibrosis. Inflammatory factors, cell growth factors, and chemokines stimulate the transition of quiescent HSCs to activated myofibroblasts3. Given the activation characteristics of HSCs, impeding their activation is an effective strategy to mitigate liver fibrosis.
TGFBI is a cellular extracellular matrix protein that regulates numerous biological functions during embryonic development and the progression of human diseases4,5,6,7,8,9. These functions include cell adhesion and bone formation, which affect the synthesis and activity of various collagens, connective tissues, and vascular and neural structures. TGFBI can promote the invasion and metastasis of various types of cancers, such as colon cancer, renal cancer, and prostate cancer10,11,12. In addition, TGFBI is closely associated with the progression of fibrotic diseases13. TGFBI plays a significant role in delaying venous thrombosis, promoting atherosclerosis14, enhancing hepatic steatosis7, amplifying inflammatory responses14,15, and facilitating glomerular6 and pulmonary fibrosis16. Interestingly, plasma proteome profiling of patients with liver cirrhosis indicated that TGFBI is upregulated in patients with liver cirrhosis13. Moreover, our previous studies have confirmed that matrix proteins such as periostin are closely linked to the development of liver fibrosis. Thus, exploring the specific functions and molecular mechanisms of TGFBI in liver fibrosis is of great significance17,18.
Additionally, it is well known that macrophages are crucial for the development, propagation, and control of hepatic inflammation and fibrosis19,20. The activation of resident Kupffer cells by key mediators leads to the production of aberrant proinflammatory cytokines and chemokines, which in turn recruit many macrophages derived from monocytes to areas of inflammation21. Recent research has revealed that many heterogeneous liver macrophages may interact differently with various cells in the liver to have varying effects on the development of liver fibrosis22,23. Interestingly, several recent studies have indicated that the TREM2+CD9+ subpopulation of macrophages is derived from monocytes and localized in fibrotic regions, where it induces myofibroblast activation and promotes liver fibrosis24,25,26,27.
Platelet-derived growth factor (PDGF), an important inflammatory mediator, stimulates the migration and proliferation of HSCs28. Clinical studies have shown that the degree of necrotic inflammation and fibrosis in patients with liver injury is closely related to the overactivation of PDGF and its downstream molecules. When liver injury occurs, PDGF is highly expressed in macrophages, injured endothelial cells, and activated hepatic stellate cells29. At present, many studies have verified that PDGF-B is one of the most potent factors associated with HSC activation. Moreover, the expression of the PDGF receptor β (PDGFRβ) on the activated HSC membrane significantly increases, which is advantageous for HSC activation30,31,32.
In this study, we aimed to elucidate the biological function of TGFBI in liver fibrosis. Furthermore, we investigated the effects and related molecular mechanisms of TGFBI on HSC activation and the macrophage phenotype.
Results
TGFBI is upregulated and mainly derived from nonparenchymal cells in fibrotic livers
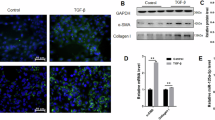
Through human chronic liver disease database analysis, we found that TGFBI expression was significantly upregulated in human cirrhotic liver tissues compared to normal liver tissues33,34. And, TGFBI expression was significantly greater in severe liver fibrosis than that in mild liver fibrosis (Fig. 1A), and through correlation analysis, it was found that the expression of TGFBI expression was positively correlated with human liver fibrosis (Fig. 1B). To further elucidate TGFBI expression in fibrotic liver tissues, we successfully constructed two types of mouse liver fibrosis models via CCl4 injection and BDL. Compared to control mice, TGFBI mRNA levels were notably increased in the livers of mice with liver fibrosis (Fig. 1C). Western blot and immunohistochemistry (IHC) also revealed significantly higher protein levels of TGFBI in fibrotic livers than in normal livers (Figs. 1D and S1A). These results clearly indicate that TGFBI is highly expressed in hepatic fibrotic tissues. As an extracellular matrix protein, it is critical to clarify the cellular origin of TGFBI for further investigation. Thus, to determine which cell type produces TGFBI in fibrotic livers, we used IF to co-label TGFBI with molecular markers of liver parenchymal cells (HNF4α), liver stellate cells (alpha smooth muscle actin, α-SMA), and macrophages (F4/80) in fibrotic livers. However, the results revealed that TGFBI was widely distributed without clear colocalization due to its characteristics of secretion (Fig. 1E). To further identify the sources of TGFBI, we isolated primary liver parenchymal cells, hepatic stellate cells, and macrophages from mouse normal and fibrotic livers and detected that TGFBI was mainly expressed in nonparenchymal cells, especially in macrophages (Fig. 1F). FISH also confirmed the expression pattern in mouse fibrotic livers (Figs. 1G and S1B). In total, TGFBI was significantly upregulated in fibrotic liver tissues and was mainly derived from hepatic stellate cells and macrophages.

A Human chronic liver disease database analysis of TGFBI expression (n = 19–41 independent samples). B Correlation analysis of expression between Tgfbi and Acta2 in the Human Chronic Liver Disease database (n = 32–41 independent samples). C, D RT-qPCR (C) and Western blot (D) analyses of TGFBI expression in mouse livers of CCl4 and BDL models. E IF staining of coexpression of TGFBI with HNF-4α, F4/80, α-SMA, and COL1A1 in the mouse livers of CCl4 and BDL models, respectively. F RT-qPCR analysis of TGFBI expression in isolated primary mouse liver parenchymal cells, HSCs, and macrophages. G FISH analysis of coexpression of Tgfbi with F4/80 and Acta2 in mouse livers, respectively. Mice samples (n = 4–6 per group). Data were presented as the mean ± SDs; scale bars, 50 µm; *p < 0.05 **p < 0.01, ***p < 0.001, ****p < 0.0001.
TGFBI deficiency markedly alleviated liver inflammation and fibrosis in mice
To investigate the biological function of TGFBI in the development of liver fibrosis, we used wild-type (WT) and Tgfbi-knockout (Tgfbi−/−) mice to induce liver fibrosis via CCl4 and BDL models. In fibrotic mice, Tgfbi−/− mice presented significantly less liver fibrosis compared with WT mice, as demonstrated by H&E, Sirius red, Masson, and IHC staining of COL1A1 (Fig. 2A). The sera from Tgfbi−/− mice presented lower alanine aminotransferase (ALT) and aspartate transaminase (AST) levels, and TUNEL staining also showed a reduction, indicating significant remission of liver damage in fibrotic Tgfbi−/− mice than in fibrotic WT mice (Fig. 2B, C). The mRNA levels of fibrosis-related molecules, including Col1a1, Col3a1, and Timp-1, were significantly lower in Tgfbi−/− mice than in WT mice (Fig. 2D). In contrast, at baseline, there was no significant difference in these indicators between the two genotypes (Fig. S2A–D). Since liver fibrosis is an evil consequence of chronic liver injury, the effect of TGFBI on inflammation was also analyzed. We found that the absence of TGFBI significantly mitigated the degree of macrophage infiltration by performing immunofluorescence (IF) staining of F4/80 (Figs. 2E and S2E) and IHC staining of CD68 in mouse fibrotic livers (Figs. 2F and S2F). The mRNA levels of inflammatory factors such as Ccl2, Il6, and Tnf-a were markedly lower in the livers of fibrotic Tgfbi−/− mice than fibrotic WT mice (Figs. 2G and S2G).

A H&E, Sirius, Masson staining, and IHC staining of COL1A1 in the fibrotic livers of WT and Tgfbi −/− mice of CCl4 and BDL models. B The measurement of serum ALT and AST in mice. C TUNEL staining and quantitative analysis of apoptosis in CCl₄ and BDL model livers. D RT-qPCR analysis of Col1a1, Col3a1, and Timp-1 expression. E IF staining of F4/80 in mouse fibrotic livers. F IHC staining of CD68 in mouse livers of CCl4 and BDL models. G RT-qPCR analysis of Ccl2, Il6, and Tnf-a expression. Mice samples (n = 4–6 per group). Data were presented as the mean ± SDs; scale bars, 50 µm; *p < 0.05 **p < 0.01, ***p < 0.001, ****p < 0.0001.
Exogenous TGFBI protein execrated mice liver fibrosis
Previous studies have confirmed that knockout of Tgfbi can alleviate liver fibrosis and inflammation. Therefore, we verified its overexpression effect by intraperitoneal injection of TGFBI recombinant protein. The results showed that exogenous TGFBI could successfully induce spontaneous fibrosis and inflammatory damage in mice (Fig. S3A–E), thereby proving that the upregulation of TGFBI expression is a sufficient condition to drive this pathological process.
