
Abstract
Despite the widespread applications of machine learning force fields (MLFFs) in solids and small molecules, there is a notable gap in applying MLFFs to simulate liquid electrolytes—a critical component of current commercial lithium-ion batteries. Here we introduce ByteDance Artificial intelligence Molecular simulation Booster (BAMBOO), a predictive framework for molecular dynamics simulations, with a demonstration of its capability in the context of liquid electrolytes for lithium batteries. We design a physics-inspired graph equivariant transformer architecture as the backbone of BAMBOO to learn from quantum mechanical simulations. Additionally, we introduce an ensemble knowledge distillation approach and apply it to MLFFs to reduce the fluctuation of observations from molecular dynamics simulations. Finally, we propose a density alignment algorithm to align BAMBOO with experimental measurements. BAMBOO demonstrates state-of-the-art accuracy in predicting key electrolyte properties such as density, viscosity and ionic conductivity across various solvents and salt combinations. The current model, trained on more than 15 chemical species, achieves an average density error of 0.01 g cm−3 on various compositions compared with experiment.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others
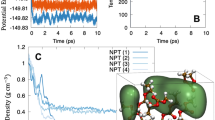

Data availability
DFT datasets of clusters are available at https://huggingface.co/datasets/mzl/bamboo (ref. 72). The input parameters, template of atomic structures for LAMMPS MD simulations and the final trained, ensemble knowledge distilled and density-aligned model of BAMBOO to reproduce the results in the paper are available via Zenodo at https://doi.org/10.5281/zenodo.14603020 (ref. 73).
Code availability
The source codes, including the GET model, the training module and the LAMMPS interface for the MD simulations, are available via GitHub at https://github.com/bytedance/bamboo and via Zenodo at https://zenodo.org/records/14603020 (ref. 73).
References
Xu, K. Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 104, 4303–4418 (2004).
Xu, K. Electrolytes, Interfaces and Interphases: Fundamentals and Applications in Batteries (Royal Society of Chemistry, 2023).
Meng, Y. S., Srinivasan, V. & Xu, K. Designing better electrolytes. Science 378, eabq3750 (2022).
Unke, O. T. et al. Machine learning force fields. Chem. Rev. 121, 10142–10186 (2021).
Frenkel, D. & Smit, B. Understanding Molecular Simulation: From Algorithms To Applications (Elsevier, 2023).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Zhang, L., Han, J., Wang, H., Car, R. & E, W. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett. 120, 143001 (2018).
Lysogorskiy, Y. et al. Performant implementation of the atomic cluster expansion (PACE) and application to copper and silicon. npj Comput. Mater. 7, 97 (2021).
Schütt, K. T., Sauceda, H. E., Kindermans, P.-J., Tkatchenko, A. & Müller, K.-R. SchNet—a deep learning architecture for molecules and materials. J. Chem. Phys. 148, 241722 (2018).
Batzner, S. et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13, 2453 (2022).
Liao, Y.-L. & Smidt, T. Equiformer: equivariant graph attention transformer for 3D atomistic graphs. In Proc. Eleventh International Conference on Learning Representations https://openreview.net/forum?id=KwmPfARgOTD (ICLR, 2023).
Thölke, P. & De Fabritiis, G. TorchMD-NET: equivariant transformers for neural network based molecular potentials. In Proc. Tenth International Conference on Learning Representations https://openreview.net/forum?id=zNHzqZ9wrRB (ICLR, 2022).
Ko, T. W., Finkler, J. A., Goedecker, S. & Behler, J. A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer. Nat. Commun. 12, 398 (2021).
Zhang, L. et al. A deep potential model with long-range electrostatic interactions. J. Chem. Phys. 156, 124107 (2022).
Anstine, D., Zubatyuk, R. & Isayev, O. AIMNet2: a neural network potential to meet your neutral, charged, organic, and elemental-organic needs. Preprint at ChemRxiv https://doi.org/10.26434/chemrxiv-2023-296ch-v3 (2023).
