
Abstract
Systematic mapping of chemical perturbation responses is revolutionizing polypharmacological drug discovery, yet remains constrained by experimental scalability. Here we introduce XPert, a biologically informed dual-branch transformer model designed to model gene-specific perturbation effects and dose–time dynamics. The dual-branch architecture separately encodes pre-perturbation and post-perturbation cellular states, allowing the model to disentangle intrinsic transcriptional patterns from regulatory shifts triggered by perturbations. By leveraging context-aware gene network modelling, XPert overcomes the over-denoising issues inherent in dominant variational-autoencoder-based approaches, achieving 36.7% higher Pearson’s correlation coefficient and 78.2% lower mean square error in cold-cell generalization under single-dose–single-time scenarios. Through extension to multidose–multitime prediction, XPert precisely resolves pharmacodynamic trajectories and uncovers key molecular events underlying the drug effects. To address real-world data scarcity, we apply knowledge transfer from large-scale preclinical screens to clinical contexts, achieving up to 15.04% improvement in patient-specific response predictions. Furthermore, XPert provides mechanistic interpretability, as evidenced by the identification of clinically validated resistance biomarkers.
Similar content being viewed by others
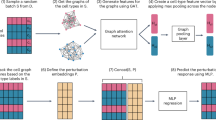

Main
Drug discovery is shifting from the traditional ‘one-drug–one-target’ paradigm to a more complex ‘one-drug–multiple-targets’ framework1, which stems from the realization that drugs often interact with multiple molecular targets and pathways, triggering complex signalling cascades that result in diverse phenotypic outcomes. This shift necessitates understanding genome-wide perturbation effects to elucidate mechanisms of action (MoA) and optimize therapy. To this end, researchers have increasingly turned to large-scale perturbational profiles across compounds, which facilitate the identification of high-affinity targets and context-specific effectors2,3. However, progress remains limited by the scarcity of high-quality perturbation data—particularly in clinical settings—and by confounding factors within perturbation profiles that obscure mechanistic insights.
Deep learning approaches have emerged as powerful tools in this domain, enabling the mapping of broader perturbation spaces from limited observations. To achieve this, a primary challenge lies in effectively representing context-specific pre- and post-perturbation states from noisy high-throughput sequencing data. Early efforts primarily focused on autoencoder-based methods. Pioneering works, such as CPA4 and chemCPA5, leveraged the autoencoder’s denoising capabilities with an adversarial network to decouple and reconstruct the perturbation profiles. Building on this, TranSiGen6 and PRnet7 introduced variational autoencoders (VAEs)8, showing improved generalization across cellular contexts. Although effective at eliminating confounders, these methods risk over-denoising, which can obscure critical biological information. Another line of research, including DeepCE9 and CIGER10, explored attention-based architectures to model gene–drug interactions but overlooked the cellular context, yielding suboptimal results. Although the advent of single-cell large models has advanced the characterization of cellular context11,12,13, they struggle with zero-shot adaptation of the post-treatment space14,15. This motivates our approach, which separately addresses pre- and post-perturbation states.
Another fundamental challenge is how to translate chemical perturbations into biological perturbation signals. State-of-the-art (SOTA) approaches typically concatenate chemical and cellular features, capturing global-cell-state alterations but failing to resolve gene-specific responses. This limitation necessitates advanced fusion strategies that integrate prior knowledge (for example, drug–target interactions (DTIs)) to bridge chemical and biological spaces. A further gap is the inadequate modelling of the well-established dose- and time-dependent nature of drug effects16,17. Previous attempts have relied on simplistic encodings (for example, one-hot encoding), which is insufficient for modelling nonlinear dose–response relationships (for example, inverted U-shaped curves)18, restricting a full understanding of transcriptional pharmacodynamics.
To address these challenges, we introduce XPert, a transformer-based19 solution that predicts drug-induced transcriptional perturbations by jointly modelling cellular contexts, multiscale drug properties and dose–time dynamics. XPert features a dual-branch architecture that concurrently captures intrinsic gene–gene interactions and extrinsic chemical perturbation effects. This architecture is further augmented by a knowledge-informed heterogeneous graph (HG)20 to bridge chemical–biological spaces and by condition tokens that encode nonlinear dose–time responses. XPert excels in diverse scenarios, including generalization to unseen drugs and cells and multi-dose–multi-time (mdmt) predictions. It also supports a pretrain–fine-tune framework that extends large-scale preclinical data for clinical predictions. By enabling precise in silico modelling of perturbation dynamics across different scales, XPert establishes a transformative tool for preclinical drug discovery and personalized therapeutic development.
Results
Overview of XPert
XPert is a transformer-based model designed to predict drug-induced transcriptional perturbation effects. Given the unperturbed cell gene expression and perturbation attributes, it simultaneously outputs the post-perturbation cell expression (xpert) and the difference between post- and pre-perturbation gene expression (xdeg = xpert – xbase).
XPert leverages a dual-branch architecture to encode both pre- and post-perturbation cell states (Fig. 1). The base encoder, built with stacked self-attention layers, models complex gene–gene interactions under diverse cellular contexts, whereas the perturb encoder branch uses cross-attention to capture cell–drug interactions and condition-dependent perturbation effects. Each cell is represented as a ‘sentence’ of gene tokens, along with a \(< \mathrm{cls} >\) token representing the global cell state. Each gene token is initialized with its functional representation and binned expression value, and is then dynamically refined based on regulatory interactions with other genes and the constraints of the perturbation (Methods).

a, Architecture of XPert, featuring a dual-branch framework composed of self-attention and cross-attention modules. XPert receives inputs from both unperturbed gene expression and multiscale drug features, and predicts both change in gene expression (xdeg) and post-perturbation gene expression (xpert). For the drug modality, chemical information is derived from the molecular representation model UniMol, whereas biological information is extracted from a pretrained heterogeneous knowledge graph, along with other tokenized variables such as drug dose and time. b, Evaluation framework used to assess XPert’s performance, which includes five types of metric: error metrics, goodness-of-fit metrics, correlation metrics, distribution metrics and precision metrics. These are applied across three blind scenarios: novel cell lines, unseen drugs and unmeasured dose–time conditions. c, Pretraining and fine-tuning pipeline designed to address data scarcity in clinical applications. XPert is pretrained on large-scale preclinical perturbation datasets (for example, L1000) and then fine-tuned on smaller, clinical datasets (for example, CDS-DB), improving the prediction accuracy for clinical applications. Illustrations in b created with BioRender.com.
XPert also models four key perturbation attributes: drug’s chemical properties, biological properties, perturbation time and dosage. All features are tokenized and fed into the model. Chemical features are extracted using UniMol21, a powerful three-dimensional (3D) molecular model. Biological tokens are derived from a knowledge-informed HG built on the drug’s MoA, encompassing three key relationships: DTI, protein–protein interaction (PPI)22 and drug–drug structure similarity (DDS). Given the sparsity of known DTIs, this graph infers potential drug–gene interactions, informed by two biological intuitions: (1) genes close in the PPI network respond similarly to perturbations and (2) structurally similar drugs often yield comparable effects23. Through unsupervised HG pretraining, XPert bridges the chemical and biological spaces of drugs, generating embeddings that reflect their biological effects. Furthermore, to account for this dose-dependent and time-dependent biological responses of drugs, XPert introduces condition tokens (for example, dose and time) to capture the nonlinear transcriptional effects to varying conditions.
By leveraging intra- and cross-modal attention mechanisms and enhanced by knowledge graph representations, XPert captures the intricate interplay between drugs and genes, leading to more accurate predictions of gene expression changes in response to perturbations.
Benchmarking drug perturbation prediction in single-dose–single-time scenario
We benchmarked XPert against existing methods on the L1000 dataset24, a major resource for studying transcriptomic responses to perturbagens. In this paper, we aim to conduct a comprehensive benchmark of existing methods and XPert based on the L1000 dataset. To ensure a fair comparison against models lacking explicit dose–time modelling, we first focused on a simpler scenario: the single-dose–single-time (sdst) prediction task (Supplementary Table 1). For this, we created the L1000_sdst subset by filtering for the most common perturbation time and dose. We then perform a strict fivefold cross-validation using three split strategies: (1) warm-start: random splits to test generalization to unseen cell–drug pairs; (2) cold-drug: excluding test drugs from training; and (3) cold-cell: excluding test cell lines from training.
Our benchmark includes four SOTA models: two VAE-based models (TranSiGen and PRnet) and two attention-based models19 (DeepCE and CIGER). In particular, both DeepCE and CIGER do not account for the cellular context. All baselines solely focus on drug chemical features. Additionally, we implemented two simple MLP baselines (MLP_UniMol and MLP_Morgan) that differ by their input drug features. Finally, three mean-based baselines (Mean, Meancell and Meandrug) were implemented to assess whether models learned beyond simple population-level averages (Methods).
To facilitate a systematic comparison, we adopted a diverse set of metrics inspired by recent single-cell perturbation benchmarks (for example, ref. 25) covering four categories: error, goodness-of-fit, correlation and distribution metrics (Methods). Metrics were separately computed for two prediction targets: perturbed profile (xpert) and gene expression changes (xdeg). Predicting xdeg is more challenging as it requires capturing subtle transcriptional shifts. To assess this, we also included precision metrics measuring the proportion of correctly predicted top up- and downregulated genes, providing a multifaceted evaluation of model performance (Supplementary Table 2).