TGFBI promotes HSC proliferation and activation directly
The activation and proliferation of hepatic stellate cells (HSCs) are the most important causes of liver fibrosis35. In this study, real-time quantitative PCR (RT-qPCR) (Fig. 3A), Western blot (Fig. 3B), and IHC staining analyses (Fig. S4A) revealed that the expression of the HSC activation marker α-SMA in the fibrotic livers of two mouse models was significantly reduced after Tgfbi knockout. On the other hand, in the recombinant TGFBI protein (rTGFBI) overexpression model, α-SMA shows consistent upregulation at both mRNA and protein levels (Fig. S4B). Connection analysis revealed that the mRNA levels of Acta2 increased with Tgfbi expression increasing (Fig. S4C), indicating a positive connection between Acta2 and Tgfbi expression. To further investigate whether TGFBI directly regulates HSC phenotypes, we isolated primary HSCs from WT and Tgfbi−/− mice for self-activation analyses. Compared with WT HSCs, Tgfbi−/− HSCs presented reduced activation and proliferation according to IF analysis of α-SMA and Ki67 expression (Fig. 3C). Furthermore, we treated mouse primary HSCs and human cell line LX2 with recombinant TGFBI protein (rTGFBI). After rTGFBI administration, α-SMA expression levels in primary HSCs and LX2 cells markedly increased, as detected by RT-qPCR (Figs. 3D and S4D) and Western blot (Figs. 3E and S4E). IF analysis also obtained the same results (Fig. 3F). CCK8 assay revealed that rTGFBI stimulation significantly increased the proliferation of primary HSCs and LX2 cells (Fig. S4F). IF assay results showed that rTGFBI treatment significantly increased the number of Ki67-positive cells in HSCs or LX2. (Fig. S4G). Transwell analysis revealed that rTGFBI treatment also increased migration ability (Fig. S4H). In brief, these results indicate that TGFBI can promote HSC activation, proliferation, and migration directly.

A, B RT-qPCR (A) and Western blot (B) analyses of α-SMA expression in WT and Tgfbi−/− mice livers of CCl4 and BDL models. C IF staining of α-SMA and Ki67 in primary HSC from WT and Tgfbi−/− mice. D, E RT-qPCR (D) and Western blot (E) analysis of α-SMA in HSC or LX2 treated with rTGFBI (ng/mL) with the indicated concentrations (n = 3 independent experiments). F IF staining of α-SMA in primary HSC or LX2 after rTGFBI (200 ng/mL) treatment for 6 h (n = 3 independent experiments). G Western blot analysis of the indicated proteins in HSCs treated with rTGFBI (200 ng/mL) for 6 h (n = 3 independent experiments). H Western blot analysis of the indicated proteins in HSCs treated with rTGFBI (200 ng/mL) alone or in combination with integrin αvβ3 or αvβ5 neutralizing antibody (n = 3 independent experiments). I Western blot analysis of the indicated proteins in HSCs treated with rTGFBI (200 ng/mL) or in combination with the indicated inhibitors for 6 h (n = 3 independent experiments). J Relative mRNA levels of Acta2 in HSCs treated with rTGFBI (200 ng/mL) alone or in combination with integrin αvβ3 or αvβ5 neutralizing antibody for 6 h (n = 3 independent experiments). K Relative mRNA levels of Acta2 in HSCs or LX2 treated with rTGFBI (200 ng/mL) or in combination with the indicated inhibitors for 6 h (n = 3 independent experiments). Mice samples (n = 4–6 per group). Data were presented as the mean ± SDs; scale bars, 50 µm; *p < 0.05 **p < 0.01, ***p < 0.001, ****p < 0.0001.
TGFBI activates HSCs through integrin αvβ3-FAK-STAT3 pathway
To determine the molecular mechanisms by which TGFBI regulates HSC activation, we explored potential signaling pathways. After treatment with rTGFBI, the levels of p-FAK, p-SRC, p-AKT, and p-STAT3 significantly increased in primary HSCs and LX2 cells, whereas the level of p-ERK did not change (Figs. 3G, S5A, and S6A). Integrins are well recognized as TGFBI receptors. Therefore, on the basis of the literature and previous studies by our research group, we used siRNA to knock out the expression of integrin αvβ3 or αvβ5, or used neutralizing antibodies against integrin αvβ3 and αvβ5 to block the binding of TGFBI to the corresponding receptors36. We found that blocking αvβ3 or knocking down αvβ3 instead of αvβ5 can inhibit the increase of the aforementioned signaling molecules induced by rTGFBI (Figs. 3H, S5B, and S6B). The upregulation of HSC activation marker α-SMA caused by TGFBI was also effectively eliminated due to the blocking or knocking down of αvβ3 (Figs. 3H, S5B, and S6B). Similar changes were observed in the mRNA levels of Acta2 (Figs. 3J and S6D). Furthermore, IF staining also revealed the increase of α-SMA and Ki67 in primary HSCs and LX2 cells were prominently inhibited by the αvβ3-neutralizing antibody (Fig. S6F). In summary, these findings indicate that the effects of TGFBI on HSC proliferation and activation can be inhibited by blocking its receptor integrin αvβ3. After determining the functional receptor of TGFBI, we further investigated the downstream signaling pathway involved in TGFBI regulating HSCs. Based on the activated signaling molecules, we used several inhibitors, including PF573228 (p-FAK inhibitor), PP2 (p-SRC inhibitor), LY294002 (p-AKT inhibitor), and Stattic3 (p-STAT3 inhibitor), to block the corresponding signaling pathway. As shown by Western blot analysis (Figs. 3I and S6C), the p-FAK inhibitor PF573228 significantly decreased p-STAT3 and α-SMA expression. And, blocking p-STAT3 by Stattic3 inhibited the increase of α-SMA expression induced by TGFBI. Similar results were obtained via RT-qPCR (Figs. 3K and S6E) and IF staining (Fig. S7A, B). Taken together, these results indicate that TGFBI affects HSC phenotypes through the integrin αvβ3-FAK-STAT3 pathway.
PDGFRβ acts as a key downstream target for HSC activation induced by TGFBI
To deeply excavate the key downstream targets regulated by TGFBI participating in HSC activation, we applied RNA sequencing technology to identify candidate differential genes in the livers of mice in the CCl4 injection model. We analyzed the original data and identified some differentially expressed genes (DEGs) whose expression levels significantly changed and whose expression strongly correlated with the development of liver fibrosis (Fig. 4A). KEGG pathway analysis revealed enrichment of the PI3K-AKT and FAK signaling pathways (Fig. S8A), which is consistent with the aforementioned signaling pathways. Among the differentially expressed genes, PDGFRβ was of interest (Fig. 4B). In human liver fibrotic samples, the expressions of TGFBI and PDGFRβ were significantly upregulated (Fig. 4C), and further analysis based on clinical databases showed a positive correlation between their expressions (Fig. 4D). In two mouse models of liver fibrosis, PDGFRβ expression was dramatically upregulated in fibrotic livers compared with normal livers, whereas its expression was significantly decreased when TGFBI was deleted, as shown by Western blot (Fig. 4E), RT-qPCR (Fig. 4F), and IHC staining (Fig. S8C). Moreover, Pdgfrb mRNA expression was positively correlated with Tgfbi mRNA expression (Fig. S8B). IHC staining revealed that PDGFRβ was distributed mainly around the region of liver fibrosis (Fig. S8C). The rTGFBI overexpression mouse model also showed that the protein and mRNA expression levels of PDGFRβ increased consistently (Fig. S8F). To further verify the main cell source of PDGFRβ, we isolated primary cells from the liver tissues from these two mouse liver fibrosis models and determined that PDGFRβ expression was the highest in HSCs (Fig. 4G). To clarify whether PDGFRβ is a downstream target of TGFBI, we treated primary HSCs and LX2 cells with rTGFBI. After rTGFBI administration, the protein and mRNA levels of PDGFRβ in these cells were significantly increased (Figs. 4H and S8G), suggesting that TGFBI upregulated PDGFRβ expression in HSCs. Also, blocking integrin αvβ3 with a neutralizing antibody evidently inhibited the increase of PDGFRβ induced by TGFBI stimulation (Figs. 4I and S8H). Subsequently, we further found that FAK-STAT3 pathway participated in TGFBI-mediated PDGFRβ expression (Figs. 4J and S8I).

