
Abstract
Invadopodia, dynamic cancer cell protrusions, deform and degrade extracellular matrix (ECM) to facilitate invasion. Intracellular calcium ions (Ca2+) are critical second messengers involved in cancer cells migration, proliferation, and apoptosis, but their role in invadopodia dynamics remains unclear. Here, we propose a chemo-mechanical model integrating Ca2+ transmembrane transport, myosin contractility, adhesion dynamics, actin polymerization, and membrane type 1 matrix metalloproteinase (MT1-MMP) hydrolysis. We find that increased invadopodia length elevates membrane tension, activating mechanosensitive channels and raising intracellular Ca2+ levels, aligning with experimental observations. Our model reveals that invadopodia oscillatory and monotonic dynamics are governed by actin polymerization and myosin recruitment, with Ca2+ transport enhancing dynamics via myosin recruitment and reciprocal effects on Ca2+ transport. Furthermore, by incorporating MT1-MMP-mediated ECM degradation in our model, we find that ECM degradation promotes invadopodia extension and elevates Ca2+ levels, which shifts the invadopodia dynamics from monotonic to oscillatory. Overall, our model offers a comprehensive theoretical framework for understanding Ca2+ transport and invadopodia dynamics in cancer cells.
Similar content being viewed by others

Introduction
Calcium ions (Ca2+) are powerful second messengers that can regulate multiple cellular behaviors, such as cell migration, proliferation, and apoptosis1,2,3. Ca2+ ions are transported across the cell membranes through Ca2+ channels and pumps4, and alterations in the individual Ca2+ channel or pump can affect disease progression5,6,7. Recently, researchers have paid special attention to the Ca2+ oncogenic pathways8,9 as well as the molecular mechanisms underlying the expression of associated proteins10,11. Dysfunction in Ca2+ channels or pumps in cancer cells alters Ca2+ transmembrane transport and results in remodeling of Ca2+ homeostasis12.
Cancer cells use invadopodia, actin-rich membrane protrusions, to explore the extracellular matrix (ECM) and navigate microenvironments, marking the first step in invasion. Invadopodia consist of an actin core surrounded by adhesion and scaffolding proteins13,14,15 and exhibit dynamic protrusion-retraction cycles16,17 driven by actin polymerization and myosin contractility18. Invadopodia play a key role in cell mechanosensing by probing ECM mechanical properties through adhesion and dynamic forces19,20,21. Invadopodia also facilitate ECM degradation by releasing matrix metalloproteinases (MMPs)22,23,24. Recent studies have shown that Ca2+ transmembrane transport regulates various cancer cell invadopodia dynamics25,26. However, how Ca2+ transport interacts with mechano-transduction factors to regulate invadopodia remains unclear.
Previous theoretical studies have revealed various modes of transmembrane ion transport to maintain intracellular ion homeostasis27,28. Jiang and Sun proposed a model for cellular volume and pressure regulation by considering the changes of intracellular ions through mechano-sensitive channels and active pumps29. Building on this framework, Yang et al. investigated how cell volume and spreading dynamics respond to substrate stiffness, combining ion transport theory with experimental data30. Li expanded on this by showing how ion movement at the cell’s front and rear drives migration under an electric field31. McEovy et al. utilized the same ion transportation framework to address the role of gap junction in cell-cell ion transportation in proliferating tumor spheroids32. Adar and Safran explored how osmotic pressure regulates cell volume through the lens of electrostatics33. Both models and experiments highlight ions regulation of cell deformation. Despite these theoretical advances34,35, these models mainly studied the transmembrane transport of general ions (including Potassium, Sodium, Calcium ions), without focusing on how fluctuations in Ca2+, crucial in mechanotransduction, influence cell dynamics. Moreover, it remains unknown how the cyclical loading induced by the protrusion and retraction of invadopodia, which periodically alters membrane tension and impacts Ca2+ transport.
Here, we develop a chemo-mechanical model for Ca2+ transmembrane transport in invadopodia with steady-state length. Our model demonstrates that longer invadopodia increase membrane tension, activating mechanosensitive channels and elevating intracellular Ca2+, consistent with optical tweezer experiments and ion channel-targeting treatments. By incorporating Ca2+ transport into our previous invadopodia dynamics model, we captured invadopodia responses to varying extracellular Ca2+ levels. Integrating Ca2+-regulated dynamics with MT1-MMP hydrolysis revealed that MT1-MMP promotes invadopodia growth, activates mechanosensitive Ca2+ channels, and induces oscillatory dynamics. Overall, our model highlights the interplay between Ca2+ homeostasis, actin polymerization, myosin recruitment, and MT1-MMP hydrolysis, offering a theoretical framework for understanding the mechanosensing and Ca2+ transport in cancer cells.
Results
Ca2+ transmembrane transport in invadopoida
Cells possess a large Ca2+ concentration gradient between their inside ( ~ 100 nM) and outside ( ~ 2 mM), maintained by Ca2+ channels4 (Fig. 1c–e). Ca2+ can be transported by passive channels along the concentration gradient36, while the active pumps transport Ca2+ against the concentration gradient by ATP hydrolysis37. The pump first binds intracellular Ca2+, undergoes a transformational change powered by ATP hydrolysis, and thereby performs Ca2+ transport. Assuming that intracellular ATP is sufficiently abundant, Ca2+ outflux through pumps per unit surface area can be described by the Hill equation38: \({\dot{n}}_{{{\rm{pump}}}}={J}_{\max }\frac{{c}_{{{\rm{in}}}}^{2}}{{H}^{2}+{c}_{{{\rm{in}}}}^{2}}\)39,40, where \({J}_{\max }\) is the maximal pumping rate, \({c}_{{{\rm{in}}}}\) is the intracellular Ca2+ concentration. \(H\) is the half activation concentration, which reflects Ca2+ affinity of the pump. The chemical potential difference between extracellular and intracellular Ca2+, denoted as \({X}_{{{\rm{calcium}}}}\), is given by: \({X}_{{{\rm{calcium}}}}={\mu }_{{{\rm{out}}}}-{\mu }_{{{\rm{in}}}}={R}_{{{\rm{g}}}}T{\mathrm{ln}}({c}_{{{\rm{out}}}}/{c}_{{{\rm{in}}}})\), where \({\mu }_{{{\rm{out}}}}\) and\(\,{\mu }_{{{\rm{in}}}}\) are Ca2+ chemical potential outside and inside, respectively. \({c}_{{{\rm{out}}}}\) represents the external Ca2+ concentration. \({R}_{{{\rm{g}}}}\) is the gas constant and \(T\) is the temperature. The ratio between the Ca2+ influx and outflux through