XPert consistently outperformed all baselines, particularly in the challenging xdeg prediction task (Fig. 2 and Supplementary Tables 13–15). We attribute this success to its dual-branch design, which effectively learns pre- to post-treatment state changes, a finding validated by ablation studies (Supplementary Note 1). For instance, XPert’s Pearson’s correlation coefficient (PCC) surpassed the next-best model, TranSiGen, by 8.2% (warm-start), 15.9% (cold-drug) and 36.7% (cold-cell). In particular, the context-specific mean baselines (Meancell and Meandrug) were highly competitive, outranking some complex models. Additionally, XPert’s performance was stable across different random seeds (Supplementary Fig. 1 and Supplementary Table 16).

a, Prediction performance for xdeg across various evaluation metrics, including correction metrics (PCC and Spearman), precision metrics (positive precision@20 (Pos P@20), negative precision @20 (Neg P@20)), goodness-of-fit metrics (R2), error metrics (r.m.s.e., m.a.e.) and distribution metrics (Wasserstein and m.m.d.). The bar heights represent the mean performance across five folds, whereas coloured squares indicate the individual values for each fold, reflecting variability across replicates. Statistical significance between XPert and the second-best model in each metric is indicated (***P ≤ 0.001; 0.001 < **P ≤ 0.01; 0.01 < *P ≤ 0.05; n.s., P > 0.05). b, Model ranking in the warm-start and cold-cell scenarios, with different shades of colour representing different metric types and sector size corresponding to model ranking. c, Distribution of xdeg for the top-ten HVGs in the cold-cell (N = 256) and cold-drug (N = 224) settings for EGFR inhibitors. Each box plot illustrates the predicted variability in gene expression across different models. The central line within each box denotes the median; the box limits represent the interquartile range (IQR; from the 25th to 75th percentile); whiskers extend to 1.5× IQR beyond the box limits; and outliers are shown as individual points beyond the whiskers. XPert exhibits the closest approximation to the ground truth, accurately capturing the expression dynamics for key genes in both scenarios.
The cold-cell setting proved the most challenging due to cell-specific drug responses, with an average performance drop of 121% versus the warm-start scenario. Although the performance varied significantly across cell lines (one-way analysis of variance, P < 1 × 10−15), XPert showed the lowest variance, indicating stable predictions (Extended Data Fig. 1 and Supplementary Table 3). This variability correlated partially with cell-line similarity to the training set, as the performance dropped from high- to low-similarity groups (mean square error (m.s.e.) ranged from 0.24 to 0.66). This underscores the difficulty of the out-of-distribution generalization and suggests cell similarity as a potential confidence proxy. Despite this, XPert still achieves an average gain of 67.54% over the current SOTA, TranSiGen, in the cold-cell setting, demonstrating a substantial advance in generalization.
Moreover, our results show an unreported limitation of VAE-based models: a lack of robustness in blind tests relative to attention-based approaches. For example, the leading VAE model, TranSiGen, performed well in warm-start tests but its performance deteriorated in cold-cell settings, scoring negative R2 values despite good correlation (Fig. 2b), suggesting a failure to adapt to unseen cellular contexts. We attribute this failure to two intrinsic VAE properties. First, the Kullback–Leibler divergence regularizer forces information compression that can lead to over-denoising, erasing critical cellular context features needed for gene-specific reconstruction. A typical example is the generation of blurry images in image generation by VAEs26,27,28. Second, VAEs are constrained by their training data, leading to low-fidelity outputs when encountering out-of-distribution samples like unseen cell lines29,30.
Plotting the predicted expression changes of the top-ten highly variable genes (HVGs) in cold settings visually confirms these findings (Fig. 2c). XPert most accurately captured the mean and range of gene expression changes and was the only model to predict correct trends for key genes like AARS and GRN. By contrast, the VAE-based TranSiGen captured the distributional shape but failed on the magnitude of the effect. These validated the strong advantage of XPert in terms of generalization ability.
Knowledge-informed XPert exhibits superior generalization and interpretability
To explore XPert’s learned latent features and mechanisms behind its performance, we further analysed its handling of batch effects31—a key challenge that hinders generalization in a high-throughput sequencing dataset. Unlike prior VAE-based models that rely on denoising, XPert explicitly distinguishes true biological signals from noise in a supervised manner, enhancing cell-specific representation and overall generalization.
We applied uniform manifold approximation and projection (UMAP)32 to project the raw post-treatment expression and the \(< \mathrm{cls} >\) token embeddings obtained from XPert in the test dataset (Fig. 3a). Compared with the raw expression, XPert partially mitigates plate-related noise, aggregating subclusters of the same cell line (for example, HCC515 and HA1E) more cohesively. Quantitative scIB benchmark confirmed its strong biological conservation ability (Supplementary Fig. 2). These highlight that XPert captures intrinsic cell identity features, concurrently preserving the perturbagens-induced biological differences. Furthermore, the model incorporates biologically relevant gene embeddings as prior knowledge, which guides it to focus on gene interrelations over sequencing noise (Fig. 3b).

a, UMAP plots of post-treatment profiles and the \(< {cls} >\) token embeddings obtained from XPert in the test dataset, coloured by cell type and batch ID. XPert’s \(< {cls} >\) embeddings effectively mitigate batch effects, leading to a more cohesive clustering of specific cell types. b, UMAP of gene token embeddings in XPert, coloured by four major Kyoto Encyclopedia of Genes and Genomes pathways. c, UMAP of drug embeddings, coloured by the drug MoA. The top plot shows the drug embeddings obtained from UniMol (representing the chemical space of drugs), whereas the bottom plot uses pretrained HG embeddings (representing the biological space). Drugs with similar MoAs cluster together in biological space rather than chemical space. d, SAR of EGFR inhibitors. The atom weights of two EGFR inhibitors—gefitinib and erlotinib—are displayed, highlighting key substructures and their consistency with the SAR.
XPert further benefits from incorporating drug–gene interaction’s prior knowledge. Although structurally similar drugs may imply similar properties, biological activity often does not directly correlate with the chemical structure. As shown in Fig. 3c, drugs with the same MoAs are dispersed in the chemical space, indicating a limitation of solely relying on chemical features. XPert overcomes this by using a pretrained-knowledge HG to create biologically coherent drug representations, where drugs with the same MoA naturally cluster together. Our ablation studies validate that this prior improves the predictive performance in the cold-drug scenario.
Furthermore, what XPert learns is inherently interpretable due to its reliance on attention mechanisms, which explicitly reveal intramolecular or intracellular interactions. Our analysis of the atom-level attention for several widely used clinical drugs shows that the model learns chemically meaningful local structures that align with known structure–activity relationships (SAR). For instance, with epidermal growth factor receptor (EGFR) inhibitors like gefitinib and erlotinib, XPert highlights the quinazoline ring core and its key hydrogen-bonding N1 and N3 atoms, which are crucial for EGFR binding (Fig. 3d and Extended Data Fig. 2). For histone deacetylase (HDAC) inhibitors, the zinc-binding group receives higher attention, as it chelates the catalytic zinc ion at the active site33. Additional case studies are provided in Supplementary Note 2.
By incorporating the multidimensional biological prior knowledge, XPert enhances its ability to capture the intricate biological mechanisms driving drug perturbations, offering a more comprehensive and interpretable model for drug-induced transcriptional responses.
XPert supports robust transcriptional responses prediction in the mdmt scenario
Understanding dose- and time-dependent responses is fundamental to pharmacodynamic research. Recent advances in an experiment-driven mdmt perturbation study enable the detailed profiling of drug-induced cellular dynamics, providing critical insights into temporal molecular drivers and potential off-target effects at high doses34,35. The complexity of such data presents a rigorous test for predictive models. We, therefore, benchmarked XPert against established baseline methods in this realistic scenario.
For the mdmt scenario, we used the L1000_mdmt subset, which contains drug–cell pairs with varied dose–time points, including 40 cell lines and 1,977 drugs (Methods). As L1000 includes many pharmacologically equivalent doses (for example, 1 μM and 1.11 μM) from minor experimental variations, we avoided coarse methods like one-hot encoding. Instead, we aggregated similar doses into ten discrete ranges and encoded them as conditional tokens, applying a parallel strategy to time attributes. By modelling the interplay between these condition tokens and gene networks, XPert’s context-aware framework captures the complex dose- and time-dependent response patterns.
Similarly, we used the three partition strategies—warm-start, cold-cell and cold-drug—to deploy comparative experiments. It is noteworthy that none of the previous models simultaneously accounted for both dose and time attributes. Some attempts were made by models such as DeepCE and CIGER to encode the perturbation dose using one-hot encoding, whereas PRnet utilized logarithmic doses as the weight of drug’s feature. To adapt the baseline models for the mdmt scenario, we concatenated the one-hot-encoded dose and time features with their standard inputs.
The results confirmed XPert’s dominant performance, as it ranks first in most metrics, followed by TranSiGen and CIGER (Fig. 4, Supplementary Fig. 3 and Supplementary Tables 17–19). For the xdeg task, XPert’s PCC improved on the next-best model by 8.34% (warm-start), 5.85% (cold-drug) and 30.54% (cold-cell). In particular, in the cold-cell scenario, only XPert and DeepCE avoided negative R2 values. A similar phenomenon was observed—although TranSiGen excelled in capturing correlations, it was readily surpassed by XPert and other attention-based models in terms of fitting ability, error and distribution metrics. CIGER’s sharp performance drop in the cold-cell setting highlights the inadequacy of its simple one-hot encoding for cell lines. This observation further underscores XPert’s notable advance in modelling cell-specific responses.