A Heat map of differentially expressed genes drawn based on RNA sequencing data. Samples were obtained from oil-treated WT mice, CCl4-induced WT mice, and Tgfbi−/− mice liver tissue lysates. B GO gene analysis of the differential proteins. C IF staining and quantitative analysis of TGFBI and PDGFRβ in human fibrotic liver tissue. D Correlation analysis of expression between Tgfbi and Pdgfrb in the Human Chronic Liver Disease database (n = 58 independent samples). E Western blot analysis of PDGFRβ expression in WT and Tgfbi−/− mice livers of CCl4 and BDL models. F RT-qPCR analysis of PDGF-B in WT and Tgfbi−/− mice livers of CCl4 and BDL models. G Relative mRNA levels of PDGFRβ in primary liver parenchymal cells, HSCs, and macrophages isolated from mice livers of CCl4 and BDL models (n = 3 independent experiments). H Western blot and RT-qPCR analysis of PDGFRβ expression in HSC treated with rTGFBI (200 ng/mL) for 6 h (n = 3 independent experiments). I Western blot and RT-qPCR analysis of α-SMA and PDGFRβ in HSC treated with rTGFBI (200 ng/mL) alone or in combination with integrin αvβ3 or αvβ5 neutralizing antibody for 6 h (n = 3 independent experiments). J Western blot and RT-qPCR analyses of PDGFRβ in HSC treated with rTGFBI (200 ng/mL) alone or in combination with the indicated inhibitors for 6 h (n = 3 independent experiments). Mice samples (n = 4–6 per group). Data were presented as the mean ± SDs; scale bars, 50 µm; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns not significant (Student’s t test).
Given that TGFBI exerts a positive regulatory effect on PDGFRβ, it was necessary to investigate the role of TGFBI-stimulated upregulation of PDGFRβ. We observed colocalization of PDGFRβ with the HSC activation marker α-SMA in mouse fibrotic livers (Fig. S8D). Under rTGFBI stimulation, α-SMA and PDGFRβ expression were simultaneously increased in primary HSCs and LX2 cells (Fig. S8E). Consistent with this finding, blockade of PDGFRβ function with the inhibitor SU11248 abrogated rTGFBI-induced activation of primary HSCs and LX2 cells, as detected by RT-qPCR (Fig. S9A and C), Western blot (Fig. S9B and D), and IF staining (Fig. S9E, F). The TGFBI-induced increases in the proliferation of primary HSCs and LX2 cells were also abrogated by SU11248 (Fig. S9E, F). Similarly, knocking down Pdgfrb by siRNA failed to upregulate α-SMA expression in primary HSCs, in spite of the presence of rTGFBI (Fig. S9G–I). These findings suggest that TGFBI promotes HSC activation by upregulating PDGFRβ, which is essential for HSC activation induced by TGFBI.
TGFBI promotes macrophage proliferation, migration, and differentiation
The aforementioned results indicated that knockdown of Tgfbi caused a severe decrease in macrophage infiltration and inflammatory factor expression levels in the two mouse liver fibrosis models (Fig. 2E–G), which implied that TGFBI has a concurrent regulatory effect on inflammatory cells. It is noteworthy that rTGFBI stimulation significantly enhanced the proliferation and migration abilities of mouse bone marrow-derived macrophages (BMDMs) and RAW264.7 cells (Fig. 5A–C). As reported, TREM2+CD9+ macrophages are a subpopulation of scar-associated macrophages (SAMs) that exert a profibrotic effect37. Very interestingly, our preceding analysis of RNA-seq data revealed that TREM2 and CD9 expression was obviously increased in fibrotic livers compared with normal livers but significantly decreased after TGFBI deficiency. Tgfbi deletion dramatically decreased the mRNA levels of Trem2 and Cd9 in fibrotic mouse livers (Fig. 5D), which confirmed the RNA sequencing results. IF staining and statistical analysis further demonstrated that the number of cells co-expressing TREM2 and CD9 was prominently reduced after Tgfbi knockout (Fig. S10A). In contrast, in the livers of mice injected with rTGFBI, not only were the mRNA levels of Trem2 and Cd9 significantly upregulated (Fig. S10C), but the immunofluorescence results also confirmed that the number of cells co-expressing TREM2 and CD9 was significantly increased (Fig. S10B). According to flow cytometry analysis, the percentage of the TREM2+CD9+ subset in the fibrotic Tgfbi−/− mice livers was significantly lower than fibrotic WT mice livers (Fig. 5E). Moreover, rTGFBI treatment of BMDMs and RAW264.7 cells resulted in considerable increases in Trem2 and Cd9 mRNA expression (Fig. 5F) and in the proportion of TREM2+CD9+ macrophages (Fig. 5G). Collectively, these findings indicate that TGFBI promotes macrophage proliferation, migration, and differentiation into the TREM2+CD9+ subpopulation.

A IF staining and quantitative analysis of Ki67 in BMDM or RAW264.7 treated with or without rTGFBI (200 ng/mL) for 6 h (n = 3 independent experiments). B CCK8 assay of BMDM or RAW264.7 treated with rTGFBI (200 ng/mL) (n = 3 independent experiments). C Transwell assay of BMDM or RAW264.7 treated with or without rTGFBI (200 ng/mL) for 24 h (n = 3 independent experiments). D RT-qPCR analysis of Trem2 and Cd9 expression in WT and Tgfbi−/− mice livers of CCl4 and BDL models. E Flow cytometry analysis and statistical analysis of the percentage of TREM2+CD9+ subpopulation in WT and Tgfbi−/− mice livers of CCl4 and BDL models (n = 3 mice per group). F RT-qPCR analysis of Trem2 and Cd9 expression in BMDM or RAW264.7 treated with or without rTGFBI (200 ng/mL) for 6 h (n = 3 independent experiments). G Flow cytometry analysis and statistical analysis of the percentage of TREM2+CD9+ subpopulation in BMDM or RAW264.7 treated with or without rTGFBI (200 ng/mL) for 24 h (n = 3 independent experiments). Data were presented as the mean ± SDs; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns not significant (Student’s t test).
TGFBI orchestrates macrophages through upregulating PDGF-B expression
It is well known that PDGF-B is not only the ligand of PDGFRβ but also a potent chemokine derived from macrophages that stimulates the migration and proliferation of inflammatory cells38,39. PDGF-B plays a biological role by activating its receptor PDGFRβ, which has been confirmed to be associated with the development of a number of diseases40,41. On the basis of these findings, we wondered whether TGFBI orchestrates macrophages by regulating PDGF-B expression. In human fibrotic livers, the expressions of TGFBI and PDGF-B are both upregulated (Fig. S11A). Consistent with this, PDGF-B expression was detected to be markedly increased in mouse fibrotic livers than that in normal livers, while it significantly decreased following Tgfbi knockout, as demonstrated by Western blot (Fig. S11C), RT-qPCR (Fig. S11D), and IHC (Fig. S11B). Conversely, the intervention of exogenous rTGFBI directly promoted the expression of PDGF-B at the mRNA and protein levels in mice fibrotic livers (Fig. S10D). Furthermore, correlation analysis revealed that the mRNA levels of Pdgfb and Tgfbi were positively correlated (Fig. S11E), suggesting that there might be a regulatory relationship between TGFBI and PDGF-B. PDGF-B was further verified to be expressed mainly by macrophages through isolation of liver cells from the two mouse models (Fig. S11F). Subsequently, through FISH experiments, it was observed that PDGF-B co-localized with the macrophage marker F4/80 (Fig. S11G), while the co-localization signals with Acta2 (HSCs) or Cd31 (endothelial cells) were weak (Fig. S11H, I), which further confirmed that macrophages were the main source of PDGF-B. Moreover, rTGFBI stimulation strikingly increased PDGF-B expression in BMDMs and RAW264.7 cells (Fig. 6A, B). Subsequently, changes related to downstream signaling pathways were further explored, with p-FAK and p-ERK showed the strongest changes (Fig. 6C). When anti-αvβ3, p-FAK, or p-ERK inhibitors were applied, the effects of TGFBI on PDGF-B upregulation and proliferation in macrophages were significantly inhibited (Figs. 6D and S10A, B, and D). Taken together, these results indicate that TGFBI regulates PDGF-B production in macrophages via integrin αvβ3-FAK-ERK pathway. Furthermore, knockdown of Pdgfb in BMDMs and RAW264.7 cells via siRNA effectively inhibited the increase of proliferation induced by TGFBI (Fig. S12E, F). Flow cytometry analysis revealed that Pdgfb knockdown notably abrogated the ability of TGFBI to promote differentiation into TREM2+CD9+ macrophages (Fig. 6E). In conclusion, these results indicate that TGFBI regulates the biological function of PDGF-B by upregulating its expression in macrophages.

