
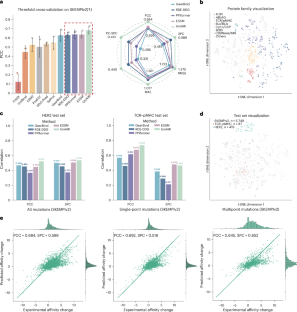
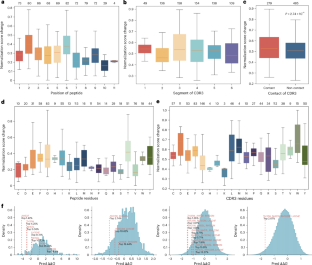
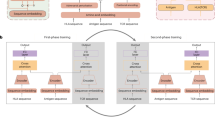
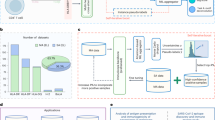
Abstract
Adaptive immunity is a central defence system essential for long-term and highly specific protection against pathogens through the precise molecular recognition of antigens by lymphocytes. However, predicting how mutations reshape these interactions remains a major challenge. Although previous computational approaches leverage large-scale pretraining for mutation-effect predictions, most are designed for specific tasks or modalities and struggle to generalize across the heterogeneous, multimodal landscape of immune recognition. Here we introduce UniAIR, a modular, multimodal framework for the accurate and generalizable prediction of mutation effects across immune recognition scenarios. UniAIR integrates a standardized data pipeline, an interface-centric sequence–structure fusion transformer that integrates evolutionary information with geometric representations, and a suite of extensions for multiexpert consensus and adaptation to predicted structure inputs. We comprehensively evaluated UniAIR through large-scale benchmarking and independent tests across immunological tasks. The evaluation covered both extracellular and intracellular immune recognition, including antibody maturation, antigen escape, TCR–pHLA optimization and analyses in which experimental structures were unavailable. Extensive experiments show that UniAIR achieves state-of-the-art performance and delivers robust predictions with minimal task-specific tuning. In particular, UniAIR successfully performed multiround peptide optimization of a TCR–pHLA complex under sparse feedback and identified key functional mutations in incomplete antibody–antigen structures. Together, UniAIR establishes a unified computational foundation for mapping mutation landscapes, advancing understanding of adaptive immune recognition and accelerating immunotherapeutic design.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
The datasets used in this work are available at https://huggingface.co/datasets/Jesse7/UniAIR_data. The curated pretraining datasets are available at SabDab60 (https://opig.stats.ox.ac.uk/webapps/sabdab-sabpred/sabdab), STCRDab61 (https://opig.stats.ox.ac.uk/webapps/stcrdab-stcrpred) and PPB-Affinity62 (https://doi.org/10.1038/s41597-024-03997-4). The curated evaluation datasets are from SKEMPIv2 (ref. 63; https://life.bsc.es/pid/skempi2) and TCRen benchmark no. 1 (ref. 56; https://doi.org/10.1038/s43588-024-00653-0). For the curated downstream datasets, the HER2 test set is from the original study42 (https://doi.org/10.1101/2023.01.08.523187). The TCR–pMHC test set is from ATLAS43 (https://atlas.ibbr.umd.edu/web/index.php). The LASSA dataset is from ref. 47 (https://doi.org/10.1016/j.immuni.2024.06.013). The SARS-CoV-2 dataset is from ref. 5 (https://doi.org/10.1073/pnas.2122954119). The TCR–pHLA structures are from ref. 50 (https://doi.org/10.1038/s42256-024-00901-y). The KRAS complex is built from the RCSB PDB (https://www.rcsb.org/) with PDB ID 6ULR.
Code availability
The deep learning models were developed and deployed using standard model libraries and the PyTorch framework. The source code and model weights of UniAIR are available via GitHub at https://github.com/hanrthu/UniAIR and Zenodo at https://zenodo.org/records/19471285 (ref. 83).
References
Rossjohn, J. et al. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 33, 169–200 (2015).
Batista, F. D. & Harwood, N. E. The who, how and where of antigen presentation to B cells. Nat. Rev. Immunol. 9, 15–27 (2009).
Paludan, S. R., Pradeu, T., Masters, S. L. & Mogensen, T. H. Constitutive immune mechanisms: mediators of host defence and immune regulation. Nat. Rev. Immunol. 21, 137–150 (2021).
Flajnik, M. F. & Kasahara, M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat. Rev. Genet. 11, 47–59 (2010).
