
Abstract
Survival analysis aims to estimate the impact of covariates on the expected time until an event occurs, which is broadly utilized in disciplines such as life sciences and healthcare, substantially influencing decision-making and improving survival outcomes. Existing methods, usually assuming similar training and testing distributions, nevertheless face challenges with real-world varying data sources, creating unpredictable shifts that undermine their reliability. This urgently necessitates that survival analysis methods should utilize stable features across diverse cohorts for predictions, rather than relying on spurious correlations. To this end, we propose a stable Cox model with theoretical guarantees to identify stable variables, which jointly optimizes an independence-driven sample reweighting module and a weighted Cox regression model. Through extensive evaluation on simulated and real-world omics and clinical data, stable Cox not only shows strong generalization ability across diverse independent test sets but also stratifies the subtype of patients significantly with the identified biomarker panels.
Similar content being viewed by others


Main
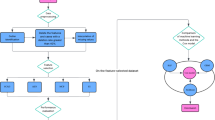
Survival analysis is a subfield of statistics that assesses the impact of covariates on the time until an event of interest occurs, widely used across various key fields such as life science to inform decision-making and predict outcomes. Among popular survival analysis methods1,2,3,4, the Cox proportional hazards (PH) model5 is the most prominent historically owing to its flexibility in handling censored data, accommodating a wide range of covariates without requiring the specification of the underlying survival distribution. While existing survival analysis methods show promising results under the assumption that training and test data share similar distributions, challenges arise when this assumption does not hold. Distribution shifts are inevitable in healthcare scenarios owing to training and test data that may be collected from different centres, standards of quantification method and subpopulation heterogeneity6. For instance, in the healthcare area, the survival data for certain diseases are plentiful in hospitals in developed areas but scarce in less developed regions. Commonly, a survival model is developed using the abundant data collected from developed areas and then applied to regions where data are lacking7. Distribution shifts inevitably occur between the two types of area owing to the high heterogeneity among populations and the differing treatment plans prevalent in these regions. More specifically, in oncology, prognostic markers play a key role in patient management and decision-making, and the identification of them is one of the major objectives in clinical research. However, we often observe that the same biomarker has been reported to show different prognostic values in different studies. For example, in studies focusing on Chinese patients with hepatocellular carcinoma (HCC), the prognostic value of epithelial cell adhesion molecule (EPCAM) expression in tumour tissues, as determined by immunohistochemistry, has shown variable outcomes8. One study identified EPCAM expression as a predictor of good prognosis9, whereas another study found that high levels of EPCAM expression were linked to poor prognosis10. We also conducted univariate Cox regression analysis on two HCC transcriptome cohorts11,12. As shown in Fig. 1a (left), consistent with the literature, there was limited overlap in the genes identified with the same prognostic value by both cohorts. Moreover, a few genes even showed completely opposing prognostic predictive values. From the data perspective, the inconsistent relationship between these biomarkers with prognosis could be caused by the distribution shifts of covariates or the real functional relationship between genes and prognosis. We generally assume that the true functional relationship a gene has with the prognosis of patients with a specific cancer type is stable and would not change across cohorts13,14,15,16. In contrast, covariate distribution could easily change owing to the expression level of some kinds of genes in different populations being different17. We further visualize the distribution of covariates for these two cohorts in Fig. 1a (right). It is evident that there is notable variance in the covariate distributions of the two cohorts, indicating the presence of distribution shifts. Distributional shifts pose serious challenges to survival analysis, potentially leading to serious declines in performance when high-risk factors are not accurately identified. The principal challenge in combating distribution shifts lies in identifying stable variables that maintain a consistent relationship with the outcome across different cohorts. It is a highly non-trivial and long-standing unsolved problem to discover such stable variables owing to the complicated time-to-event nature of survival data and correlation-driven mechanism of existing survival analysis methods18,19. Consequently, current methods might blindly learn the misleading patterns from spurious correlations present in the training set. However, such correlations are unstable and easily changed in the test set, posing a considerable risk when applying the trained model to new cohorts.

a, Left: Venn diagrams show the intersection of genes associated with prognosis in the The Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA-LIHC) cohort11 (n = 351) and the cohort of Roessler et al.12 (n = 209) of HCC transcriptome data (11,512 overlapped genes between two cohorts). Genes that meet the following criteria are considered as prognosis-related genes: (1) HR (univariate Cox analysis) larger than 1 and log-rank P value (median stratification) less than 0.05 for unfavourable; (2) HR smaller than 1 and log-rank P value less than 0.05 for favourable. Right: corresponding t-distributed stochastic neighbour embedding (t-SNE) plots of covariates of these two cohorts. b, t-SNE plots of covariates of four cohorts of breast cancer transcriptome data and lung cancer clinical data with two subpopulations. Breast cancer transcriptome data56 comprise 20,388 gene-expression messenger RNA log intensity levels profiles that serve as input for t-SNE, with each column z-score normalized. The sample sizes are as follows: cohort 1, n = 763; cohort 2, n = 521; cohort 3, n = 288; cohort 4, n = 238. Lung cancer clinic data62 have 51 clinical features, which are min–max normalized. We classify the samples into two subpopulations based on the location of the tumour: central, n = 141; peripheral, n = 255. Each dot represents a patient and is coloured by the cohorts they belong to (left) or the location of the tumour in the lung (right). These visualizations highlight the variance in covariate distributions across different cohorts and subpopulations. c, Illustration of our proposed framework compared with traditional survival analysis method. Our method is designed to eliminate spurious correlations between stable variables S and unstable variables V, resulting in getting rid of spurious correlation between V and survival outcome T (that is, P(T|V)) and focusing on stable relationship P(T|S) across environment (Envs). Consequently, stable Cox makes notable improvements over Cox PH across multiple independent test cohorts and various downstream tasks.
For instance, as shown in Fig. 1b, the distributions of the covariates differ among cohorts or subpopulations, where the ‘batch effect’ is one of the major causes of heterogeneity among cohorts20. We could assume that the shifts between cohorts or subpopulations are mainly caused by parts of covariates (that is, unstable covariates, detailed in Assumption 3 in Methods), where these unstable covariates may have spurious correlations with other stable covariates owing to selection bias. If we train a model on a particular cohort, the correlation-driven nature of current survival analysis methods substantially increases the likelihood that it will capture the spurious correlations that are specific to that cohort. Therefore, applying the identified high-risk factors as biomarkers to an unknown population carries a remarkable risk of leading to serious consequences such as wrong treatment assignment. Given the unacceptable risk in such high-stakes applications, it highlights the critical need for robust survival analysis models. These models must be capable of identifying stable features that can adapt to shifts in distribution.
The most common practice to enhance the ability of Cox PH to identify the most relevant features related to the outcome variable involves incorporating sparsity norms, including lasso21, ridge regression22, elastic net23 and smoothly clipped absolute deviation penalty24 and so on. While these methods have achieved success by promoting sparsity in the coefficients, they can only handle the scenarios without model misspecification25 and lack the capability to discern between stable and unstable variables with model misspecification. Stable learning26,27 is a branch of machine learning methods that brings causality into learning methods, aiming to bridge the gap between the tradition of precise modelling in causal-inference and black-box approaches from machine learning. Benefiting from the theoretical guarantees provided by causal-inference methods, stable learning aims to identify stable causal relationships rather than easily changeable correlations when modelling the relationships between covariates and outcomes. As a result, these kinds of methods28,29,30,31,32 show promising results on generalization, interpretability and fairness. Nevertheless, stable learning methods cannot be applied to complex time-to-event data yet.
In this Article, we propose a stable Cox regression model designed to identify stable variables for prediction, thereby ensuring strong generalization performances under distribution shifts based on these selected variables. Our approach aims to eliminate spurious correlations among covariates and focus on using stable variables for predictions. The model operates in two stages: independence-driven sample reweighting and weighted Cox regression. During the independence-driven sample reweighting stage, we employ a module to learn subject weights that render the covariate independent. In the subsequent weighted Cox regression stage, subjects are reweighted using these learned weights, leading to a weighted partial log-likelihood loss. This loss effectively isolates the effect of each variable during optimization. Theoretically, we prove that under some mild assumptions, and even with model misspecification, our stable Cox model exclusively relies on stable variables for predictions. This means that coefficients for unstable variables will be zero, provided that the learned sample weights maintain strict mutual independence among all covariates. We validate the effectiveness of the proposed method on both simulated data and two kinds of critical real-world applications: patient prognosis prediction based on omics data or clinical features. The extensive results demonstrate the generalization ability of the proposed method on unseen test cohorts or subpopulations. Notably, the coefficients derived from the method show remarkable stability and interpretability in downstream tasks. The learned coefficients can be used to discover potential biomarker panels and stratify subgroups (subtypes) with significantly different survival risks. Such applications are fundamental in guiding treatment decisions and the development of targeted drugs, which must guarantee stability in the face of population heterogeneity.