A Western blot analyses of PDGF-B in BMDM or RAW264.7 treated with rTGFBI (ng/mL) with the indicated concentrations (n = 3 independent experiments). B RT-qPCR analyses of PDGF-B in BMDM or RAW264.7 treated with rTGFBI (ng/mL) with the indicated concentrations (n = 3 independent experiments). C Western blot analysis of the indicated proteins in BMDM or RAW264.7 treated with or without rTGFBI (200 ng/mL) for 6 h. D RT-qPCR analysis of Pdgfb in BMDM or RAW264.7 treated with rTGFBI (200 ng/mL) alone or in combination with integrin αvβ3 or αvβ5 neutralizing antibody (n = 3 independent experiments). E Flow cytometry analysis and statistical analysis of the percentage of Trem2+CD9+ subpopulation in BMDM or RAW264.7 transfected with the indicated siRNAs for 24 h and treated with or without rTGFBI (200 ng/mL) for 24 h (n = 3 independent experiments). F RT-qPCR and Western blot analysis of α-SMA in HSCs cocultured with BMDMs transfected the indicated siRNAs for 24 h and treated with or without rTGFBI (200 ng/mL) for 24 h (n = 3 independent experiments). G RT-qPCR and Western blot analyses of α-SMA in HSCs treated with rTGFBI or in combination with SU11248 (n = 3 independent experiments). Mice samples (n = 4–6 per group). Data were presented as the mean ± SDs; scale bars, 50 µm; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns not significant (Student’s t test).
Macrophage-derived PDGF-B synergistically strengthens HSC activation with TGFBI
As mentioned above, TGFBI can directly induce HSC activation in a PDGFR-β-dependent manner. Infiltrating macrophages in fibrotic liver tissues can interact with HSCs, thereby activating them. Indeed, we observed that macrophages have spatial proximity with HSC by IF staining (Fig. S13A), especially TREM2+CD9+ macrophages (Fig. S13B). To further verify the interaction between macrophages and HSCs, we obtained BMDMs from WT and Tgfbi−/− mice and cocultured them with primary HSCs in vitro. Compared with Tgfbi−/− BMDMs, HSCs cocultured with WT BMDMs presented greater degrees of activation and proliferation (Fig. S13C, D). Next, we cocultured HSCs with BMDMs in which Pdgfb was knocked down using siRNA to investigate the role of PDGF-B in the interaction between macrophages and HSCs. We eventually found that BMDMs with lower Pdgfb levels failed to enhance HSC activation regardless of rTGFBI stimulation (Figs. 6F and S13E). Next, we treated HSCs individually or simultaneously with recombinant TGFBI and PDGF-B. These results demonstrated that both the recombinant proteins effectively stimulated HSC activation. Moreover, combined treatment with the two recombinant proteins had the most significant effect on HSC activation and proliferation (Figs. 6G and S13F, G), which suggests that macrophage-derived PDGF-B can synergistically strengthen HSC activation with TGFBI.
TGFBI and PDGF-B forms a positive feedback loop in macrophages
According to our foregoing data, both TGFBI and PDGF-B were mainly derived from macrophages in fibrotic livers (Figs. 1F and S11F–I). Then, IF staining also revealed that TGFBI and PDGF-B were closely related in fibrotic mouse livers (Fig. S14A). As validated previously, TGFBI orchestrates macrophages by upregulating PDGF-B. Whether macrophage-secreted PDGF-B conversely regulates TGFBI expression requires further investigation. To test this hypothesis, we isolated primary parenchymal cells, HSCs, and macrophages and stimulated them with recombinant PDGF-B protein (rPDGF-B). Finally, we found that TGFBI expression in macrophages was significantly upregulated under rPDGF-B stimulation, whereas TGFBI expression in the other two types cell did not (Fig. S14B). Subsequently, we used rPDGF-B to treat BMDMs and RAW264.7 cells at various concentrations and times. After PDGF-B stimulation, the mRNA and protein levels of TGFBI were dramatically increased in BMDMs and RAW264.7 cells, as shown by RT-qPCR (Fig. S14C, D), Western blot (Fig. S14E, F), and IF staining analysis (Fig. S14G). Furthermore, we found that there was a significant increase in p-AKT and p-ERK expression in macrophages after rPDGF-B treatment (Fig. S14H). However, treatment with the PDGFRβ inhibitor SU11248, the AKT inhibitor LY294002, and ERK inhibitor PD98059 significantly inhibited the upregulation of TGFBI induced by PDGF-B in macrophages (Fig. S14I), providing evidence that PDGF-B regulates TGFBI through the PDGFRβ-AKT-ERK pathway. The above results show that macrophage-secreted PDGF-B induced by TGFBI in turn promoted TGFBI expression, forming a positive feedback loop to sustain a profibrotic microenvironment.
Blocking PDGFRβ alleviates mouse liver inflammation and fibrosis
Our above-mentioned findings suggest PDGFRβ acts as a key downstream regulator in HSC activation, whether TGFBI directly activates HSCs by upregulating PDGFRβ expression or macrophage-derived PDGF-B synergistically activates HSCs. Upon activation of PDGFRβ, HSCs undergo proliferation and activation, exacerbating liver fibrosis. Therefore, targeting PDGFRβ is crucial for the repression of liver fibrosis. In this study, we utilized the PDGFRβ inhibitor SU11248 to administer primary HSCs in vitro and found that SU11248 impeded the exaggerated activation of HSCs induced by recombinant TGFBI and PDGF-B proteins (Fig. 6G). In addition, in the CCl4 -induced mouse liver fibrosis model, we established therapeutic and prophylactic administration regimens, respectively to evaluate the intervention effect of imatinib. After imatinib treatment, mouse liver inflammation and fibrosis were significantly reduced, as detected by H&E, Sirius Red, Masson, and IHC staining for COL1A1 and IF staining for F4/80 (Fig. 7A and D). Serological tests showed that after imatinib intervention, the levels of AST and ALT in mice were significantly reduced, suggesting that liver damage was alleviated (Fig. 7B). The expression of fibrosis- and inflammation-related genes was significantly reduced in imatinib -treated mice (Fig. 7C and E). In conclusion, blocking PDGFRβ function effectively alleviates liver inflammation and fibrosis in mice.

A H&E, Sirius, Masson staining, and IHC staining of COL1A1 and CD68 in CCl4 induced fibrotic livers of WT and Tgfbi−/− mice administered with or without imatinib treatment (TX) or prevention (PR). B The measurement of serum ALT and AST in mice. C RT-qPCR analysis of Col1a1, Col3a1, and Timp-1 expression (n = 4–5 mice per group). D IF staining and statistical analysis of F4/80 in CCl4 induced fibrotic livers of WT and Tgfbi−/− mice administrated with or without imatinib treatment (TX) or prevention (PR). E RT-qPCR analysis of Ccl2, Il6, and Tnf-a expression. Mice samples (n = 4–5 mice per group). Data were presented as the mean ± SDs; scale bars, 50 µm; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns not significant (Student’s t test).
Discussion
To the best of our knowledge, this study is the first to demonstrate that TGFBI plays a crucial role in the regulation of HSCs and macrophages during liver fibrosis development. The main findings are as follows: (i) The TGFBI expression is remarkably increased in fibrotic livers; (ii) TGFBI deletion significantly relieves mouse liver fibrosis; (iii) TGFBI not only activates HSCs by increasing PDGFRβ expression but also synergistically promotes HSC activation by increasing PDGF-B expression in the microenvironment; (iv) TGFBI regulates proliferation, migration, and differentiation of macrophages into a profibrotic TREM2+CD9+ subpopulation; (v) PDGF-B in turn upregulates TGFBI expression in macrophage, forming a positive feedback loop, exacerbating the development of liver fibrosis. Our findings highlight a key regulatory function of TGFBI in the development of liver fibrosis.