passive channels is: \({\varPi }_{{{\rm{influx}}}}/{\varPi }_{{{\rm{outflux}}}}=\exp \left({X}_{{{\rm{calcium}}}}/{R}_{{{\rm{g}}}}T\right)={c}_{{{\rm{out}}}}/{c}_{{{\rm{in}}}}\gg 1\)38. Thus, the net Ca2+ influx through passive channels per unit surface area can be expressed as \({\dot{n}}_{{{\rm{passive}}}}={f}_{{{\rm{passive}}}}({c}_{{{\rm{out}}}}-{c}_{{{\rm{in}}}}){R}_{{{\rm{g}}}}T\), where \({f}_{{{\rm{passive}}}}\) is a rate constant. Invadopodia are highly dynamic structures, and the stress on the cell membrane varies continuously. Therefore, we classify passive channels into mechanosensitive (MS) and non-mechanosensitive (non-MS) channels. Ca2+ MS channels (Fig. 1d) like Piezo and TRPV4, respond to membrane stress, with opening probability following a Boltzmann function. We applied a piecewise linear function to describe Ca2+ influx through MS channels per unit surface area as \({\dot{n}}_{{{\rm{MS}}}}={f}_{{{\rm{MS}}}}(\sigma )({c}_{{{\rm{out}}}}-{c}_{{{\rm{in}}}}){R}_{{{\rm{g}}}}T\) with \({f}_{{{\rm{MS}}}}\left(\sigma \right)=\left\{\begin{array}{cc}0, & \sigma \le {\sigma }_{{{\rm{c}}}}\\ \omega \left(\sigma -{\sigma }_{{{\rm{c}}}}\right), & \,{\sigma }_{{{\rm{c}}}} < \sigma < {\sigma }_{{{\rm{s}}}}\\ \omega \left({\sigma }_{{{\rm{s}}}}-{\sigma }_{{{\rm{c}}}}\right), & \,\sigma \ge {\sigma }_{{{\rm{s}}}}\end{array}\right.,\) where \(\omega\) is a rate constant, \({\sigma }_{{{\rm{c}}}}\) and \({\sigma }_{{{\rm{s}}}}\) are the threshold stress and saturating stress, respectively. Assuming the membrane is viscoelastic and modeled by Kelvin-Voigt model41, the membrane stress can be written as \(\sigma ={E}_{{{\rm{m}}}}\varepsilon +{\eta }_{{{\rm{m}}}}\dot{\varepsilon }={E}_{{{\rm{m}}}}\Delta l(t)/{l}_{0}+{\eta }_{{{\rm{m}}}}\Delta \dot{l}(t)/{l}_{0}\), where \({E}_{{{\rm{m}}}}\) and \({\eta }_{{{\rm{m}}}}\) are the elastic modulus and viscosity of membrane, respectively. Here, \({l}_{0}\) is the initial length of the invadopodium and \(\Delta l(t)\) is its extension length. Other than MS channels, non-MS passive channels (Fig. 1e), such as L-type, TRPV1, TRPM8, etc, also transport Ca2+ along the concentration gradient. We describe Ca2+ influx through these non-MS channels per unit surface area as \({\dot{n}}_{{{\rm{non}}}-{{\rm{MS}}}}={f}_{{{\rm{non}}}-{{\rm{MS}}}}({c}_{{{\rm{out}}}}-{c}_{{{\rm{in}}}}){R}_{{{\rm{g}}}}T\), where \({f}_{{{\rm{non}}}-{{\rm{MS}}}}\) is a rate constant. In summary, the change in Ca2+ number per unit surface area is: \(\dot{n}={\dot{n}}_{{{\rm{passive}}}}+{\dot{n}}_{{{\rm{pump}}}}={\dot{n}}_{{{\rm{MS}}}}+{\dot{n}}_{{{\rm{non}}}-{{\rm{MS}}}}+{\dot{n}}_{{{\rm{pump}}}}.\) Considering the invadopodium as a cylinder with radius of \(r\), its surface area is \({A}_{{{\rm{eff}}}}=2{{\rm{\pi }}}{rl}+{{\rm{\pi }}}{r}^{2}\approx 2{{\rm{\pi }}}{rl}\), considering \(r\,\ll \,l\), where \({l=l}_{0}+\Delta l(t)\). The total Ca2+ number change inside the cell is expressed as: \(\dot{N}={A}_{{{\rm{eff}}}}\dot{n}\).

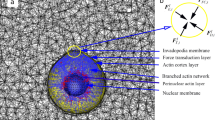
a G-actin polymerizes at the tip of F-actin with a polymerization speed \({V}_{{{\rm{p}}}}\), generating a protrusive force which is counterbalanced by cortex and ECM force. Myosin pulls F-actin toward the cell center with a retrograde flow \({V}_{{{\rm{r}}}}\), regulated by b mechanosensitive signaling pathways (Rho-ROCK pathway) and (c–e) Ca2+ transmembrane transport. Three types of Ca2+ channels are considered: c active pump, d mechanosensitive (MS) passive channel, and e non-MS passive channels. Adhesion proteins link F-actin to ECM via integrins, providing frictional resistance to retrograde flow. Invadopodia secrete MT1-MMP at their tip to degrade ECMs. f All forces are balanced in F-actin. g Open probability \({f}_{{{\rm{MS}}}}\) of mechanosensitive passive channel.
Studies have shown that Ca2+ diffusion between invadopodia and the cell body is locally regulated, leading to heterogeneity in Ca2+ concentration between invadopodia and the cell body42,43. We investigate the impact of local calcium regulation in this study (See Supplementary Note 1) and find that variations in Ca2+ diffusion speed do not affect the overall conclusion. For computational simplicity, we minimize local regulation and heterogeneity, resulting in a nearly uniform Ca2+ concentration between the invadopodium and the cell body. Thus, Ca2+ concentration can be calculated as \({c}_{{{\rm{in}}}}=N/V\), where \(V=\pi {r}^{2}l+{V}_{{{\rm{b}}}}\) is the total cell volume. We obtain the concentration change rate:
Intracellular Ca2+ is regulated by invadopodia extension length, channels activity, and extracellular Ca2+ concentration
Using the above model for Ca2+ transmembrane transport, we can simulate the intracellular Ca2+ dynamics and investigate key regulating factors. For simplicity, we consider the invadopodium reaches a steady-state length, \({{\rm{d}}}l/{{\rm{d}}}t=0\). Intracellular Ca2+ concentration increases \({{\rm{d}}}{c}_{{{\rm{in}}}}/{{\rm{d}}}t > 0\) when the concentration below the concentration homeostasis, i.e., \({c}_{{{\rm{in}}}} < {c}_{{{\rm{inss}}}}\), and decreases (\({{\rm{d}}}{c}_{{{\rm{in}}}}/{{\rm{d}}}t < 0\)) when \({c}_{{{\rm{in}}}} > {c}_{{{\rm{inss}}}}\) (Fig. 2a). As the invadopodium steady-state length increases (at constant initial invadopodium length), the intersection points move towards a higher \({c}_{{{\rm{in}}}}\), indicating the elevation of intracellular Ca2+ concentration homeostasis (Fig. 2b). This is because of the increased membrane stress, which enhances the opening of MS channels.

a Relationship between intracellular Ca2+ concentration change rate \({{\rm{d}}}{c}_{{{\rm{in}}}}/{{\rm{d}}}t\) and the intracellular Ca2+ concentration \({c}_{{{\rm{in}}}}\) (red solid line); steady-state Ca2+ concentration \({c}_{{{\rm{inss}}}}\) (red dot); passive channel contribution (green dashed line); and active pump contribution (blue dashed line). b Changes in intracellular Ca2+ concentration change rate \({{\rm{d}}}{c}_{{{\rm{in}}}}/{{\rm{d}}}t\) with increasing invadopodium extension length \(\triangle l\) along the arrow direction. Line colors: yellow, \(1\) \({{\rm{\mu }}}{{\rm{m}}}\); pink, \(3\) \({{\rm{\mu }}}{{\rm{m}}}\); purple, \(5\) \({{\rm{\mu }}}{{\rm{m}}}\). c Heatmap showing how MS channels rate constant \(\omega\) and non-MS channel rate constant \({f}_{{{\rm{non}}}-{{\rm{MS}}}}\) affect intracellular Ca2+ concentration homeostasis \({c}_{{{\rm{inss}}}}\). d Heatmap showing how maximal pumping rate \({J}_{\max }\) and the external Ca2+ concentration \({c}_{{{\rm{out}}}}\) regulate intracellular Ca2+ concentration homeostasis \({c}_{{{\rm{inss}}}}\). e The effects of Ca2+ chelators or Ca2+ blockers on simulated Ca2+ homeostasis.
