
Abstract
Language models are thriving, powering conversational agents that assist and empower humans to solve a number of tasks. Recently, these models were extended to support additional modalities including vision, audio and video, demonstrating impressive capabilities across multiple domains, including healthcare. Still, conversational agents remain limited in biology as they cannot yet fully comprehend biological sequences. Meanwhile, high-performance foundation models for biological sequences have been built through self-supervision over sequencing data, but these need to be fine-tuned for each specific application, preventing generalization between tasks. In addition, these models are not conversational, which limits their utility to users with coding capabilities. Here we propose to bridge the gap between biology foundation models and conversational agents by introducing ChatNT, a multimodal conversational agent with an advanced understanding of biological sequences. ChatNT achieves new state-of-the-art results on the Nucleotide Transformer benchmark while being able to solve all tasks at once, in English, and to generalize to unseen questions. In addition, we have curated a set of more biologically relevant instruction tasks from DNA, RNA and proteins, spanning multiple species, tissues and biological processes. ChatNT reaches performance on par with state-of-the-art specialized methods on those tasks. We also present a perplexity-based technique to help calibrate the confidence of our model predictions. By applying attribution methods through the English decoder and DNA encoder, we demonstrate that ChatNT’s answers are based on biologically coherent features such as detecting the promoter TATA motif or splice site dinucleotides. Our framework for genomics instruction tuning can be extended to more tasks and data modalities (for example, structure and imaging), making it a widely applicable tool for biology. ChatNT provides a potential direction for building generally capable agents that understand biology from first principles while being accessible to users with no coding background.
Similar content being viewed by others

Main
Understanding how cells, tissues and organisms interpret information encoded in the genome is of paramount importance for advancing our comprehension of biology. The DNA sequence of an organism comprises all the instructions to specify RNAs and proteins, but also when and in which cellular context these should be produced. Since the human genome was sequenced1, the main focus has been on identifying every genomic element, characterizing their function and assessing the impact of genetic variants on the different gene regulatory and cellular processes. Given the complexity of biological sequences and processes, and the increasing volume of genomics data, several machine learning and deep learning methods have been developed to address these questions by predicting diverse molecular phenotypes with great accuracy2,3. These tasks include predicting the binding of proteins to DNA and RNA4, DNA methylation5, chromatin features6, regulatory elements7, splicing8, gene expression9, mRNA properties such as stability10 and polyadenylation11, and protein properties such as melting point12.
Although supervised deep learning models have already significantly improved the predictive capabilities on these tasks, their performance remains often limited due to the scarcity of labelled data, given that labelling is time consuming and expensive. However, an exponentially increasing volume of raw genome data is becoming available owing to the increase in throughput and reduced cost of modern sequencing techniques, thus creating a significant opportunity for self-supervised deep learning methods to train on such unlabelled data. Through learning techniques such as masked- or next-token prediction13,14, with tokens representing one or several consecutive nucleotides, deep learning models can build powerful foundation representations of the genome during this pretraining stage, aggregating correlations between nucleotides and larger sequence patterns into rich high-dimensional vectors that capture known genomic elements and protein binding sites15. These models can later exploit these rich representations, during a fine-tuning stage, to learn faster and reach better performance on supervised tasks, that is, tasks where labels are available, despite data scarcity. Recently, several such foundation models have been built in this fashion, showing that they can be pretrained on the genomes of hundreds of species before being fine-tuned to solve a large collection of molecular phenotype prediction tasks15,16,17,18.
This being said, the performance and application domain of current DNA foundation models remains limited. In the current paradigm, foundation models require fine-tuning to each specific task individually to produce accurate representations and predictions, and are thus better characterized as narrow experts on specific tasks. This not only yields a deluge of different models as the number of tasks increases but also prevents any transfer between supervised tasks as well as to solve new tasks in a zero-shot setting (that is, without the need for further fine-tuning on some examples). Therefore, there is a need to rethink the development of genomics artificial intelligence (AI) systems with the goal of establishing general, unified models that capture the intricate relationships between all diverse biological sequences and functions. It has been shown in other fields such as natural language processing (NLP) and computer vision that training on several tasks in parallel results in knowledge transfer between tasks and improved accuracy and generalization13,19,20,21. In these domains, English language has been shown to play a wider role: a universal interface for representing various tasks and instructions and helping to guide the training of end-to-end multitask models22,23. Transferring this type of approach to biological data is a promising approach towards developing a general model that can solve all genomics tasks of interest simultaneously and with improved accuracy.
An additional important aspect of building a universal genomics AI system is its accessibility to different types of user. Most biologists do not know how to use current genomics models, let alone how to program one themselves for a given task of interest. Such models are not conversational and are, thus, of limited utility in practice to users with no coding capabilities. Here, language can play an important role as a universal interface for a general-purpose AI assistant that can solve genomics tasks through task instructions that can be explicitly represented in English language. For example, the recent success of ChatGPT24 and GPT-425 has demonstrated the power of large language models (LLMs) trained to follow human instructions, and how such tools can transform several industries owing to their ease of use. We envision the same paradigm shift for genomics and biology once we have ChatGPT-like agents that are proficient in biological tasks.
To that end, we introduce in this work an approach to build foundation models for genomics. Similarly to lines of works that emerged in NLP14,22, and inspired by recent vision and language multimodal models26,27,28,29, we propose to formulate all supervised genomics prediction tasks as text-to-text tasks and to build a multimodal DNA and language agent, dubbed the Chat Nucleotide Transformer (or ChatNT). ChatNT can be given one or several DNA sequences and is prompted in English to solve all those tasks. This formulation allows us to express all tasks with the same vocabulary, being here the concatenation of the English and DNA vocabularies, and to learn to solve them by minimizing a unified objective, similar to GPT-like models14,30, allowing seamless new task integration and generalization. Formulating tasks in English is also an easy way to provide additional metadata information to the model, such as the species, the chromosome or the cell type, that is also missing in most current DNA foundation models.
ChatNT is built to act as a generalist genomics AI system—a unified model that can interpret multiple biological sequences and handle dozens of tasks in a conversational agent setting. We created datasets of genomics instructions tasks with curated sets of questions and instructions in English for diverse classification and regression tasks. We first show that ChatNT achieves a new state-of-the-art on the Nucleotide Transformer benchmark15. We next evaluate ChatNT in additional biologically relevant tasks that cover DNA, RNA and protein processes. ChatNT achieves state-of-the-art performance across all tasks, matching the performance of several specialized models, such as APARENT2 for RNA polyadenylation11 and ESM2 for protein-related tasks31, while being able to solve a large collection of tasks at once and in English. Finally, its English conversational capabilities make its use easier than other models, widening its accessibility to scientists with no machine learning or computer science background. This framework for genomics instruction tuning can be easily extended to new tasks or biological data modalities (for example, sequencing experiments and imaging) without the need for pretraining from scratch every time, making it a widely applicable tool for biology.