Stationary hepatic stellate cells transform into activated stellate cells with a myofibroblast phenotype during the development of liver fibrosis. Activated stellate cells are the most important event causing liver fibrosis42,43. Based on the elucidation that TGFBI promotes liver fibrosis through the successful construction of two kinds of models in WT and Tgfbi−/− mice, we focused on the effect and molecular mechanism of TGFBI regulation of HSC phenotypes. Having established that TGFBI could directly promote HSC proliferation and activation, we next sought to identify the downstream targets mediating this effect. Strikingly, PDGFRβ was successfully identified by RNA sequencing and confirmed as an essential downstream mediator of TGFBI-induced HSC activation30,31,32. Another study also revealed that PDGFRβ expression is significantly elevated in activated HSCs44. Upon binding to its ligand, PDGFR immediately induces HSC activation from a quiescent state33. In addition, other studies have confirmed that activated HSCs upregulate PDGFRβ expression, whereas another isoform, PDGFRα, remains unchanged45,46. In our study, the explicit mechanism by which TGFBI upregulates PDGFRβ was the integrin αvβ3-FAK-STAT3 signaling pathway. Knocking out TGFBI in vivo or blocking TGFBI in vitro can inhibit the over-activation of PDGFRβ and its downstream signaling pathways, thereby reducing HSC activation and alleviating mice liver fibrosis. Thus, TGFBI may serve as a promising therapeutic target for liver fibrosis.
Macrophages in fibrotic liver tissue are another protagonist of the development of liver fibrosis. Liver macrophages can directly activate HSCs, thereby contributing to the progression of liver fibrosis21. During liver injury, recruited monocyte-derived macrophages (MoMacs), which express high levels of lymphocyte antigen 6 family member C (Ly6C), secrete proinflammatory and profibrogenic cytokines, sustaining the fibrotic response47,48. Additionally, relevant evidence indicates that infiltrating macrophages and tissue-resident Kupffer cells proliferate locally49,50, thereby promoting the development of fibrotic disease. In this study, we found that TGFBI depletion significantly reduced the number of macrophages infiltrating into the fibrotic region of the liver. Thus, we speculate that TGFBI plays a regulatory role in macrophages. Consistent with our hypothesis, TGFBI was found to induce macrophage proliferation and migration. More interestingly, our RNA sequencing data revealed that both TREM2 and CD9 were differentially expressed between the fibrotic livers of WT and Tgfbi−/− mice. Coincidentally, a recent study reported that a profibrotic TREM2+CD9+ macrophage subpopulation is found in human fibrotic livers by single-cell transcriptome analysis51. Several studies have shown that macrophages expressing TREM2 and CD9 are located mainly in the fibrotic region and promote the development of fibrosis52. We ultimately elucidated that TGFBI could induce macrophages to differentiate into the TREM2+CD9+ subpopulation. However, its specific role in liver fibrosis requires further investigation.
After acute or chronic liver injury, infiltrated macrophages usually produce a variety of inflammatory and fibrotic cytokines that influence themselves and their neighboring cells, including HSCs, in an autocrine and paracrine manner. Among the numerous cytokines, PDGF has been proven to be one of the most effective factors related to HSC activation53 and is the most effective proliferative factor in liver fibrosis. This study has shown that TGFBI directly activates HSCs by upregulating PDGFRβ expression. As a ligand of PDGFRβ, we investigated whether PDGF-B was also regulated by TGFBI. Notably, TGFBI not only induces the upregulation of PDGF-B through integrin αvβ3-FAK-ERK pathway, but also the upregulation of PDGF-B is required for TGFBI to regulate macrophage proliferation, migration, and differentiation into the TREM2+CD9+ subpopulation. It has been reported that PDGF-B is mainly expressed by liver cells in the early stages of various chronic liver diseases when liver fibrosis occurs54. By isolating primary liver cells from fibrotic liver tissues, we confirmed that both PDGF-B and TGFBI are derived mainly from macrophages. Since TGFBI can increase PDGF-B expression in macrophages, macrophages-secreted PDGF-B may in turn boost TGFBI expression. Indeed, rPDGF-B stimulation strongly overexpressed TGFBI in macrophages, thereby forming a positive autocrine feedback loop that further exacerbates liver inflammation and fibrosis.
Given that TGFBI can increase PDGFRβ expression to induce HSC activation and that TGFBI can increase PDGF-B expression in macrophages, we wondered whether macrophage-derived PDGF-B promotes HSC activation in coordination with TGFBI. After Pdgfb was knocked down in BMDMs by siRNA, BMDMs with normal or low Pdgfb levels were cocultured with HSCs. However, we found that HSCs cocultured with BMDMs with low Pdgfb levels did not achieve their activation and proliferation very well, despite rTGFBI stimulation. These results provide strong evidence that macrophages interact with HSCs in the process of liver fibrosis.
This study focused on hepatic stellate cells and macrophages, revealing the regulatory mechanism of TGFBI on both, but the absence of liver sinusoidal endothelial cells (LSECs) was a major limitation. As a specialized capillary endothelium of the liver, the “capillarization” caused by LSECs dysfunction is an early core event for the initiation of fibrosis. LSECs can directly activate hepatic stellate cells by secreting factors such as fibrinogen and regulate the recruitment and function of macrophages55. Therefore, ignoring this cell may prevent us from fully analyzing the global role of TGFBI in the liver cell network (especially the early endothelial-interstitial dialogue). This limitation, on the one hand, stems from the technical challenges in the in vitro culture and functional maintenance of primary LSECs56, and on the other hand, the phenotypic changes of hepatic stellate cells and macrophages in this study were more significant, which is also one of the reasons why we focused on these two types of cells. Future research will need to rely on co-culture and other models to further explore the influence of TGFBI on the phenotype of LSECs, in order to more comprehensively clarify its network of action in liver fibrosis.
In summary, we demonstrated that TGFBI plays a crucial role in liver fibrosis and that gene knockout of Tgfbi effectively improved liver fibrosis in mice. We found that TGFBI significantly upregulated PDGFRβ expression in HSCs and promoted their activation in a PDGFR-β-dependent manner. We also found that TGFBI promoted macrophage proliferation, migration, and differentiation by increasing PDGF-B expression in macrophages. Macrophages-secreted TGFBI and PDGF-B form an autocrine positive feedback loop to remodel the profibrotic microenvironment. It should be pointed out that the conclusions of this study are mainly based on animal experimental models, and their translation into clinical applications still faces challenges. Therefore, future research can not only further deepen the fine molecular mechanism of TGFBI-PDGFRβ axis in liver fibrosis, but also expand the perspective to a broader extracellular matrix network, such as exploring the synergistic effect of the “matrix protein (such as TGFBI)—integrin downstream signaling pathway” axis. Clarifying the regulatory mechanism of this network not only helps to comprehensively reveal the pathogenesis of liver fibrosis, but also provides a novel theoretical basis and direction for the development of multi-target and efficient combined treatment strategies.
Materials and methods
Animal research
Tgfbi+/− mice were initially purchased from GemPharmatech. The C57BL/6 mice were derived from the Nanjing Institute of Biomedical Sciences at Nanjing University and were raised at the Laboratory Animal Center of Xiamen University. After crossing it with wild-type C57BL/6J mice for at least ten generations, we obtained C57BL/6J Tgfbi+/− hybrid mice with pure background. The wild-type (WT) and knockout (Tgfbi+/−) mice used in the experiment were obtained through hybridization of male Tgfbi+/− hybrid and female Tgfbi+/− hybrid mice. All the mice were raised in the specific pathogen-free (SPF) environment of the Xiamen University Animal Laboratory Center, and all the experiments complied with the relevant requirements of the Animal Management Committee of Xiamen University Animal Laboratory Center. We have complied with all relevant ethical regulations for animal use. For surgical procedures (such as bile duct ligation), mice were anesthetized using isoflurane inhalation. At the experimental endpoint, mice were euthanized by cervical dislocation while under deep anesthesia.
Randomization and blinding of animal experiments
Randomization: All experimental mice were uniformly fed before grouping. Before starting the modeling or intervention, we randomly assigned the mice to the wild-type control group, model group, gene knockout group, or protein treatment group, etc., based on their body weight using a random number table method, to ensure that there was no significant difference in the initial body weight among the groups and to avoid selection bias. Blinding method: During the data collection and result evaluation stages, we implemented the single-blind principle. Specifically, researchers who conduct subsequent histological staining, image acquisition, quantitative analysis, and some molecular biology experiments (such as RT-qPCR data collection) are not aware of the specific grouping information of the samples they are processing. Unblinding and statistical comparisons are not conducted until all quantitative data analyses have been completed.
Exogenous overexpression of TGFBI mouse model
Male C57BL/6 mice aged 6 to 8 weeks were selected for this study. The mice in the experimental group were intraperitoneally injected with TGFBI recombinant protein dissolved in sterile PBS daily at a dose of 2 mg/kg per day for four consecutive weeks57,58. The control group was injected with PBS in the same way.