Either a higher MS channels rate constant \(\omega\) or \({f}_{{{\rm{non}}}-{{\rm{MS}}}}\) leads to the increase of the intracellular Ca2+ concentration homeostasis, as more Ca2+ is transported into the invadopodium via passive channels (Fig. 2c). In contrast, a larger maximal pumping rate \({J}_{\max }\) facilitates Ca2+ outflux, reducing intracellular Ca2+ homeostasis. A higher external Ca2+ concentration \({c}_{{out}}\) increases the activation energy of the active pump, reducing the inverse gradient transport (outflux) of Ca2+, thus increasing intracellular Ca2+ homeostasis (Fig. 2d). In addition to the concentration homeostasis, we also examined the timescale for Ca2+ concentration to reach the homeostasis (See Supplementary Note 2).
To validate the model, we referenced A.K. Efremov et al.’s work44, which stretched filopodia while monitoring Ca2+ concentration changes using optical tweezer. Although invadopodia and filopodia are different, their extension and Ca2+ transport mechanisms are similar. The experiment showed that longer extensions led to higher intracellular Ca2+ concentrations, aligning with Fig. 2b. Additionally, reducing external Ca2+ (via EGTA), inhibiting Ca2+ MS channels (via GSK2193874), or blocking other passive channels (via Amlodipine) decreased intracellular Ca2+ homeostasis, consistent with Fig. 2c, d. In Fig. 2e, control group simulations represent Ca2+ homeostasis under parameter settings marked by circles in Fig. 2c, d, while drug effects correspond to crosses. Overall, the agreement between the experimental and simulation validates our model.
Kinematic of invadopodia with Ca2+ transmembrane transport
Next, we introduce our chemo-mechanical model for invadopodia dynamics. Actin monomers polymerize into actin filaments (F-actin), generating the protrusive force necessary for invadopodium growth24. Based on the findings of Schumacher et al45. that invadopodia elongation depends on both filopodial and lamellipodial machinery, we assumed that actin retrograde flow is driven by myosin contractility and attenuated by substrate adhesion46. Figure 1a shows the extension speed \({{\rm{d}}}l/{{\rm{d}}}t\) is the net difference between G-actins polymerization speed \({V}_{{{\rm{p}}}}\) and retrograde flow speed \({V}_{{{\rm{r}}}}\) caused by myosin contractility, expressed as \({{\rm{d}}}l/{{\rm{d}}}t={V}_{{{\rm{p}}}}-{V}_{{{\rm{r}}}}\). As G-actin polymerizes at the invadopodium tip, the resulting protrusive force drives invadopodium extension, which is balanced by cortex tension and ECM deformation. Since the cortex exhibits viscoelastic behavior47,48, we describe the cortex with Kelvin-Voigt model. Accordingly, the deformation resists protrusive force with \({F}_{{{\rm{c}}}}=A\left({E}_{{{\rm{c}}}}\varepsilon +{\eta }_{{{\rm{c}}}}\dot{\varepsilon }\right)\), where \(A\) is the cross-sectional area, \({E}_{{{\rm{c}}}}\) and \({\eta }_{{{\rm{c}}}}\) are the elastic modulus and viscosity of cortex, respectively. The ECM resistance force is given by Hertzian contact theory \({F}_{{{\rm{e}}}}=2r{E}_{0}\Delta l\)49, where \(r\) is invadopodia radius, \({E}_{0}\) is ECM elastic modulus and \(\Delta l\) is invadopodia extension length. The contractile force from bound myosin, following Hill’s relation50, is \({F}_{{{\rm{m}}}}={F}_{{{\rm{M}}}}\left(1-{V}_{{{\rm{r}}}}/{V}_{0}\right)\), where \({V}_{0}\) and \({F}_{{{\rm{M}}}}\) are the unloaded retrograde flow speed and the maximum contraction force, respectively. Adhesion proteins bind/unbind between F-actin and ECM, generating a resistance force that decreases with the retrograde flow speed51. This relationship can be expressed as \({F}_{{{\rm{a}}}}={F}_{{{\rm{amax}}}}-{\zeta }_{{{\rm{r}}}}{V}_{{{\rm{r}}}}\), where \({F}_{{{\rm{amax}}}}\) is the intercept of the relationship between \({F}_{{{\rm{a}}}}\) and \({V}_{{{\rm{r}}}}\), and \({\zeta }_{{{\rm{r}}}}\) is the friction coefficient (See Supplementary Note 3). Balancing all forces in F-actin (Fig. 1f), we obtain the force equilibrium \({F}_{{{\rm{c}}}}+{F}_{{{\rm{e}}}}+{F}_{{{\rm{m}}}}-{F}_{{{\rm{a}}}}=0\), which gives the governing equation for the extension dynamics:
where \({k}_{{{\rm{es}}}}=A{E}_{{{\rm{c}}}}/{l}_{0}+2r{E}_{0}\) represents the equivalent stiffness of invadopodia.
Next, myosin recruits to F-actin through phosphorylation of myosin light chain (MLC) to form phosphorylated MLC (pMLC)52, applying contractile force to F-actins (Fig. 1b). The number of pMLCs varies according to53,54: \({{\rm{d}}}{m}_{{{\rm{pM}}}}/{{\rm{d}}}t={k}_{{{\rm{pM}}}}^{* }{m}_{{{\rm{M}}}}-{k}_{{{\rm{off}}}}{m}_{{{\rm{pM}}}}\), where \({k}_{{{\rm{pM}}}}^{* }\) is pMLCs formation rate, \({m}_{{{\rm{M}}}}\) is the MLCs number and \({k}_{{{\rm{off}}}}\) is pMLCs dephosphorylation rate. Rho-associated kinase (ROCK) promotes MLCs phosphorylation to generate contraction force52. Increased intracellular Ca2+ and adhesion force \({F}_{{{\rm{a}}}}\) both upregulate MLCs phosphorylation through ROCK55,56. In contrast, Rac1 induces downregulation of Rho within the ROCK pathway through Rac-mediated reactive oxygen species (ROS)57,58. Meanwhile, experimental results show a positive correlation between Rac1 expression and changes in invadopodia length59,60. Assuming a linear relationship, the pMLC formation rate can be written as18: \({k}_{{{\rm{pM}}}}^{* }={k}_{{{\rm{pM}}}}^{0}+{\alpha }_{0}{F}_{{{\rm{a}}}}-{\beta }_{0}\frac{{{\rm{d}}}\Delta l}{{{\rm{d}}}t}+{\theta }_{0}{c}_{{{\rm{in}}}};\) where \({k}_{{{\rm{pM}}}}^{0}\) is the initial formation rate of pMLCs, and \({\alpha }_{0}\), \({\beta }_{0}\), and \({\theta }_{0}\) are the linear coefficients. The maximum contractile force is \({F}_{{{\rm{M}}}}={m}_{{{\rm{pM}}}}{f}_{{{\rm{pM}}}}\), and the change in \({F}_{{{\rm{M}}}}\) is:
Here \({F}_{{{\rm{M}}}0}=\frac{{k}_{{{\rm{pM}}}}^{0}{f}_{{{\rm{pM}}}}{m}_{{{\rm{M}}}}}{{k}_{{{\rm{off}}}}}\) is the initial force, and \(\alpha =\frac{{\alpha }_{0}{F}_{{{\rm{M}}}0}}{{k}_{{{\rm{pM}}}}^{0}}\), \(\beta =\frac{{\beta }_{0}{F}_{{{\rm{M}}}0}}{{k}_{{{\rm{pM}}}}^{0}}\) and \(\theta =\frac{{\theta }_{0}{F}_{{{\rm{M}}}0}}{{k}_{{{\rm{pM}}}}^{0}}\) are coefficients for the signaling pathways affecting myosin recruitment.