a, Scatter plot compares the performance of various models across different evaluation metrics. Different colours represent distinct metric types, with darker colours and larger points indicating higher rankings. b, Box plots displaying the distribution of performance metrics (PCC, R2 and r.m.s.e.) for each model under warm-start, cold-drug and cold-cell conditions. The bar heights represent the mean performance across five folds, whereas squares indicate the individual values for each fold, reflecting variability across replicates. Statistical significance between XPert and the second-best model in each metric is indicated (***P ≤ 0.001; 0.001 < **P ≤ 0.01; 0.01 < *P ≤ 0.05; n.s., P > 0.05). c–f, mdmt analysis using vorinostat as a case study. c, Heat map of the predicted xdeg for vorinostat across the top-ten cell lines, indexed by the dose range. The top-100 most variable genes are displayed across different dose groups. d, PCA visualization of the predicted xdeg profile for vorinostat, illustrating the dose-dependent gradient. The PCC between the dose index and the principal components (PCs) is shown. The points are coloured by the dose range index, revealing a clear gradient along PC1. e, Line graphs illustrate the change in the predicted xdeg in key biological pathways (for example, apoptosis, cell-cycle arrest, and DNA damage and repair) of vorinostat across different dose ranges, highlighting the model’s ability to predict gene-specific responses. Each line represents the average predicted response of different cell contexts, whereas the shaded areas indicate the standard deviation (s.d.). f, 3D surface plot of the predicted differential expression for specific genes (HDAC6, NRIP1 and TP53) in response to varying doses and perturbation times in the A549 cell line treated with vorinostat.
To assess if XPert captures subtle gene expression changes, we conducted a case study on the vorinostat, a pan-HDAC inhibitor, which has well-documented dose- and time-dependent biological effects36. Its transcriptional response has also been extensively measured in L1000. First, with a fixed 24-h time point, we present the transcriptional impact of vorinostat at different doses across the ten cell lines with the most samples. As shown in Fig. 4c, increasing the dose of vorinostat generally leads to stronger effects on genes. A principal component analysis (PCA) further confirmed this, revealing a clear dose–response gradient along the first principal component (PC1) that strongly correlated with the increasing dose (Fig. 4d).
We also observed that changes in dosage could reverse the transcriptional effects. For instance, increasing the dose of vorinostat shifted genes like NRIP1 and ELOVL6 from upregulation to downregulation. Similar trends were consistently observed across all the cell lines analysed. Moreover, cell-type-specific expression effects were noted, where the drug had opposite effects on the same gene across different cell lines. Crucially, XPert accurately captured these nuanced patterns, consistent with experimental measurements (Supplementary Fig. 4), demonstrating its ability to model complex dose–response relationships in a cell-specific context.
Next, we examine the Δgene expression (xdeg) changes in drug biomarkers under various doses using a set of known pharmacodynamic genes for HDAC inhibitors, covering several critical cellular processes, such as proliferation, apoptosis, metastasis and immunogenicity37. We observed that transcriptional responses are not uniform across doses; different genes respond at different concentrations (Fig. 4e and Supplementary Fig. 4g). For instance, vorinostat downregulates TP53 and alters cell-cycle genes at lower doses, preceding changes in its direct targets, suggesting potential combinatorial therapies at lower doses involving HDAC inhibitors.
To jointly investigate the role of treatment time, we applied radial basis function interpolation to fit dose- and time-dependent effects of specific genes, focusing on the A549 cell line. Genes like HDAC6 and TP53 have both dose- and time-dependent responses, whereas others like NRIP1 are mainly dose-dependent (Fig. 4f and Supplementary Fig. 5). These results underscore the importance of jointly modelling the dose and time to capture transcriptional perturbation dynamics and elucidate drug mechanisms.
Few-shot learning enhances prediction in unseen dose–time condition
A practical challenge in profiling the chemical perturbation response is the high cost associated with measuring multiple time points and doses, which results in datasets that include measurements taken at only a single dose or time point. For example, our analysis of the L1000 dataset indicates that only 6.2% of the cell–drug pairs include mdmt measurements, with most containing only a single dose or time point (Fig. 5a). To address this issue, we propose leveraging large-scale mdmt datasets like L1000 for pretraining, followed by fine tuning with limited target data of specific cell–drug pairs, which can yield high-precision predictions for unmeasured dose–time conditions (Fig. 5b). This is based on that the transferability between dose and time may be easier than that between different drugs and cell contexts, ultimately aiding in the construction of dynamic drug perturbation maps and reducing experimental burdens.

a, Distribution of cell–drug pairs in the L1000 dataset, showing that 93.8% of the drug–cell pairs involve single dose or time measurements, whereas only 6.2% contain the mdmt profiles. b, Schematic of the few-shot learning strategy. The pretraining set includes drug–cell pairs with a single dose or time point, whereas the fine-tuning set involves pairs under mdmt conditions. c, Shaded error plot of PCC for various models under two training settings: training from scratch versus fine tuning, across different fine-tuning data proportions. The solid lines represent the mean PCC averaged over five folds, whereas the shaded error bands indicate the s.d. d, Waterfall plot quantifying PCC improvements of XPert, TranSiGen and CIGER models under different fine-tuning settings (one shot, 20%, 30%, 50% and 80%) compared with the zero-shot setting.
As a proof of concept, we split the complete L1000 dataset into two parts. The L1000_mdmt subset was used as the fine-tuning dataset, whereas the remaining served as the pretraining dataset. Using stratified sampling, we generated five random splits, where for each cell–drug pair, one data point was assigned to the test set and a proportion of the remaining data was used for fine-tuning.
We next compared training from scratch with pretraining–fine-tuning to assess the gains from pretraining. Under both experimental settings, XPert consistently demonstrated optimal performance across all of the metrics (Fig. 5c, Supplementary Fig. 6 and Supplementary Table 20). For xdeg prediction, XPert gained 5.64%–12.45% in PCC under various fine-tuning ratios, with improvements diminishing as the ratio increased. The power of this strategy was the most evident with one-shot fine-tuning, which substantially improved performance over the zero-shot setting for most models (Fig. 5d). For top models like XPert and TranSiGen, one-shot fine tuning matched or surpassed training from scratch on 80% of the dataset. This substantiates our hypothesis that the complete dose–time response landscape for novel cell–drug pairs can be accurately inferred from a minimal set of experimental measurements. However, models like DeepCE and CIGER gained little from pretraining, highlighting the necessity for rational model design to maximize the benefits of few-shot learning.
XPert bridges preclinical datasets to clinical prediction
Given the challenges associated with obtaining clinical perturbation data, we next investigated transferring knowledge from the large-scale L1000 preclinical dataset to smaller, high-fidelity clinical datasets via our pretrain–fine-tune framework. We hypothesized that despite its technical noise, L1000 could serve as a valuable low-fidelity pretraining resource to bolster the model performance in which clinical data are scarce, offering a promising strategy to bridge the preclinical-to-clinical gap.
To test this paradigm, we turned our focus to a clinical dataset—CDS_DB38—which includes paired pre- and post-treatment clinical transcriptomic data from cancer patients. Similar to the preclinical setting, we evaluated three different partitioning strategies: unseen-patient, unseen-drug and unseen-cancer. Given the imbalanced cancer type distribution in CDS_DB (Fig. 6 and Supplementary Fig. 7a–c), we specifically focused on two predominant cancer types: breast cancer and leukaemia.

a, Pie chart showing the proportion of different cancer types within the CDS_DB dataset, with breast cancer (58.2%) and leukaemia (32.0%) as the most prevalent types. b, t-SNE63 plot depicting the pretreatment space of breast cancer subtypes from the CDS_DB and breast cancer cell lines from the L1000 datasets, coloured by the data source and cancer subtypes. c–e, PCC comparison of various models under unseen-patient (c), unseen-drug (d) and unseen-cancer (e) evaluation scenarios. For the unseen-patient setting, results are reported under three settings: pan cancer, breast cancer and leukaemia. For each model, two training strategies are compared: training from scratch and pretraining on the L1000 dataset. Performance gains achieved by the XPert model through pretraining are highlighted in red. Box plots show the distribution of PCC values obtained from 5-fold cross-validation, with the centre line indicating the median, the box representing the IQR (25th to 75th percentile) and whiskers extending to 1.5× IQR. All individual points are shown in coloured squares. f, Violin plot showing the distribution of xdeg for the CDK1 and BUB1B genes comparing ground truth and XPert/XPert (pretrain) predictions. The width of each violin represents the kernel density estimate, and the central white dot indicates the median. g, Volcano plot showing differential attention genes identified by XPert between the non-response (NON) and response (RES) groups. The red points represent genes with significantly increased attention in the non-response group, suggesting potential drug-resistance-related genes, whereas blue points highlight those with decreased attention.
Surprisingly, despite a notable domain shift between preclinical and clinical data (Supplementary Fig. 7d–f), pretraining consistently enhanced the prediction for unseen patients. Specifically, XPert achieved performance gains of 2.51% for pan cancer, 15.04% for breast cancer and 12.58% for leukaemia (Fig. 6c–e, Supplementary Fig. 8 and Supplementary Tables 21–25). The limited gain in pan cancer probably reflects that most cancer types had very few samples (<20) for fine tuning, constraining overall performance.
Both XPert and XPert (pretrain) accurately predict the distribution of xdeg in unseen patients; however, XPert (pretrain) demonstrates a more precise capture of extreme values, exhibiting lower error on genes with large expression changes, as confirmed by our stratified analysis (Fig. 6f, Supplementary Fig. 9 and Supplementary Table 4). This demonstrates that deep learning can learn transferable representations from preclinical data that are effectively refined using only a few clinical profiles. This strategy is, therefore, promising for developing specialized models for specific cancer types.