Results
General framework of stable Cox regression model
Let \(X=({X}_{1},{X}_{2},\ldots ,{X}_{p})\in {{\mathbb{R}}}^{p}\) be the p-dimensional subjects’ features, T ∈ [0, ∞) be the possibly censored failure times, and δ ∈ {0, 1} be the indicators for censoring. Suppose we get n independent and identically distributed (iid) survival data \({\left\{{x}^{(i)},{t}^{\,(i)},{\delta }^{(i)}\right\}}_{i = 1}^{n}\) drawn from a training distribution Ptr on the random variables X, T and δ, where x(i) and t(i) means the feaures (covariates) and possibly censored failure time of subject i, respectively. Let Pte denote the unknown test distribution.
In survival analysis problems involving multiple covariates, typically only a small subset notably influences the survival outcomes, while the remaining covariates may represent noise or show spurious correlations with the outcomes that are unstable across unseen testing distributions. For example, in omics data, some tumour genes show a causal role if a gene whose high expression leads to aggressive forms of certain types of cancer, such as the ERBB2 (Erb-B2 receptor tyrosine kinase 2, also known as HER2)-positive breast cancers that tend to be more aggressive33. However, the expression of some genes (for example, genes for lactase persistence) may highly correlate with the location where the person lives17, and the development level of the hospital in their city may determine their prognosis. The relationship of these genes with their prognosis is unstable across cities. In this way, two kinds of genes would be spuriously correlated owing to the location. To formalize this scenario, we make a structural assumption of covariates by splitting them into stable variables S and unstable variables V, where the failure time T only depends on the stable variables S. Stable variables are real predictors of the outcome, while unstable variables are associated with the outcome through their correlation with stable variables, which can vary across different study populations. Such an assumption can be guaranteed by T ⊥ V|S (detailed discussion in Assumption 3 in Methods).
In covariate-shift scenarios, we usually suppose probabilty P(T|X) remains unchanged while P(X) may change between training and test sets. For example, in survival analysis, some genes consistently show a stable trend associated with unfavourable prognosis across different cohorts, while others do not show such a stable trend. As shown in Fig. 1c, owing to selection bias, there may exist spurious correlations between stable covariates S and unstable covariates V, resulting in changes in P(X). Thus unexpected correlation would mislead the model to learn the spurious correlation between V and T. This correlation P(T|V) is unstable across possible testing distributions, which would result in the degradation of generalization performance. To get rid of the unstable correlation and capture the stable relationships between S and T, we propose to learn a group of sample weights to remove the correlations among covariates in observational data, and then optimize the Cox model in the weighted distribution. It is theoretically guaranteed that the stable Cox model utilizes only stable variables for prediction (detailed in ‘Theoretical results’ in Methods).
Our stable Cox regression model consists of two stages. In the first stage, we propose to utilize a sample reweighting module to learn sample weights so that X are statistically independent in the weighted distribution (Fig. 2a). In the implementation, we utilize the typical independence-driven algorithm, namely, Sample Reweighted Decorrelation Operator (SRDO)31. A previous study31 proposed to learn weighting function w(X) by estimating the density ratio of the training distribution Ptr and a specific weighted distribution \(\tilde{P}\). They define \(\tilde{P}\) through a process of random resampling across each feature, resulting in \(\tilde{P}({X}_{1},{X}_{2},\ldots ,{X}_{p})=\mathop{\prod }\nolimits_{j = 1}^{p}{P}^{{\mathrm{tr}}}({X}_{j})\). Consequently, the weighting function w(X) is given by
The density ratio in equation (1) can be effectively addressed through class probability estimation problems34. As a result, SRDO can guarantee statistical independence between covariates X if the density ratio is estimated accurately.

a, Stage 1: independence-driven sample reweighting. The sample reweighting module decorrelates the spurious correlations among covariates. b, Stage 2: weighted Cox regression. The module utilizes the learned sample weights W to reweight the event of each subject in the partial log-likelihood loss. The weighted partial log-likelihood loss could drive the model to utilize stable variables S to make predictions rather than unstable variables V (that is, \({\hat{\beta }}_{w}(V\,)=0\)).
In the second stage, as shown in Fig. 2b we propose to reweight the partially log-likelihood loss of the Cox PH model by the learned weights as follows:
where β is the coefficients of covariates to be learned. λ0(t(i)) represents the value of baseline hazard function at time t(i). \({L}_{w = 1}^{(i)}(\,\beta )\) denotes the unweighted likelihood of the event to be observed occurring for subject i at time t(i). The likelihood considers the sum over all subjects j for whom the event has not yet occurred by time t(i) (including subject i itself). In the process of likelihood maximization, the model’s predictive probability that the event occurs for subject i before any subject j is optimized. \({L}_{w}^{(i)}(\,\beta )\) is the weighted version of the likelihood of each subject, where each subject happens w(X) times. Under mild assumptions, we could prove that the estimated coefficients of unstable covariates \({\hat{\beta }}_{w}(V)\) will approach zeros with high probability (detailed in ‘Theoretical results’ in Methods).
Evaluation on the simulated survival data
Experimental set-up
To evaluate stable Cox in a controllable manner, we generate three kinds of survival data with different hazard functions that usually occur in real-world applications, following the generation process of refs. 25,35. In addition, to simulate the spurious correlation between stable and unstable variables, we design the sample selection process to introduce such correlation. In the simulation study, we are aware of which variables are stable or unstable, allowing us to rigorously assess whether our method successfully identifies stable variables. The detailed data generation process and experimental set-up are in ‘Simulated survival data’ in Methods. Baselines are introduced in ‘Baseline approaches’ in Methods.
Results
The results of the full setting are depicted in Fig. 3a. In general, all the baselines demonstrate satisfactory performance when test bias rate rtest ∈ (1, 3]; however, their performances drop dramatically when the rtest ∈ [−3, −1). This is because the correlations between Vb and T are similar between training data (training bias rate rtrain = 1.7) and test data when rtest > 1 and that correlation can be exploited in prediction. In such cases, V is useful to proxy for the misspecification form and omitted function of S. However, then rtest < −1, the correlation between Vb and T reverses compared with the training set, leading to excessive instability when Vb is used for predictions. Figure 3c depicts the box plots of each method on the Cox-exp dataset. Our stable Cox method shows low variance across testing sets. Importantly, compared with the Cox PH model, our method improves its average performance of Concordance index (C-index), the worst case of testing environments and the variances across environments remarkably. As our model is built on the Cox PH model, we could safely attribute the notable improvement to the seamless joint of the proposed framework with Cox PH model.

a, The results of methods on four data generation models (Cox-exp, Cox–Weibull, Poly and Log T). We fix p = 10 and n = 10,000. The rtrain is set as 2.5. rtest varies from {−3.0, −2.0, −1.7, −1.5, −1.3, 1.3, 1.5, 1.7, 2.0, 3.0}. b, The results of Cox PH trained on the top-five features selected by the P value of coefficients of the corresponding methods. The P value is derived from a two-sided Wald test, with the null hypothesis being that the coefficient (β) is zero. c, The box plots of each method on the Cox-exp dataset (n = 10,000). Box plots show the median (centre lines), interquartile range (hinges) and 1.5 times the interquartile range (whiskers). d, The −log2P value calculated by a two-sided Wald test of the coefficients of stable Cox and Cox PH for stable variables (S) and unstable variables (V) respectively (n = 10,000). Box plots show the median (centre lines), interquartile range (hinges) and 1.5 times the interquartile range (whiskers). Any data points beyond the whiskers are marked as outliers, providing a clear visual indication of deviations from the main distribution. Vb represents a spurious variable exhibiting a strong correlation with the outcome T in training set. However, such correlation is unstable on the test sets. e, The covariate-shift generalization results with respect to the number of selected features (n = 10,000). The error bars represent mean ± s.d. of all testing sets. The marker represent the results on each testing set. f, Left: \(| | \hat{\beta }(V)| |\) with varying sample size n. Right: the performance with varying sample size n. The shaded area represents mean ± s.d. of all testing sets. All the above results were performed ten times with different seeds to generate different datasets and we report the mean results.