Unke, O. & Meuwly, M. PhysNet: a neural network for predicting energies, forces, dipole moments and partial charges. J. Chem. Theory Comput. 15, 3678–3693 (2019).
Unke, O. et al. SpookyNet: learning force fields with electronic degrees of freedom and nonlocal effects. Nat. Commun. 12, 12 (2021).
Yu, H. et al. Spin-dependent graph neural network potential for magnetic materials. Phys. Rev. B 109, 144426 (2023).
Chen, C. & Ong, S. A universal graph deep learning interatomic potential for the periodic table. Nat. Comput. Sci. 2, 718–728 (2022).
Deng, B. et al. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat. Mach. Intell. 5, 1031–1041 (2023).
Merchant, A. et al. Scaling deep learning for materials discovery. Nature 624, 80–85 (2023).
Zhang, D. et al. DPA-2: towards a universal large atomic model for molecular and material simulation. npj Comput. Mater. https://doi.org/10.1038/s41524-024-01493-2 (2024).
Batatia, I. et al. A foundation model for atomistic materials chemistry. Preprint at https://arxiv.org/abs/2401.00096 (2024).
Kovács, D. P. et al. MACE-OFF23: transferable machine learning force fields for organic molecules. Preprint at https://arxiv.org/abs/2312.15211v1 (2023).
Wang, H. & Yang, W. Force field for water based on neural network. J. Phys. Chem. Lett. 9, 3232–3240 (2018).
Zhang, J., Pagotto, J., Gould, T. & Duignan, T. T. Accurate, fast and generalisable first principles simulation of aqueous lithium chloride. Preprint at https://arxiv.org/abs/2310.12535v1 (2023).
Magdău, I. B. et al. Machine learning force fields for molecular liquids: ethylene carbonate/ethyl methyl carbonate binary solvent. npj Comput. Mater. 9, 146 (2023).
Montes-Campos, H., Carrete, J., Bichelmaier, S., Varela, L. M. & Madsen, G. K. H. A differentiable neural-network force field for ionic liquids. J. Chem. Inf. Model 62, 88–101 (2022).
Wang, F. & Cheng, J. Understanding the solvation structures of glyme-based electrolytes by machine learning molecular dynamics. Chinese J. Struct. Chem. 42, 100061 (2023).
Dajnowicz, S. et al. High-dimensional neural network potential for liquid electrolyte simulations. J. Phys. Chem. B 126, 08 (2022).
Jacobson, L. et al. Transferable neural network potential energy surfaces for closed-shell organic molecules: extension to ions. J. Chem. Theory Comput. 18, 03 (2022).
Fu, X. et al. Forces are not enough: benchmark and critical evaluation for machine learning force fields with molecular simulations. Trans. Mach. Learn. Res. https://openreview.net/pdf?id=A8pqQipwkt (2023).
Ormeño, F. & General, I. Convergence and equilibrium in molecular dynamics simulations. Commun. Chem. 7, 02 (2024).
Wang, X. et al. DMFF: an open-source automatic differentiable platform for molecular force field development and molecular dynamics simulation. J. Chem. Theory Comput. 19, 5897–5909 (2023).
Greener, J. G. & Jones, D. T. Differentiable molecular simulation can learn all the parameters in a coarse-grained force field for proteins. PLoS ONE 16, e0256990 (2021).
Asif, U., Tang, J. & Harrer, S. Ensemble knowledge distillation for learning improved and efficient network. In 24th European Conference on Artificial Intelligence http://ecai2020.eu/papers/521_paper.pdf (ECAI, 2020).
Chandler, D., Weeks, J. D. & Andersen, H. C. Van der Waals picture of liquids, solids, and phase transformations. Science 220, 787–794 (1983).
Kontogeorgis, G. M., Maribo-Mogensen, B. & Thomsen, K. The Debye-Hückel theory and its importance in modeling electrolyte solutions. Fluid Ph. Equilib. 462, 130–152 (2018).