Moreover, XPert is the only model benefitting from pretraining in both the unseen-drug and unseen-cancer settings. We attribute this advantage to XPert’s mechanistic foundation in learning fine-grained drug–gene interaction patterns, which enables a seamless transfer of pretrained pharmacological knowledge across the preclinical-to-clinical domain by focusing on conserved interaction mechanisms rather than context-specific patterns. This highlights XPert’s inherent strength in navigating clinical heterogeneity and facilitating the transfer learning from preclinical-to-clinical applications.
Further, we explored the link between drug-induced transcriptomic changes and clinical responses. Our focus was on a subset of CDS_DB, specifically GSE20181, which provides records of patient responses to letrozole treatment. In particular, responders exhibited a stronger transcriptomic response than non-responders, characterized by a more pronounced long-tail distribution of xdeg and a greater number of enriched HVGs (17 for the response group versus 6 for the non-response group; Supplementary Fig. 11a–c). This motivated our exploration of patients’ pretreatment states to identify the key drivers of drug resistance.
To explore this, we conducted additional analyses using the gene-level attention scores captured by the base encoder of XPert. Only a subset of genes showed notable intersample variability and attention patterns remained stable across folds (Supplementary Fig. 10). We next performed a differential attention analysis between two response groups, comparing it with conventional differential expression analysis. FGFR2 was enriched in both analyses (Fig. 6g and Supplementary Fig. 11d), consistent with its reported role in enhancing the PI3K/AKT pathway and promoting antioestrogen resistance in breast cancer39. More importantly, our attention-based analysis uniquely identified other key resistance biomarkers, such as TIAM1 (ref. 40), RPCP41, HK1 (ref. 42) and CDKN1B43 that were invisible to the expression-level analysis. These results underscore the power of attention-based methods to reveal latent gene–phenotype associations beyond mere expression changes, thereby providing a new layer of insight into drug resistance mechanisms.
Discussion
In this study, we introduce XPert, a knowledge-guided, dual-branch attention framework for predicting drug-induced transcriptional responses. Through a comprehensive evaluation on both xpert and xdeg prediction tasks across multidimensional metrics, our results highlight the exceptional capability of attention-based frameworks in context-aware cellular modelling. When tasked with inferring responses in unseen cellular states, XPert outperformed the next-best model by an average of 67.54% across all metrics in the sdst scenario. Moreover, our analysis reveals a previously underappreciated limitation of the dominant VAE-based approaches—excessive overcorrection that obscures cellular context in blind-test scenarios—resulting in substantial deficits in both error metrics and expression-change distribution fidelity.
Another pivotal contribution of this work lies in addressing dose–time dynamics in drug-induced transcriptional effects. We propose a universal encoding method for perturbation attributes (for example, dose and time), enabling the interpretable modelling of nonlinear pharmacodynamic relationships. In this regard, XPert represents the most effective framework currently available for mdmt scenarios. We demonstrated this by generating gene-specific 3D dose–time response maps (for example, for vorinostat) that reveal dynamic gene network reorganization induced by chemical perturbations. Furthermore, our proof-of-concept experiments establish that with few-shot learning strategies, deep learning algorithms can assist in constructing more comprehensive dynamic maps by interpolating unmeasured dose–time conditions. This approach promises to substantially reduce the experimental burden and accelerate the construction of large-scale perturbation omics landscapes.
Beyond that, we extend the XPert framework by applying transfer learning to overcome clinical data scarcity and translational roadblocks in perturbation studies. By modelling conserved drug–gene interactions, it enables reliable knowledge transfer from larger-scale preclinical screens to patient transcriptomes, supporting personalized response prediction and biomarker identification. To ensure completeness, we extended this strategy to PANACEA44, an independent preclinical dataset. Despite discrepancies in measured perturbation signals that can induce negative transfer, XPert demonstrated superior robustness than other models (Supplementary Note 3).
Although XPert demonstrates strong performance, we identify key avenues for future development. One limitation is its computational cost, which could be mitigated by memory-efficient training strategies (for example, DeepSpeed45) and scalable architectures like Hyena46 designed for long-range dependencies (Supplementary Fig. 12 and Supplementary Table 27). Biologically, extending the framework’s scope is the primary goal. This includes transitioning from bulk-level to single-cell-level predictions as large-scale datasets become available47,48,49, and broadening the model beyond small-molecule transcriptional effects to encompass biologics, genetic perturbations and multiomics integration, contingent on future data availability2,50.
In summary, XPert represents a substantial step forward in modelling drug-induced perturbation effects through an interpretable and generalizable deep learning framework. With further development, XPert holds substantial promise as a core component of the next-generation in silico drug discovery pipelines and precision medicine platforms.
Methods
Dataset preprocessing
To systematically evaluate the performance of XPert and SOTA models in drug perturbation prediction, we utilized three benchmark datasets, including two preclinical datasets—LINCS L1000 (referred to as L1000) and PANACEA—as well as one clinical dataset, namely, the cancer-drug-induced gene expression signature database (CDS-DB).
LINCS L1000 dataset
The L1000 dataset24, a widely used resource for studying thousands of perturbagens in human cells, contains gene expression profiles resulting from various drug treatments across different cell lines. The LINCS L1000 data are organized into five levels at different stages of the analysis pipeline. In line with previous studies, we extracted the gene expression data of drug-induced perturbations and control samples from the L1000 level-3 data. The L1000 platform measures the mRNA transcript abundance of 978 ‘landmark’ genes, which are believed to capture approximately 80% of the information in the entire transcriptome. The transcriptional changes in these 978 genes serve as the prediction target in this study.
Data cleaning was performed to remove low-quality data, following several key steps: (1) perturbations with missing or ambiguous information were excluded; (2) profiles with low-frequency perturbation time points were removed, retaining only those with perturbation times of 3 h, 6 h or 24 h; (3) profiles that did not pass quality control were filtered out.
Subsequently, we matched each expression profile with a randomly selected dimethyl sulfoxide control sample from the same plate to create paired pre-/post-treatment profiles. Then, replicate-collapsed z-score vectors were computed to derive the unique features for each perturbation condition.
On the basis of the experimental setup, we performed further data cleaning on the L1000 dataset, resulting in several subsets, as described below. More details are provided in Supplementary Table 1:
-
(1)
L1000_full: the complete L1000 dataset after the aforementioned cleaning process
-
(2)
L1000_sdst: a subset retaining only the most common condition, with a perturbation dose of 10 µM and a perturbation time of 24 h
-
(3)
L1000_mdmt: a subset that includes profiles with multiple perturbation times and doses for each cell–drug pair
-
(4)
L1000_mdmt_pretrain: derived from L1000_full by excluding the profiles in L1000_mdmt
In particular, due to the presence of thousands of perturbation doses in the raw L1000 dataset, we grouped these doses into ten discrete dose intervals. This step was taken to facilitate standardization, unifying highly similar doses that are biologically indistinguishable (for example, 10 µM and 10.01 µM). Although such binning is advantageous for data harmonization and cross-dataset alignment, a potential limitation is that it may obscure subtle, fine-grained dose–response relationships; therefore, the choice of binning granularity should be tailored to the specific downstream task and research objective. The mapping between original doses and their corresponding dose intervals is provided in Supplementary Table 6.
PANACEA
PANACEA44 is a resource developed by the Columbia Cancer Target Discovery and Development Center, which includes dose–response and RNA-sequencing profiles for 25 cell lines exposed to approximately 400 clinical oncology drugs. The dataset focuses on understanding tumour-specific drug MoA. It includes perturbational profiles for 32 kinase inhibitors and 11 distinct cell lines representing molecularly diverse tumour subtypes, with each perturbation performed in triplicates. The experimental conditions are standardized with each drug administered at its IC20 dose for 24 h.
For RNA-sequencing raw counts, the data are processed by calculating log2[TPM + 1], and the final features are filtered based on the 978 landmark genes from the L1000 database. To ensure consistency, the dose of each small-molecule drug is mapped to one of the ten predefined dose ranges in the L1000 database, with the corresponding dose-to-intervals mapping provided in Supplementary Table 5. All biological replicates are averaged to generate unique profiles for each perturbation condition.
CDS-DB
CDS-DB38 is a unique and comprehensive resource that provides patient-derived paired pre- and post-treatment clinical transcriptomic data. It encompasses 78 treatment-specific transcriptomic datasets, covering 85 therapeutic regimens, 39 cancer subtypes and 3,628 patient samples. The CDS-DB contains data from two different sequencing technologies—microarray and RNA-sequencing—which undergo distinct data preprocessing methods and batch effect removal procedures. To mitigate potential biases introduced by platform differences, we retained only the microarray data, which had a larger sample size.
Then, we excluded samples involving combination therapies or non-chemical drugs to maintain focus on single-agent treatments. Finally, we obtained a final dataset consisting of 613 paired profiles, representing 14 cancer subtypes and 14 different drugs. All profiles were restricted to the 978 landmark genes from the L1000 database.
Given the noteworthy variability in clinical treatment protocols, we standardized the administration dosage and treatment time into unified intervals for different therapeutic regimens. This step reduces heterogeneity in the dataset and ensures comparability across different studies. The mapping details are provided in Supplementary Tables 6 and 7.
Transcript profile embedding
Inspired by the application of transformer architectures in single-cell large language models, we adopt a similar strategy to encode gene expression profiles for pre-perturbation cells. In this context, each cell is analogous to a ‘sentence’ composed of genes, together with a special token \(< \mathrm{cls} >\) that captures the global state of each cell. Specifically, we define a transcriptomic data structure as a tensor \({X} \in {{R}}^{{N} \times ({M}+{1}) \times {d}}\), where N is the number of cells, M is the number of genes and \(d\) is the embedding dimension. For each cell i, the structure consists of two components: (1) input gene embeddings (\(\in {R}^{M\times d}\)), where each element xi, j encodes the embedding of gene j in cell i, and (2) cell embedding (\(\in {R}^{1\times d}\)), represented by the \(< \mathrm{cls} >\) token. Concatenating these two parts yields the final input representation for cell i (\({C}_{i}\in {R}^{(M+1)\times d}\)), as detailed in the following subsections.