Furthermore, we demonstrate the significance of the coefficients derived from our model, highlighting the use of P values for each coefficient as a tool for feature selection. Figure 3d summarizes the −log2P value of the coefficients of S and V for stable Cox and Cox PH. The P value is derived from a two-sided Wald test, with the null hypothesis being that the coefficient (β) is zero. The larger −log2P values for stable Cox (S) compared with stable Cox (V) suggest that coefficients for stable variables S are more statistically significant than those for unstable variables V. However, the −log2P value of Vb of the Cox PH model is substantially higher than those for stable variables S, demonstrating that Cox PH regards Vb as a highly important feature in predictions. On the basis of this, we can leverage the P-value rankings to select the top-N significant features, an approach particularly beneficial in applications requiring minimal feature usage for predictions. For example, measuring key genes to predict patient survival is more cost-effective than assessing an entire genome. Figure 3b presents the outcomes of this feature selection strategy, where we use the top-five features selected by each method to train a new corresponding prediction model. Note that the new prediction model for stable Cox is Cox PH. It yields results akin to the full model setting (as shown in Fig. 3a). Moreover, as shown in Fig. 3f, when the number of selected features exceeds five, the stability of the model drops significantly. These phenomena demonstrate that our model could learn distinct P values between S and V, enabling a reduction in the necessary number of covariates.
In addition to prediction performance, we illustrate the effectiveness of our method in weakening spurious correlation in Fig. 3f. We calculate \(| | \hat{\beta }(V)| {| }_{1}\) to quantify the residual correlation between unstable variables V and the outcome T. Across varying sample sizes, the stable Cox model consistently shows lower residual correlations. With a larger sample size, \(| | \hat{\beta }(V)| {| }_{1}\) will be reduced (Fig. 3f, left), and the gap between our method and Cox PH will be larger (Fig. 3f, right). This finding confirms that with a larger number of samples, the independence module more effectively reduces statistical dependence, thereby enhancing model stability. More experiments on the sensitivity of the independence assumption among covariates after reweighing can be found in Supplementary Information, section C.2.
Evaluation on multiple cancer transcriptome survival data
Experimental set-up
Transcriptomics is a crucial and rapidly developed field in biology, providing extensive information on disease states, biomarker discovery and new drug development through the analysis of gene expression36. Scientists can identify several key genes associated with the prognostic outcome for a specific disease, which are referred to as prognostic biomarkers. These biomarkers are instrumental in predicting a patient’s prognosis, enabling a more tailored and effective treatment approach. By focusing on these disease-specific genes, targeted therapies can be developed, offering a more personalized and potentially more effective treatment strategy. To comprehensively evaluate our method, we have constructed the following transcriptome survival datasets: HCC transcriptome dataset, breast cancer transcriptome dataset and melanoma transcriptome dataset, where each dataset has one training cohort and three independent testing cohorts. The data details and experimental set-up are in ‘Baseline approaches’ in Methods.
Results
The performance of all the methods on each testing cohort and average performance across testing cohorts of each dataset are reported in Fig. 4. First, we observe that the Cox PH model outperforms the corresponding univariate Cox PH model across three datasets in terms of overall performance. The major difference between them is that the Cox PH model considers the relationship between genes to select the final biomarker panel, whereas the univariate Cox PH considers only the importance of each gene separately. This indicates that it is necessary to consider the relationship between genes to discover the biomarker panel. Second, for the HCC transcriptome dataset, parametric methods show competitive results on the Fujimoto et al. cohort37 and the Roessler et al. cohort12, but they fail on the Hoshida et al. cohort38 and show high standard error. The reason is that the Fujimoto et al. cohort and the Roessler et al. cohort may share a similar data distribution with the training set and the parametric assumption may align well with this distribution. However, the Hoshida et al. cohort may have larger distribution shifts with the training set. Nevertheless, our stable Cox model consistently shows a high average C-index and low standard error on the three independent testing cohorts, indicating robustness and reliability in various unseen testing conditions. Third, our model outperforms Cox PH across three datasets by a large margin (from 10.4% to 13.9% overall improvements in the top-10 panel), indicating that making covariates independent could help the model get rid of spurious correlation and focus on relevant variables. Fourth, in the melanoma transcriptome dataset, it is shown that the performance deteriorates when the number of selected genes exceeds ten. This decline may occur because the optimal number of biomarkers varies across different diseases39. Incorporating too many genes into the biomarker panel can introduce noise or unstable features, adversely affecting performance. This phenomenon is consistent with the results in our simulation experiments (Fig. 3e), that is, involving more irrelevant features in the model will damage the stability of the model.

a, The quantitative results of the Fujimoto et al. cohort (n = 203), the Roessler et al. cohort (n = 209) and the Hoshida et al. cohort (n = 80) with respect to the top-5, -10, -15 and -20 selected features by each method trained on the TCGA-LIHC cohort (n = 351) of HCC transcriptome data (left three columns). The mean value of the C-index across three test cohorts, specifically for the top-5, -10, -15 and -20 features selected by each model, is reported in the ‘Overall’ column. The error bars represent mean ± s.d. of all test cohorts. b, The quantitative results of the Curtis et al. cohort 1 (n = 521), cohort 2 (n = 288) and cohort 3 (n = 238) with respect to top-5, -10, -15 and -20 selected features by each method trained on the training cohort (n = 763) of the breast cancer transcriptome dataset (left three columns). The mean value of the C-index across 3 test cohorts, specifically for the top-5, -10, -15 and -20 features selected by each model, is reported in the ‘Overall’ column. The error bars represent mean ± s.d. of all testing cohorts. c, The quantitative results of the Gide et al. cohort (n = 91), the Riaz et al. cohort (n = 54) and the Van et al. cohort (n = 41) with respect to the top-5, -10, -15 and -20 selected features by each method trained on the Liu et al. cohort (n = 120) of the melanoma transcriptome dataset (left three columns). The mean value of the C-index across three test cohorts, specifically for the top-5, -10, -15 and -20 features selected by each model, is reported in the ‘Overall’ column. The error bars represent mean ± s.d. of all testing cohorts. The average improvement on three independent testing cohorts shows the stable performance of our method.
Accurately inferring survival subgroups among patients with cancer can significantly aid in clinical decision-making and enhance the patient’s survival outcomes. We use the median value of \({\hat{\beta }}^{T}X\) in each cohort to divide the patients into the high-risk group (above median) and the low-risk group (below median). The difference between the survival curves of the two subgroups is measured by the P value of a two-sided log-rank test. The hazard ratio (HR) is from the univariate Cox regression of subgroup separation results. The Kaplan–Meier plots of subgroups stratified by Cox PH and stable Cox on three testing cohorts of the breast cancer transcriptome dataset are shown in Fig. 5a. The P value of stable Cox is lower than that of Cox PH on the corresponding cohort and lower than the significance level 0.05, and the HR of stable Cox is significantly larger than that of Cox PH. This demonstrates that the genes screened by our model can assist in stratifying patients into respective subgroups and offer precise prognostic stratification and treatment strategies. The Kaplan–Meier plots of the HCC transcriptome and melanoma transcriptome datasets are shown in Supplementary Fig. C5. Moreover, we conduct univariate Cox regression analyses on the subgroups divided by the key clinical indicators (that is, age, estrogen eeceptors (ER) status encoded by the ESR1 and ESR2 genes, HER2 status and progesterone receptor (PR) status encoded by the PGR gene) and report their HR value and the corresponding 95% confidence interval (CI) in Fig. 5b. The higher HR value means the method could identify high-risk and low-risk subgroups well under this clinical variable subgroup. Overall, compared with the Cox PH model, the stable Cox model demonstrates higher HR values in most clinical variable subgroups. This suggests that the stable Cox model offers superior prognostic prediction performance in subgroup analyses. It is worth noting that in the HER2 status positive and PR status negative subgroup, stable Cox can also effectively identify those with a worse prognosis among these patients commonly regarded as having a high malignant grade and unfavourable prognosis in clinical practice40. This contributes to improving the precision of patient stratification, facilitating enhanced patient management and treatment.

a, Kaplan–Meier plot of two subgroups (above and below the median of βTX) separated by the top-ten genes identified by the Cox PH and stable Cox for three test cohorts. The P value is calculated using a two-sided log-rank test. HR is calculated by a univariate Cox regression of the subgroup assignment variable on outcomes. b, Univariate Cox regression analyses among clinical subgroups of patients. The subgroups are defined according to the clinical variables of patients. Box and error bars represent the HR value and its 95% CI, respectively. The subgroups for HR calculation are separated based on optimal cut-off value defined as the minimal proportion of observations per subgroup of at least 30% to further separate the clinical subgroup into more refined subgroups. The detailed sample size of each subgroup can be found in Supplementary Table A2. c, The favourable/unfavourable consistency of individual genes across training and test cohorts.