Competition of actin polymerization and myosin recruitment drives the invadopodia oscillations
Considering that the system consisting of Eqs. (1)–(3), obtains a fixed point (\({c}_{{{\rm{ins}}}},\,{F}_{{{\rm{Ms}}}},\,\Delta {l}_{{{\rm{s}}}}\)) under the condition \({{\rm{d}}}{c}_{{{\rm{in}}}}/{{\rm{d}}}t=0\) \({{\rm{d}}}{F}_{{{\rm{M}}}}/{{\rm{d}}}t=0\) and \({{\rm{d}}}\Delta l/{{\rm{d}}}t=0\), where \({c}_{{{\rm{ins}}}}={c}_{{{\rm{inss}}}}\), \({F}_{{{\rm{Ms}}}}={F}_{{{\rm{M}}}0}+\alpha {F}_{{{\rm{amax}}}}-{\alpha \zeta }_{{{\rm{r}}}}{V}_{{{\rm{p}}}}+\theta {c}_{{{\rm{inss}}}}\) and \(\Delta {l}_{{{\rm{s}}}}=\big[{F}_{{{\rm{amax}}}}-{\zeta }_{{{\rm{r}}}}{V}_{{{\rm{p}}}}+{F}_{{{\rm{Ms}}}}\cdot \big({V}_{{{\rm{p}}}}/{V}_{0}\,-1\big)\big]/{k}_{{{\rm{es}}}}\). Compared to invadopodium extension dynamics (with timescale ~270 s) and myosin contractility generation (with timescale ~250 s), intracellular Ca2+ concentration adjusts rapidly (timescale ~0.5 s). Note that the timescale \(\tau\) (that is defined in the equation \(\tau \frac{{{\rm{d}}}x}{{{\rm{d}}}t}=f(x,y,z)\) for sub-cellular process) represents the characteristic time required for reaching equilibrium in the system. Therefore, according to Tikhonov’s theorem, we assume that Ca2+ homeostasis \({c}_{{{\rm{inss}}}}\) is reached before steady states of extension and myosin recruitment. Note that the necessary conditions for Tikhonov’s theorem have been verified in the Supplementary Note 4. Next, we analyze the stability of the fixed point using linear perturbation method (see Method). By applying small perturbations \(\delta {l}_{s}\) and \({\delta F}_{{Ms}}\), we obtain the eigenvalues of the Jacobian matrix:
where \({k}_{{{\rm{l}}}}\) is the rate of reaching \(\Delta {l}_{{{\rm{s}}}}\) and \({k}_{{{\rm{F}}}}\) is the rate of signal associated myosin contraction to reaching \({F}_{{{\rm{Ms}}}}\). Here, \({k}_{{{\rm{off}}}}\) represents not only pMLCs dephosphorylation rate, but also the rate that characterizes intrinsic myosin contraction (independent of signaling feedback). Stability is determined by the real part of eigenvalues \({\mathrm{Re}}\left({\lambda }_{1,2}\right)\), and oscillations by the imaginary part \({{\rm{Im}}}\left({\lambda }_{1,2}\right)\). When \({\mathrm{Re}}\left({\lambda }_{1,2}\right) < 0\), the invadopodium exhibits stable growth (Regions I and II in Fig. 3a–c), whereas \({\mathrm{Re}}\left({\lambda }_{1,2}\right) > 0\) leads to unstable growth (Region III). Within the stable regime, if \({{\rm{Im}}}\left({\lambda }_{1,2}\right)=0\), the invadopodium exhibits monotonic growth (Region I); however, if \({{\rm{Im}}}\left({\lambda }_{1,2}\right)\ne 0\), its growth becomes oscillatory (Region II).

a \({k}_{{{\rm{l}}}}\) and \({k}_{{{\rm{F}}}}\) judge the mode of invadopodia growth. Regions: I (dark blue), II (light blue), and III (pink). b Invadopodia extension dynamics in the three regions (I, II, III). c Contraction force-extension curve corresponding to region I and II. d Heatmap showing how ROCK feedback parameter \(\alpha\) and adhesion proteins frictional coefficient \({\zeta }_{{{\rm{r}}}}\) regulate the oscillation period of invadopodia \(T\). e Heatmap showing how pMLCs dephosphorylation rate \({k}_{{{\rm{off}}}}\) and ECM elastic modulus \({E}_{0}\) regulate the oscillation period of invadopodia \(T\). f Dynamics of invadopodium extension length \(\Delta l\) at stiff (red solid line) and soft (green solid line) ECM; the green dashed line indicates the average \(\Delta l\) for soft ECM.
We next investigate the oscillatory growth of invadopodia and how factors such as adhesion, myosin, and ECM stiffness affect the oscillation period. Figure 3d shows a heatmap of the invadopodia oscillation period \(T=2{{\rm{\pi }}}/{{\rm{Im}}}({\lambda }_{{\mathrm{1,2}}})\) as a function of the ROCK feedback parameter \(\alpha\) and adhesive coefficient \({\zeta }_{{{\rm{r}}}}\). An increase in the ROCK parameter enhances pMLC formation, leading to a stronger steady-state myosin contractile force and thereby extending the oscillation period. In contrast, a higher adhesion coefficient \({\zeta }_{{{\rm{r}}}}\) weakens adhesion forces at the same instantaneous retrograde speed, resulting in a reduced oscillation amplitude. Since the polymerization rate and the maximum retrograde speed remain unchanged, the oscillation period decreases. Similarly, a heatmap of the oscillation period as a function of ECM elastic modulus \({E}_{0}\) and pMLCs dephosphorylation rate \({k}_{{{\rm{off}}}}\) (Fig. 3e) reveals that both higher \({E}_{0}\) and \({k}_{{{\rm{off}}}}\) contribute to a shorter oscillation period. A higher \({k}_{{{\rm{off}}}}\) reduces the steady-state pMLC number and myosin contraction force. A higher \({E}_{0}\) increases ECM resistance, reducing steady-state invadopodium length (see Fig. 3f). Experimental results indicate that stiffer substrates limit invadopodia extension and inhibit cancer cell invasion61,62, aligning with our model’s prediction that higher \({E}_{0}\) results in shorter invadopodium length. Chang et al. observed that the invadopodia oscillation period is shorter in stiffer ECMs61, consistent with our model (indicated by the arrow in Fig. 3e). In short, our model explains the experimental observations of invadopodia behavior on substrates with varying stiffness.
