
Abstract
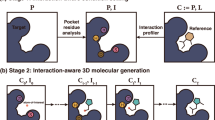
Optimizing the chemical structure of promising drug candidates through systematic modifications to improve potency and physiochemical properties is a vital step in the drug discovery pipeline. In contrast to the well-established de novo generation schemes, computational methods specifically tailored for lead optimization remain largely underexplored. Prior models are often limited to addressing specific subtasks, such as generating two-dimensional molecular structures, while neglecting crucial protein–ligand interactions in three-dimensional space. To overcome these challenges, we propose Delete (Deep lead optimization enveloped in protein pocket), a one-stop solution for lead optimization by combining generative artificial intelligence and structure-based approaches. Our model can handle all subtasks of lead optimization through a unified deleting (masking) strategy, and it accounts for intricate pocket–ligand interactions through an equivariant network design. Statistical assessments and retrospective studies across individual subtasks demonstrate that Delete has an outstanding ability to craft molecules with superior protein-binding energy and reasonable drug-likeness using given fragments or atoms. Subsequently, we utilize Delete to design inhibitors targeting the previously identified LTK protein. Among the ligands designed by Delete, CA-B-1 is successfully validated as a potent (1.36 nM) and selective inhibitor by in vitro and in vivo experiments. This work represents a successful implementation of the powerful structure-based lead optimization model, Delete, for rapid and controllable rational drug design.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
The datasets used in this study are available via Zenodo at https://doi.org/10.5281/zenodo.14586176 (ref. 64). Source data are provided with this paper.
Code availability
The source code of this study is freely available at GitHub (https://github.com/HaotianZhangAI4Science/Delete) to allow replication of the results.
References
Grabley, S. & Thiericke, R. Drug Discovery from Nature (Springer Science & Business Media, 1998).
Wong, F. et al. Discovery of a structural class of antibiotics with explainable deep learning. Nature 626, 177–185 (2024).
Liu, G. et al. Deep learning-guided discovery of an antibiotic targeting Acinetobacter baumannii. Nat. Chem. Biol. 19, 1342–1350 (2023).
Stokes, J. M. et al. A deep learning approach to antibiotic discovery. Cell 180, 688–702.e613 (2020).
Godinez, W. J. et al. Design of potent antimalarials with generative chemistry. Nat. Mach. Intell. 4, 180–186 (2022).
Zhavoronkov, A. et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 37, 1038–1040 (2019).
Li, Y. et al. Generative deep learning enables the discovery of a potent and selective RIPK1 inhibitor. Nat.Com. 13, 6891 (2022).
Aronson, J. K. & Green, A. R. Me-too pharmaceutical products: History, definitions, examples, and relevance to drug shortages and essential medicines lists. Br. J. Clin. Pharmacol. 86, 2114–2122 (2020).
Zhang, O. et al. ResGen is a pocket-aware 3D molecular generation model based on parallel multiscale modelling. Nat. Mach. Intell. 5, 1020–1030 (2023).
Lin, H. et al. DiffBP: generative diffusion of 3D molecules for target protein binding. Chem. Sci. 16, 1417–1431 (2025).
Schneuing, A. et al. Structure-based drug design with equivariant diffusion models. Nat. Comput. Sci. 4, 899–909 (2024).
Peng, X. et al. Pocket2mol: efficient molecular sampling based on 3d protein pockets. In Proc. 39th International Conference on Machine Learning 17644–17655 (PMLR, 2022).
Davidson, M. et al. Comparison of one-year efficacy and safety of atorvastatin versus lovastatin in primary hypercholesterolemia. Am. J. Cardiol. 79, 1475–1481 (1997).
Becnel Boyd, L. et al. Relationships among ciprofloxacin, gatifloxacin, levofloxacin, and norfloxacin MICs for fluoroquinolone-resistant escherichia coli clinical isolates. Antimicrob. Agents Chemother. 53, 229–234 (2009).
Bethke, E. et al. From type I to type II: design, synthesis, and characterization of potent pyrazin-2-ones as DFG-out inhibitors of PDGFRβ. ChemMedChem 11, 2664–2674 (2016).
Green, H., Koes, D. R. & Durrant, J. D. DeepFrag: a deep convolutional neural network for fragment-based lead optimization. Chem. Sci. 12, 8036–8047 (2021).
Igashov, I. et al. Equivariant 3D-conditional diffusion model for molecular linker design. Nat. Mach. Intell. 6, 417–427 (2024).
Jin, J. et al. FFLOM: a flow-based autoregressive model for fragment-to-lead optimization. J. Med. Chem. 66, 10808–10823 (2023).
