
Abstract
Hydroxide exchange membrane electrolyzers (HEMELs) enable hydrogen production using low-cost, earth-abundant materials. Improving electrode fabrication is integral to enhancing device performance, and ionomer—responsible for transporting hydroxide and mechanically supporting the catalyst—is a major component. Here, we use experiments and computation to study the effects of relative humidity (RH) during the drying process of poly(aryl piperidinium) ionomer films on HEMEL electrodes. Broadly, the drying environments determine the physical structure and electrochemical traits of the ionomer network. High RH drying yields a highly porous network with excessive water uptake, structural defects, washout, and 64% reduction in hydroxide conductivity. Extremely low RH drying produces an overly compact pore network that hinders hydroxide mobility. In contrast, moderately low RH drying (9% RH) creates an ionomer film with well-balanced traits: excellent mechanical stability and connectivity needed for catalyst retention and hydroxide transport, which improves HEMEL performance by 40% at 1.8 V compared to suboptimal RHs. This research advances HEMEL manufacturing by providing a simple, scalable, and low-cost approach to optimize electrode ionomer films.

Similar content being viewed by others
Introduction
Hydrogen is utilized in diverse applications such as ammonia synthesis, methanol production, petroleum refining, and steel manufacturing1,2,3,4. Conventionally, hydrogen is sourced from energy- and carbon-intensive steam methane reforming5,6,7,8. Electrochemical water splitting driven by renewable electricity, however, offers an alternative for hydrogen production with low carbon emissions5,9. Among the various types of water electrolyzers available, hydroxide exchange membrane electrolyzers (HEMELs) have garnered attention due to their high efficiencies, good dynamic performance, and utilization of earth-abundant, cost-effective catalyst materials and bipolar plates10,11,12,13. The latter is a major concern for proton exchange membrane electrolyzers (PEMELs), which require expensive and scarce platinum group metals to maintain integrity in an acidic environment14,15. Substitution of such rare components with earth-abundant materials in HEMELs accounts for up to a 75% and 89.6% reduction in bipolar plate and catalyst cost, respectively16. Despite reservations about the long-term feasibility of widespread PEMEL adoption, they represent a more mature technology with well-established manufacturing processes that have been extensively studied and commercialized17. The same attention has not yet been paid to HEMELs, as they postdate PEMELs by several decades. As a result, there remains a gap in understanding the most effective methods for reliably manufacturing HEMELs, which hinders the development of optimized performance, durability, reliability, and cost.
In this work, we advance the knowledge of membrane electrode assembly (MEA) manufacturing by demonstrating that the relative humidity (RH) during the drying of ionomer onto an electrode critically influences the stability and structure of the ionomer film network, which in turn affects bulk hydroxide conductivity (HC), integrity of the catalyst layer, and overall electrolyzer performance. This advancement quantifies the ideal drying environment of ionomer films onto electrodes and remedies swelling, conductivity, and ionomer/catalyst washout challenges presently associated with membranes and ionomer films18,19,20,21,22,23,24.
We focus on the poly(aryl piperidinium) polymer family (PiperIon®, specifically), which has shown to be among the most promising and ubiquitous hydroxide exchange ionomer materials due to its excellent HC and chemical stability22. As with most ionomers, PiperIon® is hygroscopic due to its cationic groups, which leads to water uptake (WU) and swelling that, in excess, can decrease ionic conductivity and mechanical robustness25,26,27. When ionomer is incorporated throughout an electrode via dip-coating, the solvent used to dissolve the ionomer (in this case, ethanol) must evaporate to allow for an ionomer film to develop over the catalyst layer. During this process, the polymer network will naturally intake water vapor from the local atmosphere as the ethanol evaporates, leading to polymer/ethanol/water mixtures at various concentrations. The amount of water incorporated into the polymer matrix will be reflected by the available water in the surrounding environment (i.e. the RH)28,29. At sufficiently low ethanol/water concentrations, the solvated polymer chains coalesce into a mechanically robust film, ideally containing a well-connected pore network that enables rapid hydroxide exchange while minimizing catalyst and ionomer washout. We found that subjecting solvated ionomer to elevated RH conditions caused the polymer chains to swell and irreversibly alter the network’s structure, which led to mechanical instability and compromised HC. Conversely, exceedingly low RH environments created an overly dense ionomer film atop the catalyst layer that resisted hydroxide transport. Between these extremes, a moderately low drying-RH of 9% yielded an ionomer-coated electrode that was highly ionically conductive (nearly 3-fold higher HC than its counterparts), resistant to catalyst washout, and electrochemically superior as demonstrated by in situ performance tests.