Results
Transforming DNA foundation models into conversational agents with ChatNT
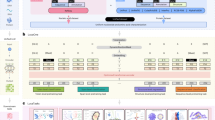
ChatNT is a framework for genomics instruction tuning, extending instruction-tuning agents to the multimodal space of biology and biological sequences. Our framework is designed to be modular and trainable end to end. It combines (1) a DNA encoder model, pretrained on genome sequencing data and that provides DNA sequence representations; (2) an English decoder, typically a pretrained GPT-style LLM, to comprehend the user instructions and produce responses; and (3) a projection layer that projects the representations extracted by the DNA encoder into the embedding space of the input English words, such that both can be used by the English decoder (Fig. 1c and Methods). In contrast to most multimodal works (for example, ref. 26) that would typically freeze the encoder and train only the projection, and sometimes the decoder, we decided in this work to backpropagate the gradients in the encoder in addition to the projection to allow supervised knowledge propagation at the DNA model level. As the English decoder is kept frozen, ChatNT benefits from its entire initial conversational capabilities, ensuring these do not degrade during training. In this work, we use the Nucleotide Transformer v2 (500-million-parameter model pretrained on genomes from 850 species) as the DNA encoder15 and Vicuna-7b (instruction-fine-tuned LLaMA model with 7 billion parameters) as the English decoder32 to build the conversational agent ChatNT. Keeping this modular architecture allows one to use constantly improving encoders and decoders in the future without changing the model architecture.

a, An illustration of the different categories of downstream tasks included during training. UTR, untranslated region. b, Statistics on the number of English and DNA tokens available for each task in our genomics instructions dataset. English question–answer instructions are tokenized with the LLaMA tokenizer30, while DNA sequences are tokenized using the Nucleotide Transformer tokenizer15. c, The ChatNT approach to build a multimodal and multitask genomics AI system. The ChatNT conversational agent can be prompted in English to solve various tasks given an input question and nucleotide sequence. In this example, the user inputs a DNA sequence (fasta file) and asks the agent to evaluate the degradation rate of the given RNA sequence. The question tokens are combined with the projected DNA representations before passing through the English language model decoder. The pretrained decoder writes the answer through next-token prediction, in this case predicting the degradation rate of the input sequence.
To train and evaluate ChatNT, we converted datasets of genomics tasks into instructions datasets by framing each task in English (Supplementary Fig. 1; see Methods and the following ‘Results’ sections). We created for every task a train and test file each containing the respective DNA sequences combined with curated questions and answers in English. See Fig. 1c for an example of question and answer for predicting RNA degradation levels: ‘User: Determine the degradation rate of the human RNA sequence @myseq.fna on a scale from −5 to 5. ChatNT: The degradation rate for this sequence is 1.83.’, where the projected embeddings of the candidate DNA sequence are inserted at the @myseq.fna position. We keep the same train–test splits as the original sources of each task and use different questions for train and test to assess the English generalization capabilities of the model. This allows one to evaluate not only the agent capability to generalize between DNA sequences but also its robustness to the English language used. We also provide a flexible way to interleave English and DNA sequences through the usage of positional tags (@myseq.fna), allowing users to refer to several sequences in the same question.
ChatNT is trained to solve all tasks simultaneously, with a uniform sampling over tasks per batch. Multitasking is achieved by ChatNT by prompting in natural language, where the question asked by the user will guide the agent towards the task of interest. Given a text prompt and one or multiple DNA sequences as input, ChatNT is trained to minimize a unified objective for all tasks, which takes the form of the cross-entropy loss between ChatNT predictions and the target answer tokens, as in other instruction fine-tuning works32,33. This single objective allows one to learn seamlessly across tasks without introducing conflicting gradients or scale issues coming from different objectives and loss functions (for example, cross-entropy for classification versus mean squared error (MSE) for regression). In addition, it allows us to extend the model with additional tasks in the future without requiring changes in the model architecture or training it from scratch. In summary, ChatNT provides a general genomics AI system that solves multiple tasks in a conversational manner, thus providing a different paradigm for genomics models.
In addition to seamlessly integrating multiple types of labelled and experimental data into a single general foundation model, ChatNT is designed to be conversational to enable users to easily interact with it and to use it without requiring a programming background (see examples in Supplementary Fig. 1). We rely on a frozen English language model, Vicuna-7b32, that has been instruction fine-tuned from LLaMA30. ChatNT keeps all the intrinsic conversational capabilities of the language model. Interestingly, we observed that, as the training dataset used to build LLaMA already contained a large set of life sciences papers, our agent is also capable to answer multiple questions about genomics, including defining regulatory elements such as promoters and enhancers, zero shot, that is, without any additional training data. In addition, ChatNT can answer numerous non-biology-related questions and solve tasks such as summarizing or writing simple programming code. As our approach is general and builds on top of any pretrained English language model, ChatNT capabilities can improve organically with new and more powerful open-sourced language models. While the conversational capability is an important aspect of ChatNT but is already provided by the respective language model, we focused in this work on demonstrating that the conversational agent ChatNT can solve a wide range of advanced genomics tasks in English with high accuracy.
ChatNT achieves improved performance on a genomics benchmark
To develop ChatNT and optimize its architecture, we created an instructions version of the Nucleotide Transformer benchmark15 (Methods and Supplementary Table 1). This collection of genomic datasets is suitable for fast iteration during model experimentation as it contains a varied panel of small-sized datasets and has been extensively evaluated in multiple studies of DNA foundation models15,17. We trained ChatNT to solve all 18 tasks at once and in English and evaluated its performance on test set DNA sequences and questions.
We first used this benchmark to systematically compare the performance of ChatNT with two different projection architectures. The classical way of aggregating information from the encoder in previous multimodal models is to use a trainable projection to convert the encoder embeddings into language embedding tokens, which have the same dimensionality of the word embedding space in the language model26,27,34. In ChatNT, we used the Perceiver resampler from Flamingo27 based on gated cross-attention as the projection layer (Supplementary Fig. 2a). Using this projection layer and fine-tuning both the DNA encoder and the projection on all 18 tasks, ChatNT obtained a new state-of-the-art accuracy on this benchmark with an average Matthew’s correlation coefficient (MCC) of 0.71, 2 points above the previous state-of-the-art Nucleotide Transformer v2 (500M) model (Fig. 2a and Supplementary Figs. 2d and 3).

a, The average performance of ChatNT, ChatNT with no English-aware projection and 13 different genomics foundation models across all 18 tasks of the Nucleotide Transformer benchmark15. The bar plots represent mean MCC values ± s.e.m. (n = 18). b, A radar plot depicting the performance of ChatNT in each of the 18 tasks compared with specialized Nucleotide Transformer v2 models fine-tuned individually on each task.
However, similar to all other projection layers26,34,35, the current implementation of the Perceiver resampler generates the same fixed set of embeddings for the encoder tokens independently of the question asked, and therefore it needs to capture in this set of embeddings all relevant information for every downstream task. We hypothesized that this feature can create an information bottleneck in genomics when scaling the model for multiple downstream tasks given the diversity of potential sequences, from different lengths and species, and biological properties. Therefore, we developed an English-aware Perceiver projection that extracts representations from the input sequence dependent on the English question asked by the user, which allows one to leverage contextual information encoded in the input DNA sequences that are relevant for the specific question (Methods and Supplementary Fig. 2b). We observed significantly improved performance by accounting for the question when projecting the DNA embeddings into the English decoder space (average MCC of 0.77 versus 0.71; Fig. 2a and Supplementary Fig. 2c,d). This can be explained by the very context- and task-specific information in DNA sequences that we must retain in order to tackle diverse genomics tasks. Because the decoder remains frozen, the projection layer needs to not only bring the sequence embeddings into the embedding space of the English decoder but also to perform the operations to extract the relevant information from the embedding to answer the question. Our results show that making the projection aware of the question facilitates both aspects, thus achieving a better performance and transfer across tasks.