Input gene embedding
The input for gene j consists of two components: (1) gene token (\({g}_{j}\)) and (2) binned expression value (\({e}_{j}\)).
Gene tokens (\({g}_{j}\)): similar to word tokens in natural language processing51, in the XPert framework, we utilize biologically meaningful gene embeddings as gene tokens (functional representation of gene signatures). Specifically, we leverage predefined gene token embeddings from the CellLM52 model, which uses GraphMAE53 to extract these gene embeddings from the PPI network, forming a gene vocabulary in a biologically meaningful manner. Although we focus on 978 landmark genes in this study, this method offers flexibility and can harmonize gene sets across multiple studies, enabling broad application across different datasets.
Binned expression values (\({e}_{j}\)): to address the challenges posed by variability in absolute magnitudes across different sequencing protocols, we apply a value binning technique, as proposed in scGPT12, to convert all expression counts into relative values. For each non-zero expression value in each cell, we calculate the raw absolute values and assign them to B consecutive intervals \(\left[{b}_{k},{b}_{k+1}\right]\), where \(k\in \{1,2\ldots B\}\). Since large datasets like L1000 have already undergone transformation and batch removal steps, the bin edges are shared across all cells in the dataset, rather than varying across individual cells. However, to account for differences across datasets, bin edges should be recalculated when applying the method to new datasets. Through this binning technique, the semantic meaning of \({e}_{i}\) remains consistent across cells from different datasets.
We then introduce PyTorch embedding layers (https://pytorch.org/docs/stable/generated/torch.nn.Embedding.html) to represent the gene tokens and binned expression values, denoted as embg and embe, respectively. Each token is mapped to a fixed-length embedding vector of dimension \(d\).
The gene embedding for gene \(j\) can, thus, be expressed as
Cell embedding
In addition to the gene tokens, we introduce a special \(\bf < \mathrm{cls} >\) token to represent the overall cell state, which aggregates the learned gene-level representations during model training. The \(< \mathrm{cls} >\) token is initialized with a Gaussian distribution and is appended to the beginning of the sequence of gene tokens.
Therefore, the final input embedding for the entire cell \({C}_{i}\in {R}^{(M+1)\times d}\) is constructed by concatenating the embeddings of \(< \mathrm{cls} >\) token (Ccls) and gene tokens:
where M is the fixed number of genes for each profile.
Drug tokenization
The transformer architecture requires tokenized features as input. For drugs, we consider two intrinsic features—chemical properties and biological effects—as well as additional condition tokens to represent perturbation covariates (for example, dose and time).
Chemical tokens
UniMol21 is a universal 3D molecular pretraining framework aimed at enhancing the representation capacity and broadening applications in drug design. It leverages a transformer-based model trained on 209 million molecular 3D conformations, outperforming SOTA methods. The model processes atom types and coordinates as inputs, using a self-attention mechanism to enable effective communication between representations, ultimately yielding robust molecular features.
Given the superiority of UniMol in representing 3D chemical structures, in the XPert architecture, we use UniMol to derive chemical tokens for each drug. Specifically, for each drug molecule, we first convert its SMILES string into canonical SMILES using the RDKit54 package. Atom types are extracted via RDKit’s GetAtoms function as UniMol inputs. The pretrained molecular model generates a mol token (global representation) and atom tokens (local representations), both encoded as 512-dimensional vectors. These are projected onto \(d\) dimensions via a linear transformation layer. For drug \(j\), the chemical tokens form a matrix \(X\in {{{R}}}^{\left(N+1\right)\times d}\), where N denotes the preset maximum atom count (default: 120).
Although UniMol serves as the default chemical representation in XPert, we additionally evaluated the model with two alternative, widely used molecular features: Morgan Fingerprints (two-dimensional molecular descriptors, 1,024 dimensions) and KPGT molecular fingerprints (one-dimensional/two-dimensional neural fingerprints, 2,304 dimensions). Fivefold cross-validation on the L1000_sdst subset showed that XPert’s performance remained robust across different molecular features (Supplementary Table 26). This indicates that users can flexibly customize the choice of chemical representations, still fully leveraging the advantages of the XPert architecture.
Biological token based on prior-knowledge HG
There is a gap between drugs’ chemical space and their biological effect space. Chemical tokens are limited to representing features at the biological aspect. Given that DTIs are a reliable source of drug MoAs, we propose incorporating DTI information as prior knowledge to enhance the biological token representation. However, known DTIs are sparsely annotated (only 12,890 known interactions among 8,981 drugs obtained in our datasets and 19,392 proteins)24,55. Inspired by recent studies56,57, which constructed heterogeneous knowledge graphs to capture hidden relationships between drugs and proteins/genes, we adopt a similar methodology.
In addition to DTIs, we consider two other relationships: DDS and PPI. For DDS, we compute the Tanimoto similarity between all pairs of drugs using the RDKit package. Drug nodes with a Tanimoto similarity above 0.5 are connected, with the similarity value used as the edge weight. For PPIs, we obtain data from the STRING database22, retaining high-confidence edges (with a score greater than 700) and transforming the score \((\frac{\mathrm{score}}{1,000})\) as the edge weight. Drug nodes are initialized with UniMol \(< \mathrm{mol} >\) token embeddings, whereas protein nodes use the PPI-derived gene embeddings. To provide a clear overview of the graph structure, we report quantitative statistics of the knowledge HG, including the number of nodes, edges per relation type (DTI, DDS and PPI) and overall graph sparsity (Supplementary Table 8).
Next, we leverage a commonly used heterogeneous graph neural network model under an unsupervised contrastive learning framework to learn latent relationships between heterogeneous nodes. The heterogeneous graph neural network model consists of three HeteroConv layers constructed with SAGEConv in PyTorch Geometric, allowing message passing across different edge types. For training, we adopt a mini-batch neighbour sampling strategy to balance memory efficiency and coverage; here for each target node, a fixed number of neighbours is sampled per layer (25, 10 and 5 for the first, second and third layers, respectively). The model is optimized using Adam, with an early stopping criterion based on the validation loss. The full set of hyperparameters and training configurations is provided in Supplementary Table 9.
Positive and negative edge pairs are constructed for each relation type to enable contrastive learning, where connected pairs are treated as positives and randomly sampled non-neighbours serve as negatives. The contrastive loss was implemented following the InfoNCE formulation, with the training objective to maximize the similarity between embeddings of positive pairs and minimizing it for negative pairs.
Specifically, for each positive edge \({\left(u,v\right)}^{+}\), we sampled multiple negative pairs \({\left(u,{v}^{-}\right)}^{-}\) by replacing the target node \(v\) with non-neighbours of the source node \(u\). Let \(\mathbf{h}_{u}\) and \(\mathbf{h}_{v}\) denote the embeddings of nodes \(u\) and \(v\), respectively. The cosine similarity is scaled by a temperature parameter \(\tau\):
The probability of a positive pair being correctly identified is then
The overall loss is defined as
which encourages the embeddings of connected nodes to be close, explicitly pushing apart negative pairs.
Here N denotes the number of positive edges, \({\left(u,v\right)}^{+}\) indicates a positive node pair connected in the HG and \({\left(u,{v}^{-}\right)}^{-}\) represents negative samples obtained by randomly sampling non-neighbour nodes. \(\mathbf{h}_{u}\) and \(\mathbf{h}_{v}\) are the embedding vectors of nodes \(u\) and \(v\), respectively, and the trained model outputs \(d\)-dimensional biological token vectors for drugs.
Condition tokens
Condition tokens encode other perturbation covariates (for example, dose and time). One challenge lies in the diversity of drug dosages and protocol variability across datasets. To propose a unified tokenization strategy, we discretize raw values into predefined ranges (Supplementary Tables 5 and 6), preserving relative differences and reducing complexity. This discretization enables cross-dataset covariate normalization and mitigates scale inconsistencies. For example, preclinical and clinical doses are mapped by aligning their minimum effective ranges.
Integration of tokens
For drug \(j\), all tokens are concatenated as
For each drug, these tokens are arranged in fixed order. We then introduce learnable positional embeddings to preserve sequential relationships of each token. Using PyTorch embedding layers, positional embeddings \({E}^{\mathrm{pos}}\in {{{R}}}^{L\times d}\) (where L is the total token length) are summed in an element-wise manner with the drug tokens to produce the final input features D. Although XPert uses learnable embeddings by default, we note that fixed alternatives, such as sinusoidal positional encoding, achieve comparable performance (Supplementary Table 10).
XPert architecture overview
The XPert model is a transformer-based architecture designed to predict drug-induced transcriptional perturbations. This architecture is composed of two primary encoder branches: the base encoder branch and the Perturbation (Pert) encoder branch, designed to simultaneously encode pretreatment cellular states and drug-induced perturbation effects on gene expression.
Base encoder branch
The base encoder captures the unperturbed state of the cell by learning the dependencies between genes within the cell. It utilizes stacked self-attention layers to iteratively process the initial gene expression representation of the unperturbed cell. Given the initial representation \({C}^{\mathrm{base}}\in {{{R}}}^{\left(k+1\right)\times d}\), the encoder sequentially applies self-attention blocks across \(n\) layers:
The final output \({{C}_{n}}^{\mathrm{base}}\in {{{R}}}^{\left(k+1\right)\times d}\) represents the unperturbed cell state after \(n\) layers of self-attention.