The broad finding of this work is that control over the ionomer drying environment RH is a simple, scalable, and low-cost method to tailor ionomer films to exhibit ideal electrochemical, material, and mechanical properties. Although this is a seemingly obvious cause-and-effect relationship between polymer hygroscopicity and humidity, it is one that has been routinely overlooked in the electrolyzer field. Our study suggests that the RH should be strictly controlled during the drying process, as even slight fluctuations in humidity from HVAC systems have been found to significantly impact HEMEL performance. The RH in this lab-scale study was controlled with the use of saturated salt solutions, but industrial-scale RH control technologies are readily available to facilitate mass-production of consistently high-performing MEAs.
Results and discussion
Computational insight of ionomer film drying in varyingly hydrated environments
To grasp why drying ionomer films under varying RHs can be responsible for substantial enhancements in HC, washout mitigation, and MEA performance, we must first examine how the ionomer behaves during its drying process across different RH environments. To gain further insight into the atomic scale structure of the PiperIon® polymer during drying, we used classical molecular dynamics to predict structure snapshots for different specified amounts of water and solvent (ethanol) in the system. The structures studied here span the expected range of conditions present during the entire drying process. λ values of 0–12 H2O per cationic repeating unit modeled the water composition of the polymer network under different RH drying environments by representing the amount of water that entered the network from the ambient humidity. Previous work shows that this hydration range is expected for poly(aryl piperidinium) in different RH environments30. Varying weight fractions of ethanol from 95 to 4 wt% modeled different times during the evaporation process from the starting to the final concentration of ethanol in the polymer network. These structure snapshots are thus expected to span the range of anticipated states of the ionomer during drying and capture trends in the ionomer properties.
Figure 1a shows PiperIon® structures for a subset of the modeled conditions, with the full set shown in Supplementary Fig. 1. Figure 1b quantifies all relative structure volumes and shows that ionomer films containing the least ethanol are approximately 60% of the volume of films with the most ethanol. Analysis of the water clusters in these structures in Fig. 1c shows that for intermediate hydrations of 4 < λ < 8 with <32 wt% ethanol, a percolation threshold occurs where above this threshold all water molecules belong to the same water cluster that spans the polymer volume. Percolation by water is substantially delayed when more ethanol is present in the pores, as the systems with 64 and 95 wt% ethanol do not begin to form a fully connected water network until at least a λ of 12. The lack of a fully percolating pore network for low amounts of water and ethanol is consistent with the scanning electron microscopy (SEM) image of Fig. 2e, showing low RH drying leads to overly dense ionomer films, and suggests a minimum RH is needed to allow connected pore channels to form that enable good HC. Figure 1d shows that for the polymer systems with 4 wt% (representative of the end of the drying process) and 16 wt% ethanol, the hydration level has a substantial impact on the pore channel width distribution. The average pore channel diameter increased for λ = 8 hydration compared to λ = 4, suggesting that ionomer dried at higher hydration levels creates a network favorable for facile transport. However, these larger pore channels could lead to ionomer/catalyst washout if the polymer does not form a sufficiently robust network during drying.

a Structure snapshots of PiperIon® systems. As ethanol evaporates and water enters the membrane due to RH the structures will transition from (λ = 0, 95 wt% EtOH) conditions to have a higher hydration level and lower amount of ethanol. The isosurfaces denote where the atomic radii-weighted Gaussian density of each set of atom species is 0.5. The trends are not sensitive to the isosurface threshold used. Blue regions contain water and hydroxide, gray regions contain ethanol, and orange regions contain PiperIon® atoms. The sizes of the structures are scaled relative to the size of the (λ = 0, 95 wt% EtOH) structure. b Volumes of the PiperIon® structures relative to the volume of the (λ = 0, 95 wt% EtOH) structure. c Fraction of water molecules contained within the largest water cluster as a function of hydration level. Water cluster connectivity was calculated using the first coordination shell of the water molecules for each structure, which originates from the water oxygen-water oxygen radial distribution functions (RDFs) shown in Supplementary Fig. 3. d Pore channel width distributions of the PiperIon® structures.