Poier, P., Lagardère, L., Piquemal, J.-P. & Jensen, F. Molecular dynamics using nonvariational polarizable force fields: theory, periodic boundary conditions implementation, and application to the bond capacity model. J. Chem. Theory Comput. 2019, 09 (2019).
Schröder, H., Creon, A. & Schwabe, T. Reformulation of the D3(Becke–Johnson) dispersion correction without resorting to higher than C6 dispersion coefficients. J. Chem. Theory Comput. 11, 3163–3170 (2015).
Torres-Sánchez, A., Vanegas, J. M. & Arroyo, M. Geometric derivation of the microscopic stress: a covariant central force decomposition. J. Mech. Phys. Solids 93, 224–239 (2016).
Vaswani, A. et al. Attention is all you need. In Advances in Neural Information Processing Systems Vol. 30 (eds Guyon, I. et al.) https://proceedings.neurips.cc/paper_files/paper/2017/file/3f5ee243547dee91fbd053c1c4a845aa-Paper.pdf (Curran, 2017).
Gong, S. et al. Examining graph neural networks for crystal structures: limitations and opportunities for capturing periodicity. Sci. Adv. 9, eadi3245 (2023).
Thompson, A. P. et al. LAMMPS—a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comp. Phys. Comm. 271, 108171 (2022).
Musaelian, A. et al. Learning local equivariant representations for large-scale atomistic dynamics. Nat. Commun. 14, 579 (2023).
Wang, Y. et al. Enhancing geometric representations for molecules with equivariant vector-scalar interactive message passing. Nat. Commun. https://doi.org/10.1038/s41467-023-43720-2 (2024).
Batatia, I., Kovacs, D. P., Simm, G., Ortner, C. & Csányi, G. MACE: higher order equivariant message passing neural networks for fast and accurate force fields. Adv. Neural Inf. Process. Syst. 35, 11423–11436 (2022).
Pelaez, R. P. et al. TorchMD-Net 2.0: fast neural network potentials for molecular simulations. Preprint at https://arxiv.org/abs/2402.17660 (2024).
Martius, G. & Lampert, C. H. Extrapolation and learning equations. In Proc. 35th International Conference on Machine Learning https://proceedings.mlr.press/v80/sahoo18a.html (PMLR, 2018).
Dave, A. R. Automated Design and Discovery of Liquid Electrolytes for Lithium-Ion Batteries. PhD thesis, Carnegie Mellon Univ. (2023).
Hagiyama, K. et al. Physical properties of substituted 1,3-dioxolan-2-ones. Chem. Lett. 37, 210–211 (2008).
Sasaki, Y. in Fluorinated Materials for Energy Conversion (eds Nakajima, T. & Groult, H.) 285–304 (Elsevier Science, 2005).
Jänes, A., Thomberg, T., Eskusson, J. & Lust, E. Fluoroethylene carbonate and propylene carbonate mixtures based electrolytes for supercapacitors. ECS Trans. 58, 71 (2014).
Gores, H. J. et al. in Handbook of Battery Materials 525–626 (John Wiley & Sons, 2011).
Dave, A. et al. Autonomous optimization of non-aqueous Li-ion battery electrolytes via robotic experimentation and machine learning coupling. Nat. Commun. 13, 5454 (2022).
Logan, E. R. et al. A study of the transport properties of ethylene carbonate-free Li electrolytes. J. Electrochem. Soc. 165, A705–A716 (2018).
Zhu, S. et al. Differentiable modeling and optimization of non-aqueous Li-based battery electrolyte solutions using geometric deep learning. Nat. Commun. 15, 8649 (2024).
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996).
Zheng, T. et al. Data-driven parametrization of molecular mechanics force fields for expansive chemical space coverage. Chem. Sci. 16, 2730–2740 (2025).
Niblett, S. P., Kourtis, P., Magdău, I. B., Grey, C. P. & Csányi, G. Transferability of datasets between machine-learning interaction potentials. Preprint at https://arxiv.org/abs/2409.05590 (2024).
Zhang, H., Juraskova, V. & Duarte, F. Modelling chemical processes in explicit solvents with machine learning potentials. Nat. Commun. 15, 6114 (2024).