Pert encoder branch
The Pert encoder is responsible for integrating drug molecular features with cellular context through cascaded cross-attention and self-attention layers. The cross-attention module explicitly models gene-level perturbation effects by aligning the multimodal drug representation with cellular-state features. Subsequent self-attention layers refine these interaction patterns and maintain the positional awareness of key regulatory genes.
In the cross-attention layers, the cell representation is treated as the query, and tokenized drug representation serves as the key and value matrix. This allows the model to learn gene-level perturbation effects induced by the drug. After \(m\) layers of cross-attention and self-attention, the final perturbed cell state \({{C}_{m}}^{\mathrm{pert}}\) is obtained:
Multiobjective learning
XPert uses a multiobjective learning approach, where three distinct prediction tasks are jointly optimized, including two gene-level tasks and one cell-level task.
Perturbation gene expression prediction (\({{x}}_{\mathrm{pert}}\)): the perturbation predictor is a multilayer perceptron (MLP) that uses the perturbed representation \({{C}_{n}}^{\mathrm{pert}}\) to predict the gene expression values \({x}_{\mathrm{pert}}\) after drug treatment:
The optimization objective is to minimize the mean square error (m.s.e.) loss between the ground-truth (\({x}_{\mathrm{pert}}\)) and predicted gene expression (\({\hat{x}}_{\mathrm{pert}}\)) after perturbation:
where \(\alpha\) is a weighting coefficient.
Gene expression delta prediction (xdeg): the gene expression delta predictor uses the difference between the pre-perturbation and post-perturbation gene representations \({{C}_{n}}^{\mathrm{pert}}-{{C}_{n}}^{\mathrm{base}}\) to estimate the differential gene expression values: xdeg = xpert – xbase. Here xdeg denotes the differential gene expression vector that captures the element-wise difference between the post-perturbation expression profile xpert and the baseline profile xbase of all the profiled genes. The loss for this task is a combination of m.s.e. and PCC losses. By incorporating the PCC loss, the model is encouraged to not only minimize the absolute differences between predictions and ground truth but also to capture the underlying correlation structure, leading to more accurate and biologically meaningful predictions.
where \(\beta\) and \(\gamma\) are weighting coefficients, and \({\hat{x}}_{{\text{deg}}}\) is the predicted differential gene expression value.
Cell-type classification: to alleviate batch effects and enhance the model’s ability to distinguish cell contexts, we introduce an auxiliary task that aims to classify the cell type based on the \(< \mathrm{cls} >\) token representations of \({{C}_{n}}^{\mathrm{pert}}\) and \({{C}_{n}}^{\mathrm{base}}\) via an added classifier. The classification task is guided by a multiclass cross-entropy loss58:
where \({y}_{\mathrm{true}}\) represents the true cell-type labels and \({y}_{\mathrm{pred}}\) are the predicted labels; \(\delta\) is the weight of the multiclass task loss.
We further performed ablation experiments to examine the effect of individual loss components (Supplementary Note 1).
Training and testing
The training objective of XPert is to minimize the weighted sum of the losses for each task:
XPert is implemented in a PyTorch framework. For optimization, we use the Adam optimizer with an initial learning rate of 4 × 10−3 and a weight decay of 1 × 10−5. To facilitate more stable convergence, we use a learning rate scheduler (LambdaLR) that adjusts the learning rate dynamically. Specifically, the learning rate is reduced by a factor of 0.5 after a predetermined number of warm-up epochs. Early stopping59 is also adopted, where training is terminated if the validation loss plateaus for 50 consecutive epochs to avoid overfitting.
Additionally, we leverage flash attention to speed up attention computation and optimize the GPU memory. This optimization is particularly advantageous for transformer-based models like XPert, especially when handling long input sequences of gene tokens, enabling seamless scalability to larger-scale gene modelling tasks.
We perform random hyperparameter search on the training set to identify the optimal combination of parameters. Supplementary Table 11 outlines the range of values and default values for each hyperparameter. The same set of hyperparameters is consistently applied across all dataset splits and datasets. On the basis of empirical evidence, usually, the default values yield satisfactory results for XPert. However, when adapting XPert to new datasets, we recommend considering larger batch sizes and more attention layers for larger datasets, reducing these parameters for smaller datasets. Additionally, experimenting with different learning rates and learning rate schedulers is advised, as XPert exhibits sensitivity to these settings.
To train and test XPert, all datasets are strictly split using fivefold cross-validation based on different perturbation attributes. A total of four split strategies are adopted:
-
(1)
warm-start: random splitting of the dataset, with a training-to-testing ratio of 4:1 for profiles
-
(2)
cold-drug: grouping the datasets by drug categories, with a training-to-testing ratio of 4:1 for drug types
-
(3)
cold-cell: grouping the datasets by cell line for each profile, with a training-to-testing ratio of 4:1 for cell lines or disease types
-
(4)
cold-dose–time: for each unique drug–cell line pair, partitioning the data based on dose–time attributes
For the L1000_sdst, PANACEA and CDS_DB datasets, the warm-start, cold-drug and cold-cell strategies are applied. For the L1000_mdmt dataset, all four split strategies are utilized.
Pretraining and fine-tuning
The pretraining step aims to equip the model with the ability to learn generalizable patterns related to cellular states, drug properties and perturbation effects using a large-scale dataset. In our setup, two datasets were used for pretraining. To assess the model’s ability to generalize across unseen dose–time conditions, we utilized the L1000_mdmt_pretrain dataset. For evaluating the model’s adaptability to independent datasets (PANACEA and CDS-DB), we used the complete L1000 dataset (L1000_mdmt_full) for pretraining. To ensure a fair comparison, all the evaluated models underwent full-parameter fine-tuning. Once pretrained, the model was fine-tuned on downstream datasets to adapt its learned representations to the specific context of the target dataset.
Implementation details
The XPert model was implemented using PyTorch (v. 2.1) as the deep learning framework. Data handling and preprocessing were performed with Scanpy. Key dependencies include torch-geometric (v. 2.6.1), torchmetrics (v. 1.6.0) and flash_attn (v. 2.6.0.post1), among others. The model was trained on an NVIDIA 4090 GPU to ensure efficient computation and faster convergence. Training on the L1000_sdst dataset took approximately 10 h, whereas the L1000_full dataset required around 60 h to fully converge.
Mean baseline models
To establish a fundamental performance benchmark and to contextualize the contributions of more complex deep learning architectures, we incorporated three mean-based baseline models. These simple yet informative baselines are designed to assess whether a model learns to predict perturbation-specific gene expression changes under multiple cell contexts beyond capturing an average expression profile, either globally or conditioned on a specific context (that is, cell line or drug).
Specifically, we considered three mean baselines:
-
(1)
Global mean baseline (Mean): following the implementation in prior work60, the prediction for each test sample is given by the mean expression profile across all training data, including both perturbed and control samples.
-
(2)
Cell-specific mean baseline (Meancell): for a given test sample, the prediction is the average expression profile of all training samples belonging to the same cell line.
-
(3)
Drug-specific mean baseline (Meandrug): for a given test sample, the prediction is the average expression profile of all training samples treated with the same drug.
For the warm-start setting, all three baselines were included. For the cold-cell (cold-cancer) setting, only Mean and Meandrug were applicable. For the cold-drug setting, only Mean and Meancell were used.
Evaluation metrics
To facilitate a systematic and comprehensive comparison of XPert with other SOTA models, we refer to benchmark studies such as ref. 25, which evaluate performance using a variety of metrics. In this work, we consider a total of ten evaluation metrics, classified into four categories: error metrics (for example, mean squared error (m.s.e.), root mean squared error (r.m.s.e.) and mean absolute error (m.a.e.)), goodness-of-fit metrics (for example, R2), correlation metrics (for example, PCC and Spearman’s correlation (Spearman)) and distributional similarity metrics (for example, Wasserstein distance (Wasserstein) and maximum mean discrepancy (m.m.d.)). These metrics collectively provide a robust assessment of model performance in terms of prediction accuracy, statistical alignment and distributional consistency (Supplementary Table 2 lists the abbreviations of all metrics).
Error metrics
-
1.
m.s.e.: m.s.e. measures the average squared differences between the actual and predicted values. The formula is defined as
$$\begin{array}{l}{\rm{m}}.{\rm{s}}.{\rm{e}}.=\frac{1}{n}\mathop{\sum }\limits_{i=1}^{n}{\left({y}_{i}-{\hat{y}}_{i}\right)}^{2}\end{array},$$(17)where \({y}_{i}\) is the actual value, \({\hat{y}}_{i}\) is the predicted value and \(n\) is the number of samples. Lower m.s.e. values indicate that the model’s predictions are closer to the true values.
r.m.s.e.: r.m.s.e. is the square root of the m.s.e., providing a measure of prediction accuracy in the same units as the original data. It penalizes larger errors more heavily due to the squaring of differences. The formula is
$$\begin{array}{l}\text{r.m.s.e.}=\sqrt{\frac{1}{n}{\sum }_{i=1}^{n}{\left({y}_{i}-{\hat{y}}_{{{i}}}\right)}^{2}}\end{array}.$$(18) -
2.
m.a.e.: m.a.e. computes the average of the absolute differences between the actual and predicted values. m.a.e. provides a straightforward measure of the average magnitude of errors in the predictions. The formula is
Goodness-of-fit metrics
-
1.
R2: R2 quantifies the proportion of variance in the dependent variable that is predictable from the independent variables, which measures how well the predicted values fit the actual data. It is a dimensionless number between 0 and 1, where higher values indicate a better fit of the model to the data. It is calculated as
where \(\bar{y}\) is the mean of the actual values.
Correlation metrics
-
1.