Furthermore, we compare the favourable/unfavourable consistency of the correlation of individual proteins of the top-ten important genes across different cohorts. The univariate Cox regression analysis was used to calculate the predictive ability for the survival of the identified genes in the training and three test cohorts, respectively. Genes that meet the following criteria are considered as prognosis-related genes: (1) HR larger than 1 and log-rank P value less than 0.05 for unfavourable; (2) HR smaller than 1 and log-rank P value less than 0.05 for favourable; (3) otherwise is no trend. The results are shown in Fig. 5c. As we can see, no genes screened by stable Cox show both favourable and unfavourable relationships with the survival outcome. In addition, 2 genes (WEE2-AS1 and RPGRIP1) screened by stable Cox show a high-frequency (over or equal to 75%) favourable relationship with survival outcome, and 4 genes (CLEC3A, LSG1, SRGAP2 and F2RL1) have a high-frequency unfavourable relationship across 4 cohorts. However, Cox PH has 3 genes showing both favourable and unfavourable relationships with the survival outcome, and 4 genes (MCEMP1, RPGRIP1, F2RL1 and CXCL2) show a 75% frequency favourable/unfavourable relationship with survival outcome. This phenomenon indicates that genes screened by our method also show more stable prognostic relationships at the individual gene level. Moreover, we noticed that three of the top-ten genes identified by both the Cox PH and stable Cox methods are the same (OR5M11, RPGRIP1 and F2RL1), while the others differ. The gene panel identified by our method demonstrates superior predictive performance (Fig. 4b), more significant stratification of subgroups (Fig. 5a,b), and greater consistency in distinguishing favourable and unfavourable genes across both training and test cohorts (Fig. 5c). These encouraging findings substantiate our method’s capability to identify promising candidate biomarkers for prognosis, which is crucial for guiding treatment decisions and developing targeted therapies.
Evaluation on lung and breast cancer clinical survival data
Experimental set-up
Clinical data provide abundant information to characterize patients, including patient demographics, disease stage, treatment methods and so on. This information has a strong correlation with the patient’s survival outcome. In this section, we conduct experiments on lung cancer data and breast cancer data. The data details and experimental set-up are in ‘Clinical survival data’ in Methods.
Results
Figure 6a shows the performance of various methods across six subpopulations for OS and DFS. The results clearly show that the stable Cox model significantly surpasses the baseline methods in each subpopulation for both overall survival (OS) and disease-free survival (DFS). Notably, stable Cox achieves a performance improvement of 4.5% and 17.7% over the Cox PH model in OS and DFS tasks, respectively. This demonstrates that the coefficients of our model can ensure stability across diverse subpopulations and tasks, markedly reducing the risk and bias when applied in real-world scenarios. Furthermore, the results of OS and recurrence-free survival (RFS) prediction of breast cancer data are shown in Fig. 6b. As we can see, stable Cox demonstrates superiority over Cox PH on both test cohort 1 and cohort 2 and shows good stability overall, regardless of whether it is for OS (6.58% improvement) or RFS (6.5% improvement) tasks. We find that the parametric methods achieve better results than Cox PH, demonstrating that their assumptions on survival time are closer to the underlying distribution. Nevertheless, our model could still outperform them taking Cox PH as a base model. Following the procedure of omics study, Kaplan–Meier plots of high-risk and low-risk subgroups separated by the Cox PH model or the stable Cox model for breast cancer dataset are shown in Fig. 6c. The differences between the two subgroups generated by stable Cox on all test scenarios are quite significant (lower than the significance level of 0.05). Nevertheless, the variance of the P values or HR value of Cox PH is quite large. Notably, Cox PH is very significant on test cohort 1 of OS prediction, but the difference between the survival curves of the two subgroups is not significant. The phenomenon demonstrates that the coefficients learned by our model can be used as a reliable index to guide decision-making and improve the patient’s postoperative prognosis. The Kaplan–Meier plots of the lung cancer clinical dataset for OS and DFS outcomes are shown in Supplementary Figs. C8 and C9, respectively.

a, The performance of methods on the six test subpopulations of the lung cancer dataset (OS and DFS). We randomly select 40% of the patients (n = 240) as the training set, and age ≤ 60 (n = 43) or age > 60 (n = 113), tumour location was classified as ‘central’ (n = 47) or ‘peripheral’ (n = 107), and obstructive pneumonitis/atelectasis is ‘present’ (n = 80) or ‘absent’ (n = 76) as the test set. ‘Overall’ is the mean value of all testing sets. The error bars represent mean ± s.d. of all test cohorts. The average improvement on multiple independent testing subpopulations shows the stable performance of our method. b, The performance of methods trained on the training cohort (n = 394) on two testing cohorts, cohort 1 (n = 273) and cohort 2 (n = 177), of breast cancer (OS and RFS). ‘Overall’ is the mean value of all test sets. The error bars represent mean ± s.d. of all test cohorts. The average improvement on multiple independent testing cohorts shows the stable performance of our method. c, Kaplan–Meier plot of two subgroups separated by the coefficients of the Cox PH and stable Cox model for two testing cohorts of breast cancer dataset (OS and RFS). d, Top-ten clinical variables ranked by the P value derived by a two-sided Wald test of coefficients learned by Cox PH and stable Cox. The numbers in brackets are the −log2P value of the corresponding coefficients. The clinical variables in red colour are the most common biomarkers for breast cancer prognosis. IHC, immunohistochemistry.
Furthermore, we present the top-ten clinical variables with the most significant coefficients of Cox PH and stable Cox in Fig. 6d. ER, PR and HER2 status are the most common biomarkers for breast cancer prognosis. Their joint status could stratify patients into subgroups with significantly different prognosis situations40. As observed, these clinical indices are among the top-ten clinical variables identified by the stable Cox model, whereas the Cox PH model includes only the PR status. This suggests that our model has a superior capability in discovering clinically significant biomarker panels.
Discussion
Stable Cox is a theoretical-guaranteed model to deal with distribution shifts and discover biomarker panels in survival analysis. Compared with traditional survival analysis methods, stable Cox offers three levels of advantages for survival time prediction and important variable identification. (1) Feature selection under covariate shift. Feature selection aims to construct a diagnostic or predictive model for a given regression or classification task via selecting a minimal-size subset of variables that show the best performance41. Selecting significant features and their combination for survival analysis could help discover biomarker panels, which is important for both decision-making and drug development. Stable Cox can be viewed as an embedded feature selection method under distribution shifts, which seeks to minimize the size of the selected feature subset while maximizing the prediction performance simultaneously. (2) Causal implication. Recent studies have shown that data-driven approaches are often mistakenly used to draw causal effects, with neither their parameters nor their predictions inherently offering a causal interpretation42,43,44,45. Therefore, the foundation that data-driven predictive models yield reliable decisions for precision medicine is questionable. A promising and practical pathway is to build a predictive model that is located at the common ground between machine learning and causal inference26. The key idea of our approach is to eliminate the spurious correlation among covariates. When viewing this method from the causal-inference perspective, we regard each input variable as the treatment iteratively and all remaining input variables as its corresponding confounder, and thus the learned sample weights realize confounder balancing46 globally for whichever input variable acts as the treatment through the independence between treatment and corresponding confounder. Therefore, the learned correlation of our model would approximately represent the causal effects of the covariates on survival probabilities that are invariant across different domains. From this perspective, our model has inherent causal implications, representing a significant stride towards reliable survival analysis. (3) Easy to implement. The independence module supplies the weighted Cox model with a collection of sample weights. The additional time complexity in our method, compared with the standard Cox model, originates from the independence-driven sample reweighting module. The independence module could be optimized independently and has low time complexity, which is \({\mathcal{O}}(tn(ph+hg+2g))\), where (h, g) are the dimensions of hidden layers of the multilayer perceptron and t is the number of iterations for training.
Methods
Preliminaries
In this subsection, we present notations, weighting function and the base model employed in our approach.
Notations
Let \({\mathcal{X}}\) denote the support of the feature set X. For a vector \(x\in {\mathcal{X}}\), we define the following tensor powers. x⊗0 represents the scalar value 1, x⊗1 denotes the original vector x, and x⊗2 denotes the matrix xxT.