Lei Ba, J., Kiros, J. R. & Hinton, G. E. Layer normalization. Preprint at https://arxiv.org/abs/1607.06450 (2016).
Breneman, C. M. & Wiberg, K. B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 11, 361–373 (1990).
Paszke, A. et al. PyTorch: an imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. 32 (2019).
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. Preprint at https://arxiv.org/abs/1412.6980 (2017).
Becke, A. D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Hellweg, A. & Rappoport, D. Development of new auxiliary basis functions of the Karlsruhe segmented contracted basis sets including diffuse basis functions (def2-SVPD, def2-TZVPPD, and def2-QVPPD) for RI-MP2 and RI-CC calculations. Phys. Chem. Chem. Phys. 17, 1010–1017 (2015).
Weigend, F. Hartree–Fock exchange fitting basis sets for H to Rn. J. Comput. Chem. 29, 167–175 (2008).
Wu, X. et al. Python-based quantum chemistry calculations with GPU acceleration. Preprint at https://arxiv.org/abs/2404.09452v1 (2024).
Shao, Y. et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 113, 184–215 (2015).
Mistry, A., Yu, Z., Cheng, L. & Srinivasan, V. On relative importance of vehicular and structural motions in defining electrolyte transport. J. Electrochem. Soc. 170, 110536 (2023).
Mu, Z. mzl/bamboo. Hugging Face https://doi.org/10.57967/hf/3971 (2025).
Mu, Z. muzhenliang/bamboo: v0.1. Zenodo https://doi.org/10.5281/zenodo.14603020 (2025).
Simeon, G. & De Fabritiis, G. TensorNet: Cartesian tensor representations for efficient learning of molecular potentials. Adv. Neural Inf. Process. Syst. 36, 37334–37353 (2024).
Acknowledgements
We acknowledge insightful discussion on ion transport theory with A. Mistry, an Assistant Professor at the Colorado School of Mines. We also acknowledge the experimental data points provided by A. Dave, a former PhD student at Carnegie Mellon University, upon request. H.W. and M.C. worked as interns at ByteDance Research during this study.
Author information
Authors and Affiliations
Contributions
Conceptualization: Y.Z., W.Y., W.G., Z.M., Z. Yu, S.G., T.Z., X.H., Z. Yang, Z.W., L.C., X.W., S.S. and L.X. Methodology: S.G., Y.Z., Z.M., Z.P., H.W., M.C., W.Y. and W.G. Investigation: S.G., Y.Z., Z.M., Z.P., H.W., M.C., X.H., Z. Yu, W.Y. and W.G. Supervision: W.G., W.Y. and L.X. Writing: S.G., Y.Z., Z.M., Z.P., H.W., W.Y. and W.G.
Corresponding authors
Ethics declarations
Competing interests
All authors were employees of ByteDance when conducting this research project. ByteDance holds intellectual property rights pertinent to the research presented here. Furthermore, the innovations described here have resulted in the filing of a patent application in China (application no. 202311322469.2), which is currently pending.
Peer review
Peer review information
Nature Machine Intelligence thanks Jiayu Peng and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Notes 1–11, Figs. 1–16, Tables 1–16 and Equations (1)–(68).
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gong, S., Zhang, Y., Mu, Z. et al. A predictive machine learning force-field framework for liquid electrolyte development. Nat Mach Intell 7, 543–552 (2025). https://doi.org/10.1038/s42256-025-01009-7
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s42256-025-01009-7
This article is cited by
-
A dynamic routing-guided interpretable framework for salt–solvent chemistry
Nature Computational Science (2026)
-
Foundation models for atomistic simulation of chemistry and materials
Nature Reviews Chemistry (2026)
-
A unified predictive and generative solution for liquid electrolyte formulation
Nature Machine Intelligence (2026)
-
Machine learning of charges and long-range interactions from energies and forces
Nature Communications (2025)
-
Machine-learning-accelerated mechanistic exploration of interface modification in lithium metal anode
npj Computational Materials (2025)