Cell culture
The cells used in this study included: the mouse mononuclear macrophage leukemia cell line RAW264.7, the mouse fibroblast cell line L-929, and the human hepatic stellate cell line LX-2, all of which were purchased from the American Type Culture Collection (ATCC). The generations used in all cell line experiments were controlled between the 5th and 15th generations. In addition, this study also utilized primary cells isolated from mice, including: bone marrow-derived macrophages (BMDMs), primary hepatic stellate cells (HSCs), hepatic macrophages, and hepatic parenchymal cells. All cells were routinely cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (P/S) in a humidified incubator containing 5% CO2 and 95% air at 37 °C.
Reagent
The following reagents and inhibitors were applied: recombinant mouse TGFBI protein (#2559-BG-050, R&D Systems); recombinant human TGFBI protein (#3409-BG-050, R&D Systems); PF573228, 10 μM (FAK inhibitor, #HY-10461, MedChemExpress); PP2, 10 μM (Src inhibitor, #S7008, Selleck); LY294002, 10 μM (AKT inhibitor, #S1105, Selleck); Stattic, 2.5 μM (STAT3 inhibitor, #S7024, Selleck)59; PD98059 20 μM (ERK inhibitor, #HY-12028)60; αvβ3 or αvβ5 blocking antibody, 0.1 mg/ml (Merck Millipore). SU11248 (PDGFR inhibitor, sunitinib, #HY-10255A, MedChemExpress) and Imatinib Mesylate (PDGFR inhibitor, #HY-50946, MedChemExpress)61.
Construction of a hepatic fibrosis model with carbon tetrachloride
In previous studies, male mice at 8–10 weeks of age received intraperitoneal injections of carbon tetrachloride (CCl4; Sigma‒Aldrich) to induce liver fibrosis. WT and Tgfbi−/− mice were injected with CCl4 (diluted in corn oil at v: v = 1:4), whereas the control group was administered corn oil alone (2.5 mL/kg body weight) twice a week for 8 weeks. The mice were euthanized 48 h after the final CCl4 injection for subsequent analysis. Liver and serum samples were collected for molecular, histological, and serological assessment.
Construction of a hepatic fibrosis model with bile duct ligation
In accordance with previous studies, male mice aged 8–10 weeks received bile duct ligation (BDL) surgery and were fasted for 12 h before surgery. The control group underwent a sham operation. The mice were fed normally for 3 weeks after BDL or sham surgery and were sacrificed 3 weeks later for analysis. Liver and serum samples were collected for molecular, histological, and serological analysis.
Construction of animal models for imatinib
In this study, a mouse liver fibrosis model was established using carbon tetrachloride. The grouping and administration are as follows: Treatment group: After 6 weeks of carbon tetrachloride modeling, mice received daily intraperitoneal injections of 50 mg/kg imatinib for two weeks, while continuing weekly carbon tetrachloride injections to maintain the fibrosis model. At the end of the experiment (week 8), animals were sacrificed under anesthesia, and liver tissues were collected. Prevention group: To assess the preventive effect of imatinib, mice in this group received daily intraperitoneal injections of 20 mg/kg imatinib starting with the first carbon tetrachloride injection. The sacrifice timing and tissue collection method were identical to those in the treatment group.
Preparation of mouse bone marrow-derived macrophages
Mouse bone marrow-derived macrophages (BMDMs) were isolated from the bone marrow of mouse femurs62. After the mice were killed, the femur was cut, the bone marrow was washed with a 5 mL syringe, filtered through a 70 μm filter membrane, and centrifuged at 1200 rpm for 5 min. The supernatant was discarded, 3 mL of red cell lysate was added, the mixture was cracked at room temperature for 2 min, and the mixture was centrifuged at 1200 rpm for 5 min. The cell precipitates obtained from the above steps were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 20% FBS, 1% P/S, and 30% L-929 cell line-conditioned medium for seven days on a resuspension plate, and BMDM cells were obtained for subsequent experiments.
Mouse liver cell isolation experiment
Primary hepatic stellate cells (HSCs), liver macrophages, and hepatic parenchymal cells were isolated via density gradient centrifugation with Percoll (Sigma‒Aldrich) solution according to a previous experimental method63. The inferior vena cava was incubated, buffer SC1 was injected until the blood cells in the liver were rinsed, and buffer SC2 containing collagenase P was injected at 37 °C for 10 min. The liver was cut and placed in a 6 cm dish containing 5 mL of digestive fluid SC2 containing collagenase P (Roche, CH) and pronase (Roche, CH). The cut liver was blown away, the liquid GBSS-B was stored at 35 mL, filtered through a 70 μm filter membrane, and centrifuged at room temperature at 50 × g for two minutes, after which the supernatant was collected for subsequent use. The resulting cell precipitate consisted of liver parenchymal cells, which were then recultured in DMEM for subsequent experiments. The supernatant was centrifuged at 250 × g at room temperature for 10 min, the supernatant was discarded, 10 mL of GBSS-B mixture was resuspended, the mixture was centrifuged at 250 × g again, and 2 mL of the suspended cells was added to the upper layer of 35% Percoll solution at the lower layer and 15% Percoll solution at the upper layer. The mixture was centrifuged at 900 × g at 4 °C with an acceleration of 9 and a deceleration of 0 for 15 min. After centrifugation, the intermediate layer cells of 35% and 15% Percoll solution were absorbed, and the same volume of liquid GBSS-B was added for resuspension. Centrifugation at 900 × g was performed to maximize acceleration and velocity reduction. After centrifugation, the supernatant was discarded, the mixture was resuspended in DMEM, and the medium was changed two hours later. Primary liver stellate cells were obtained and used for subsequent experiments. The liver macrophages were then separated. The centrifuged nonparenchymal cell suspension was added to the top layer of 50% Percoll solution in the lower layer and 25% Percoll solution in the upper layer. Centrifugation was performed at 4 °C at 1500 × g with an acceleration of 9 and a reduction of 0 for 20 min. The intermediate layer cells of 50% and 25% Percoll solution were absorbed, the same volume of liquid GBSS-B was added, and the centrifugation at 1500 × g maximized both acceleration and deceleration. After centrifugation, the supernatant was discarded, the pellet was resuspended in DMEM, and the medium was changed 30 min later to obtain primary liver macrophages, which were subsequently used for subsequent experiments.
BMDMs were isolated, L-929 cells were cultured in advance, and the supernatant was collected to induce BMDMs. C57BL/6 wild-type mice aged 6–8 weeks were sacrificed, the bone marrow of the femur was washed away with PBS, the mixture was filtered, the mixture was centrifuged at 1200 rpm for 5 min, the supernatant was discarded, and 3 mL of red cell mixture was added; the mice were split at room temperature for 2 min and centrifuged at 1200 rpm for 5 min. The cells were subsequently suspended in DMEM (20% fetal bovine serum, 30% L-929 cell supernatant, 50% DMEM, and 1% penicillin/streptomycin) and cultured for 7 days. Cell experiments were subsequently performed after digestion and passage.
Histological analysis
Liver tissue sections were stained with hematoxylin and eosin (H&E), Sirius, and Masson’s trichrome. The sections were stained with hematoxylin staining solution for 20 s. After the water turned blue again, they were re-stained with eosin staining solution for 8 s. Finally, it is successively dehydrated by gradient ethanol, transparent with xylene, and sealed with neutral gum. The sections were directly stained with Sirius Red staining solution for 10 min. After dyeing is completed, it is rapidly decomposed and dehydrated with anhydrous ethanol, made transparent with xylene, and then sealed with neutral gum. The experimental procedures were carried out in accordance with the manufacturer’s instructions (G1006-100ML, Wuhan Saiwei Technology Co., LTD.). The key steps of the main process include: staining with nuclear dye (iron hematoxylin), followed by staining the cytoplasm with brilliant spring red solution, and finally staining the collagen fibers with aniline blue. Images were obtained via a Leica DM4B microscope.
Serum biochemistry
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured via standard enzymatic methods according to the manufacturer’s instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Immunohistochemistry (IHC) and immunofluorescence (IF)
The embedded liver tissue was cut into 5 μm tissue sections for routine dewaxing and rehydration, and antigen repair was performed with sodium citrate buffer, followed by DAB staining according to the manufacturer’s instructions for the immunohistochemical kit (Maixin Biotechnologies, KIT-9710), followed by hematoxylin staining for 20 s. Tissue sections were sealed with neutral gum after dehydration. A DM4B light microscope was used to select the position of interest to take pictures and convert them into pictures. For IF staining, fresh liver tissue was dehydrated in phosphate buffer (PBS) containing 30% sucrose and then quickly frozen in liquid nitrogen after OCT encapsulation. The frozen tissue was cut into 6 μm slices and incubated at 37 °C for 30 min. The primary antibody was added, the secondary antibody with a fluorescent label was coupled, and nuclear staining was performed with DAPI. Finally, the stained sections were observed and photographed via a confocal microscope.