Shan, S. et al. Deep learning guided optimization of human antibody against SARS-CoV-2 variants with broad neutralization. Proc. Natl Acad. Sci. USA 119, e2122954119 (2022).
Pappas, L. et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature 516, 418–422 (2014).
Reddehase, M. J. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2, 831–844 (2002).
Vázquez-García, I. et al. Ovarian cancer mutational processes drive site-specific immune evasion. Nature 612, 778–786 (2022).
Willmann, K. L. et al. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat. Commun. 5, 5360 (2014).
Wang, G. et al. Deep-learning-enabled protein–protein interaction analysis for prediction of SARS-CoV-2 infectivity and variant evolution. Nat. Med. 29, 2007–2018 (2023).
Wiehe, K. et al. Mutation-guided vaccine design: a process for developing boosting immunogens for HIV broadly neutralizing antibody induction. Cell Host Microbe 32, 693–709.e697 (2024).
Cheng, J., Liang, T., Xie, X.-Q., Feng, Z. & Meng, L. A new era of antibody discovery: an in-depth review of AI-driven approaches. Drug Discov. Today 29, 103984 (2024).
Cho, S. et al. Structural basis of affinity maturation and intramolecular cooperativity in a protein-protein interaction. Structure 13, 1775–1787 (2005).
Homola, J. Surface plasmon resonance sensors for detection of chemical and biological species. Chem. Rev. 108, 462–493 (2008).
Lequin, R. M. Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). Clin. Chem. 51, 2415–2418 (2005).
Fowler, D. M., Stephany, J. J. & Fields, S. Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat. Protoc. 9, 2267–2284 (2014).
Frank, F. et al. Deep mutational scanning identifies SARS-CoV-2 Nucleocapsid escape mutations of currently available rapid antigen tests. Cell 185, 3603–3616.e3613 (2022).
Boder, E. T. & Wittrup, K. D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557 (1997).
McMahon, C. et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 25, 289–296 (2018).
Gerasimavicius, L., Livesey, B. J. & Marsh, J. A. Correspondence between functional scores from deep mutational scans and predicted effects on protein stability. Protein Sci. 32, e4688 (2023).
Mason, D. M. et al. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nat. Biomed. Eng. 5, 600–612 (2021).
Brandes, N., Goldman, G., Wang, C. H., Ye, C. J. & Ntranos, V. Genome-wide prediction of disease variant effects with a deep protein language model. Nat. Genet. 55, 1512–1522 (2023).
Hie, B., Zhong, E. D., Berger, B. & Bryson, B. Learning the language of viral evolution and escape. Science 371, 284–288 (2021).
Shanker, V. R., Bruun, T. U., Hie, B. L. & Kim, P. S. Unsupervised evolution of protein and antibody complexes with a structure-informed language model. Science 385, 46–53 (2024).
van Beusekom, B. et al. Homology-based hydrogen bond information improves crystallographic structures in the PDB. Protein Sci. 27, 798–808 (2018).
Waman, V. P. et al. CATH v4.4: major expansion of CATH by experimental and predicted structural data. Nucleic Acids Res. 53, D348–D355 (2024).
McMaster, B., Thorpe, C., Ogg, G., Deane, C. M. & Koohy, H. Can AlphaFold’s breakthrough in protein structure help decode the fundamental principles of adaptive cellular immunity?. Nat. Methods 21, 766–776 (2024).
Han, W. et al. Predicting the antigenic evolution of SARS-COV-2 with deep learning. Nat. Commun. 14, 3478 (2023).
Taft, J. M. et al. Deep mutational learning predicts ACE2 binding and antibody escape to combinatorial mutations in the SARS-CoV-2 receptor-binding domain. Cell 185, 4008–4022.e4014 (2022).
Hie, B. L. et al. Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol. 42, 275–283 (2024).
Cai, H. et al. Pretrainable geometric graph neural network for antibody affinity maturation. Nat. Commun. 15, 7785 (2024).
Visani, G. M. et al. T-cell receptor specificity landscape revealed through de novo peptide design. Proc. Natl Acad. Sci. USA 122, e2504783122 (2025).
O’Donnell, T. J. et al. Reading the repertoire: progress in adaptive immune receptor analysis using machine learning. Cell System 15, 1168–1189 (2024).
Xie, L. et al. AI developments for T and B cell receptor modeling and therapeutic design. Preprint at https://arxiv.org/abs/2601.17138 (2026).