PCC: PCC measures the linear relationship between two variables. It ranges from –1 to 1, where 1 indicates a perfect positive linear correlation, –1 indicates a perfect negative linear correlation and 0 indicates no linear correlation. The formula is
$$\begin{array}{l}\mathrm{PCC}=\frac{{\sum }_{i=1}^{n}\left({y}_{i}-\bar{y}\right)\left(\hat{{y}_{i}}-\overline{\hat{y}}\right)}{\sqrt{{\sum }_{i=1}^{n}{\left({y}_{i}-\bar{y}\right)}^{2}{\sum }_{i=1}^{n}{\left(\hat{{y}_{i}}-\overline{\hat{y}}\right)}^{2}}},\end{array}$$(21)where \(\bar{y}\) and \(\bar{\hat{y}}\) are the means of the actual and predicted values, respectively.
-
2.
Spearman’s rank correlation coefficient (Spearman’s \(\rho\)): Spearman evaluates the monotonic relationship between two variables by ranking the data points and computes the Pearson correlation on the ranks. It is defined as
where \({d}_{i}\) is the difference between the ranks of corresponding values \({y}_{i}\) and \({\hat{y}}_{i}\), and \(n\) is the number of samples.
Distributional similarity metrics
-
1.
m.m.d. quantifies the difference between two distributions based on their embeddings in a reproducing kernel Hilbert space. It is suitable for assessing distributional differences in high-dimensional spaces. The formula for m.m.d. is
$$\begin{array}{l}{\text{m.m.d.}}^{2}={E}_{{y}_{i},{y}_{i}^{{\prime} }}\left[k\left({y}_{i},{y}_{i}^{{\prime} }\right)\right]+{E}_{{\hat{y}}_{i},{\hat{y}}_{i}^{{\prime} }}\left[k\left({\hat{y}}_{i},{\hat{y}}_{i}^{{\prime} }\right)\right]-2{E}_{{y}_{i},{\hat{y}}_{i}}\left[k\left({y}_{i},{\hat{y}}_{i}\right)\right]\end{array},$$(23)where \({y}_{i}\) and \({y}_{i}^{{\prime} }\) are samples from the actual and predicted distributions; \({\hat{y}}_{i}\) and \({\hat{y}}_{i}^{{\prime} }\) are samples from the predicted distributions. \(k\left({y}_{i},{\hat{y}}_{i}\right)\) is a kernel function, and we use the radial basis function kernel in this study. Smaller m.m.d. values indicate that the distributions of actual and predicted values are more similar.
-
2.
Wasserstein: the Wasserstein distance measures the difference between two probability distributions. In the context of model evaluation, it measures the ‘cost’ of transforming the predicted distribution into the actual distribution. For two probability distributions \(P\) and \(Q\), the formula is given by
where \(P\) and \(Q\) are the probability distributions of the actual and predicted values, respectively, and \(\Pi (P,Q)\) represents the set of all possible joint distributions with marginals \(P\) and \(Q\).
Precision metrics
To evaluate the model’s ability to capture differentially expressed genes (xdeg), we use precision metrics, including both positive and negative precision@K (Pos/Neg P@K), which measures the fraction of intersection between the top-K up- or downregulated genes predicted by the model and the ground truth. The formulas are as follows:
where \({G}_{K}\) represents the sets of top-K up- or downregulated genes in the ground truth and \({G\prime}_{K}\) represents the predicted top-K up- or downregulated genes. \(|\cdot |\) denotes the cardinality of a set.
UMAP and t-distributed stochastic neighbour embedding visualizations
For visualization, we first applied PCA to reduce the profile dimensionality to 40, followed by UMAP or t-distributed stochastic neighbour embedding (t-SNE) to project data into two dimensions, enabling interpretation by cell types, batch indices or other labels. For UMAP, a k-nearest-neighbour graph was constructed on principal components using k = 15 neighbours.
Statistics and reproducibility
For model performance evaluation, a paired t-test was conducted to compare the differences between XPert and baseline models under different experimental conditions. For differential gene expression analysis, a two-sample t-test was used to assess the significance of the differences between two groups (treatment versus control, response versus non-response). Detailed descriptions are provided in the figure legends. The significance level was set as ***P ≤ 0.001; 0.001 < **P ≤ 0.01; 0.01 < *P ≤ 0.05; n.s., P > 0.05.
Data availability
For the L1000 dataset, we obtained its level-3 data from the raw expanded CMap LINCS Resource 2020 (ref. 24) available at https://clue.io/data/CMap2020#LINCS2020. For PANACEA44, raw data are obtained from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE186341. For CDS_DB38, data are collected from the official website at http://cdsdb.ncpsb.org.cn/. Our processed datasets can be accessed via figshare (https://doi.org/10.6084/m9.figshare.28955141)61 and Zenodo (https://doi.org/10.5281/zenodo.15357711)62. Furthermore, we also provide a minimal dataset on GitHub (https://github.com/GSanShui/XPert) to facilitate interpretation, verification and extension of the method. Source data are provided with this paper.
Code availability
The source code of XPert, including scripts for data preprocessing, model training and evaluation, and the code for reproducing the main results and figures presented in this study are publicly available via Zenodo (https://doi.org/10.5281/zenodo.15357711)62, figshare (https://doi.org/10.6084/m9.figshare.28955141)61 and GitHub (https://github.com/GSanShui/XPert).
References
Kabir, A. & Muth, A. Polypharmacology: the science of multi-targeting molecules. Pharmacol. Res. 176, 106055 (2022).
Zhao, W. et al. Large-scale characterization of drug responses of clinically relevant proteins in cancer cell lines. Cancer Cell 38, 829–843.e4 (2020).
More, P. et al. Transcriptional response to standard AML drugs identifies synergistic combinations. Int. J. Mol. Sci. 24, 12926 (2023).
Lotfollahi, M. et al. Predicting cellular responses to complex perturbations in high-throughput screens. Mol. Syst. Biol. 19, e11517 (2023).
Hetzel, L. et al. Predicting cellular responses to novel drug perturbations at a single-cell resolution. Adv. Neural Inf. Process. Syst. 35, 26711–26722 (2022).
Tong, X. et al. TranSiGen: deep representation learning of chemical-induced transcriptional profile for phenotype-based drug discovery. Nat. Commun. 15, 5378 (2024).
Qi, X. et al. PRnet: predicting transcriptional responses to novel chemical perturbations using deep generative model for drug discovery. Nat. Commun. 15, 9256 (2024).
Kingma, D. P. & Welling, M. Auto-encoding variational Bayes. In Proc. 2nd International Conference on Learning Representations (ICLR) (eds Courville, A. et al.) 1–14 (OpenReview.net, 2014).
Pham, T.-H., Qiu, Y., Zeng, J., Xie, L. & Zhang, P. A deep learning framework for high-throughput mechanism-driven phenotype compound screening and its application to COVID-19 drug repurposing. Nat. Mach. Intell. 3, 247–257 (2021).
Pham, T.-H. et al. Chemical-induced gene expression ranking and its application to pancreatic cancer drug repurposing. Patterns 3, 100441 (2022).
Theodoris, C. V. et al. Transfer learning enables predictions in network biology. Nature 618, 616–624 (2023).
Cui, H. et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nat. Methods 21, 1470–1480 (2024).
Hao, M. et al. Large-scale foundation model on single-cell transcriptomics. Nat. Methods 21, 1481–1491 (2024).
Wu, Y. et al. PerturBench: benchmarking machine learning models for cellular perturbation analysis. In Proc. 39th Annual Conference on Neural Information Processing Systems (NeurIPS) Track on Datasets and Benchmarks (eds Risdal, M. et al.) 1–41 (Curran Associates, Inc., 2025).
Wenteler, A. et al. PertEval-scFM: benchmarking single-cell foundation models for perturbation effect prediction. In Proc. 42nd International Conference on Machine Learning (ICML) (eds Singh, A. et al.) 66633–66677 (PMLR, 2025).
Levy, R. H. Time-dependent pharmacokinetics. Pharmacol. Ther. 17, 383–397 (1982).
Browne, T. R. et al. Pharmacokinetics: dose-dependent changes. J. Clin. Pharmacol. 26, 463–468 (1986).
Rezvani, N., Bolduc, D. L. & Dourson, M. L. in Encyclopedia of Toxicology 3rd edn (ed. Wexler, P.) 870–877 (Elsevier, 2014).
Vaswani, A. et al. Attention is all you need. Adv. Neural Inf. Process. Syst. 30, 261–272 (2017).
Zhang, C., Song, D., Huang, C., Swami, A. & Chawla, N. V. Heterogeneous graph neural network. In Proc. 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining (eds Teredesai, A. et al.) 793–803 (ACM, 2019).
Zhou, G. et al. Uni-Mol: a universal 3D molecular representation learning framework. In Proc. 11th International Conference on Learning Representations (ICLR) (eds Nickel, M. et al.) 15076–15106 (OpenReview.net, 2023).
Mering, C. V. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261 (2003).
Kubinyi, H. Chemical similarity and biological activities. J. Braz. Chem. Soc. 13, 717–726 (2002).
Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452.e17 (2017).
Li, L. et al. A systematic comparison of single-cell perturbation response prediction models. Preprint at bioRxiv https://doi.org/10.1101/2024.12.23.630036 (2024).
Huang, H., Li, Z., He, R., Sun, Z. & Tan, T. IntroVAE: introspective variational autoencoders for photographic image synthesis. Adv. Neural Inf. Process. Syst. 31, 10236–10245 (2018).
Bredell, G., Flouris, K., Chaitanya, K., Erdil, E. & Konukoglu, E. Explicitly minimizing the blur error of variational autoencoders. In Proc. 11th International Conference on Learning Representations (ICLR) (eds Nickel, M. et al.) 29682–29697 (OpenReview.net, 2023).