Ca2+ transmembrane transportation enhances invadopodia oscillatory growth by mediating myosin recruitment
We then investigated how intracellular Ca2+ transmembrane transport mediates invadopodium dynamics. By integrating the Ca2+ transport model with invadopodium kinematics, we simulated the dynamics of invadopodium length, myosin contractile force, and intracellular Ca2+ concentration. Figure 4a shows that higher \({c}_{{{\rm{out}}}}\) promotes myosin contractile force, which in turn reduces the invadopodia length. To further examine the interplay between intracellular Ca2+ and invadopodium extension/retraction cycles, we analyzed the time evolution of invadopodium extension length, intracellular Ca2+ concentration, and myosin contractile force, both with and without intracellular Ca2+ feedback (Supplementary Fig. 4), which is further outlined as two key stages:
-
(i)
As the invadopodium extends, its membrane gets stretched, opening the MS channels and allowing Ca2+ influx and raising intracellular Ca2+ levels. The elevated intracellular Ca2+ concentration enhances myosin contraction.
-
(ii)
A higher myosin contractile force shortens invadopodia. This shortening reduces the membrane tension and makes MS channels closed, which lowers intracellular Ca2+ levels and decreases Ca2+ feedback.

a Dynamics of invadopodium extension length \(\triangle l\), myosin contraction force \({F}_{{{\rm{m}}}}\), and intracellular Ca2+ concentration \({c}_{{{\rm{in}}}}\) at high (red solid line) and low (green solid line) external Ca2+ concentrations \({c}_{{{\rm{out}}}}\); dashed lines show average \(\triangle l\) and \({F}_{{{\rm{m}}}}\) for high (red) and low (green) \({c}_{{{\rm{out}}}}\). b Schematic representation of invadopodia in retraction and protrusion states. Retraction: shortened invadopodia reduce membrane tension, closing MS channels, lowering intracellular Ca2+ levels, and weakening Ca2+ feedback (thin black arrows). Protrusion: extended invadopodia stretch the membrane, opening MS channels, increasing Ca2+ influx and intracellular Ca2+ level, and enhancing Ca2+ feedback (thick black arrows). c Changes in intracellular Ca2+ homeostasis with increasing \(\theta\) along the arrow direction. Line colors: orange, \(1\times {10}^{4}\); green, \(5\times {10}^{4}\); purple, \(9\times {10}^{4}\). d By varying the parameters, invadopodia dynamic regions were divided according to theoretical calculations. The three regions correspond to the monotonical invadopodium growth (region I, dark blue), oscillatory growth (region II, light blue), and unstable growth (region III, pink).
This feedback loop weakens myosin contraction, allowing the invadopodium to re-extend and initiate a new cycle (Fig. 4b). In short, the cyclic change in the opening and closing of MS channels influences invadopodium dynamics.
Next, considering that Ca2+-mediated phosphorylation coefficient \(\theta\) affects myosin recruitment, our model shows that a larger \(\theta\) enhances myosin recruitment, decreasing steady state intracellular Ca2+ levels (Fig. 4c). We further varied \(\theta\) and the pMLCs dephosphorylation rate \({k}_{{{\rm{off}}}}\) and repeated stability analysis using linear perturbation methods. The theoretical calculations (lines in Fig. 4d) confirm that the increased Ca2+-mediated phosphorylation coefficient \(\theta\) promote oscillatory growth.
MT1-MMP facilitates invadopodia extension and oscillatory dynamics by softening the ECM and activating mechanosensitive Ca2+ channels
During cancer cell invasion, invadopodia secrete MT1-MMP to degrade surrounding ECMs, facilitating migration63. MT1-MMP accumulates at invadopodia tips and degrades matrix fibers through proteolysis17,24,64,65,66. We model ECM degradation as a first-order reaction with matrix density \(\rho \left(t\right)\) evolving as:
where \({k}_{{{\rm{MMP}}}}\) is the rate constant and \({\rho }_{{{\rm{s}}}}\) is the steady state ECM density. As proposed by David Boal67, an entropic polymer network deforms in response to external stress. Each chain in the network follows a conventional Gaussian probability distribution and undergoes affine deformation. By calculating the entropy change of the network before and after deformation, it can be shown that, under constant temperature, the microscopic polymer chain density is directly proportional to the elastic modulus. We express the elastic modulus as \(E\left(t\right)={E}_{0}\rho (t)/{\rho }_{0}\) with the initial elastic modulus \({E}_{0}\) and initial ECM density \({\rho }_{0}\).
By integrating the MT1-MMP degradation process with Ca2+-mediated invadopodia dynamics, our model predicts MT1-MMP’s effect on invadopodia oscillation. MT1-MMP increases both invadopodia extension length and myosin contractile force at steady state (Fig. 5a). Time-dependent analysis reveals that MT1-MMP promotes longer invadopodia and higher Ca2+ level with more oscillations (Fig. 5b, c). Three timestamps (phases i, ii and iii) in Fig. 5d, e show that MT1-MMP degrades ECM, softening it and allowing invadopodia to extend longer. The longer invadopodia length elevates the membrane tension and further promotes the opening of MS Ca2+ channels, leading to a higher intracellular Ca2+ concentration. Meanwhile, a higher intracellular Ca2+ concentration promotes myosin recruitment, eventually facilitating the invadopodia oscillations (Fig. 5d). In the contrary, without MT1-MMP, invadopodia reach steady state faster (Fig. 5e). MT1-MMP hydrolysis reduces ECM density at steady state. In Fig. 5f, linear stability analysis shows that increased MT1-MMP activity softens ECM, shifting invadopodia growth from monotonic to oscillatory (black arrows in Fig. 5f).

a Myosin contraction force \({F}_{{{\rm{M}}}}\) versus invadopodium extension length \(\triangle l\) with MT1-MMP (light blue) and without MT1-MMP (pink). Blue and red dots indicate the steady states with and without MT1-MMP. b, c Comparison of invadopodium extension and intracellular Ca2+ dynamics with MT1-MMP (blue) and without MT1-MMP (red). Schematic of invadopodia and ECM at positions (i), (ii), and (iii). d With MT1-MMP: ECM degradation softens the substrate, allowing enhanced invadopodium extension, opening of MS channels, elevating intracellular Ca2+ levels, and promoting invadopodium oscillation. e Without MT1-MMP: no ECM degradation and unchanged substrate stiffness lead to earlier stabilization of invadopodia. f The phase diagram for monotonic (region I, dark blue) and oscillatory growth (region II, light blue) in response to ECM density change ratio \({\rho }_{{{\rm{s}}}}/{\rho }_{0}\) and reciprocals of pMLCs dephosphorylation rate \(1/{k}_{{{\rm{off}}}}\).
Discussion
In this study, by proposing a chemo-mechanical model on Ca2+ regulated invadopodia growth, we investigate how Ca2+ acts as a second messenger to regulate invadopodia dynamics. Our model integrates key factors like actin polymerization, myosin recruitment, mechanosensitive signaling (Rho-ROCK pathway), Ca2+ transmembrane transport, and MT1-MMP secretion, offering a theoretical understanding of invadopodia dynamics. Our model indicates that increased invadopodia length elevates membrane tension, activating mechanosensitive channels and raising intracellular Ca2+ levels, in line with optical tweezer experiments and ion channel-targeting treatments. By integrating Ca2+ transport with our model of invadopodia dynamics, we captured how invadopodia adapt to variations in extracellular Ca2+ concentrations. Furthermore, incorporating MT1-MMP degradation into the model revealed that elevated MT1-MMP level enhances invadopodia growth and increases intracellular Ca2+ level, shifting growth from monotonic to oscillatory.