Mhanna, V. et al. Adaptive immune receptor repertoire analysis. Nat. Rev. Methods Primers 4, 6 (2024).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Guo, H. et al. Advancing expert specialization for better MoE. Adv. Neural Inf. Process. Syst. 38, 48767–48809 (2026).
Luo, S. et al. Rotamer density estimator is an unsupervised learner of the effect of mutations on protein–protein interaction. In The Eleventh International Conference on Learning Representations (2023).
Bushuiev, A. et al. Learning to design protein-protein interactions with enhanced generalization. In International Conference on Learning Representations 21010–21035 (2025).
Su, J. et al. SaProt: protein language modeling with structure-aware vocabulary. In International Conference on Learning Representations 6987–7009 (2024).
Li, M. et al. ProSST: protein language modeling with quantized structure and disentangled attention. Adv. Neural Inf. Process. Syst. 37, 35700–35726 (2024).
Shanehsazzadeh, A. et al. Unlocking de novo antibody design with generative artificial intelligence. Preprint at bioRxiv https://doi.org/10.1101/2023.01.08.523187 (2024).
Borrman, T. et al. ATLAS: a database linking binding affinities with structures for wild-type and mutant TCR-pMHC complexes. Proteins Struct. Funct. Bioinf. 85, 908–916 (2017).
Gao, Y. et al. Pan-peptide meta learning for T-cell receptor–antigen binding recognition. Nat. Mach. Intell. 5, 236–249 (2023).
Sidney, J., Peters, B., Frahm, N., Brander, C. & Sette, A. HLA class I supertypes: a revised and updated classification. BMC Immunol. 9, 1 (2008).
Garry, R. F. Lassa fever—the road ahead. Nat. Rev. Microbiol. 21, 87–96 (2023).
Carr, C. R. et al. Deep mutational scanning reveals functional constraints and antibody-escape potential of Lassa virus glycoprotein complex. Immunity 57, 2061–2076.e2011 (2024).
Zhang, Y. et al. Epitope-anchored contrastive transfer learning for paired CD8+ T cell receptor–antigen recognition. Nat. Mach. Intell. 6, 1344–1358 (2024).
Sim, M. J. W. et al. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc. Natl Acad. Sci. USA 117, 12826–12835 (2020).
Feng, Z. et al. Sliding-attention transformer neural architecture for predicting T cell receptor–antigen–human leucocyte antigen binding. Nat. Mach. Intell. 6, 1216–1230 (2024).
Ullanat, V., Jing, B., Sledzieski, S. & Berger, B. Learning the language of protein-protein interactions. Nat. Commun. 17, 1199 (2025).
Bran, A. M. et al. Augmenting large language models with chemistry tools. Nat. Mach. Intell. 6, 525–535 (2024).
Liu, A. et al. DeepSeek-V3.2: pushing the frontier of open large language models. Preprint at https://arxiv.org/abs/2512.02556 (2025).
OpenAI. Introducing GPT-5.2. https://openai.com/index/introducing-gpt-5-2/ (2025).
Korpela, D. et al. EPIC-TRACE: predicting TCR binding to unseen epitopes using attention and contextualized embeddings. Bioinformatics 39, btad743 (2023).
Karnaukhov, V. K. et al. Structure-based prediction of T cell receptor recognition of unseen epitopes using TCRen. Nat. Comput. Sci. 4, 510–521 (2024).
Springer, I., Tickotsky, N. & Louzoun, Y. Contribution of T cell receptor alpha and beta CDR3, MHC typing, V and J genes to peptide binding prediction. Front. Immunol. 12, 664514 (2021).
Facundo, D. & Batista, M. S. N. Affinity dependence of the B cell response to antigen: a threshold, a ceiling, and the importance of off-rate. Immunity 8, 751–759 (1998).
Irving, M. et al. Interplay between T cell receptor binding kinetics and the level of cognate peptide presented by major histocompatibility complexes governs CD8+ T cell responsiveness. J. Biol. Chem. 287, 23068–23078 (2012).
Dunbar, J. et al. SAbDab: the structural antibody database. Nucleic Acids Res. 42, D1140–1146 (2014).
Leem, J., de Oliveira, S. H. P., Krawczyk, K. & Deane, C. M. STCRDab: the structural T-cell receptor database. Nucleic Acids Res. 46, D406–D412 (2018).