Real-time fluorescence quantitative PCR
Total RNA was extracted from liver tissues or cells via the TRIzol reagent (Thermo Fisher Scientific). cDNA was subsequently synthesized via a ReverTra Ace qPCR RT kit (FSQ-201, TOYOBO, Japan). Quantitative real-time fluorescent PCR (RT‒qPCR) was performed via a CFX96 real-time fluorescent quantitative PCR system (Bio-Rad, Hercules, CA, USA). The sequences of the PCR primers used are listed in the accompanying table. Gene expression levels were normalized to those of Gapdh, which served as an internal reference gene. The Supplementary Table 2 provides the qPCR primers and siRNA sequences used in this study.
Western blot analysis
SDS‒PAGE gel at an appropriate concentration was selected according to the detected protein molecular weight, and the separated protein molecules were transferred to a PVDF membrane, which was closed and incubated with primary antibody at 4 °C overnight. The anti-mouse/rabbit secondary antibody with the membrane coupled with horseradish peroxidase (HRP) was further incubated, detected via the enhanced chemiluminescence (ECL) method, and developed on X-ray film. The antibodies used in this study are all listed in Supplementary Table 1. The untrimmed and unedited images with size markings printed on blot/gel images are listed as supplementary figures in Supplementary Fig. 16.
RNA interference
Primary HSCs or BMDMs were transfected with specific siRNAs (Sangon Biotech, China) against PDGFRβ and PDGF-B, which were predesigned according to the manufacturer’s instructions. The transfection efficiency of the transfected cells was verified by RT‒qPCR 24 h after transfection.
RNA sequencing (RNA-seq)
The RNA sequencing process extracted from the total lysate of liver tissue is as follows: total RNA extraction, quality inspection, library construction and sequencing, bioinformatics analysis: FastQC was used to assess the quality of the original sequencing data, and Trimmomatic software was used to remove low-quality read segments and linkers to obtain high-quality clean data. Align the clean data to the mouse reference genome. The expression levels of genes were quantified using software. Differential expression analysis: Perform differential expression analysis using the R package. The significance threshold (FDR) we set is adj. p-value < 0.05, and the absolute log2× change is > 1. Only genes that meet both of these conditions simultaneously are recognized as differentially expressed genes.
Flow cytometry
Treated primary cells or cell lines were prepared as single-cell suspensions, incubated with fluorescently labeled antibodies, and subsequently analyzed via flow cytometry. The gating strategies for each of the flow cytometry experiments are detailed in Supplementary Fig. 15. The primary antibodies used in this study are listed in Supplementary Table 1.
Fluorescence in situ hybridization (FISH)
The embedded paraffin-embedded tissue was cut into 5 μm thick sections, dewaxed, and rehydrated following the experimental procedures according to the instruction manual of the fluorescence in situ hybridization kit (Genepharma, China). Finally, the samples were nucleated with DAPI, sealed with an anti-fluorescence quencher, and imaged with a confocal microscope.
Cell migration experiment
The LX-2 and RAW264.7 cells, as well as the primary HSCs and BMDMs, were counted, and then 5 × 105 cell suspensions in 100 μL of serum-free DMEM were added to the transwell chamber (8 μm). Complete medium (600 μL), with or without recombinant TGFBI protein, was added to the lower chamber. After the cells migrated down the chamber for 48 h, they were fixed with 4% paraformaldehyde, and the transwell chamber was stained with crystal violet and washed with PBS. After the chamber was dried, photographs were taken via an inverted microscope, and the differences were statistically analyzed.
Cell counting kit-8 (CCK8) assay
The LX-2 and RAW264.7 cell lines and the primary HSC and BMDM cells were counted in 96-well Petri dishes with 1000 cells per 100 μL of DMEM. The 0, 24, 48, and 72 h time points were set, CCK8 reagent (Beyotime, C0039, China) was added, and 405 nm wavelength detection was carried out with the enzyme-labeled instrument two hours later. After the detection of all the time nodes was completed, the data were processed and statistically analyzed.
Multiple immunofluorescence staining
The embedded liver tissue was cut into 5 μm tissue sections for routine dewatering and rehydration. Antigen repair was performed with sodium citrate buffer, and the primary and secondary antibodies were used to culture and stain the cells according to the instructions of the TSA fluorescence triple-staining kit (ABclonal Technology, RK05903, China). Then, the nuclei were stained with DAPI. Finally, the sections were sealed with glycerin, observed, and photographed via a confocal microscope.
Statistics and reproducibility
Quantitative data (mean ± SDs) were analyzed using Student’s t-test, one-way ANOVA (for multiple comparisons), or two-way ANOVA (performed using GraphPad Prism 8 software, GraphPad, San Diego, California, USA) for multiple comparisons. The data distribution was assumed to be normal, but no formal test was conducted. Statistical significance was defined as P < 0.05. The specific test methods used in each experiment are described in the corresponding legends. The sample size (n) represents the number of biological independent samples or animals, as defined in the legends.
Ethical compliance
All animal experiments were performed in accordance with the animal study protocols approved by the Animal Care and Use Committee of Xiamen University.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated in this study have been made publicly available to ensure verifiability. The RNA sequencing data are stored in the Science Data Bank of the Chinese Academy of Sciences (ScienceDB) and can be accessed publicly through the persistent identifier (DOI: 10.57760/sciencedb.35038). The quantitative source data of all figures in the text can be found in Supplementary Data 1. The untrimmed original blot images are included in Supplementary Fig. 16 of the supplementary information file. If there are any other material requirements, you can contact the corresponding author for assistance.
References
Kisseleva, T. & Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 18, 151–166 (2021).
Bataller, R. & Brenner, D. A. Liver fibrosis. J. Clin. Investig. 115, 209–218 (2005).
Tsuchida, T. & Friedman, S. L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411 (2017).
Nielsen, N. S. et al. Biochemical mechanisms of aggregation in TGFBI-linked corneal dystrophies. Prog. Retin Eye Res. 77, 100843 (2020).
Ruiz, M. et al. TGFBI secreted by mesenchymal stromal cells ameliorates osteoarthritis and is detected in extracellular vesicles. Biomaterials 226, 119544 (2020).
Leibing, T. et al. Deficiency for scavenger receptors Stabilin-1 and Stabilin-2 leads to age-dependent renal and hepatic depositions of fasciclin domain proteins TGFBI and periostin in mice. Aging Cell 22, e13914 (2023).
Lee, S. G. et al. TGFBI remodels adipose metabolism by regulating the Notch-1 signaling pathway. Exp. Mol. Med. 55, 520–531 (2023).
Lee, J., Lee, J., Sim, W. & Kim, J. H. Soluble TGFBI aggravates the malignancy of cholangiocarcinoma through activation of the ITGB1 dependent PPARgamma signalling pathway. Cell Oncol. 45, 275–291 (2022).
Han, K. E. et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Prog. Retin Eye Res. 50, 67–88 (2016).
Corona, A. & Blobe, G. C. The role of the extracellular matrix protein TGFBI in cancer. Cell Signal 84, 110028 (2021).
Lecker, L. S. M. et al. TGFBI production by macrophages contributes to an immunosuppressive microenvironment in ovarian cancer. Cancer Res. 81, 5706–5719 (2021).
Zhou, J. et al. A novel role of TGFBI in macrophage polarization and macrophage-induced pancreatic cancer growth and therapeutic resistance. Cancer Lett. 578, 216457 (2023).
Niu, L. et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 15, e8793 (2019).
Bochenek, M. L. et al. Endothelial overexpression of TGF-β-induced protein impairs venous thrombus resolution. JACC: Basic Transl. Sci. 9, 100–116 (2024).
Shrestha, S., McFadden, M. J., Gramolini, A. O. & Santerre, J. P. Proteome analysis of secretions from human monocyte-derived macrophages post-exposure to biomaterials and the effect of secretions on cardiac fibroblast fibrotic character. Acta Biomater. 111, 80–90 (2020).
Yang, K. et al. Transforming growth factor-beta induced protein regulates pulmonary fibrosis via the G-protein signaling modulator 2 /Snail axis. Peptides 155, 170842 (2022).