In contrast to previous models29,32,68 that focused on general ion transport and volume changes, our model addressed the Ca2+ transport across the membrane and its associated effects on invadopodia dynamics. For Ca2+ transmembrane transport, passive Ca2+ channels allow Ca2+ influx along the gradient, and active Ca2+ pumps push Ca2+ outflux against the gradient. During invadopodia dynamics, protrusion and retraction cycles alter membrane tension, which triggers the opening and closing of the mechano-sensitive Ca2+ channels. Our model considers the active pump in terms of energy, which is released from ATP hydrolysis enabling intracellular Ca2+ to overcome the concentration barrier. The rate parameters of Ca2+ channels in our model are close in order of magnitude consistent with the rate parameters of ion channels in previous studies29,32. In our model, Ca2+ transmembrane transport describes intracellular Ca2+ number changes caused by various Ca2+ channels. Importantly, our model predictions have been verified by the existing experimental results, including external Ca2+ concentration change and invadopodia dynamics.
For individual invadopodium, actin polymerization provides the primary driving force for invadopodia extension, while myosin recruitment pulls the invadopodium to contract. Experiments using blebbistatin or Y27632 to inhibit myosin contractility or the Rho-ROCK pathway observed monotonic invadopodia growth and significantly increased extension length18. These findings align with our model, which suggests that invadopodia dynamics, whether monotonic or oscillatory, are governed by the interplay between actin polymerization and myosin recruitment. Recent experiments have shown that actin polymerization is sensitive to the applied mechanical load69,70, future studies can further incorporate the force sensitivity of actin polymerization into our framework to study the role of specific mechanosensitive protein in invadopodia dynamics.
To study the relationship between intracellular Ca2+ concentration and invadopodia more clearly, we investigate not only the role of intracellular Ca2+ on invadopodia steady state, but also the feedback effect of invadopodia dynamic on intracellular Ca2+ concentration. We first discuss intracellular Ca2+ concentration changes in response to a perturbation on invadopodia steady state, which is determined by passive channels and pumps. Subsequently, we focus on the whole process that considers the dynamics of invadopodia. Increasing Ca2+ effect on myosin recruitment \(\theta\), invadopodium extension length at steady state reduce. In turn, Ca2+ MS channels are limited, resulting in a decrease in intracellular Ca2+ concentration homeostasis.
Our chemo-mechanical model demonstrates the process of matrix degradation and deformation. The growth of the invadopodia squeezes and deforms the ECM at the tip, and the MT1-MMP secreted by the invadopodia degrades the ECM. Most previous studies investigated the mechanical and chemical properties of ECM separately16,64, indicating that both the mechanical and chemical properties of the ECM are critical for invadopodia invasion. Our simulation suggests that increasing ECM elastic modulus reduces invadopodia extension length, consistent with existing experimental observations61. By assuming a linear relationship between the ECM’s elastic modulus and matrix density, our model shows that the invadopodia extend longer and become more oscillatory for the case of MT1-MMP. We further demonstrate that MT1-MMP hydrolysis elevates intracellular Ca2+ concentration. Although our model simplified the ECM as a purely elastic material and considered ECM degradation as first-order reactions, our model provides a clear understanding of how the coupling of both the mechanical and chemical properties of the ECM plays a role in invadopodia dynamics. These insights underscore the need to clearly distinguish invadopodia―actin-rich cancer cell protrusions that coordinate protrusive and matrix-degrading functions―from general cell protrusions. Different experimental conditions17,45,71 have revealed a continuum of protrusion types with diverse morphologies but overlapping functions. A better definition of invadopodia is required to separate them from other protrusions in future studies. Our model also does not consider the positive feedback loops between invadopodia and ECM degradation72,73; however, future studies could incorporate such positive signaling feedback into our theoretical framework to systematically examine their coupling.
Alterations in Ca2+ transmembrane transport have been widely observed in cancer74,75, with Ca2+ homeostasis linked to the regulation of calcium-sensitive oncogenic pathways and related proteins12. Previous studies have shown that the expression of specific Ca2+ channels is altered in various cancers76,77. Our model specifically addresses Ca2+ dynamics, showing how passive Ca2+ channels and active pumps maintain intracellular Ca2+ homeostasis during invadopodia cycles. Invadopodia facilitate ECM degradation, enabling cancer cell migration and invasion. By linking Ca2+ transmembrane transport to invadopodia dynamics, our model provides insights into cancer cell motility. Our Ca2+ transmembrane transport model can be integrated with other cell and tissue models to provide a more comprehensive understanding of complex metastatic processes. For instance, the localized regulation of Ca2+, including the spatial association of the Ca2+ channels TRPV4 and Orai1-STIM42,43, shapes intracellular Ca2+ distribution and constitutes an important area of research in tumor cell invasion. Moreover, during cancer cell migration, Ca2+ levels are typically elevated at the trailing edge, facilitating cell movement by activating motor proteins26,78. Future theoretical and computational models may build upon our model, which accounts for Ca2+ redistribution in cancer cell invasion, leading to a new insight of Ca2+ related targeted cancer therapy.
Method
Linear stability analysis
According to Tikhonov’s theorem, we assume that Ca2+ homeostasis \({c}_{{{\rm{inss}}}}\) is reached before steady states of invadopodia extension and myosin recruitment. The three ODEs (Eqs. 1–3) can be simplified as:
By setting \({{\rm{d}}}\Delta l/{{\rm{d}}}t={{\rm{d}}}{F}_{{{\rm{M}}}}/{{\rm{d}}}t=0\), we obtain the fix point (\({F}_{{{\rm{Ms}}}},\,\Delta {l}_{{{\rm{s}}}}\)), where \({F}_{{{\rm{Ms}}}}={F}_{{{\rm{M}}}0}+\alpha {F}_{{{\rm{amax}}}}-{\alpha \zeta }_{{{\rm{r}}}}{V}_{{{\rm{p}}}}+\theta {c}_{{{\rm{inss}}}}\) and \(\Delta {l}_{{{\rm{s}}}}=\left[{F}_{{{\rm{amax}}}}-{\zeta }_{{{\rm{r}}}}{V}_{{{\rm{p}}}}+{F}_{{{\rm{Ms}}}}\cdot \right.\left.\left({V}_{{{\rm{p}}}}/{V}_{0}\,-1\right)\right]/{k}_{{{\rm{es}}}}\). Applying a small perturbation \(({\delta F}_{{{\rm{Ms}}}},\,\delta \Delta {l}_{{{\rm{s}}}})\) at the fix point, we derive:
The eigenvalues of the Jacobian matrix:
Here \({k}_{{{\rm{l}}}}=\frac{{k}_{{{\rm{es}}}}}{\frac{{F}_{{{\rm{Ms}}}}}{{V}_{0}}+\frac{A{\eta }_{{{\rm{c}}}}}{{l}_{0}}-{\zeta }_{{{\rm{r}}}}}\) represents the rate of approaching \(\Delta {l}_{{{\rm{s}}}}\) while \({k}_{F}=\frac{{k}_{{{\rm{es}}}}}{({\alpha \zeta }_{{{\rm{r}}}}-\beta )\left(\frac{{V}_{{{\rm{p}}}}}{{V}_{0}}-1\right)}\) represents the rate of signal-associated myosin contraction to reaching \({F}_{{{\rm{Ms}}}}\). Additionally, \({k}_{{{\rm{off}}}}\) not only represents pMLCs dephosphorylation rate, but also characterizes intrinsic myosin contraction (independent of signaling feedback).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the figures and other findings of this study are available within the article and its Supplementary Information, or from the corresponding author upon reasonable request.