In summary, ChatNT with an English-aware projection (from now on called just ChatNT) achieves a new state-of-the-art accuracy on this benchmark (average MCC of 0.77) in addition to solving all 18 tasks at once (Fig. 2a). ChatNT improves the average performance by 8 points over the previous state-of-the-art Nucleotide Transformer v2 (500M) model, which was used as the DNA encoder within ChatNT (average MCC of 0.77 versus 0.69; Fig. 2a,b). In addition to generalizing across DNA sequences and tasks, ChatNT also generalizes across questions, as demonstrated by the low variability in the accuracy of the predictions based on the variability of the language prompt (Supplementary Fig. 4). Our results demonstrate that a single unified objective formulated in natural language triggers transfer learning between multiple downstream tasks and helps to deliver improved performance.
A curated instructions dataset of biologically relevant tasks
Although the Nucleotide Transformer benchmark15 was very suitable for model experimentation and to debug the system, it misses many tasks of great biological relevance in genomics related to more complex biological processes as well as more recent experimental techniques and tasks that involve quantitative predictions. Therefore we curated a second genomics instructions dataset containing 27 genomics tasks framed in English derived from different studies that cover several regulatory processes (Methods and Supplementary Table 2). These include tasks related to DNA (21 tasks), RNA (3) and protein sequences (3) from multiple species framed as both binary/multilabel classification and regression tasks. The final instructions dataset contains a total of 605 million DNA tokens, that is, 3.6 billion base pairs, and 273 million English tokens (including an average of 1,000 question–answer pairs per task) (Fig. 1b).
This collection includes a non-redundant subset of tasks from the Nucleotide Transformer15 and the BEND36 benchmarks, complemented with relevant tasks from the plant AgroNT benchmark37 and human ChromTransfer38. These benchmarks have been extensively used in the literature, come from different research groups and represent diverse DNA processes and species. These selected tasks include binary and multilabel classification tasks covering biological processes related to histone and chromatin features, promoter and enhancer regulatory elements, and splicing sites.
We further added state-of-the-art and challenging regression tasks related to promoter activity37, enhancer activity7, RNA polyadenylation11 and degradation10, and multiple protein properties39. These are reference datasets in the respective fields and are related to very complex properties of biological DNA, RNA and protein sequences. All RNA and protein tasks are predicted from the corresponding DNA and coding sequences (CDS) instead of the RNA and protein sequences, respectively. Getting the matching DNA sequence is trivial for RNA sequences but more challenging for protein sequences owing to the complexity of codon usage. Therefore, we used the CDS annotations for protein tasks curated by Boshar et al.39.
See Fig. 3 and Supplementary Fig. 1 for examples of questions and answers for different types of genomics tasks used in our dataset (see also Supplementary Figs. 5–7). For instance, a training example for an enhancer classification task would be ‘User: Is there an enhancer from human cells present in this sequence @myseq.fna, and can you characterize as weak or strong? ChatNT: Yes, a weak enhancer is present within the DNA sequence that you provided.’, where the projected embeddings of the candidate DNA sequence are inserted at the @myseq.fna position. Regression tasks are also framed in English, and the agent needs to write the digits corresponding to the requested quantity: for example, ‘User: Determine the degradation rate of the mouse RNA sequence @myseq.fna on a scale from −5 to 5. ChatNT: The measured degradation rate for this sequence is 2.4.’ (see Methods for details on the quantitative scale). The loss is equally computed as the cross-entropy loss between the predicted and the target answer tokens, treating scalar values as digit tokens. This approach worked well in our setting and yielded results comparable to traditional MSE loss because ChatNT is autoregressive, allowing it to capture the sequential structure of numbers. Errors in earlier digits, such as the order of magnitude, result in higher loss, prompting the model to focus on predicting the most significant digits first before refining the less significant ones, thus effectively introducing a hierarchical decomposition of numbers. For performance evaluation, we extract the digits from each answer and test their correlation with the ground-truth values.

a,d,e, Left: an example of a conversation for promoter (a), DNA methylation (d) and splice sites tasks (e). Right: a heatmap displaying the confusion matrix comparing the predicted labels of ChatNT and observed labels. The performance metric is reported. b,c,f, Left: an example of a conversation for promoter strength in tobacco leaf (b), RNA degradation (c) and protein meltome tasks (f). Right: a scatter plot comparing the predictions of ChatNT and observed values. The PCC is reported.
In summary, this curated set of tasks provides a general perspective of the capabilities and usefulness of our model in different biological sequence domains. We train ChatNT as a general agent to solve all 27 genomics tasks at once and in English, and compare its performance with the state-of-the-art specialized model for each task (Methods).
Evaluation on tasks from various genomics processes and species
We first evaluated the performance of ChatNT on the 21 tasks related to different DNA processes from yeast, plants, fly, mouse and human. ChatNT is competitive with the performance of the different specialized models that were fine-tuned directly on each of these individual tasks, being also robust to the variability of the language prompt (Figs. 3a,b,d,e and 4a,c). In particular, we obtained an improved performance on the detection of human enhancer types. Still, we observed significantly reduced performance for enhancers from plant species when compared with the state-of-the-art AgroNT model fine-tuned specifically on this task37. As AgroNT was pretrained on genomes from 48 diverse plant species, improving the encoder used in ChatNT might lead to improved performance on this type of task.

a, Bar plots with the performance of ChatNT compared with respective baselines per task. The metric used for each task is the same used in the respective baseline study (Supplementary Table 2). Data are presented as mean ± s.d. (n > 10 per task). Chrom., chromatin; Dev., developmental, Hk., housekeeping. b–d, A comparison between ChatNT and baselines for all tasks (n = 27) (b), classification tasks (n = 17) (c) and regression tasks (n = 10) (d). Metrics are the same as in a. The box plots mark the median, upper and lower quartiles and 1.5× interquartile range (whiskers); outliers are shown individually.
As ChatNT solves the tasks in English, it can seamlessly handle binary and multilabel classification tasks. By extracting the term predicted by ChatNT in the answer, we can quantify its predictive performance. As we show for some examples in Fig. 3, ChatNT accurately identifies input sequences with human or mouse promoters (Fig. 3a), with CpG sites methylated in human embryonic stem cells (HUES64 cell line; Fig. 3d) and with splice acceptor and donor sites (Fig. 3e).
ChatNT is also able to solve quantitative tasks by writing the digits of the predicted score. We observed competitive performance on predicting promoter activity in plants, namely tobacco leaves (Fig. 3b) and maize protoplasts, but significantly reduced performance on Drosophila enhancer activity over the state-of-the-art DeepSTARR model7 (Fig. 4a). Importantly, the distributions of the predicted digits correlate well with the original scores (Fig. 3b). This capability to proficiently address regression tasks is of paramount importance in biology and is particularly significant in light of the acknowledged limitations and unreliability of numerical processing in language models40,41. Still, we observed a reduced average performance on regression tasks over classification ones, probably due to the difference in complexity and classification tasks being more represented in the training set. We assume that this might be solved by improving the balance between classification and regression tasks during training, through either a weight loss or a task sampling frequencies curriculum42.
ChatNT solves transcriptomics and proteomics tasks
ChatNT is built with a flexible architecture that allows it to handle any type of biological sequence that can be processed with our DNA encoder, the Nucleotide Transformer15. To showcase its generalization, we have included in the new genomics instructions dataset three RNA and three protein regression tasks (Supplementary Figs. 6 and 7). These include predicting RNA polyadenylation and degradation rates as well as different protein features. Examples of conversations used for model training are: ‘User: What is the measured polyadenylation ratio of the proximal site of the RNA sequence @myseq.fna in human HEK293 cells, considering a range from 0 to 1? ChatNT: That sequence has a polyadenylation ratio of the proximal site of 0.69.’ and ‘User: Specify the melting point of the protein with the given coding sequence (CDS) @myseq.fna within the 0 to 100 range. ChatNT: This protein demonstrates a melting point of 80.81.’. The performance of ChatNT was compared with the state-of-the-art specialized models APARENT2 for polyadenylation11, Saluki for RNA degradation10 and ESM2 for the protein tasks31 (Supplementary Table 2).