Liu, J. Research on the application of variational autoencoder in image generation. ITM Web Conf. 70, 02001 (2025).
Ran, X., Xu, M., Mei, L., Xu, Q. & Liu, Q. Detecting out-of-distribution samples via variational auto-encoder with reliable uncertainty estimation. Neural Netw. 145, 199–208 (2022).
Sundar, V. K., Ramakrishna, S., Rahiminasab, Z., Easwaran, A. & Dubey, A. Out-of-distribution detection in multi-label datasets using latent space of β-VAE. In Proc. 2020 IEEE Security and Privacy Workshops (SPW) (ed. Takabi, H.) 250–255 (IEEE, 2020).
Leek, J. T. et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 11, 733–739 (2010).
McInnes, L., Healy, J. & Melville, J. UMAP: uniform manifold approximation and projection for dimension reduction. J. Open Source Softw. 3, 861 (2018).
Sangwan, R., Rajan, R. & Mandal, P. K. HDAC as onco target: reviewing the synthetic approaches with SAR study of their inhibitors. Eur. J. Med. Chem. 158, 620–706 (2018).
Zecha, J. et al. Decrypting drug actions and protein modifications by dose- and time-resolved proteomics. Science 380, 93–101 (2023).
Eckert, S. et al. Decrypting the molecular basis of cellular drug phenotypes by dose-resolved expression proteomics. Nat. Biotechnol. 43, 406–415 (2025).
Yan, W. et al. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene 32, 599–609 (2013).
Mottamal, M., Zheng, S., Huang, T. & Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 20, 3898–3941 (2015).
Liu, Z. et al. CDS-DB, an omnibus for patient-derived gene expression signatures induced by cancer treatment. Nucleic Acids Res. 52, D1163–D1179 (2024).
Servetto, A., Formisano, L. & Arteaga, C. L. FGFR signaling and endocrine resistance in breast cancer: challenges for the clinical development of FGFR inhibitors. Biochim. Biophys. Acta 1876, 188595 (2021).
Qiao, J. et al. TIAM1 upregulation confers NVP-BEZ235 resistance to breast cancer cells through FGFR/STAT3 pathway. Biochem. Genet. 58, 953–965 (2020).
Duan, L. et al. Prolylcarboxypeptidase regulates proliferation, autophagy, and resistance to 4-hydroxytamoxifen-induced cytotoxicity in estrogen receptor-positive breast cancer cells. J. Biol. Chem. 286, 2864–2876 (2011).
Ma, X. et al. ErbB2-upregulated HK1 and HK2 promote breast cancer cell proliferation, migration and invasion. Med. Oncol. 40, 154 (2023).
Hoogstraat, M. et al. Comprehensive characterization of pre- and post-treatment samples of breast cancer reveal potential mechanisms of chemotherapy resistance. NPJ Breast Cancer 8, 60 (2022).
Douglass, E. F. et al. A community challenge for a pancancer drug mechanism of action inference from perturbational profile data. Cell Rep. Med. 3, 100492 (2022).
Rasley, J., Rajbhandari, S., Ruwase, O. & He, Y. DeepSpeed: system optimizations enable training deep learning models with over 100 billion parameters. In Proc. 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining (eds Gupta, R. et al.) 3505–3506 (ACM, 2020).
Poli, M. et al. Hyena hierarchy: towards larger convolutional language models. In Proc. 40th International Conference on Machine Learning (ICML) (eds Krause, A. et al.) 28043–28078 (PMLR, 2023).
Wei, Z. et al. PerturBase: a comprehensive database for single-cell perturbation data analysis and visualization. Nucleic Acids Res. 52, gkae858 (2024).
Zhang, J. et al. Tahoe-100M: a giga-scale single-cell perturbation atlas for context-dependent gene function and cellular modeling. Preprint at bioRxiv https://doi.org/10.1101/2025.02.20.639398 (2025).
Peidli, S. et al. scPerturb: harmonized single-cell perturbation data. Nat. Methods 21, 531–540 (2024).
Qian, L. et al. AI-empowered perturbation proteomics for complex biological systems. Cell Genom. 4, 100691 (2024).
Choo, S. & Kim, W. A study on the evaluation of tokenizer performance in natural language processing. Appl. Artif. Intell. 37, 2175112 (2023).
Zhao, S., Zhang, J. & Nie, Z. Large-scale cell representation learning via divide-and-conquer contrastive learning. Preprint at http://arxiv.org/abs/2306.04371 (2023).
Hou, Z. et al. GraphMAE: self-supervised masked graph autoencoders. In Proc. 28th ACM SIGKDD Conference on Knowledge Discovery and Data Mining (eds Zhang, A. & Rangwala, H.) 594–604 (ACM, 2022).
Landrum, G. RDKit: Open-Source Cheminformatics (2006); https://www.rdkit.org
Chen, H. et al. Drug target prediction through deep learning functional representation of gene signatures. Nat. Commun. 15, 1853 (2024).
Ye, Q. et al. A unified drug–target interaction prediction framework based on knowledge graph and recommendation system. Nat. Commun. 12, 6775 (2021).
Su, X., Hu, P., You, Z.-H., Yu, P. S. & Hu, L. Dual-channel learning framework for drug-drug interaction prediction via relation-aware heterogeneous graph transformer. Proc. AAAI Conf. Artif. Intell. 38, 249–256 (2024).
Mao, A., Mohri, M. & Zhong, Y. Cross-entropy loss functions: theoretical analysis and applications. In Proc. 40th International Conference on Machine Learning (ICML) (eds Krause, A. et al.) 23803–23828 (PMLR, 2023).
Caruana, R., Lawrence, S. & Giles, C. Overfitting in neural nets: backpropagation, conjugate gradient, and early stopping. Adv. Neural Inf. Process. Syst. 13, 402–408 (2000).
Kernfeld, E. et al. A comparison of computational methods for expression forecasting. Genome Biol. 26, 388 (2025).
Guo, Y. et al. XPert: modelling drug-induced cellular perturbation responses with a biologically informed dual-branch transformer. figshare https://doi.org/10.6084/m9.figshare.28955141 (2025).
Guo, Y. et al. XPert v1.1: modelling drug-induced cellular perturbation responses with a biologically informed dual-branch transformer. Zenodo https://doi.org/10.5281/zenodo.17182939 (2025).
Van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (U21A20420 to B.Y.), the ‘Pioneer’ and ‘Leading Goose’ R&D Program of Zhejiang under grant number 2024C03003 (to B.Y.), Zhejiang Provincial Natural Science Foundation of China (LR22H310002 to J.C.), the Fundamental Research Funds for the Central Universities (226-2024-00047 to J.C.) and the Fundamental Research Funds for the Central Universities (226-2025-00065 to B.Y.).
Author information
Authors and Affiliations
Contributions
Y.G. conceived and designed the project, implemented the computational framework, and conducted the benchmark experiments and case studies under the supervision of B.Y., C.-Y.H. and J.C. Y.G. drafted the paper, which was critically reviewed and revised by B.Y., C.-Y.H. and J.C. H.Z., H.H. and J.W. provided constructive feedback during the study and contributed to the revision of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Mingyue Zheng and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Generalization performance of XPert and baseline models across cell lines under the cold-cell setting.
(a) Boxplots of predictive metrics (MSE, Pearson (xpert), and Pearson (xdeg)) for each cell line on the L1000_sdst dataset. Individual cell lines are shown as colored scatter points (N = 164). The central line within each box denotes the median; the box limits represent the interquartile range (IQR, from the 25th to 75th percentile); whiskers extend to 1.5× IQR beyond the box limits. (b) Scatter plot of the maximum Spearman correlation between test and training cell lines versus the predictive performance (Pearson (deg)). The gray line denotes the linear regression fit. (c) Distribution of maximum Spearman correlation values across test cell lines, with group boundaries defined by mean ± standard deviation (purple dashed lines). Cell lines were stratified into Low (N = 22), Medium (N = 110), and High (N = 31) similarity groups. (d) Box-and-whisker plots comparing the prediction performance distribution (MSE, left; Pearson (deg), right) across the three cell similarity groups (Low (N = 22), Medium (N = 110), and High (N = 31)). The central line within each box denotes the median; the box limits represent the interquartile range (IQR, from the 25th to 75th percentile); whiskers extend to 1.5× IQR beyond the box limits; and outliers are shown as individual points beyond the whiskers.
Extended Data Fig. 2 Visualization of XPert’s learned atomic attention for EGFR and HDAC inhibitors.
For each drug, the figure displays three panels: (left) the chemical structure with atoms colored by their mean attention score, highlighting key pharmacophoric groups; (middle) a heatmap showing the attention weight distribution for each atom across different cell line samples; and (right) a scatter plot of mean attention scores across five random seeds, used to quantify pattern similarity (Mean Cosine Similarity) and variance (Mean CV). (a) For EGFR inhibitors Gefitinib and Erlotinib, attention is consistently concentrated on the quinazoline ring scaffold, which is crucial for their binding activity. (b) For HDAC inhibitors Belinostat and Vorinostat, the model places higher attention on the Zinc-Binding Group (ZBG), the moiety responsible for chelating the catalytic zinc ion in the enzyme’s active site.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–17, Tables 1–12 and Notes 1–3.
Supplementary Tables (download XLSX )
Supplementary Tables 13–30.
Source data
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, Y., Zhang, H., Hu, H. et al. Modelling drug-induced cellular perturbation responses with a biologically informed dual-branch transformer. Nat Mach Intell 8, 96–112 (2026). https://doi.org/10.1038/s42256-025-01165-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s42256-025-01165-w