Code availability
MATLAB files used in this work are openly available on Github (https://github.com/zhaojunbo-ustc/calcium-transport-invadopodia).
References
Marchi, S., Giorgi, C., Galluzzi, L. & Pinton, P. Ca2+ Fluxes and Cancer. Mol. Cell 78, 1055–1069 (2020).
Monteith, G. R., Prevarskaya, N. & Roberts-Thomson, S. J. The calcium–cancer signalling nexus. Nat. Rev. Cancer 17, 373–380 (2017).
Demaurex, N. & Distelhorst, C. Apoptosis—the Calcium Connection. Science 300, 65–67 (2003).
Clapham, D. E. Calcium Signaling. Cell 131, 1047–1058 (2007).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Zamponi, G. W., Striessnig, J., Koschak, A. & Dolphin, A. C. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev. 67, 821–870 (2015).
Aung, C. S. et al. Plasma membrane calcium ATPase 4 and the remodeling of calcium homeostasis in human colon cancer cells. Carcinogenesis 30, 1962–1969 (2009).
Feng, M. et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 143, 84–98 (2010).
Zhu, H. et al. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 5, 3455–3471 (2014).
Bolanz, K. A., Hediger, M. A. & Landowski, C. P. The role of TRPV6 in breast carcinogenesis. Mol. Cancer Ther. 7, 271–279 (2008).
Dhennin-Duthille, I. et al. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: correlation with pathological parameters. Cell. Physiol. Biochem. 28, 813–822 (2011).
Monteith, G. R., Davis, F. M. & Roberts-Thomson, S. J. Calcium channels and pumps in cancer: changes and consequences. J. Biol. Chem. 287, 31666–31673 (2012).
Murphy, D. A. & Courtneidge, S. A. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 12, 413–426 (2011).
Linder, S., Cervero, P., Eddy, R. & Condeelis, J. Mechanisms and roles of podosomes and invadopodia. Nat. Rev. Mol. Cell Biol. 24, 86–106 (2023).
Yamaguchi, H. et al. Molecular mechanisms of invadopodium formation. J. Cell Biol. 168, 441–452 (2005).
Wisdom, K. M. et al. Matrix mechanical plasticity regulates cancer cell migration through confining microenvironments. Nat. Commun. 9, 4144 (2018).
Magalhaes, M. A. O. et al. Cortactin phosphorylation regulates cell invasion through a pH-dependent pathway. J. Cell Biol. 195, 903–920 (2011).
Gong, Z. et al. Recursive feedback between matrix dissipation and chemo-mechanical signaling drives oscillatory growth of cancer cell invadopodia. Cell Rep. 35, 109047 (2021).
Alexander, N. R. et al. Extracellular matrix rigidity promotes invadopodia activity. Curr. Biol. 18, 1295–1299 (2008).
Dalaka, E. et al. Direct measurement of vertical forces shows correlation between mechanical activity and proteolytic ability of invadopodia. Sci. Adv. 6, eaax6912 (2020).
Elosegui-Artola, A. et al. Rigidity sensing and adaptation through regulation of integrin types. Nat. Mater. 13, 631–637 (2014).
Artym, V. V., Zhang, Y., Seillier-Moiseiwitsch, F., Yamada, K. M. & Mueller, S. C. Dynamic interactions of Cortactin and Membrane Type 1 Matrix Metalloproteinase at Invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 66, 3034–3043 (2006).
Wolf, K. et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 9, 893–904 (2007).
Ferrari, R. et al. MT1-MMP directs force-producing proteolytic contacts that drive tumor cell invasion. Nat. Commun. 10, 4886 (2019).
Prevarskaya, N., Skryma, R. & Shuba, Y. Calcium in tumour metastasis: new roles for known actors. Nat. Rev. Cancer 11, 609–618 (2011).
Tsai, F.-C. et al. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 16, 133–144 (2014).
Cadart, C., Venkova, L., Recho, P., Lagomarsino, M. C. & Piel, M. The physics of cell-size regulation across timescales. Nat. Phys. 15, 993–1004 (2019).
Recho, P., Hallou, A. & Hannezo, E. Theory of mechanochemical patterning in biphasic biological tissues. Proc. Natl. Acad. Sci. USA 116, 5344–5349 (2019).
Jiang, H. & Sun, S. X. Cellular pressure and volume regulation and implications for cell mechanics. Biophys. J. 105, 609–619 (2013).
Xie, K., Yang, Y. & Jiang, H. Controlling cellular volume via mechanical and physical properties of substrate. Biophys. J. 114, 675–687 (2018).
Li, Y., Mori, Y. & Sun, S. X. Flow-driven cell migration under external electric fields. Phys. Rev. Lett. 115, 268101 (2015).
McEvoy, E., Han, Y. L., Guo, M. & Shenoy, V. B. Gap junctions amplify spatial variations in cell volume in proliferating tumor spheroids. Nat. Commun. 11, 6148 (2020).
Adar, R. M. & Safran, S. A. Active volume regulation in adhered cells. Proc. Natl. Acad. Sci. 117, 5604–5609 (2020).
Venkova, L. et al. A mechano-osmotic feedback couples cell volume to the rate of cell deformation. eLife 11, e72381 (2022).
Adar, R. M., Vishen, A. S., Joanny, J.-F., Sens, P. & Safran, S. A. Volume regulation in adhered cells: Roles of surface tension and cell swelling. Biophys. J. 122, 506–512 (2023).
Carafoli, E. Calcium signaling: A tale for all seasons. Proc. Natl. Acad. Sci. USA 99, 1115–1122 (2002).
Strehler, E. E. & Zacharias, D. A. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol. Rev. 81, 21–50 (2001).
Hill, T. L. Free Energy Transduction and Biochemical Cycle Kinetics. (Springer New York, New York, NY, 1989) https://doi.org/10.1007/978-1-4612-3558-3.
Caride, A. J. et al. The plasma membrane calcium pump displays memory of past calcium spikes. J. Biol. Chem. 276, 39797–39804 (2001).
Graupner, M., Erler, F. & Meyer-Hermann, M. A theory of plasma membrane calcium pump stimulation and activity. J. Biol. Phys. 31, 183–206 (2005).
Shimolina, L. E. et al. Mapping cisplatin-induced viscosity alterations in cancer cells using molecular rotor and fluorescence lifetime imaging microscopy. J. Biomed. Opt. 25, 126004 (2020).
Sun, J. et al. STIM1- and Orai1-mediated Ca2+ oscillation orchestrates invadopodium formation and melanoma invasion. J. Cell Biol. 207, 535–548 (2014).