In the context of probabilistic expectations, \({{\mathbb{E}}}_{Q}[\cdot ]\) and \({{\mathbb{E}}}_{Q}[\cdot | \cdot ]\) are used to represent the expectation and conditional expectation under a distribution Q, respectively. To simplify notation, when referring to expectations under the training distribution Ptr, we omit the subscript and use \({\mathbb{E}}[\cdot ]\) and \({\mathbb{E}}[\cdot | \cdot ]\).
Weighting function
Consider the set \({\mathcal{W}}\), defined as the collection of weighting functions that satisfy the condition:
For every \(w\in {\mathcal{W}}\), the associated weighted distribution \({\tilde{P}}_{w}\) is uniquely determined by its probability density function:
To simplify notation, \({{\mathbb{E}}}_{w}[\cdot ]\) denotes the expectations under the distribution \({\tilde{P}}_{w}\). Furthermore, for any measurable function f(X), it holds that \({\mathbb{E}}[w(X\,)f(X\,)]={{\mathbb{E}}}_{w}[\;f(X\,)]\).
In addition, independence-driven sample reweighting algorithms focus on learning a subset \({{\mathcal{W}}}_{\perp }\subseteq {\mathcal{W}}\), rather than the entire set \({\mathcal{W}}\). A weighting function in \({{\mathcal{W}}}_{\perp }\) ensures that the features X are mutually independent in the corresponding weighted distribution \({\tilde{P}}_{w}\), that is
Cox PH model
In survival analysis5,47, the key variables T and δ are generated through the following process:
Here, Tfailure denotes the time of failure (event occurrence), and Tcensored represents the censoring time. Consequently, the observed variable T corresponds to the failure time Tfailure if it occurs before the censoring time Tcensored, and it equals the censoring time otherwise.
In the Cox PH model, also known as Cox regression5, the hazard function of the failure time Tfailure, denoted as λ(u), measures the instantaneous rate at which events occur at time u, conditional on no occurrence until time u. Mathematically, it is defined as: \(\lambda (u;X)=\mathop{\lim }\nolimits_{h\to {0}^{+}}{P}^{{\mathrm{tr}}}({T}^{\,{\rm{failure}}}\le u+h| {T}^{\,{\rm{failure}}}\ge u,X)/h\). For an individual characterized by covariates X, the standard Cox model posits that the individual’s hazard function is of the form:
Here, λ0(u) represents the baseline hazard function, reflecting the time-dependent risk of an event when covariates are at their baseline levels. The vector β embodies the effect parameters, indicating how the hazard varies in response to the explanatory covariates. According to the PH assumption48, there exists a multiplicative relationship between the covariates and the hazard. For example, in a scenario with constant coefficients, a drug treatment might consistently reduce the subject’s hazard by a factor of two at any time t, irrespective of variations in the baseline hazard.
Cox5 proposed estimating the parameter β using the log partial likelihood function, focusing solely on the impact of the covariates:
It is noteworthy that Breslow’s method provides an approach to estimate the baseline hazard function, enabling the full hazard function to be deduced as the product of the baseline hazard and the exponential term.
Stable Cox regression
A previous study49 has shown that if the assumption in equation (7) accurately represents the true hazard function, the estimator \(\hat{\beta }\) from equation (8) will converge to the actual β in equation (7) as the sample size approaches infinity. However, model misspecification, where the parameterization in equation (7) fails to capture the true model, frequently challenges the Cox model, as documented in refs. 25,50,51. Moreover, in practical scenarios, the test data may come from different centres or populations than the training data, resulting in distribution shifts. This is exemplified by the biology function analyses in Fig. 5, where the standard Cox model tends to rely on features with spurious correlations to the outcome, leading to suboptimal performance on test data.
In response to these challenges, we propose the stable Cox regression method. This approach focuses on leveraging features that show greater consistency across different environments for prediction, rather than depending on fragile, spurious correlations found in the training data. Specifically, the stable Cox regression method integrates a sample reweighting module with a weighted Cox regression module for robust performance across varied datasets.
Sample reweighting module
The primary objective of this module is to minimize the dependency of covariates. Initially, we take the original covariate matrix M and generate a column-decorrelated version \(\tilde{M}\) through random, column-wise resampling. This procedure disrupts the joint distribution of variables in X, leading to p independent marginal distributions. Thus, while the original feature distribution is Ptr(X), the resampled distribution becomes \(\tilde{P}(X)={P}^{{\mathrm{tr}}}({X}_{1}){P}^{{\mathrm{tr}}}({X}_{2})\cdots {P}^{{\mathrm{tr}}}({X}_{p})\).
Next, we employ density ratio estimation techniques as per ref. 34. We construct a joint distribution \({P}^{{\prime} }(X,Z)\) over \(X\in {\mathcal{X}}\) and Z ∈ {0, 1}, with \({P}^{{\prime} }(Z=0)={P}^{{\prime} }(Z=1)=1/2\), \({P}^{{\prime} }(X| Z=1)=\tilde{P}(X)\) and \({P}^{{\prime} }(X| Z=0)={P}^{{\mathrm{tr}}}(X\,)\). By applying Bayes’ theorem, the density ratio is formulated as:
The probabilities \({P}^{{\prime} }(Z=1| X)\) and \({P}^{{\prime} }(Z=0| X)\) are estimated by training a multilayer perceptron to differentiate between the samples from the original matrix M and the column-decorrelated version \(\tilde{M}\). To normalize the sample weights to have a unit mean, w(x(i)) is scaled by dividing it by the average \(\frac{1}{n}\mathop{\sum }\nolimits_{i = 1}^{n}w\left({x}^{(i)}\right)\).
Weighted Cox regression module
In this module, the sample weights obtained earlier are applied to reweight the event of each subject in the Cox PH loss function. Specifically, the log partial likelihood function Lw(β) and the estimator \({\hat{\beta }}_{w}\) are defined as follows:
Characterizing weighted Cox regression with counting processes
Following refs. 25,49, we employ counting processes to model events for different individuals. For individual i, let N(i)(u) (u ≥ 0) denote the counting process, with its intensity function given by:
where \({Y}^{\,(i)}(u)={\mathbb{I}}\left[{t}^{(i)} > u\right]\) is a predictable process, taking values in {0, 1} and indicating active observation of the individual. \(\lambda \left(u;{x}^{(i)}\right)\) is the true hazard function for individual i with features x(i) at time u. The loss function can then be expressed as:
Equations (10) and (12) represent the empirical losses with respect to a finite sample size n. To extrapolate to the population level, we consider the expected values over these empirical terms, replacing the counting process N(i)(t) with the hazard functions. The population-level loss function and the corresponding population-level solution are given by:
Here, for any r ∈ {0, 1, 2}
As established in Theorem 1, under certain assumptions, the empirical solution \({\hat{\beta }}_{w}\) converges to the population-level solution βw in probability as the sample size n approaches infinity.
Theoretical analysis
In this subsection, we present a theoretical analysis of our proposed stable Cox regression method.
Assumptions
To derive our theoretical results, we first establish several necessary assumptions.
Regularity assumptions
Consider w(X), the learned weighting function from the first stage of our method. We posit several regularity assumptions on the problem setting.
Assumption 1 (bounded parameter assumptions)
The following bounded parameter assumptions are made:
-
There exists a constant τ such that T ≤ τ almost surely.
-
There exists a constant C such that ∥X∥2 ≤ C almost surely.
-
There exists a constant B > 0 such that w(X) ≤ B almost surely.
-
For any u ≥ 0, \({\mathbb{E}}[\lambda (u;X)] < \infty\).
Remark 1
These assumptions are standard in Cox regression and weighted regression literature. Specifically, the first assumption, used in ref. 49, holds when all individuals have a finite censored time. The second assumption, similar to ref. 25, is reasonable as individual features are finite. The third assumption, in line with ref. 27, ensures a plausible weighting function for theoretical analysis. Lastly, the fourth assumption ensures the expected hazard rate for all individuals at any specific time u is finite, a condition easily met when the first two points are valid and the hazard function is continuous.
We also make an assumption regarding the population-level loss function, as defined in equation (13).
Assumption 2 (existence and uniqueness of the population-level solution)
There exists a unique solution to \(\arg \mathop{\min }\limits_{\beta }{\tilde{L}}_{w}(\,\beta )\).
Remark 2
This assumption, also adopted in previous studies25,49, ensures the existence and uniqueness of the population-level solution. It is a feasible condition as \({\tilde{L}}_{w}(\,\beta )\) is a convex function, a claim substantiated in Supplementary Information, section B.1.
S–V structure assumption
In line with previous studies27,29,30, we posit that the targets (specifically, the failure time Tfailure and censoring time Tcensored) are influenced only by a subset of variables, denoted as S. This is formalized in the following assumption.