(a–h) Images of ionomer films (roughly 40 µm thick, no accompanying electrode or catalyst layer) cast in 0% (a, e), 9% (b, f), 40% (c, g), and 94% RH (d, h) conditions. Images (a–d) and (e–h) were captured at 20 K and 50 K magnification, respectively. “Divots” reference the artifact in (h). “Treads” are the crack-like structures that run along the majority of the films, highlighted in (g).
Our modeling results elucidated how variations in ambient humidity influenced the drying behavior of the ionomer network, affecting the pore structure and density. To investigate how these differences manifested in the film’s structure, SEM was employed to examine the morphological variations of films in their dried form (after the solvent had evaporated). At 20 K magnification, the morphologies of films dried at 0% and 9% RH (Fig. 2a, b) appeared nearly identical, apart from some slight unevenness on the 0% RH sample. However, when the RH was increased to 40%, noticeable divots ~0.3 µm in diameter appeared (Fig. 2c) that were absent at lower RHs. At 94% RH, these divots grew larger, as seen in Fig. 2d, reaching about 1 µm in diameter, suggesting that the size of this type of defect intensifies with greater amounts of available water.
The divots are an artifact of water vapor absorption into the polymer from ambient humidity. In the presence of water, the ionomer is known to agglomerate, as seen in Supplementary Fig. 4. This agglomeration occurs primarily on the topmost layer of the polymer film, known as the skin, where ambient water vapor first interacts with the ionomer. We hypothesize that the polymer agglomerates act as nucleation centers that draw other polymer chains toward them and cause tension along the skin, which adds to the inherent tension caused by the drying process. Skin regions with high tension can potentially tear, which then exposes the solvated ionomer layer beneath and leads to a localized divot as the wet ionomer falls downward.
Morphological differences between the 0% RH and 9% RH-dried films became clear at 50 K magnification. The 0% RH film (Fig. 2e) exhibited regions of high polymer density that appeared solid and slab-like after ethanol evaporation because the absence of water prevented any degree of swelling. Additionally, isolated raised regions developed, further indicating heterogeneous drying. However, Fig. 2f shows that the low-humidity film had a homogeneous morphology with no signs of uneven drying. These images indicate that drying ionomer with 0% RH is too extreme and creates an overly dense polymer network that, as discussed in the following section, will lead to transport limitations. It is important to note that the defects discussed here are permanent once the ethanol evaporates from the ionomer films, as the films maintained their structure even after full hydration (to remove them from the casting plate) and then pulled to a vacuum for SEM imaging.
Additionally, we observed that the treads (highlighted in the Fig. 2g inset) were affected by the drying environment. As the humidity increased, the average tread width increased by 17%—15 to 17.5 nm—between the 9% and 40% RH samples, indicating that higher RH drying conditions result in a generally more swollen, open morphology (also determined with porosity analysis in Fig. 1d). This characteristic is reflected in the differing WU properties among ionomer films dried in varying environments, shown in Fig. 3. WU in the films climbed progressively with humidity. At 80 °C, for example, the WU values were found to be 65%, 66%, 80%, and 94% uptake for the four RH conditions, in increasing order. Films cast at high RHs likely had a greater propensity to intake water because of their swollen structure, which could provide vacancies that readily accommodate water. By contrast, the smaller tread widths of the 9% RH sample and the compact network of the 0% RH sample indicate that films dried at no or low humidity promote densely dried polymer networks.

Water uptake measurements of ionomer films (~40-µm thick) dried at four RH conditions; recorded at 20 °C, 50 °C, and 80 °C. Note that the 50 °C and 80 °C points overlap at 40% RH.