Vellino, S. et al. Cross-talk between the calcium channel TRPV4 and reactive oxygen species interlocks adhesive and degradative functions of invadosomes. J. Cell Biol. 220, e201910079 (2021).
Efremov, A. K. et al. Application of piconewton forces to individual filopodia reveals mechanosensory role of L-type Ca2+ channels. Biomaterials 284, 121477 (2022).
Schoumacher, M., Goldman, R. D., Louvard, D. & Vignjevic, D. M. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 189, 541–556 (2010).
Chan, C. E. & Odde, D. J. Traction dynamics of Filopodia on compliant substrates. Science 322, 1687–1691 (2008).
Alcaraz, J. et al. Microrheology of Human Lung Epithelial Cells Measured by Atomic Force Microscopy. Biophys. J. 84, 2071–2079 (2003).
Cartagena, A. & Raman, A. Local Viscoelastic Properties of Live Cells Investigated Using Dynamic and Quasi-Static Atomic Force Microscopy Methods. Biophys. J. 106, 1033–1043 (2014).
Johnson, K. L. Contact Mechanics. (Cambridge University Press: Cambridge, 1985).
Hill, T. L. Theoretical formalism for the sliding filament model of contraction of striated muscle Part I. Prog. Biophys. Mol. Biol. 28, 267–340 (1974).
Liu, P. et al. Elastic properties of force-transmitting linkages determine multistable mechanosensitive behaviour of cell adhesion. Nat. Phys. https://doi.org/10.1038/s41567-025-02964-z (2025).
Somlyo, A. P. & Somlyo, A. V. Ca 2+ Sensitivity of Smooth Muscle and Nonmuscle Myosin II: Modulated by G Proteins, Kinases, and Myosin Phosphatase. Physiol. Rev. 83, 1325–1358 (2003).
McEvoy, E., Deshpande, V. S. & McGarry, P. Transient active force generation and stress fibre remodelling in cells under cyclic loading. Biomech. Model. Mechanobiol. 18, 921–937 (2019).
Yin, X., Liu, Y.-Q., Zhang, L.-Y., Liang, D. & Xu, G.-K. Emergence, Pattern, and Frequency of Spontaneous Waves in Spreading Epithelial Monolayers. Nano Lett. 24, 3631–3637 (2024).
Jerrell, R. J. & Parekh, A. Matrix rigidity differentially regulates invadopodia activity through ROCK1 and ROCK2. Biomaterials 84, 119–129 (2016).
Lee, S. H. et al. A molecular clock controls periodically driven cell migration in confined spaces. Cell Syst. 13, 514–529.e10 (2022).
Nimnual, A. S., Taylor, L. J. & Bar-Sagi, D. Redox-dependent downregulation of Rho by Rac. Nat. Cell Biol. 5, 236–241 (2003).
Clark, K., Langeslag, M., Figdor, C. G. & Van Leeuwen, F. N. Myosin II and mechanotransduction: a balancing act. Trends Cell Biol. 17, 178–186 (2007).
Jeannot, P. & Besson, A. Cortactin function in invadopodia. Small GTPases 11, 256–270 (2020).
Revach, O.-Y., Winograd-Katz, S. E., Samuels, Y. & Geiger, B. The involvement of mutant Rac1 in the formation of invadopodia in cultured melanoma cells. Exp. Cell Res. 343, 82–88 (2016).
Chang, J., Pang, E. M., Adebowale, K., Wisdom, K. M. & Chaudhuri, O. Increased Stiffness Inhibits Invadopodia Formation and Cell Migration in 3D. Biophys. J. 119, 726–736 (2020).
Zaman, M. H. et al. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc. Natl. Acad. Sci. USA 103, 10889–10894 (2006).
Poincloux, R., Lizárraga, F. & Chavrier, P. Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J. Cell Sci. 122, 3015–3024 (2009).
Pourfarhangi, K. E., Bergman, A. & Gligorijevic, B. ECM cross-linking regulates Invadopodia dynamics. Biophys. J. 114, 1455–1466 (2018).
Hoshino, D. et al. Establishment and validation of computational model for MT1-MMP-dependent ECM degradation and intervention strategies. PLoS Comput. Biol. 8, e1002479 (2012).
Palmulli, R., Jackson, H. K. & Edgar, J. R. Tethered Exosomes Containing the Matrix Metalloproteinase MT1-MMP contribute to extracellular matrix degradation. J. Extracell. Vesicles 14, e70122 (2025).
Boal D. Mechanics of the Cell. 2nd edn. (Cambridge University Press, Cambridge, 2012).
Mori, Y. Mathematical properties of pump-leak models of cell volume control and electrolyte balance. J. Math. Biol. 65, 875–918 (2012)
Yu, M. et al. Effects of mechanical stimuli on profilin- and formin-mediated actin polymerization. Nano Lett. 18, 5239–5247 (2018).
Yu, M. et al. mDia1 senses both force and torque during F-actin filament polymerization. Nat. Commun. 8, 1650 (2017).
Branch, K. M., Hoshino, D. & Weaver, A. M. Adhesion rings surround invadopodia and promote maturation. Biol. Open 1, 711–722 (2012).
Colombero, C. et al. mTOR repression in response to amino acid starvation promotes ECM degradation through MT1-MMP Endocytosis arrest. Adv. Sci. 8, 2101614 (2021).
Remy, D. et al. TFEB triggers a matrix degradation and invasion program in triple-negative breast cancer cells upon mTORC1 repression. Dev. Cell 60, 1018–1035.e8 (2025).
Fixemer, T., Wissenbach, U., Flockerzi, V. & Bonkhoff, H. Expression of the Ca2+ -selective cation channel TRPV6 in human prostate cancer: a novel prognostic marker for tumor progression. Oncogene 22, 7858–7861 (2003).
Peleg, S., Sellin, J. H., Wang, Y., Freeman, M. R. & Umar, S. Suppression of aberrant transient receptor potential cation channel, subfamily V, member 6 expression in hyperproliferative colonic crypts by dietary calcium. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G593–G601 (2010).
Chodon, D. et al. Estrogen regulation of TRPM8 expression in breast cancer cells. BMC Cancer 10, 212 (2010)
Guilbert, A. et al. Expression of TRPC6 channels in human epithelial breast cancer cells. BMC Cancer 8, 125 (2008).
Brundage, R. A., Fogarty, K. E., Tuft, R. A. & Fay, F. S. Calcium Gradients Underlying Polarization and Chemotaxis of Eosinophils. Science 254, 703–706 (1991).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants No. 12472323, No. 12202439, No. 12025207, No. 11872357), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB1150000), and the Fundamental Research Funds for the Central Universities. This work was partially carried out at the University of Science and Technology of China Center for Micro and Nanoscale Research and Fabrication.
Author information
Authors and Affiliations
Contributions
J.Z., Z.G. and H.J. conceived the chemo-mechanical model. J.Z., H.Z., Z.G. and H.J performed the theoretical and numerical analyses. Z.G., H.J. supervised the research. J.Z. and Z.G. led the drafting of the manuscript, with H.Z., Y.Y., R.X., and H.J. contributing to revisions and edits.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Physics thanks Artem K. Efremov, Stefan Linder and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, J., Zhang, H., Yang, Y. et al. Ca2+ transmembrane transport enhances oscillatory growth of cancer cell invadopodia. Commun Phys 8, 346 (2025). https://doi.org/10.1038/s42005-025-02268-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42005-025-02268-x