Assumption 3
Assume that the feature set X can be partitioned into two disjoint subsets S and V, such that S ∪ V = X and \(S\cap V={{\emptyset}}\). Within the training distribution Ptr, it holds that:
Remark 3
It’s important to note that for the failure time Tfailure, the condition Tfailure ⊥ V|S is satisfied if and only if there exists a function \({\lambda }^{{\prime} }(u;S)\) such that for any time u and features X, \({\lambda }^{{\prime} }(u;S)=\lambda (u;X)\). The substantiation of this claim is detailed in ‘Proposition B.1’ in Supplementary Information, section B.2, highlighting the relevance of S in characterizing the hazard functions.
Moreover, in scenarios involving covariate shifts—where Pte(Tfailure, Tcensored|X) = Ptr(Tfailure, Tcensored|X), yet Pte(X) ≠ Ptr(X)—the conditional independence relationship (Tfailure, Tcensored) ⊥ V|S continues to hold within the test distribution. In addition, it follows that Pte(Tfailure, Tcensored|S) = Ptr(Tfailure, Tcensored|S). This crucial aspect is further elaborated in ‘Proposition B.2’ presented in Supplementary Information, section B.2. This proposition demonstrates that the set S can provide a stable and robust estimator for Cox regression, even in the presence of covariate shifts.
Theoretical results
Building on the previously stated assumptions, we now present a theoretical analysis of our stable Cox regression model.
Characterizing the solution of the weighted Cox regression
We first establish that the solution \({\hat{\beta }}_{w}\) to the empirical loss, as defined in equation (10), converges to the solution βw of the population-level loss, as specified in equation (13).
Theorem 1
Given a weighting function \(w\in {\mathcal{W}}\), and under Assumptions 1 and 2, it holds that
where \(\mathop{\to }\limits^{\,{P}\,}\) denotes convergence in probability.
Remark 4
The detailed proof of this theorem is provided in Supplementary Information, section B.3. While previous research25,49 primarily focused on the asymptotic properties of standard Cox regression, Theorem 1 extends this analysis to weighted Cox regression, which presents a novel area of interest.
Eliminating irrelevant variables via stable Cox regression
Building on Theorem 1, it can be demonstrated that stable Cox regression effectively eliminates irrelevant variables V, as per Assumption 3.
Theorem 2
Consider a weighting function \(w\in {{\mathcal{W}}}_{\perp }\) that ensures mutual independence of X in the weighted distribution. Under Assumptions 1, 2 and 3, the following holds:
where \({\hat{\beta }}_{w}(V\,)\) represents the coefficients of \({\hat{\beta }}_{w}\) corresponding to V.
Remark 5
The proof is available in Supplementary Information, section B.4. As noted in Remark 3, S can serve as an invariant predictor under covariate shift, whereas V may show spurious correlations with the outcome T, as evidenced by the biological function analyses in Fig. 5. Consequently, our stable Cox regression method effectively eliminate the influence of irrelevant variables V, ensuring stable prediction in data with covariate shift.
Experimental set-up details
Simulated survival data
We first generate all the covariates X = (S, V). In our experiments, the dimension of X is fixed to p = 10, and the dimensions of S and V are specified as ps = pv = 0.5 × p = 5. We generate covariate X by the following process:
N(0, 1) represents the standard Gaussian distribution. The survival time T is generated by the survival time function in Supplementary Table A2 and the corresponding transformation function between survival time and hazard function is \(T={H}_{0}^{-1}[-\log (U\,)]\exp (-{\beta }^{T}S)\) (ref. 25). The generation models Cox-exp and Cox–Weibull are on the omission of nonlinear term g(S) from Cox models, where g(S) = S1 × S2 × S3 is a nonlinear model misspecification term that is used to simulate the nonlinear generation process in the real world. Poly is on the misspecification of regression forms with the omission of the nonlinear term, and Log T is on non-PH models also with the omission of the nonlinear term.
Afterwards, we generate various environments by constructing spurious correlations between V and S, further leading to the change of P(T|V). Among all the unstable variables V, we simulate unstable correlation P(Vb|S) on a subset Vb ∈ V. We vary P(Vb|S) through different strengths of selection bias with a bias rate r ∈ [−3, −1) ∪ (1, 3] to generate multiple testing environments, and set rtr as 2.5 for the training set. For each sample, we select it with probability \({\mathrm{Pr}}={\prod }_{{V}_{i}\in {V}_{{\mathrm{b}}}}| r{| }^{-5{D}_{i}}\), where Di = |f(S) − sign(r)Vi|, where sign(⋅) denotes the sign function. In our experiments, we set \({p}_{{v}_{{\mathrm{b}}}}=0.1\times p\). Moreover, we randomly select 10% of subjects as censored events, where their censored survival time is generated by Tcensored ∼ U(0, T). We train our models on data from one single environment generated with a bias rate rtrain and test on data from multiple environments with bias rates rtest ranging in [−3, −1) ∪ (1, 3]. By adjusting the parameter rtest, we can modulate the strength of spurious correlations, thereby enabling a clear evaluation of the underlying mechanisms and performance of our method across different selection bias scenarios. Each model is trained ten times independently with different training datasets from the same bias rate rtrain. Likewise, for each rtest, we generate ten test datasets with different random seeds. We utilize the widely used C-index metric in survival analysis, evaluating the model’s ability to correctly provide a relative ranking of the predicted risk or time to event52. The metrics we report are the mean results of these ten times.
Transcriptome survival data
To comprehensively evaluate our method, the following selection criteria are guided by several considerations. First, omics data are typically generated from various cohorts around the world. Discovering omics biomarkers is a complex and costly process that involves identifying candidate biomarkers, qualification, verification and clinical assay development53. Consequently, it is impractical to develop a new biomarker panel for each cohort. A practical path is to discover biomarkers that could be stably generalized on unseen testing cohorts. Therefore, it is critical to assess the generalization ability of discovered biomarkers across multiple cohorts. Consequently, the dataset should encompass multiple cohorts for the same disease. Second, it is necessary that the dataset includes survival information, which is often scarce in high-quality omics literature. Third, the presence of clinical information is preferable to enable detailed studies based on clinical subgroups. On the basis of these criteria, we construct three datasets from multiple cohorts to evaluate survival models under distribution shifts. In each cohort, comprehensive transcriptome data and the corresponding OS information of individual patients are available. The distribution of covariates is thought of as being naturally different. The first dataset is the HCC transcriptome dataset, which is constructed by the five gene-expression datasets in HCCDB (a database of hepatocellular carcinoma expression atlas)54. In particular, we utilize the The Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA-LIHC) cohort (n = 351)11 as the training set, the Grinchuk et al. cohort (n = 115)55 as the validation dataset, and the Fujimoto et al. cohort (n = 203)37, the Roessler et al. cohort (n = 209)12 and the Hoshida et al. cohort (n = 80)38 as three independent testing cohorts. The second dataset is a breast cancer transcriptome dataset, which is constructed from ref. 56. This study56 has the transcriptome and OS information of 1,980 patients from 5 cohorts. We use 1 cohort (n = 763) as the training set, 1 cohort (n = 170) as the validation set, and the other 3 as the testing cohorts, termed as the Curtis et al. cohort 1 (n = 521), cohort 2 (n = 288) and cohort 3 (n = 238). Furthermore, the third dataset is the melanoma transcriptome dataset. Melanoma is a serious form of skin cancer that arises from pigment-producing cells known as melanocytes, often due to excessive exposure to ultraviolet radiation. We treat the data from the Liu et al. cohort (n = 120)57 as the training set and the Hugo et al. cohort (n = 26)58 as the validation set. Moreover, the Gide et al. cohort (n = 91)59, the Riaz et al. cohort (n = 54)60 and the Van et al. cohort (n = 41)61 are treated as the test sets. First, to reduce the number of candidate genes, we utilize a univariate Cox model to calculate the HR value of each gene and select the top 100 as the candidate genes. On the basis of these candidate genes, to discover as few genes as possible for constructing a survival analysis model, we select the top-5, -10, -15 and -20 significant genes learned by each model as the biomarker panel. Then, based on the selected biomarker panel by each method, we construct a new corresponding model for predictions. Moreover, in this section, we also compare with a Cox PH model, termed as univariate Cox PH, which directly trains a Cox PH model on the top-N genes ranked by the previous univariate Cox PH model. The detailed data preprocessing process and the pipeline are in Supplementary Information, section A.1.