Overall, we observed good performance for ChatNT on the test sets of the six RNA and protein tasks, with Pearson correlation coefficients (PCCs) between 0.62 and 0.91 (Figs. 3c,f and 4a). ChatNT outperformed the specialized models for the prediction of proximal polyadenylation site ratio (PCC of 0.91 versus 0.90) and protein melting points (PCC of 0.89 versus 0.85). Regarding the RNA degradation tasks in human and mouse, ChatNT obtained a PCC of 0.62 and 0.63, 10 points below the specialized Saluki model10 (PCC of 0.74 and 0.71). ChatNT also obtained competitive performance with the state-of-the-art protein language model ESM231 on the two other protein tasks related to protein fluorescence and stability. Although ChatNT cannot yet outperform every specialized model on RNA and protein tasks, we show that it can already handle such tasks and achieve high performance using the DNA foundation model Nucleotide Transformer as a DNA encoder. ChatNT’s flexible architecture allows one to plug in different encoders, such as language models specialized for RNA43,44 and protein domains31, which should reduce the gap to specialized deep learning models in the transcriptomics and proteomics fields and improve the capabilities and generalization of ChatNT towards a unified model of biology.
Assessing the confidence of ChatNT answers
ChatNT is built to assist and augment scientists and researchers in their daily research. As such, its performance and reliability are paramount. However, in contrast to standard machine learning models that return probabilities or quantitative scores, ChatNT directly answers questions, preventing the user from getting a sense of its confidence and, thus, reducing its practical value for sensitive applications. This is an important challenge and common to all current conversational agents24,26. To address this, we investigated a way to assess the confidence of our agent for binary classification tasks. Instead of directly generating answers to the binary classification question for a given sequence, we compute the model perplexity for that question over examples of both positive and negative answers. We make sure that these selected answers were not included in the model training dataset. Those perplexity values towards positive and negative answers are then used to derive logits and probabilities for each class for the candidate question. This method allows us to derive probabilities from ChatNT for each question example, similar to standard classifiers, and we refer to it as the perplexity-based classifier (Fig. 5a). Currently, this process is studied as a post-hoc analysis, not yet integrated into the ChatNT tool, but it could be incorporated into future versions once fully developed to provide this information to the user.

a, A cartoon describing the perplexity-based classifier based on ChatNT answers. b, A calibration plot for the task of human chromatin (chrom.) accessibility (cell line HepG2). The scatter plot compares the predicted probability and fraction of positives over ten bins for the original (green) and calibrated (purple) perplexity-based classifiers. c, A histogram with the predicted probability over ten bins for the original perplexity-based classifiers. d, A histogram with the predicted probability over ten bins for the calibrated perplexity-based classifiers. e, A comparison of the performance (MCC) of the ChatNT answers (yes versus no) and its derived perplexity-based probabilities for all binary classification tasks.
Computing probabilities enables us to assess the calibration of the model, that is, the correlation between the predicted probability, its confidence and the accuracy of its prediction. We say that a model is well calibrated when a prediction of a class with confidence p is correct 100% of the time. We computed the ChatNT perplexity-based probabilities for all binary classification tasks. In Fig. 5b–d, we show an example of a calibration plot based on the predictions for the chromatin accessibility task. We observe that our model is well calibrated for low- and high-confidence areas, but less in medium-confidence ones. For instance, examples predicted with a probability of 0.9 are correctly predicted 90% of the time, while examples predicted with probability 0.5 are correctly predicted only 25% of the time. To improve this, we show that we can calibrate our model by fitting on the training set a Platt’s model45, to improve the confidence of the model across all ranges of predictions (Fig. 5b–d). This calibration step is performed for all binary classification tasks. Overall, we achieve the same performance for ChatNT across tasks using these perplexity-based predictions (Fig. 5e) but with improved calibration. As a consequence, our approach can accurately measure the predictive performance of a language model in addition to effectively assessing its uncertainty level. This technique, while being general, should also be beneficial to other language model fields.
Model interpretation reveals learned DNA sequence features
A key application of language models for biological sequences has been to uncover the underlying code or grammar of DNA, RNA and protein sequences. To evaluate whether ChatNT’s predictions rely on biologically relevant sequence features and could be used for further discoveries, we used model interpretation tools across hundreds of sequences from different tasks in our dataset. While these techniques have been widely applied for sequence models, notably in the case of DNA7,46, this has never been done for multimodal models where the output is expressed in natural language such as ChatNT. To do that, we quantified the contribution of each DNA token, that is, 6-mer, within the input DNA sequence to the predicted English tokens answered by ChatNT (Fig. 6a). More specifically, we calculated the gradient of the predicted token with respect to the input DNA tokens47, backpropagating through the English decoder, the perceiver projection and the DNA encoder. We applied this approach to sequences from three different genomics tasks for which clear predictive sequence features are known in the literature: splice donor sites that usually overlap a GT dinucleotide, splice acceptor sites that overlap a AG dinucleotide and promoters that rely on the TATA-box motif.

a, A cartoon describing the interpretation of ChatNT predictions with regard to the input sequence. For a given input question and DNA sequence, we compute the gradients of each predicted English token towards every input DNA token. w.r.t., with respect to. b–d, Sequence features extracted from ChatNT for the tasks splice donors (b), splice acceptors (c) and TATA promoter (d). Shown are the consolidated sequence motifs with the y axis as bits (top) and the k-mer spectrum across sequences per task (bottom).
For each task, we computed the contribution of input DNA tokens with respect to the ‘yes’ or ‘no’ token answered by ChatNT. We performed this analysis across a subset of test set sequences per label per task, identifying the highest attributed DNA tokens per input sequence. Displaying the frequency of important 6-mers across all sequences revealed that positively labelled sequences were enriched for known sequence features such as the splice donor and acceptor dinucleotides (Fig. 6b–d). Indeed, combining the top-scoring tokens across all positive sequences and calculating the nucleotide frequencies per position recovers the main sequence pattern per task that matches the known splice donor GT dinucleotide, splice acceptor AG dinucleotides and promoter TATA motif. Altogether, these results demonstrate that ChatNT’s answers are based on biologically coherent features and that the model can be used to interpret the underlying language of DNA. It is important to emphasize that ChatNT accomplishes this with a single, unified model. Typically, such analysis usually relies on specialized or fine-tuned models tailored to individual tasks. The fact that our results reveal consistent and meaningful features extracted across diverse tasks underscores the capacity of ChatNT to learn a generalized grammar of DNA and to connect it to natural language.
Discussion
We presented ChatNT, a multimodal conversational agent that can handle DNA, RNA and protein sequences and solve multiple biologically relevant downstream tasks. We built and curated datasets of genomics instructions tasks including binary and multilabel classification and regression tasks spanning different species and genomics processes. Tasks relative to transcriptomic and proteomic processes were also included to demonstrate the versatility and generality of this approach across domains. ChatNT achieves a new state-of-the-art on the Nucleotide Transformer benchmark15 and demonstrates a performance on par with specialized models on our set of 27 tasks. Importantly, unlike conventional approaches requiring a specialized model for each task, ChatNT solves all tasks within a unified model in addition to offering a simple and natural chatbot interface for people to use the model. We also introduced a technique to probe the confidence of language models for binary classification tasks and used it to calibrate them when needed. Through model interpretation analyses, we have also shown that ChatNT has learned to associate biologically relevant sequence features when answering about different genomics tasks. Altogether, ChatNT is a proof that natural language LLMs can be extended to process biosequence modalities, not only displaying conversational capabilities but also answering accurately multiple biologically relevant questions.