Hu, C. et al. ScaffoldGVAE: scaffold generation and hopping of drug molecules via a variational autoencoder based on multi-view graph neural networks. J. Cheminform. 15, 91 (2023).
Loeffler, H. H. et al. Reinvent 4: modern AI–driven generative molecule design. J. Cheminform. 16, 20 (2024).
Liu, X., Ye, K., van Vlijmen, H. W. T., Ijzerman, A. P. & van Westen, G. J. P. DrugEx v3: scaffold-constrained drug design with graph transformer-based reinforcement learning. J. Cheminf. 15, 24 (2023).
Neves, M. A. C., Totrov, M. & Abagyan, R. Docking and scoring with ICM: the benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 26, 675–686 (2012).
Adeshina, Y. O., Deeds, E. J. & Karanicolas, J. Machine learning classification can reduce false positives in structure-based virtual screening. Proc. Natl Acad. Sci. USA 117, 18477–18488 (2020).
Deng, C. et al. Vector neurons: a general framework for SO(3)-equivariant networks. In Proc. IEEE/CVF International Conference on Computer Vision 12200–12209 (IEEE, 2021).
Jing, B., Eismann, S., Suriana, P., Townshend, R. J. & Dror, R. Learning from protein structure with geometric vector perceptrons. Preprint at https://arxiv.org/abs/2009.01411 (2020).
Mahmood, O., Mansimov, E., Bonneau, R. & Cho, K. Masked graph modeling for molecule generation. Nat. Commun. 12, 3156 (2021).
Hu, Z., Dong, Y., Wang, K., Chang, K.-W. & Sun Y. Gpt-gnn: generative pre-training of graph neural networks. In Proc. 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining 1857–1867 (ACM, 2020).
Izumi, H. et al. The CLIP1–LTK fusion is an oncogenic driver in non‐small‐cell lung cancer. Nature 600, 319–323 (2021).
Ferla, M. P. et al. Fragmenstein: predicting protein–ligand structures of compounds derived from known crystallographic fragment hits using a strict conserved-binding–based methodology. J. Cheminform. 17, 4 (2025).
Imrie, F., Bradley, A. R., van der Schaar, M. & Deane, C. M. Deep generative models for 3D linker design. J. Chem. Inf. Model. 60, 1983–1995 (2020).
Langevin, M., Minoux, H., Levesque, M. & Bianciotto, M. Scaffold-constrained molecular generation. J. Chem. Inf. Model. 60, 5637–5646 (2020).
Zhang, O. et al. Learning on topological surface and geometric structure for 3D molecular generation. Nat. Comput. Sci. 3, 849–859 (2023).
Clark, D. E. & Pickett, S. D. Computational methods for the prediction of ‘drug-likeness’. Drug Discov. Today 5, 49–58 (2000).
Ertl, P. & Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 1, 8 (2009).
Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 12, 306–317 (2008).
Sangster, J. Octanol‐water partition coefficients of simple organic compounds. J. Phy. Chem. Ref. Data 18, 1111–1229 (1989).
Gaulton, A. et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–D1107 (2012).
Irwin, J. J. & Shoichet, B. K. ZINC - a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 45, 177–182 (2005).
Francoeur, P. G. et al. Three-dimensional convolutional neural networks and a cross-docked data set for structure-based drug design. J. Chem. Inf. Model. 60, 4200–4215 (2020).
Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010).
Alexandar, S. P., Yennamalli, R. M. & Ulaganathan, V. Coarse grained modelling highlights the binding differences in the two different allosteric sites of the human kinesin EG5 and its implications in inhibitor design. Comput. Biol. Chem. 99, 107708 (2022).
Zheng, S. et al. Deep scaffold hopping with multimodal transformer neural networks. J. Cheminform. 13, 87 (2021).
Cooper, A. J., Sequist, L. V. & Lin, J. J. Third-generation EGFR and ALK inhibitors: mechanisms of resistance and management. Nat. Rev. Clin. Oncol. 19, 499–514 (2022).
Roll, J. D. & Reuther, G. W. ALK-activating homologous mutations in LTK induce cellular transformation. PLoS ONE 7, e31733 (2012).
Roskoski, R. ROS1 protein-tyrosine kinase inhibitors in the treatment of ROS1 fusion protein-driven non-small cell lung cancers. Pharmacol. Res. 121, 202–212 (2017).
Luo, S., Guan, J., Ma, J. & Peng, J. A 3D generative model for structure-based drug design. Preprint at https://arxiv.org/abs/2203.10446 (2022).
Ragoza, M., Masuda, T. & Koes, D. R. Generating 3D molecules conditional on receptor binding sites with deep generative models. Chem. Sci. 13, 2701–2713 (2022).