Clinical survival data
The lung cancer clinical data62 have the patient characteristics and clinical outcomes for 382 patients. We utilize clinical information as covariates and follow-up data including OS and DFS as targets for two tasks. For these data, we aim to simulate the scenario that the training data were collected randomly and the test data have several patient subgroups with different characteristics or clinical values. We hope that the trained model can achieve stable performance on different subgroups. In particular, we randomly select 40% of the patients (n = 240) as the training set and the remaining patients are divided into the following subgroups according to their covariates, age ≤60 (n = 43) or age > 60 (n = 113), female (n = 4) or male (n = 152), tumour location classified as ‘central’ (n = 47) or ‘peripheral’ (n = 107), and obstructive pneumonitis/atelectasis is ‘present’ (n = 80) or ‘absent’ (n = 76). Female (n = 4) and male (n = 152) subgroups are used as the validation set and the remaining subgroups are used as testing subgroups. The breast cancer clinical dataset was collected from https://www.kaggle.com/datasets/gunesevitan/breast-cancer-metabric/data, which has the clinical and follow-up data for patients from several cohorts56. After filtering low-quality samples, we utilize 1 cohort as the training set (n = 394) and 1 cohort (n = 14) as the validation set, and the other 2 cohorts as the test sets, termed as test cohort 1 (n = 273) and test cohort 2 (n = 177). The survival outcomes of this dataset are OS and RFS. These datasets enable us to validate the effectiveness of our model across two generalization scenarios frequently encountered in real-world settings, namely, generalization on subpopulations and cohorts. The detailed data preprocessing process and the pipeline are in Supplementary Information, section A.1.
Baseline approaches
We compare our model with two mainstream kinds of methods in survival analysis: semi-parametric model (Cox PH model5) and parametric models (Weibull accelerated failure time (AFT) model, log-logistic AFT model and log-normal model63). For all the methods, we add the l2 norm to reduce the multicollinearity problem. Here we introduce three parametric survival compared in the experiments. In survival analysis, a parametric model assumes that the underlying survival times follow a known probability distribution, such as the Weibull, log-normal or log-logistic distributions. AFT models63 are the most frequently used parametric models. Unlike the Cox PH model, which models the hazard rate (the risk of the event occurring at a particular time), AFT models directly model the survival time itself. The general form of an AFT model is:
where ϵ is a random error term. Different distributions of ϵ imply different distributions of the survival time.
The Weibull AFT model
The Weibull AFT model adopts the Weibull distribution for ϵ, and it has the following cumulative hazard rate:
where ρ controls the shape of distribution, and \(\lambda (X)=\)\(\exp ({\beta }_{0}+{\beta }_{1}{X}_{1}+\cdots +{\beta }_{p}{X}_{p}),\) represents accelerating or decelerating hazards by covariates.
The log-logistic AFT model
A log-logistic AFT model assumes that the error follows the log-logistic distribution and it has the following cumulative hazard rate:
The log-normal AFT model
A log-normal AFT model assumes that the error follows the log-normal distribution and it has the following cumulative hazard rate:
The AFT model directly interprets the effects on survival time and is suitable when the exact survival time predictions are needed, but it relies on the correct specification of the underlying distribution, which can be a limitation. The Cox PH model is robust and widely used, not requiring the assumption of a specific survival time distribution and can handle time-varying covariates, but it assumes PH, and it is less straightforward for predicting absolute survival times.
Evaluation metrics
The metrics used closely reflect the accuracy of survival prediction and the difference of the subgroups identified. Three sets of evaluation metrics were used.
Concordance index
The C-index quantifies the ability of the survival model to correctly rank patient outcomes, which can be calculated as the ratio of all individual pairs whose predicted survival times are accurately ranked64:
where N is the total number of comparable pairs of individuals, t(i) and t(j) are the observed times of events for individuals i and j, respectively, and \(\hat{H}(i)\) and \(\hat{H}(j)\) are the corresponding predicted risk scores (hazards). The C-index score takes values between 0 and 1, and 0.5 means a random guess. A larger value indicates that the predicted survival ranking is closer to the ground-truth survival ranking.
The log-rank P value
We plot the Kaplan–Meier survival curves of the two risk groups and calculate the log-rank P value of the survival difference between them according to ref. 65. A log-rank P value lower than 0.05 means the survival curves of the two subgroups are significantly different.
Hazard ratio
The HR is the ratio of the hazard rates corresponding to the conditions characterized by two distinct levels of a treatment variable of interest. For Cox regression, the HR value of jth covariate can be directly calculated by:
HRi > 1 means that the covariate i indicates a higher hazard of death from the treatment. HRi < 1 suggests a reduced hazard, and HRi = 1 implies no difference in risk. To calculate the HR value for two risk subgroups, we first create a group variable g. In this variable, patients in the high-risk subgroup are assigned a value of 1, while all others are assigned a value of 0. Next, we use g as the univariate in a Cox PH model to regress the survival outcome. The HR value for the subgroup separation is then given by exp(βg), where βg is the coefficient of the group variable in the Cox PH model.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All benchmark datasets used in this paper are publicly available. For the processed RNA-sequencing data and corresponding survival outcome of HCC transcriptome dataset, the TCGA-LIHC cohort data were downloaded from https://portal.gdc.cancer.gov/projects/TCGA-LIHC. The Grinchuk et al. cohort data were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76427. The Fujimoto et al. cohort data were downloaded from https://docs.icgc-argo.org/docs/data-access/icgc-25k-data. The Roessler at al. cohort data were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse14520. The Hoshida et al. cohort data were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE10143. For the breast cancer dataset, the RNA-sequencing, survival outcome, cohort and clinical information can be downloaded from https://www.cbioportal.org/study/summary?id=brca_metabric. For the melanoma transcriptome dataset, the Liu et al. cohort data were download from https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000452.v3.p1. The Hugo et al. cohort data were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse78220. The Gide et al. cohort data were downloaded from https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJEB23709. The Riaz et al. cohort data were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE91061. The Van et al. cohort data were downloaded from https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000452.v2.p1. For the lung cancer clinical dataset, the clinical and survival outcome data can be downloaded from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6777828/. All data sources used in this paper are listed in Supplementary Table A1. The simulated data were generated when running the source code. The preprocessed real-world data are available on GitHub at https://github.com/googlebaba/StableCox and on Zenodo at https://doi.org/10.5281/zenodo.13852489 (ref. 66).
Code availability
The implementation code is available on GitHub at https://github.com/googlebaba/StableCox and on Zenodo at https://doi.org/10.5281/zenodo.13852489 (ref. 66).
References
Anderson, K. M. A nonproportional hazards Weibull accelerated failure time regression model. Biometrics 47, 281–288 (1991).
Friedman, M. Piecewise exponential models for survival data with covariates. Ann. Stat. 10, 101–113 (1982).
Ishwaran, H., Kogalur, U. B., Blackstone, E. H. & Lauer, M. S. Random survival forests. Ann. Appl. Stat. 2, 841–860 (2008).
Wang, P., Li, Y. & Reddy, C. K. Machine learning for survival analysis: a survey. ACM Comput. Surv. 51, 110 (2019).
Cox, D. R. Regression models and life-tables. J. R. Stat. Soc. Ser. B 34, 187–202 (1972).
Guo, L. L. et al. Evaluation of domain generalization and adaptation on improving model robustness to temporal dataset shift in clinical medicine. Sci. Rep. 12, 2726 (2022).
Chaudhary, K., Poirion, O. B., Lu, L. & Garmire, L. X. Deep learning–based multi-omics integration robustly predicts survival in liver cancer. Clin. Cancer Res. 24, 1248–1259 (2018).
Zhou, L. & Zhu, Y. The epcam overexpression is associated with clinicopathological significance and prognosis in hepatocellular carcinoma patients: a systematic review and meta-analysis. Int. J. Surg. 56, 274–280 (2018).
Liang, J. et al. Expression pattern of tumour-associated antigens in hepatocellular carcinoma: association with immune infiltration and disease progression. Br. J. Cancer 109, 1031–1039 (2013).
Xu, M. et al. Expression of epithelial cell adhesion molecule associated with elevated ductular reactions in hepatocellar carcinoma. Clin. Res. Hepatol. Gastroenterol. 38, 699–705 (2014).
Zhu, Y., Qiu, P. & Ji, Y. TCGA-assembler: open-source software for retrieving and processing TCGA data. Nat. Methods 11, 599–600 (2014).
Roessler, S. et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 70, 10202–10212 (2010).
Thorgeirsson, S. S., Lee, J.-S. & Grisham, J. W. Molecular prognostication of liver cancer: end of the beginning. J. Hepatol. 44, 798–805 (2006).