To extract the complex information from DNA sequences that is needed to solve all tasks in a single unified model, we introduced an architecture based on the Perceiver resampler27 to resample and project DNA embeddings into the natural language embedding space. We identified an information bottleneck issue that arises from the diversity of tasks, species and biological processes encoded in DNA sequences, and we showed how to solve it by conditioning the projection on the question asked. This conditioning allows the projection module to extract from the DNA embeddings the right amount of information to solve the task at hand, as we show by the improved performance over a projection module that is not conditioned on the question.
In this work, we decided to focus on situations where a user, such as a researcher or scientist, is interested in detecting molecular phenotypes or computing quantitative properties for a given DNA sequence. While we believe this encompasses an already significant number of practical use cases, it would be interesting to expand the agent capabilities to handle other typical bioinformatics pipelines. Such pipelines could include calling tools to compute statistics about the sequences, aligning the sequences to a reference database to compute multiple sequence alignments, query external databases for additional information about the sequences, or recursively call the ChatNT model over a FASTA file containing multiple sequences and generating a summarized table results with its corresponding analysis. This is supported by the success of external tools in LLMs such as Toolformer48, LLaVA-Plus34, geneGPT49 or GPT-425. Such pipelines could also benefit from ChatNT’s capability to handle several sequences at the same time in order to reduce the inference compute cost. Replacing ChatNT’s current English decoder by larger models and/or models fine-tuned using reinforcement learning human feedback such as Llama2-chat 70B50 could also help to extend the model capabilities in these directions as well as improve its overall usefulness.
Regarding the length of sequences that ChatNT can process, the agent has the inherent limitations of its DNA encoder. The current version uses the Nucleotide Transformer v2 model as a DNA encoder, which has been trained on sequences up to 12 kb long. However, there exist different techniques that can be used to expand its context, with various works showing improved performance of Nucleotide Transformer models up to 50-kb sequences18,51. In addition, one can change the DNA encoder in this framework, for example, using Borzoi that can process sequences of 524 kb (ref. 52), and use the long context needed for the task of interest. ChatNT can be improved in the future through the use of newly developed DNA sequence encoders.
ChatNT processes species and cell type information implicitly through natural language prompts rather than as structured inputs. When a query specifies a species or cell type, the model extracts and integrates this contextual information directly from the text. This approach leverages the pretrained language model’s ability to interpret and condition predictions based on linguistic context. This capability is facilitated by our instruction-tuning process, which includes task-specific examples featuring diverse species and cell types. Incorporating more data with diverse prompts should enable the model to generalize across a broad range of biological contexts. Once we further scale the number of tasks and data, an additional use case for the agent in the future will be the zero-shot prediction of the functional impact of genomic variants. This can be prompted using the positional tags introduced in this work that allow users to refer to several sequences in the same question. For example, the user can provide both the reference and mutated sequences to ChatNT using two different positional tags and let the model use the full projected embeddings of the two sequences to make the final evaluation of the mutation impact. An example to illustrate this case would be ‘User: I have a mutation that changes the sequence @wildtype.fna to @mutation.fna. ChatNT: Yes, this genetic variant is pathogenic.’. This will open new possibilities to compare different sequences and assess the impact of genetic variants through the learned representations of DNA sequences.
The capabilities of ChatNT have been demonstrated for DNA sequences using a pretrained DNA foundation model, the Nucleotide Transformer15. As shown in our experiments, working with DNA sequences allows one to tackle tasks not only in genomics but also in transcriptomics and proteomics, the latter using the corresponding CDS region. However, our approach could be easily extended to integrate encoders from other omics modalities such as RNA43,44,53,54 and protein31,55 language models to work natively with RNA and amino acid sequences. Through our positional tag system that supports multiple sequences, one could simply add an arbitrary number of encoders and train their respective projections to combine different omics and modalities within the same questions. We envision that such an approach could expand even further the capabilities and performance of our model by achieving superior transfer learning across modalities.
Finally, we note that, for now, ChatNT does not have mechanisms in place to inform the user about the tasks and cell types covered (for example, those included in the training data) or when the user asks questions out of that training distribution. Following current research in LLMs, this type of mechanisms can be easily done by adding to ChatNT’s system message the types of tasks and datasets it was trained on and how it should behave if asked questions out of that range. These mechanisms should also help to prevent misuse and ensure the safe use of this type of tool.
This work serves as a proof of concept that it is possible to build multimodal biosequence and English conversational agents that can solve advanced, biologically relevant tasks and is meant to lay a first set of foundations to build future highly capable agents that understand biological sequences and principles. Similar to the developments in NLP50,56,57,58 and multimodal models59, we expect new capabilities such as zero-shot performance to emerge through developments on two main fronts: (1) scaling the number of tasks by including examples from diverse biological processes, tissues, individuals and species60,61; and (2) integrating more data modalities, such as RNA and protein sequences, imaging data and health records from individuals. When such capabilities emerge, it will be of the highest importance to carefully assess model safety and robustness, for instance through red teaming62. As such, ChatNT represents an important step along the trajectory towards general-purpose AI for biology and medicine63.
Methods
ChatNT model
Architecture
ChatNT is a multimodal agent that takes as input one or multiple DNA sequences and an English prompt and returns a distribution over English words that is used to autoregressively produce an answer in English. We introduce a DNA English token placeholder <DNA> that is added in the input English prompt for the user to refer to the DNA sequence. The architecture is also extended to handle several DNA sequences. In this case, each DNA sequence is processed independently by the DNA encoder, and the input English prompt is expected to contain as many DNA English token placeholders as sequences are inputted.
The ChatNT architecture is made of three parts: a pretrained DNA encoder, a projection model that projects the DNA embeddings into the English tokens embedding spaces and a pretrained English decoder. While our architecture is general and could work with any choice of DNA encoder and English decoder, we decided to use the pretrained Nucleotide Transformer v2 (500-million-parameter model pretrained on genomes from 850 species)15 and Vicuna-7b (instruction fine-tuned Llama model with 7 billion parameters)32 models, respectively. During training, we keep the English decoder frozen and update only the weights of the DNA encoder and the projection model. The projection model is initialized from scratch at the beginning of the training.
The DNA encoder processes the DNA sequence and returns one embedding vector per input token, one token representing a nucleotide 6-mer in the case of the Nucleotide Transformer model. We note L the number of nucleotides in the DNA sequence and N the number of DNA tokens (with roughly \(N\approx \frac{L}{6}\)). Every input DNA sequence was padded if needed until a final length of 2,048 tokens, representing approximately 12 kb. As the output embedding dimension of the DNA encoder can be different from the words embedding dimensions of the English language model, we first use a dense neural network to project each DNA token embedding to the English word dimension. In a second phase, we use a Perceiver resampler architecture27 that uses cross-attention between the projected DNA tokens embeddings and learnable queries, to resample the N DNA tokens embedding to K embedding vectors (Supplementary Fig. 2a). We have adapted this Perceiver resampler to include an additional cross-attention step between the learnable queries and the English question in order to extract context-dependent representations from the DNA sequence (Supplementary Fig. 2b).
Meanwhile, the English prompt is tokenized and English tokens embeddings are produced for each tokens. The K resampled DNA embedding vectors are then inserted in place of the DNA sequence placeholder tokens in the English input sequence. In the case of multiple input DNA sequences, these operations are applied consecutively and independently for each DNA sequence. We experimented with several values of K in practice and observed that low values such as 1 or 4 are not enough for the DNA encoder to impact the behaviour of the frozen English decoder. We found K = 64 to provide a good trade-off between the input length of the English decoder and the performance in practice.