Hussain, J. & Rea, C. Computationally efficient algorithm to identify matched molecular pairs (MMPs) in large data sets. J. Chem. Inf. Model. 50, 339–348 (2010).
Bemis, G. W. & Murcko, M. A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 39, 2887–2893 (1996).
Degen, J., Wegscheid-Gerlach, C., Zaliani, A. & Rarey, M. On the art of compiling and using ‘drug-like’ chemical fragment spaces. ChemMedChem 3, 1503–1507 (2008).
Varin, T., Schuffenhauer, A., Ertl, P. & Renner, S. Mining for bioactive scaffolds with scaffold networks: improved compound set enrichment from primary screening data. J. Chem. Inf. Model. 51, 1528–1538 (2011).
Gainza, P. et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 17, 184–192 (2020).
Liu, T., Lin, Y., Wen, X., Jorissen, R. N. & Gilson, M. K. BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 35, D198–D201 (2007).
Svetnik, V. et al. Boosting: an ensemble learning tool for compound classification and QSAR modeling. J. Chem. Inf. Model. 45, 786–799 (2005).
Rogers, D. & Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754 (2010).
Ho, T. K. Random decision forests. In Proc. 3rd International Conference on Document Analysis and Recognition 278–282 (IEEE, 1995).
Chen, T. Xgboost: extreme gradient boosting. R package version 0.4-2 (2015).
Dorogush, A. V., Ershov, V. & Gulin, A. CatBoost: gradient boosting with categorical features support. Preprint at https://arxiv.org/abs/1810.11363 (2018).
Ke, G. et al. Lightgbm: a highly efficient gradient boosting decision tree. In Proc. Advances in Neural Information Processing Systems 3149–3157 (ACM, 2017).
Guo, G., Wang, H., Bell, D., Bi, Y. & Greer, K. KNN model-based approach in classification. In Proc. On The Move to Meaningful Internet Systems 2003: CoopIS, DOA, and ODBASE (eds Meersman, R. et al.) 986–996 (Springer, 2003).
Devillers, J. Neural Networks in QSAR and Drug Design (Academic, 1996).
Mordelet, F. & Vert, J. P. A bagging SVM to learn from positive and unlabeled examples. Pattern Recog. Lett. 37, 201–209 (2014).
Hu, Y. et al. Silicon photonic MEMS switches based on split waveguide crossings. Nat.Commun. 16, 331 (2025).
Zhang, H. Delete code. Zenodo https://doi.org/10.5281/zenodo.14586175 (2025).
Acknowledgements
This study was supported by the National Key Research and Development Program of China (grant no. 2021YFE0206400) (to T.H.), the National Natural Science Foundation of China (grant nos. 92370130 (to T.H.), 22220102001 (to T.H.), 82204279 (to P.P.) and 82373791 (to Y.K.)), the Fundamental Research Funds for the Central Universities (grant no. 226-2022-00220) (to P.P.) and the Shenzhen Science and Technology Innovation Commission (grant no. KCXFZ20201221173404013) (to J.C.). This work was financially supported by Baidu Scholarship. We thank C. Guo from the Core Facilities, Zhejiang University School of Medicine for their technical support.
Author information
Authors and Affiliations
Contributions
S.C. designed and performed the in vitro and in vivo experiments. O.Z. contributed to the Delete idea and code. H.Z. contributed to the retrospective studies. C.J., M.C., Y.L., Y.A., G.W., Q.Y., J.C. and Y.H. contributed to the synthesis of molecules. X.Z. contributed to the comparative model reproduction. Z.W. contributed to the ADMET analysis of generated molecules. Q.S., X.W., W.Q., Y.Y., X.C., N.W., T.W., H.L. and D.L. provided advice on experimental design. W.X., C.-Y.H. and Y.K. contributed to the manuscript revision and experimental design. T.H. and P.P. contributed to the essential financial support and conception and were responsible for the overall quality.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Ramil Nugmanov and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Discussion, Figs. 1–11, Tables 1–3 and Methods.
Source data
Source Data Figs. 4 and 5 (download XLSX )
Souce data, unprocessed western blots, statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, S., Zhang, O., Jiang, C. et al. Deep lead optimization enveloped in protein pocket and its application in designing potent and selective ligands targeting LTK protein. Nat Mach Intell 7, 448–458 (2025). https://doi.org/10.1038/s42256-025-00997-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s42256-025-00997-w
This article is cited by
-
ECloudGen: leveraging electron clouds as a latent variable to scale up structure-based molecular design
Nature Computational Science (2025)
-
Bridging chemical space and biological efficacy: advances and challenges in applying generative models in structural modification of natural products
Natural Products and Bioprospecting (2025)