Xiao, H. et al. Periostin deficiency reduces diethylnitrosamine-induced liver cancer in mice by decreasing hepatic stellate cell activation and cancer cell proliferation. J. Pathol. 255, 212–223 (2021).
Zhang, Y. et al. Periostin protects against alcohol-related liver disease by activating autophagy by interacting with protein disulfide isomerase. Cell Mol. Gastroenterol. Hepatol. 15, 1475–1504 (2023).
Pellicoro, A., Ramachandran, P., Iredale, J. P. & Fallowfield, J. A. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 14, 181–194 (2014).
Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 66, 1300–1312 (2017).
Tacke, F. & Zimmermann, H. W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 60, 1090–1096 (2014).
Olona, A. et al. Adipoclast: a multinucleated fat-eating macrophage. BMC Biol. 19, 246 (2021).
Coelho, I., Duarte, N., Macedo, M. P. & Penha-Goncalves, C. Insights into macrophage/monocyte-endothelial cell crosstalk in the liver: a role for trem-2. J. Clin. Med. 10, https://doi.org/10.3390/jcm10061248 (2021).
Xiong, X. et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell 75, 644–660.e645 (2019).
Remmerie, A. et al. Osteopontin expression identifies a subset of recruited macrophages distinct from Kupffer cells in the fatty liver. Immunity 53, 641–657.e614 (2020).
Joshi, N. et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur. Respir. J 55, https://doi.org/10.1183/13993003.00646-2019 (2020).
Hou, J. et al. TREM2 sustains macrophage-hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis. J. Clin. Invest 131, https://doi.org/10.1172/JCI135197 (2021).
Okada, H. et al. Acyclic retinoid targets platelet-derived growth factor signaling in the prevention of hepatic fibrosis and hepatocellular carcinoma development. Cancer Res. 72, 4459–4471 (2012).
Ying, H. Z. et al. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (Review). Mol. Med Rep. 16, 7879–7889 (2017).
Wang, R. et al. Gomisin D alleviates liver fibrosis through targeting PDGFRbeta in hepatic stellate cells. Int. J. Biol. Macromol. 235, 123639 (2023).
Kocabayoglu, P. et al. beta-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 63, 141–147 (2015).
Liu, F. et al. Salvianolic acid B inhibits hepatic stellate cell activation and liver fibrosis by targeting PDGFRβ. Int. Immunopharmacol. 122, https://doi.org/10.1016/j.intimp.2023.110550 (2023).
Zhang, W. et al. Heterogeneity of hepatic stellate cells in fibrogenesis of the liver: insights from single-cell transcriptomic analysis in liver injury. Cells 10, 2129 (2021).
Gijbels, E. et al. Robustness testing and optimization of an adverse outcome pathway on cholestatic liver injury. Arch. Toxicol. 94, 1151–1172 (2020).
Higashi, T., Friedman, S. L. & Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 121, 27–42 (2017).
Han, B. et al. The role of TGFBI (betaig-H3) in gastrointestinal tract tumorigenesis. Mol. Cancer 14, 64 (2015).
Seidman, J. S. et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity 52, 1057–1074.e1057 (2020).
Mooring, M. et al. Hepatocyte CYR61 polarizes profibrotic macrophages to orchestrate NASH fibrosis. Sci. Transl. Med. 15, eade3157 (2023).
Miller, C. R. & Hjelmeland, A. B. Breaking the feed forward inflammatory cytokine loop in the tumor microenvironment of PDGFB-driven glioblastomas. J. Clin. Invest. https://doi.org/10.1172/JCI175127 (2023).
Xie, H. et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat. Med. 20, 1270–1278 (2014).
Zaidi, M., Lizneva, D. & Yuen, T. The role of PDGF-BB in the bone-vascular relationship during aging. J. Clin. Invest. 131, https://doi.org/10.1172/JCI153644 (2021).
Czochra, P. et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J. Hepatol. 45, 419–428 (2006).
Hui, A. Y. & Friedman, S. L. Molecular basis of hepatic fibrosis. Expert Rev. Mol. Med. 5, 1–23 (2003).
Kocabayoglu, P. et al. β-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 63, 141–147 (2015).
Yang, J. et al. A positive feedback loop between Inactive VHL-triggered histone lactylation and PDGFRbeta signaling drives clear cell renal cell carcinoma progression. Int. J. Biol. Sci. 18, 3470–3483 (2022).
Cui, Z. et al. Endothelial PDGF-BB/PDGFR-beta signaling promotes osteoarthritis by enhancing angiogenesis-dependent abnormal subchondral bone formation. Bone Res. 10, 58 (2022).
Mabire, M. et al. MAIT cell inhibition promotes liver fibrosis regression via macrophage phenotype reprogramming. Nat. Commun. 14, 1830 (2023).
Cai, B. et al. Macrophage MerTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab. 31, 406–421.e407 (2020).
Davies, L. C. et al. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat. Commun. 4, 1886 (2013).
Robbins, C. S. et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 19, 1166–1172 (2013).
Ramachandran, P. et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518 (2019).
Guilliams, M., Thierry, G. R., Bonnardel, J. & Bajenoff, M. Establishment and maintenance of the macrophage niche. Immunity 52, 434–451 (2020).
Borkham-Kamphorst, E. et al. Pro-fibrogenic potential of PDGF-D in liver fibrosis. J. Hepatol. 46, 1064–1074 (2007).
Wang, Y. et al. Hepatocyte Ninjurin2 promotes hepatic stellate cell activation and liver fibrosis through the IGF1R/EGR1/PDGF-BB signaling pathway. Metabolism 140, 155380 (2023).
Mo, H. et al. The role of liver sinusoidal endothelial cells in metabolic dysfunction-associated steatotic liver diseases and liver cancer: mechanisms and potential therapies. Angiogenesis 28, https://doi.org/10.1007/s10456-025-09969-5 (2025).
Winkler, M. et al. Endothelial GATA4 controls liver fibrosis and regeneration by preventing a pathogenic switch in angiocrine signaling. J. Hepatol. 74, 380–393 (2021).
Knorr, J. et al. Interleukin-18 signaling promotes activation of hepatic stellate cells in mouse liver fibrosis. Hepatology 77, 1968–1982 (2023).
Xia, P. et al. Therapeutic effects of recombinant human S100A6 and soluble receptor for advanced glycation end products(sRAGE) on CCl(4)-induced liver fibrosis in mice. Eur. J. Pharm. 833, 86–93 (2018).
Ma, H. et al. Periostin promotes colorectal tumorigenesis through integrin-FAK-Src pathway-mediated YAP/TAZ activation. Cell Rep. 30, 793–806.e796 (2020).
Lee, H. R., Lee, J. & Kim, H. J. Differential effects of MEK inhibitors on rat neural stem cell differentiation: Repressive roles of MEK2 in neurogenesis and induction of astrocytogenesis by PD98059. Pharm. Res. 149, 104466 (2019).
Neef, M. et al. Oral imatinib treatment reduces early fibrogenesis but does not prevent progression in the long term. J. Hepatol. 44, 167–175 (2006).
Shi, R., Wang, J., Zhang, Z., Leng, Y. & Chen, A. F. ASGR1 promotes liver injury in sepsis by modulating monocyte-to-macrophage differentiation via NF-kappaB/ATF5 pathway. Life Sci. 315, 121339 (2023).
Mederacke, I., Dapito, D. H., Affo, S., Uchinami, H. & Schwabe, R. F. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 10, 305–315 (2015).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (82370623, 82571812, 82170600, 82070607, and 82273416) and the Natural Science Foundation of Fujian Province of China (2022J01015).
Author information
Authors and Affiliations
Contributions
H.W. designed and conducted the experiments, analyzed and interpreted the data, and drafted the manuscript. X.Y. and Yan.Z2. conducted the experiments and collected the data. L.K. and Yue.Z3. conducted the experiments. R.H. and S.Y. provided technical support. G.O. inputs critical intellectual property. T.W. analyzed and interpreted the data. Fan L. and Y.L. designed the study, analyzed and interpreted the data, critically revised the manuscript, obtained funding, and supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Biology thanks Martí Ortega-Ribera, Andrew Leask, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Dr Toshiro Moroishi and Dr Ophelia Bu.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, H., Yan, X., Kuang, L. et al. TGFBI promotes liver fibrosis through remodeling the profibrotic microenvironment by a positive feedback regulatory loop. Commun Biol 9, 355 (2026). https://doi.org/10.1038/s42003-026-09601-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-026-09601-2