During inference, the DNA encoder embeddings for the DNA sequences are computed only once. The inference is done autoregressively by predicting sequentially each new token until an end of sequence token is predicted. The key, queries and values of the English decoder are cached during generation to avoid computing unnecessary operations. We use temperature sampling with a temperature of τ = 0.001.
In our unified approach, all tasks are framed as text (with additional DNA input)-to-text transformations, where the model generates output as an unconstrained text sequence. For regression tasks, numerical values are produced within this sequence and optimized using cross-entropy loss, without any predefined structural constraints. This flexibility, however, makes standard regression metrics such as MSE not directly applicable, as computing MSE requires a structured numerical output format with a designated placeholder. Enforcing such a format would limit the generality and adaptability of our method. Still, despite its broad scope, our objective achieves competitive results on regression tasks.
The whole code base of the ChatNT has been developed in Jax64 using Haiku65 for neural network implementation. All trainings were performed on a cluster of 8 graphics processing unit (GPU) H100 instances, and evaluations of the model can be done in a single GPU A100-80gb. All trained parameters from the DNA encoder and perceiver projection as well as optimizer accumulators and all frozen parameters from the English decoder are stored and updated in float32.
Training
We describe all hyperparameters for the different parts of ChatNT in Supplementary Table 3. ChatNT was trained using Adam optimizer66 with lr = 3e−5 and default settings for other hyperparameters: β1 = 0.9, β2 = 0.999, ϵ = 1e−8, ϵroot = 0.0. We used gradient clipping of 1 and accumulated gradients over a batch size of 65,536 tokens, equivalent to 256 samples. We used an uniform sampling over tasks per batch such that each batch has the same proportion of samples per task. We trained the model on the 27-task dataset for 2 billion tokens (7.8 million samples) on a cluster of 8 GPU H100 over 4 days.
Evaluation
Evaluating the performance of ChatNT can be done in a single GPU A100 in batches of 32 samples and takes 1:40 min to generate a maximum of 40 tokens per sample (13 tokens per second). For each task, we evaluated ChatNT on the upmost 5,000 sampled test samples and report the metric used in the respective benchmark study (Supplementary Tables 1 and 2).
Genomics instructions datasets
Instructions for the Nucleotide Transformer benchmark
We created an instructions version of the Nucleotide Transformer benchmark15 (Supplementary Table 1). To convert the DNA sequence datasets into instructions datasets, we curated dozens of English questions and answers for each task and sampled a question–answer pair per input DNA sequence. We used the DNA token placeholder <DNA> in the question when referring to the input DNA sequences. The answer contains the classification label for the respective input sequence. We converted all 18 binary and multilabel classification datasets into diverse question–answer instructions for each DNA sequence. We provide for each task train and test sets containing different DNA sequences as well as different questions to assess the performance and English generalization capabilities of the model. We kept the same train and test sets as in the original dataset.
New curated genomics instructions dataset of biologically relevant tasks
The new genomics instructions dataset created here contains a set of 27 tasks framed in English derived from different studies (more details in Supplementary Table 2). It covers several regulatory processes related to DNA (21 tasks), RNA (3) and protein sequences (3). These tasks are derived from multiple species, including human, mouse, fly and plants. Among all tasks, there are 15 binary classification, 2 multilabel classification and 10 regression tasks. The number of training examples per task ranges from 5,500 to 3 million. See Supplementary Information for all details on the data references and processing for each specific task.
We converted the DNA sequence datasets into instructions datasets as described above for the Nucleotide Transformer benchmark. The answer contains the classification label or regression score (up to decimal cases) for the respective input sequence. In addition to simple examples with a single turn of question–answer with a single sequence, we also added more complex examples with multiple turns with consecutive questions that can be related or not, and exchanges where the question refers to multiple sequences. The final genomics instructions dataset contains a total of 605 million DNA tokens, that is, 3.6 billion base pairs, and 273 million English tokens (including questions and answers).
We obtain for each task train and test sets containing different DNA sequences as well as different questions to assess the performance and English generalization capabilities of the model.
Baselines for the genomics tasks
For each of the 27 genomics tasks, we compared the performance of ChatNT with the state-of-the-art method for the respective dataset. These included the convolutional neural networks DeepSTARR7, ChromTransfer38, APARENT211 and Saluki10 and the fine-tuned foundation models based on Nucleotide Transformer15, agroNT37, DNABERT67 and ESM231. We used different performance metrics per task to follow the same metric used in the respective studies. Details on the baseline method and performance metric per task can be found in Supplementary Table 2. Most baseline performance metrics were directly retrieved from the respective papers. Only for ESM2 did we have to rerun them on the updated dataset versions.
Calibration of ChatNT predictions
We developed an approach to assess and calibrate the confidence of ChatNT answers for binary classification tasks.
For a given binary classification task, we select N examples of positive and negative answers each, selected from the respective task’s test set. We note these examples respectively \({{\bf{y}}}_{i}^{{\rm{pos}}}\) and \({{\bf{y}}}_{i}^{{\rm{neg}}}\) where 0 ≥ i > N. Then, for a given question x and DNA sequence s, we compute the average perplexity of the model over the positive and negative examples respectively. We denote these two values as \(p{p}_{\theta }^{{\rm{pos}}}({\bf{x}},{\bf{s}})\) and \(p{p}_{\theta }^{{\rm{neg}}}({\bf{x}},{\bf{s}})\), respectively, where θ represents the ChatNT weights tensor. We compute them as follows:
where \({({{\bf{y}}}_{i}^{{\rm{pos}}})}_{j}\) denotes the jth token of answer \({{\bf{y}}}_{i}^{{\rm{pos}}}\) and \({p}_{\theta }({({{\bf{y}}}_{i}^{{\rm{pos}}})}_{j}| ({\bf{x}},{\bf{s}},{{\bf{y}}}_{i}^{{\rm{pos}}}))\) returns the probability of token j given the question, DNA sequence and tokens from the answers up to the jth one according to ChatNT. The negative perplexity values are computed similarly over negative answers.
Those perplexity values towards positive and negative answers represent a measure of how well the model aligns the question to those answers. We interpret them directly as logits and use a softmax transformation to compute probabilities for the respective class for the input question. This method allows one to derive probabilities from ChatNT for each question example. We applied this approach to 1,000 test examples per task.
To calibrate those predictions, we first compute perplexity-based probabilities to 10,000 training examples as our calibration dataset and use them to fit a Platt’s model45. More specifically, we use logistic regression from scikit-learn68 as the calibrator model and train it with the following parameters with an inverse regularization factor C = 0.1 and with the lbfgs solver. The logistic regression model learns to map the perplexity-based probabilities from ChatNT onto a more accurate scale. We then apply this model to calibrate the probabilities of the 1,000 test examples mentioned above.
As metrics, we computed both area under the receiver operating characteristic curve (AUROC) and MCCs for both the original perplexity-based probabilities and the calibrated ones.
Attribution map calculation
We used gradient-based methods to compute saliency maps for attribution analysis47, using a gradient ⊙ input approach. For each sequence, the gradient of the model’s first predicted output token (either ‘yes’ or ‘no’) was calculated with respect to the input sequence, producing a position-specific map that highlights the contribution of each k-mer to the model’s prediction.