Jiang, G. et al. CD146 promotes metastasis and predicts poor prognosis of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 35, 38 (2016).
Jiang, Y. et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 567, 257–261 (2019).
Liu, F., Liu, Y. & Chen, Z. Tim-3 expression and its role in hepatocellular carcinoma. J. Hematol. Oncol. 11, 126 (2018).
Tishkoff, S. A. et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nat. Genet. 39, 31–40 (2007).
Curth, A. & Schaar, M. Understanding the impact of competing events on heterogeneous treatment effect estimation from time-to-event data. In International Conference on Artificial Intelligence and Statistics 7961–7980 (PMLR, 2023).
Curth, A., Lee, C. & Schaar, M. SurvITE: learning heterogeneous treatment effects from time-to-event data. Adv. Neural Inf. Process. Syst. 34, 26740–26753 (2021).
Goh, W. W. B., Wang, W. & Wong, L. Why batch effects matter in omics data, and how to avoid them. Trends Biotechnol. 35, 498–507 (2017).
Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 16, 385–395 (1997).
Verweij, P. J. & Van Houwelingen, H. C. Penalized likelihood in Cox regression. Stat. Med. 13, 2427–2436 (1994).
Simon, N., Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for Cox’s proportional hazards model via coordinate descent. J. Stat. Softw. 39, 1–13 (2011).
Fan, J. & Li, R. Variable selection for Cox’s proportional hazards model and frailty model. Ann. Stat. 30, 74–99 (2002).
Lin, D. Y. & Wei, L.-J. The robust inference for the Cox proportional hazards model. J. Am. Stat. Assoc. 84, 1074–1078 (1989).
Cui, P. & Athey, S. Stable learning establishes some common ground between causal inference and machine learning. Nat. Mach. Intell. 4, 110–115 (2022).
Xu, R., Zhang, X., Shen, Z., Zhang, T. & Cui, P. A theoretical analysis on independence-driven importance weighting for covariate-shift generalization. In International Conference on Machine Learning 24803–24829 (PMLR, 2022).
Kuang, K., Cui, P., Athey, S., Xiong, R. & Li, B. Stable prediction across unknown environments. In Proc. 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining 1617–1626 (ACM, 2018).
Kuang, K., Xiong, R., Cui, P., Athey, S. & Li, B. Stable prediction with model misspecification and agnostic distribution shift. In Proc. AAAI Conference on Artificial Intelligence Vol. 34, 4485–4492 (AAAI Press, 2020).
Shen, Z., Cui, P., Kuang, K., Li, B. & Chen, P. Causally regularized learning with agnostic data selection bias. In Proc. 26th ACM International Conference on Multimedia 411–419 (ACM, 2018).
Shen, Z., Cui, P., Zhang, T. & Kunag, K. Stable learning via sample reweighting. In Proc. AAAI Conference on Artificial Intelligence Vol. 34, 5692–5699 (AAAI Press, 2020).
Fan, S., Wang, X., Shi, C., Cui, P. & Wang, B. Generalizing graph neural networks on out-of-distribution graphs. IEEE Trans. Pattern Anal. Mach. Intell. 46, 322–337 (2024).
Hsu, J. L. & Hung, M.-C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 35, 575–588 (2016).
Sugiyama, M., Suzuki, T. & Kanamori, T. Density Ratio Estimation in Machine Learning (Cambridge Univ. Press, 2012).
Bender, R., Augustin, T. & Blettner, M. Generating survival times to simulate Cox proportional hazards models. Stat. Med. 24, 1713–1723 (2005).
Mertins, P. et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 (2016).
Fujimoto, A. et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 48, 500–509 (2016).
Hoshida, Y. et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N. Engl. J. Med. 359, 1995–2004 (2008).
Van't Veer, L. J. et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536 (2002).
Onitilo, A. A., Engel, J. M., Greenlee, R. T. & Mukesh, B. N. Breast cancer subtypes based on ER/PR and HER2 expression: comparison of clinicopathologic features and survival. Clin. Med. & Res. 7, 4–13 (2009).
Guyon, I. & Elisseeff, A. An introduction to variable and feature selection. J. Mach. Learn. Res. 3, 1157–1182 (2003).
Prosperi, M. et al. Causal inference and counterfactual prediction in machine learning for actionable healthcare. Nat. Mach. Intell. 2, 369–375 (2020).
Zhang, K., Schölkopf, B., Muandet, K. & Wang, Z. Domain adaptation under target and conditional shift. In International Conference on Machine Learning 819–827 (PMLR, 2013).
Zhao, H., Des Combes, R. T., Zhang, K. & Gordon, G. On learning invariant representations for domain adaptation. In International Conference on Machine Learning 7523–7532 (PMLR, 2019).
Ahuja, K., Shanmugam, K., Varshney, K. & Dhurandhar, A. Invariant risk minimization games. In International Conference on Machine Learning 145–155 (PMLR, 2020).
Hainmueller, J. Entropy balancing for causal effects: a multivariate reweighting method to produce balanced samples in observational studies. Polit. Anal. 20, 25–46 (2012).
Kalbfleisch, J. D. & Prentice, R. L. The Statistical Analysis of Failure Time Data (Wiley, 2011).
Breslow, N. E. Analysis of survival data under the proportional hazards model. Int. Stat. Rev. 43, 45–57 (1975).
Andersen, P. K. & Gill, R. D. Cox’s regression model for counting processes: a large sample study. Ann. Stat. 10, 1100–1120 (1982).
Gail, M. H., Wieand, S. & Piantadosi, S. Biased estimates of treatment effect in randomized experiments with nonlinear regressions and omitted covariates. Biometrika 71, 431–444 (1984).
Lagakos, S. The loss in efficiency from misspecifying covariates in proportional hazards regression models. Biometrika 75, 156–160 (1988).
Harrell Jr, F. E., Lee, K. L. & Mark, D. B. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat. Med. 15, 361–387 (1996).
Rifai, N., Gillette, M. A. & Carr, S. A. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol. 24, 971–983 (2006).
Lian, Q. et al. HCCDB: a database of hepatocellular carcinoma expression atlas. Genomics Proteomics Bioinformatics 16, 269–275 (2018).
Grinchuk, O. V. et al. Tumor-adjacent tissue co-expression profile analysis reveals pro-oncogenic ribosomal gene signature for prognosis of resectable hepatocellular carcinoma. Mol. Oncol. 12, 89–113 (2018).
Curtis, C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012).
Liu, D. et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 25, 1916–1927 (2019).
Hugo, W. et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44 (2016).
Gide, T. N. et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell 35, 238–255 (2019).
Riaz, N. et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171, 934–949 (2017).
Van Allen, E. M. et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015).
Gu, K. et al. Integrated evaluation of clinical, pathological and radiological prognostic factors in squamous cell carcinoma of the lung. PLoS ONE 14, 0223298 (2019).
Wei, L.-J. The accelerated failure time model: a useful alternative to the Cox regression model in survival analysis. Stat. Med. 11, 1871–1879 (1992).
Steck, H., Krishnapuram, B., Dehing-Oberije, C., Lambin, P. & Raykar, V. C. On ranking in survival analysis: bounds on the concordance index. Adv. Neural Inf. Process. Syst. 20, 1209–1216 (2007).
Bland, J. M. & Altman, D. G. The logrank test. BMJ 328, 1073 (2004).
Fan, S. et al. Stable Cox regression for survival analysis under distribution shifts. Zenodo https://doi.org/10.5281/zenodo.13852489 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (numbers 62425206 and 62141607 to P.C., 32088101 to C.C. and 62402263 to S.F.) and the National Key Research and Development Program of China (2021YFA1301603 to C.C.). S.F. was supported by China Postdoctoral Science Foundation (numbers 2023M741946, 2024T170494 and GZB20230345). Y.H. was supported by China National Postdoctoral Program for Innovative Talents (number BX20230195).
Author information
Authors and Affiliations
Contributions
S.F., C.C. and P.C. conceived of the project. S.F. designed the proposed model. R.X. performed the theoretical analysis and discussed with S.F. S.F. mainly performed experiments and analysed the results, assisted by Q.D. S.F., R.X., Q.D. and Y.H. wrote the paper. P.C. and C.C. revised the paper and supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Deepti Gurdasani and Jiguang Wang for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. A1 and C2–C9, Discussion, omitted proofs and Tables A1–A4.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fan, S., Xu, R., Dong, Q. et al. Stable Cox regression for survival analysis under distribution shifts. Nat Mach Intell 6, 1525–1541 (2024). https://doi.org/10.1038/s42256-024-00932-5
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s42256-024-00932-5