Liu, H. et al. PPB-Affinity: protein-protein binding affinity dataset for AI-based protein drug discovery. Sci. Data 11, 1316 (2024).
Jankauskaitė, J., Jiménez-García, B., Dapkūnas, J., Fernández-Recio, J. & Moal, I. H. SKEMPI 2.0: an updated benchmark of changes in protein–protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics 35, 462–469 (2018).
Rodrigues, C. H. M., Myung, Y., Pires, D. E. V. & Ascher, D. B. mCSM-PPI2: predicting the effects of mutations on protein–protein interactions. Nucleic Acids Res. 47, W338–W344 (2019).
Nahta, R. & Esteva, F. J. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res. 8, 215 (2006).
Huang, X., Pearce, R. & Zhang, Y. EvoEF2: accurate and fast energy function for computational protein design. Bioinformatics 36, 1135–1142 (2020).
Pearce, R., Huang, X., Setiawan, D. & Zhang, Y. EvoDesign: designing protein–protein binding interactions using evolutionary interface profiles in conjunction with an optimized physical energy function. J. Mol. Biol. 431, 2467–2476 (2019).
Ahdritz, G. et al. OpenFold: retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. Nat. Methods 21, 1514–1524 (2024).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Berendsen, H. J. C., van der Spoel, D. & van Drunen, R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 (1995).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
MacKerell, A. D. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Giovanni Bussi, D. D. Michele Parrinello canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Jorgensen, W. L. Free energy calculations: a breakthrough for modeling organic chemistry in solution. Acc. Chem. Res. 22, 184–189 (1989).
Deng, Y. & Roux, B. Calculation of standard binding free energies: aromatic molecules in the T4 lysozyme L99A mutant. J. Chem. Theory Comput. 2, 1255–1273 (2006).
Boresch, S., Tettinger, F., Leitgeb, M. & Karplus, M. Absolute binding free energies: a quantitative approach for their calculation. J. Phys. Chem. B 107, 9535–9551 (2003).
Mobley, D. L., Chodera, J. D. & Dill, K. A. On the use of orientational restraints and symmetry corrections in alchemical free energy calculations. J. Chem. Phys. 125, 084902 (2006).
Han, R. hanrthu/UniAIR: Initial Release of UniAIR v1.0.0. Zenodo https://doi.org/10.5281/zenodo.19471285 (2026).
Acknowledgements
This study was supported by grants from the National Science Foundation of China (T2541010 to T.C.; T2522008 to G.W.; 82522048, 62501406 to X.L.), the National Key R&D Program of China (2024YFF1207100, 2024YFF1207103 to T.C.; 2025YFF0515300, 2025YFF0515301 to S.W. and G.W.), Shenzhen Medical Research Fund (E250200620, E250200622 and E250200623 to S.W. and G.W.), the National Health and Medical Research Council of Australia (APP1127948, APP1144652 and APP2036864 to J.S.), Australian Research Council (LP220200614 to J.S.), the Scientific Research Innovation Capability Support Project for Young Faculty (SRICSPYF-ZY2025015 to G.W.) and the Fundamental Research Funds for the Beijing University of Posts and Telecommunications (grant number 2025AI4S18 to G.W.). This work was also supported by the Beijing National Research Center for Information Science and Technology (BNRist) and the Major and Seed Inter-Disciplinary Research projects awarded by Monash University. This study was also funded by New Cornerstone Science Foundation through the XPLORER PRIZE. The funders had no roles in the study design, data collection and analysis, publication decisions or manuscript preparation.
Author information
Authors and Affiliations
Contributions
R.H., Y.Z., L.F., T.P., X.W., J.X., P.Z. and X.C. collected and analysed the data. R.H., J.X., X.W., T.P., W.L. and C.J. developed the models and downstream applications. S.C. provided high-performance computational resources and infrastructure support. G.W., T.C., J.S., S.W. and X.L. conceived of and supervised the project. R.H., Y.Z., G.W., X.L., J.S., T.C., T.P., J.X., X.W. and J.L. wrote and revised the paper. All authors discussed the results and reviewed the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Miaozhe Huo, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–7, Tables 1–6, baseline implementations and iDist embedding for dataset analysis.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Han, R., Zhang, Y., Liu, X. et al. Generalizable mutation-effect prediction across adaptive immune recognition via unified multimodal framework. Nat Mach Intell (2026). https://doi.org/10.1038/s42256-026-01243-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42256-026-01243-7