This analysis was performed on 500 test set sequences for each label for the tasks splice donors and splice acceptors, and for 200 sequences per label for TATA promoters. Input sequences were tokenized into k-mers of length 6, with a one-hot encoded vector created for each token. These one-hot encoded tokens were then multiplied by the model’s embedding matrix, enabling the computation of gradients relative to the input in its embedding space rather than their discrete entities. These gradients were multiplied element-wise by the one-hot encoded input sequence, isolating the gradient sensitivity of each k-mer at its respective position, following the gradient ⊙ input approach. The absolute values of the gradients were taken to capture the magnitude of each k-mer’s contribution to the predicted output.
To summarize the k-mer contributions, attribution maps were used to generate sequence logos per sequence, visualizing the relative importance of each k-mer. We used Logomaker69 to scale the height of each k-mer based on its relative importance in the attribution scores. In addition, for each attribution map, we identified the five highest attributed k-mers and compiled them, allowing us to calculate global k-mer frequencies by aggregating these across all identified positions. This provided insight into the most highly attributed k-mers and their frequency distribution across the set of sequences used.
Finally, we computed sequence logos for each task by averaging the attribution maps across sequences and selecting the k-mer at the maximum attribution position. The subsequence surrounding this k-mer, including its left and right flanks, was converted into a position weight matrix and visualized as a sequence logo using Logomaker69.
Data availability
All input data are freely available from public sources referenced in the respective Methods sections. All genomics instruction datasets prepared for training ChatNT are available in Supplementary Information, including the DNA sequences, questions and answers for the train and test sets of each dataset. We also provide all questions and ChatNT answers on the sequences of the test set used to evaluate its performance on the different tasks. Source data are provided with this paper.
Code availability
Model code and weights of ChatNT in PyTorch are available via HuggingFace at https://huggingface.co/InstaDeepAI/ChatNT. We also provide the pseudocode of the algorithmic steps and key concepts underlying our multimodal model in Supplementary Information.
References
Consortium, I. H. G. S. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Eraslan, G., Avsec, Ž., Gagneur, J. & Theis, F. J. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 20, 389–403 (2019).
Yue, T. et al. Deep learning for genomics: from early neural nets to modern large language models. Int. J. Mol. Sci. 24, 15858 (2023).
Alipanahi, B., Delong, A., Weirauch, M. T. & Frey, B. J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 33, 831–838 (2015).
Angermueller, C., Lee, H. J., Reik, W. & Stegle, O. Deepcpg: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 18, 67 (2017).
Chen, K. M., Wong, A. K., Troyanskaya, O. G. & Zhou, J. A sequence-based global map of regulatory activity for deciphering human genetics. Nat. Genet. 54, 940–949 (2022).
de Almeida, B. P., Reiter, F., Pagani, M. & Stark, A. Deepstarr predicts enhancer activity from DNA sequence and enables the de novo design of synthetic enhancers. Nat. Genet. 54, 613–624 (2022).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548 (2019).
Avsec, Ž. et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 18, 1196–1203 (2021).
Agarwal, V. & Kelley, D. R. The genetic and biochemical determinants of mRNA degradation rates in mammals. Genome Biol. 23, 245 (2022).
Linder, J., Koplik, S. E., Kundaje, A. & Seelig, G. Deciphering the impact of genetic variation on human polyadenylation using APARENT2. Genome Biol. 23, 232 (2022).
Dallago, C. et al. FLIP: benchmark tasks in fitness landscape inference for proteins. In Proc. Neural Information Processing Systems Track on Datasets and Benchmarks (eds Vanschoren, J. & Yeung, S.) (NeurIPS, 2021); https://datasets-benchmarks-proceedings.neurips.cc/paper_files/paper/2021/file/2b44928ae11fb9384c4cf38708677c48-Paper-round2.pdf
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. Preprint at https://arxiv.org/abs/1810.04805 (2018).
Brown, T. et al. Language models are few-shot learners. Adv. Neural Inf. Process. Syst. 33, 1877–1901 (2020).
Dalla-Torre, H. et al. Nucleotide Transformer: building and evaluating robust foundation models for human genomics. Nat. Methods https://doi.org/10.1038/s41592-024-02523-z (2025).
Zhou, Z. et al. DNABERT-2: efficient foundation model and benchmark for multi-species genome. In Proc. International Conference on Learning Representations (ICLR, 2024); https://openreview.net/pdf?id=oMLQB4EZE1
Nguyen, E. et al. HyenaDNA: Long-range genomic sequence modeling at single nucleotide resolution. In Proc. Advances in Neural Information Processing Systems 43177–43201 (Curran Associates, Inc., 2023).
de Almeida, B. P. et al. SegmentNT: annotating the genome at single-nucleotide resolution with DNA foundation models. Preprint at bioRxiv https://doi.org/10.1101/2024.03.14.584712 (2024).
Bao, H., Dong, L., Piao, S. & Wei, F. BEiT: BERT pre-training of image transformers. In Proc. International Conference on Learning Representations (ICLR, 2022); https://openreview.net/pdf?id=p-BhZSz59o4
Gidaris, S., Singh, P. & Komodakis, N. Unsupervised representation learning by predicting image rotations. In International Conference on Learning Representations (ICLR, 2018); https://openreview.net/forum?id=S1v4N2l0-
Radford, A. et al. Learning transferable visual models from natural language supervision. In Proc. 38th International Conference on Machine Learning 8748–8763 (PMLR, 2021).
Raffel, C. et al. Exploring the limits of transfer learning with a unified text-to-text transformer. J. Mach. Learn. Res. 21, 5485–5551 (2020).
Roberts, A. et al. Scaling up models and data with t5x and seqio. J. Mach. Learn. Res. 24, 1–8 (2023).
ChatGPT. OpenAI https://openai.com/blog/chatgpt/ (2023).
GPT-4 Technical Report (OpenAI, 2023).
Liu, H., Li, C., Wu, Q. & Lee, Y. J. Visual instruction tuning. In Proc. Advances in Neural Information Processing Systems 34892–34916 (Curran Associates, Inc., 2023).
Alayrac, J.-B. et al. Flamingo: a visual language model for few-shot learning. Adv. Neural Inf. Process. Syst. 35, 23716–23736 (2022).
Li, C. et al. LLaVA-MED: training a large language-and-vision assistant for biomedicine in one day. In Proc. Advances in Neural Information Processing Systems (eds Oh, A. et al) 28541–28564 (Curran Associates, Inc., 2023).
Huang, Z., Bianchi, F., Yuksekgonul, M., Montine, T. J. & Zou, J. A visual-language foundation model for pathology image analysis using medical twitter. Nat. Med. 29, 2307–2316 (2023).
Touvron, H. et al. LLaMA: open and efficient foundation language models. Preprint at https://arxiv.org/abs/2302.13971 (2023).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Peng, B., Li, C., He, P., Galley, M. & Gao, J. Instruction tuning with GPT-4. Preprint at https://arxiv.org/abs/2304.03277 (2023).
Taori, R. et al. Alpaca: a strong, replicable instruction-following model. Stanford Center for Research on Foundation Models https://crfm.stanford.edu/2023/03/13/alpaca.html (2023).
Liu, S. et al. LLaVA-Plus: learning to use tools for creating multimodal agents. Preprint at https://arxiv.org/abs/2311.05437 (2023).
Li, J., Li, D., Savarese, S. & Hoi, S. BLIP-2: bootstrapping language-image pre-training with frozen image encoders and large language models. In Proc. 40th International Conference on Machine Learning (eds Krause, A. et al) 19730–19742 (PMLR, 2023).
Marin, F. I. et al. BEND: benchmarking DNA language models on biologically meaningful tasks. In Proc. International Conference on Learning Representations (ICLR, 2024); https://openreview.net/pdf?id=uKB4cFNQFg
Mendoza-Revilla, J. et al. A foundational large language model for edible plant genomes. Commun. Biol. https://doi.org/10.1038/s42003-024-06465-2 (2023).
Salvatore, M., Horlacher, M., Marsico, A., Winther, O. & Andersson, R. Transfer learning identifies sequence determinants of cell-type specific regulatory element accessibility. Genome Biol. 5, lqad026 (2023).
Boshar, S., Trop, E., de Almeida, B. P., Copoiu, L. & Pierrot, T. Are genomic language models all you need? Exploring genomic language models on protein downstream tasks. Bioinformatics 40, btae529 (2024).
Nogueira, R., Jiang, Z. & Lin, J. Investigating the limitations of transformers with simple arithmetic tasks. In Proc. 1st Mathematical Reasoning in General Artificial Intelligence Workshop (ICLR, 2021).
Hendrycks, D. et al. Measuring mathematical problem solving with the math dataset. In Proc. Neural Information Processing Systems Track on Datasets and Benchmarks (eds Vanschoren, J. & Yeung, S.) vol. 1 (Curran, 2021); https://datasets-benchmarks-proceedings.neurips.cc/paper_files/paper/2021/file/be83ab3ecd0db773eb2dc1b0a17836a1-Paper-round2.pdf
Song, X. et al. OmniPred: language models as universal regressors. In Transactions on Machine Learning Research (TMLR, 2024); https://openreview.net/pdf?id=t9c3pfrR1X
Chen, J. et al. Interpretable RNA foundation model from unannotated data for highly accurate RNA structure and function predictions. Preprint at https://arxiv.org/abs/2204.00300 (2022).
Li, S. et al. CodonBERT large language model for mRNA vaccines. Genome Res. 34, 1027–1035 (2024).
Platt, J. et al. Probabilistic outputs for support vector machines and comparisons to regularized likelihood methods. Adv. Large Margin Classif. 10, 61–74 (1999).
Avsec, Ž. et al. Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet. 53, 354–366 (2021).
Simonyan, K. Deep inside convolutional networks: visualising image classification models and saliency maps. Preprint at https://arxiv.org/abs/1312.6034 (2013).
Schick, T. et al. Toolformer: Language models can teach themselves to use tools. In Proc. Advances in Neural Information Processing Systems (eds Oh, A. et al) 68539–68551 (Curran Associates, Inc., 2023).
Jin, Q., Yang, Y., Chen, Q. & Lu, Z. GeneGPT: augmenting large language models with domain tools for improved access to biomedical information. Bioinformatics 40, btae075 (2024).
Touvron, H. et al. Llama 2: open foundation and fine-tuned chat models. Preprint at https://arxiv.org/abs/2307.09288 (2023).
Trop, E. et al. Advancing DNA language models: the genomics long-range benchmark. In LLMs4Bio AAAI Workshop 2024 (2024); https://openreview.net/forum?id=8O9HLDrmtq
Linder, J., Srivastava, D., Yuan, H., Agarwal, V. & Kelley, D. R. Predicting RNA-seq coverage from dna sequence as a unifying model of gene regulation. Nat. Genet. https://doi.org/10.1038/s41588-024-02053-6 (2025).
Akiyama, M. & Sakakibara, Y. Informative RNA base embedding for RNA structural alignment and clustering by deep representation learning. NAR Genomics Bioinf. 4, lqac012 (2022).
Zhang, Y. et al. Multiple sequence alignment-based RNA language model and its application to structural inference. Nucleic Acids Res. 52, e3 (2023).
Brandes, N., Ofer, D., Peleg, Y., Rappoport, N. & Linial, M. ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics 38, 2102–2110 (2022).
Hoffmann, J. et al. Training compute-optimal large language models. In Proc. Advances in Neural Information Processing Systems (NeurIPS, 2022); https://proceedings.neurips.cc/paper_files/paper/2022/file/c1e2faff6f588870935f114ebe04a3e5-Paper-Conference.pdf
Jiang, A. Q. et al. Mistral 7b. Preprint at https://arxiv.org/abs/2310.06825 (2023).
Chung, H. W. et al. Scaling instruction-finetuned language models. J. Mach. Learn. Res. 25, 1–53 (2024).
Li, C. et al. Multimodal foundation models: from specialists to general-purpose assistants. Found. Trends Comput. Graph. Vis. 16, 1–214 (2024).
ENCODE. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Roadmap Epigenomics. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Perez, E. et al. Red teaming language models with language models. In Proc. 2022 Conference on Empirical Methods in Natural Language Processing (eds Goldberg, Y. et al.) 3419–3448 (Association for Computational Linguistics, 2022).
Moor, M. et al. Foundation models for generalist medical artificial intelligence. Nature 616, 259–265 (2023).
Bradbury, J. et al. JAX: composable transformations of Python+NumPy programs. GitHub http://github.com/google/jax (2018).
Hennigan, T., Cai, T., Norman, T., Martens, L. & Babuschkin, I. Haiku: sonnet for JAX. GitHub http://github.com/deepmind/dm-haiku (2020).
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. In Proc. International Conference on Learning Representations (ICLR, 2015).
Ji, Y., Zhou, Z., Liu, H. & Davuluri, R. V. DNABERT: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics 37, 2112–2120 (2021).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Tareen, A. & Kinney, J. B. Logomaker: beautiful sequence logos in Python. Bioinformatics 36, 2272–2274 (2020).
Acknowledgements
We thank A. Kayantao for his help improving the figure panels and B. Mallouli and his team for help on the ChatNT interface. We thank E. Trop, J. Mendoza-Revilla, M. Roller, N. Lopez Carranza, L. Copoi and M. Skwark for helpful discussions. We also thank L. Rosseló for help on the project management side of this research project.
Author information
Authors and Affiliations
Contributions
T.P. conceived the research idea. G.R., B.P.d.A., H.D.-T., C.B., L.H., P.P., S.L. and C.R. performed the analyses. M. Lopez, A.L., M. Lang, U.Ş., K.B. and T.P. provided advice on study design and analyses. G.R., B.P.d.A. and T.P. wrote the paper with input from all co-authors.
Corresponding author
Ethics declarations
Competing interests
G.R., B.P.d.A., H.D-T., M. Lopez, A.L., K.B. and T.P. are employees of InstaDeep. C.B., L.H., P.P., M. Lang and U.Ş. are employees of BioNTech. The other authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Anya Korsakova, Fan Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–6, Tables 1–3 and pseudocode.
Supplementary Code 1
Supplementary pseudocode—pseudocode that describes the model training and inference.
Source data
Source Data (download ZIP )
All genomics instructions datasets prepared for training ChatNT, including the DNA sequences, questions and answers for the train and test sets of each dataset. We also provide all questions and ChatNT answers on the test set sequences used to evaluate its performance on the different tasks.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
de Almeida, B.P., Richard, G., Dalla-Torre, H. et al. A multimodal conversational agent for DNA, RNA and protein tasks. Nat Mach Intell 7, 928–941 (2025). https://doi.org/10.1038/s42256-025-01047-1
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s42256-025-01047-1
This article is cited by
-
The DNA dialect: a comprehensive guide to pretrained genomic language models
Molecular Systems Biology (2026)
-
Computational frameworks for enhanced extracellular vesicle biomarker discovery
Experimental & Molecular Medicine (2026)
-
Multimodal foundation transformer models for multiscale genomics
Nature Methods (2026)
-
Deep generative optimization of mRNA codon sequences for enhanced mRNA translation and therapeutic efficacy
Nature Communications (2025)
-
Benchmarking pre-trained genomic language models for RNA sequence-related predictive applications
Nature Communications (2025)