RH impact on ionomer film conductivity
Ionic conductivity is a critical parameter that quantifies the efficiency of ion transport through ionomer and substantially impacts device performance by lowering the overpotentials associated with hydroxide transport22,31. Broadly, ionic conductivity is increased by a larger ionomer charge group density, which occurs when the polymer network is densely packed. However, in conditions of high RH drying, the ionomer network swells. This leads to a reduction in polymer density, which dilutes charge group concentration, subsequently reducing the ionic conductivity.
The HC of ionomer films dried at various RHs was analyzed with a four-electrode conductivity sensor and displayed in Fig. 4. The 9% RH film exhibited superior HC (272 mS cm−1) compared to its counterparts due to optimal charge density where smaller distances between charge groups existed, allowing for sufficient hydroxide transport with lower overpotentials, assuming a Nernst-Einstein behavior. As the RH of the drying condition further increased, a clear relationship between itself and HC evolved. Increasing beyond the 9% RH condition substantially lowered the HC by as much as 64%: 272 to 98 mS cm−1 between 9% and 94% RH, respectively. The overly spacious polymer chain arrangement with low charge group concentration in the high RH condition induced this nearly 3-fold difference in conductivity.

HC measurements of ionomer films (roughly 40-µm thick) dried at four RH conditions; recorded at 80 °C with a four-electrode conductivity sensor.
For 0% RH drying, our data revealed a critical limit wherein high polymer density becomes counterproductive. While it is true that charge density is maximized when the polymer is compact, there comes a threshold where the pore-limiting diameter can no longer accommodate sufficient water and hydroxide diffusion. As seen in Fig. 1d, the pore channel width shrinks as the effective-RH decreases, limiting the available space for water and hydroxide molecules. When the mobility of hydroxide ions is restricted, there is an effective choke on ionic conductivity. This effect may be evidenced by the 116 mS cm−1 drop in HC between the 9% and 0% RH cases. The limited movement could also reduce the access of ions to active sites. Maintaining an optimal balance between polymer density and swelling is essential to achieve sufficient ionic conductivity without compromising hydroxide ion mobility in HEMELs.
RH impact on the structural integrity of ionomer films
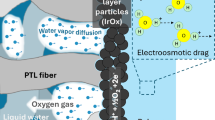
Aside from assisting with ion mobility, maintaining mechanical integrity (e.g. preventing ionomer and catalyst retention) is arguably the most important role of ionomer at the electrode. With dip-coating, the catalyst is enveloped by an ionomer film that deters washout from mechanical perturbations during electrolyzer operation such as water flow and bubble formation24,32,33. To sufficiently mitigate catalyst washout, the ionomer network must imperviously bind to both itself and the catalyst layer. However, drying ionomer under too high of an RH can lead to weakly interlocked polymer chains that produce an ionomer film with poor mechanical properties (Supplementary Fig. 5 for tensile testing). This results in a less structured network inadequate for catalyst anchoring, ultimately impacting the performance and longevity of the ionomer/catalyst system. In severe situations, this structural deficiency will lead to a total washout of the protective ionomer film from the electrode, leaving the catalyst entirely exposed and susceptible to possible dissolution reactions. This extreme can be seen in Fig. 5a, b, which compares the effect of RH on ionomer and catalyst washout with energy dispersive X-ray (EDX) mapping of ionomer and catalyst-coated anodes at their end-of-life (EoL) (a 1.5-h catalyst break-in procedure and 3, 1-h polarization curves; detailed information in the ‘methods’ section). The C atoms in the ionomer that remain at the EoL are represented by magenta dots. As seen in Fig. 5a, the 9% RH anode demonstrates higher ionomer retention at its EoL than the 94% RH case (Fig. 5b), which has little remaining ionomer due to weak and inadequate network formation. Supplementary Fig. 6 shows the EDX mapping for all RH conditions, which illustrates a qualitative loss of ionomer while increasing from 9% to 94% RH as well as an intense loss at 0% RH. Although long-term durability testing was not conducted, we anticipate that the 0% and 94% RH-dried electrodes, in which ionomer washout left a substantial portion of the catalyst exposed, would suffer premature catalyst loss due to the absence of a protective ionomer layer.

(a, b) EDX mapping demonstrating ionomer washout. The ionomer is represented by purple dots and the samples are two anodes [nickel felt with a self-supported catalyst layer and 1.25-µm thick dip-coated ionomer film (Supplementary Fig. 9)] at their EoL (a break-in procedure and 3 polarization curves—roughly 4.5 h total). Sample a is the 9% RH-dried anode, b is 94% RH. c–f Images of catalyst washout at EoL using a spray catalyst/ionomer slurry at the anode (as opposed to a self-supported catalyst with a dip-coated ionomer layer). The containers are holding effluent from the reservoir tank that feeds water into the anode inlet and receives water from the anode outlet. Image (c), (d), (e), and (f) represent 0%, 9%, 40%, and 94% RH, respectively.
In addition to dip-coating a self-supported catalyst in ionomer, spray-coating a catalyst/ionomer slurry onto an electrode was also tested at the same RHs to assess the impacts of RH-drying on sprayed-electrodes. Catalyst washout was exacerbated with spray-coating, where it is clear that the drying RH affected the ionomer in its role as a binding agent (Fig. 5c–f). Total catalyst washout occurred for the 0% RH sample, likely due to the polymer network becoming brittle, which would disrupt the connected, durable matrix that encapsulates the catalyst particles. Notably, however, there was no visible washout for the 9% RH case, which is unsurprising given its superior mechanical stability (Supplementary Fig. 5 for dynamic mechanical analysis). Echoing the trends discussed above, an increasing amount of washout resulted when panning from 9% to 94% RH environments due to the ionomer’s compromised binding abilities.
Implications of RH on MEA performance
Building on the insights gained from investigating the RH effects on the physical and electrochemical characteristics of ionomer films while drying, we can now explore how these factors influence the holistic performance of ionomer at the anode in a HEMEL. The electrochemical performance was examined with full-cell polarization curves, which are displayed in Fig. 6. By far, the top-performing MEA was that which employed the 9% RH-dried anode. At 1.8 V, the current density was 554 mA cm−2, outperforming the worst MEA by 42%. Mirrored by the characterization techniques discussed above, performance decreased as RH increased from 9% to 40% (451 mA cm−2) and then 94% (390 mA cm−2). Additionally, both the 40% and 94% RH MEAs exhibited a larger high-frequency resistance (HFR) than the 9% RH MEA, which is expected since some of the ionomer had likely washed out of the electrode (as seen in the EDX mapping), creating interfacial resistance. The 94% RH MEA exhibited a particularly large HFR and yielded an overpotential that was 109% greater than that of the 9% RH MEA. This likely resulted from a major loss of ionomer at the anode due to washout, meaning that contact resistances heightened and the only viable point of reaction was at the membrane-anode interface, which becomes especially problematic at high current densities.

a HEMEL polarization curves for a full MEA wherein the anode ionomer was dip-coated and dried at a specific RH. Current controlled testing was swept from 0 to 1300 mA cm−2. b The current density achieved at 1.8 volts for each of the RH cases—extracted from (a)—with the corresponding HFR for that sample (recorded at EoL at 500 mA cm−2), as represented by the dark line.
The 0% RH MEA suffered high performance losses which is consistent with the poor ionic conductivity observed in the corresponding ionomer film. As a result, the 0% RH MEA produced a low current density (389 mA cm−2 at 1.8 V) during HEMEL operation, similar to the 94% RH MEA. Although they yielded similar current densities, the 0% RH MEA had a significantly lower HFR than the 94% RH MEA, which indicates that ionomer loss was not as severe for the 0% RH case. However, the overly dense ionomer film on the electrode still likely prevented effective hydroxide transport to active sites. Akin to the material characterization, these findings reveal a limit to the benefits of low RH drying conditions.
Although we have discussed PiperIonA® (PAP-TP-85) in detail thus far, two other hydroxide exchange ionomers in the PiperIon® family, PAP-TP-100 and PAP-TP-85-MQN, were also evaluated and reported in the supplemental information (Supplementary Fig. 7). These ionomers exhibited similar trends to those discussed above for PiperIonA®. Broadly, we hypothesize that RH-driven drying conditions would similarly affect other anion exchange ionomers used in HEMEL electrodes due to their inherent hygroscopicity.
Overall, to obtain an optimal performing ionomer-incorporated electrode, it is recommended to dry the ionomer coating at a relatively low humidity of 9% RH. As detailed in Supplementary Fig. 8, this can be simply done using an airtight vessel and a saturated potassium hydroxide solution in secondary containment, both of which are low-cost and readily available, making this a serviceable procedure for those in the MEA production sector.
Conclusions
In summary, this study highlights the interplay between the RH during PiperIon® ionomer film drying and its effects on ionomer functionality in electrochemical systems. Drying ionomer film onto an electrode at 9% RH was found to yield a well-connected polymer network that balances the availability of cationic groups for sufficient hydroxide diffusion and mechanical integrity for ionomer/catalyst washout mitigation, leading to superior electrochemical performance. The MEA constructed using an ionomer film dried at this RH achieved a 40% higher current density at 1.8 V than MEAs constructed using ionomer dried at 0 or 94% RH, indicating that an optimal drying RH exists to maximize performance. Ionomer films dried with 0% RH exhibited overly dense and brittle ionomer networks that hinder hydroxide mobility and thus decrease ionic conductivity. High RH drying conversely results in excessive swelling and diminished charge density, negatively impacting ion transport and mechanical stability, as evidenced by ionomer washout and HC analysis. The drying procedure we recommend is low-cost and can be readily applied to a variety of MEA production processes. It can also be used to explain variability in performance that might occur for electrodes fabricated in environments that are not strictly controlled. Although it is recommended to dry ionomer at an electrode using 9% RH, further fine-tuning of the RH for different ionomer families might yield even greater performance improvements.
Methods
Molecular dynamics simulations
Each copolymer chain was modeled using a 17:3 stoichiometric ratio of the repeating monomer cationic piperidinium and terphenyl (TP) units (average IEC = 2.4 meq g−1). All systems were built using the Enhanced Monte Carlo (EMC) software package34. Each system was built using 50 polymer chains and contained 1 OH- counterion per piperidinium group. Four different hydration levels (λ) were simulated, with 0, 1, 4, 6, 8, and 12 solvent molecules added per piperidinium group. The humified environment is expected to lead to lower PAP hydration levels than those during AEMWE operation (approximately λ = 20)30. Six different ethanol concentrations were simulated, with ethanol constituting 4, 8, 16, 32, 64, and 95 wt% of solvent molecules. The 95 wt% ethanol and λ = 0 structure represents the initial state of the membranes. The amount of water (ethanol) in the polymer is expected to increase (decrease) as water vapor in the humidified air exchanges with liquid ethanol present from the membrane casting. A small amount of ethanol (~4 wt%) is expected to remain in the membranes even after long drying times.
All polymer systems were simulated using the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) software package using the OPLS force field and the SPC/E water model35,36,37,38,39,40. Hydroxide ions were modeled as rigid ions using parameters from Bonthuis et al.41. The charges and parameters used to parameterize the force field are the same as those given in previous work, which have been shown to accurately predict PAP polymer properties42. The calculation parameters are also the same as those used in previous work42. Briefly, temperature and pressure were held constant in the NVT and NPT ensembles using a Nose-Hoover thermostat and barostat with time constants of 0.1 ps. A short-range cutoff of 12 Å was used for nonbonded interactions, while long-range electrostatic interactions were implemented using the particle-particle particle-mesh (PPPM) technique with a relative error accuracy of 10−4. Water bonds and angles and OH- bonds were constrained using the SHAKE algorithm43. The systems were all annealed following the same four step procedure using a 1 fs timestep, which involved sequential MD simulations using: 1) the NVT ensemble for 1 ns at 1000 K and 1 bar, 2) the NPT ensemble at 300 K for 100 ps at each pressure of 100, 1000, 10000, 5000, 1000, 500, 100, 10, and 1 bar, followed by the NPT ensemble at 300 K for 2 ns at 1 bar, 3) the NVT ensemble at 1 bar for 1 ns at 2000 K, followed by the NVT ensemble at 1 bar for 100 ps at each temperature of 1800, 1600, 1400, 1200, 1000, 900, 800, 700, 600, 500, and 400 K, followed by the NVT ensemble at 1 bar for 1 ns at 300 K, and 4) the NVT ensemble for 20 ns at 300 K and 1 bar for a final equilibration. This procedure has been previously shown to result in annealed polymer systems that accurately reflect experiment44,45.
All data analysis of the molecular dynamics calculations was performed using Python with accompanying plots made using the matplotlib package and structures visualized using VMD46. The channel width distribution (CWD) calculations were made using the PoreBlazer software package47. The PoreBlazer calculations were performed on structure snapshots obtained after annealing and removing the water, ethanol, and hydroxide molecules. The first coordination shell of an atom type pair was defined as the r value where the first trough occurs in the RDF.
Anode and cathode synthesis
Self-supported catalyst with dip-coating
Iron-nickel oxyhydroxide (FexNiyOOH-20F) self-supported catalyst was grown onto a 5 cm2 nickel felt (Bekaert 2Ni18-0.25, 250 μm, 60% porosity) substrate which was cleaned with acetone in a sonicator, deoxidized with 1.0 M sulfuric acid, and rinsed with deionized water. The pre-catalyst solution was comprised of iron nitrate hexahydrate [Fe(NO3)3·6H2O] and sodium fluoride (NaF); diluted with deionized water, the solution was amalgamated and then soddened with oxygen gas48. The cleaned nickel substrate was suspended vertically into the pre-catalyst solution, in an oxygen-rich environment, and let to sit for 18 h. After the growing period concluded, the catalyst-covered substrate was gently rinsed with deionized water and allowed to dry before being coated in ionomer. The average catalyst loading by mass was 6.84 mg cm−2.
PiperIonA ionomer solution from Versogen–5 wt. % in ethanol, 2.4 mmol g−1 ion exchange capacity–was saturated into the pores of the catalyst-coated nickel felt with dip-coating: where the felt is submerged into the ionomer solution for 75 min. Once done, the excess ionomer is removed and the felt is set upright to dry. Traditionally, after dip-coating, the substrate would be let to dry at ambient conditions (room temperature, pressure, and RH). The RH conditions in this study were controlled and the methods for this are detailed subsequently.
Spray-coated electrode
The catalyst/ionomer ink slurry was made with 50 mg of NiFe2O4 nanopowder catalyst (20 nm), 200 mg of PiperIonA ionomer (this ratio reflects the catalyst/ionomer loading from the self-supported catalyst with ionomer dip-coating), 200 mg of deionized water, and 800 mg of isopropanol. The slurry was sonicated in an ice bath for 1 h and then sprayed onto a 5 cm2 nickel felt (Bekaert 2Ni18-0.25, 250 μm, 60% porosity) substrate with an Iwata airbrush. The spraying was expedited (taking less than 10 min), to ensure the ink would not dry prematurely.
Cathode synthesis
The cathode-side GDE was made from Toray 060 carbon paper (5 wt. % PTFE). A catalyst slurry was sprayed onto a PiperIonA membrane, creating a catalyst-coated membrane (CCM). The catalyst slurry was made from 20 mg of Pt/C (47 wt. % TKK), 171 mg of PiperIonA 5 wt% in ethanol ionomer, 200 mg of deionized water, and 800 mg of isopropanol. The slurry was sonicated in an ice bath for 1 h and then sprayed onto 40-μm thick PiperIonA membrane in bicarbonate form with an Iwata airbrush at ambient conditions.
Thick ionomer film synthesis for material characterization
Approximately 40-μm thick ionomer films were created with 5 wt% PiperIonA in 200 proof ethanol. Varying dimensions of ionomer films were cast, and so the amount of ionomer needed varied. To obtain close to 40 μm, a trial-and-error system was employed to perfect the amount of ionomer needed for that given area. Regardless of the size, the film was cast onto acrylic with an acetate lining on top. The ionomer was applied with a micropipette and allowed to level. It was then put into the RH control box and let dry between 1 and 4 days, depending on the RH condition. Once dry, the ionomer film water submerged in deionized water (fully hydrated) to be soaked off the acetate backing.
Controlled relative humidity environment
Various controlled relative humidity (RH) environments were created with the use of saturated salt solutions, a vacuum oven, and a dry nitrogen environment. For 9%, 40%, and 94% RH, potassium hydroxide (KOH), sodium iodide (NaI), and potassium nitrate (KNO3) were used, respectively. These salts were selected with the goal of creating a broad range of RH conditions. The average RH-controlled building will keep the humidity near 40% RH, which is why we selected that condition. Two extremes (0% and 94% RH) were selected as well as a moderate deviation from 40% RH (chosen to be 9% RH). We chose 9, 40, and 94% RH specifically because the salts used to create those conditions were not excessively hazardous or unfamiliar. The salt solutions were over-saturated to allowed for excess water absorption/desorption if needed by the environment. In an airtight container, the salt solutions (in their own, secondary, open container), along with the freshly dip-coated anode were added and let to sit overnight. An RH probe was fully contained within the system to monitor the humidity and ensure equilibrium was reached. The (near) 0% RH environment utilized a vacuum oven at ambient temperature. After the dip-coating process was completed, the ionomer-coated anode was transferred into a vacuum oven and allowed to dry overnight. The (near) 0% RH environment was also achieved with dry nitrogen air that had a slow, continuous flow over the drying anode and proved to have similar results as the vacuum oven.
Cell assembly and running conditions
Assembly for electrochemical characterization
Within the 5 cm2-active-area MEA, a cathode, anode, and membrane are enveloped by serpentine flow fields and fastened together by end-plates. The CCM and anode, since containing ionomer in bicarbonate form, were ion-exchanged into hydroxide form with two KOH baths–one 30 min and the other overnight. Once sufficiently exchanged, the CCM and anode were moved from the KOH baths and the excess solution was gently removed from the substrates. The MEA had a graphite end plate with triple serpentine channels on the cathode side, and a platinized titanium end plate with triple serpentine channels on the anode side. The hardware was torqued to 60 in-lbs. Deionized water heated to 80 °C was fed to the anode at 3 mL min–1, with the cell also being heated to 80 °C. Due to scarce residual KOH from the ion exchange, the resulting conductivity of the effluent was <100 μS cm−1. Arbin battery testing equipment was used to provide the voltage and current.
Electrochemical characterization (polarization curves + EIS)
Polarization curves were conducted to quantify cell performance the of HEMELs. All reported polarization curves used anodes with a self-supported catalyst. Catalyst break-in/activation was first carried out as the current density was increased stepwise from 0.5 to 2.5 A cm−2 in 0.5 A cm−2 increments every 16.7 min. The polarization curves (current density vs. cell voltage) were then recorded at 80 °C by stepping the current density from 0 to 1.3 A cm−2 with an increment of 0.1 A cm−2; each current density was held for one minute. Three concurrent polarization curves were recorded for each cell. The polarization curves reported in this study are the second of three.
Electrochemical impedance spectroscopy (EIS) measurements were taken using a Solartron SI 1287 electrochemical interface and a SI 1260 impedance/Gain-phase analyzer with an AC oscillation of 10 mV amplitude over frequencies from 100 kHz to 0.1 Hz. The high frequency resistance (HFR) was directly obtained from the x-intercept of the Nyquist plot, as this is where the ohmic region is represented. EIS was recorded at open circuit potential (OCV) and 0.5 A cm−2.
Structural and material characterization
Scanning electron microscopy (SEM) & energy dispersive X-ray diffraction (EDX)
SEM and EDX were conducted with an Auriga 60 Crossbeam at an accelerating voltage of 3 kV and magnification levels between 20,000X and 100,000X. To enhance the sample conductivity for SEM, gold/palladium was coated onto the polymer samples with 60 s of sputtering.
Water uptake (WU) was calculated using Eq. (1):
Dry weights of membranes were measured in bicarbonate form. The wet mass of the membranes (Wwet) was measured after submerging the sample into deionized water at a specified temperature for 30 min, then dabbing the excess water from the surface. Wet masses were recorded at 20 °C, 50 °C, and 80 °C; a low-speed stir bar unified the temperature throughout the bath. The dry mass of the membrane (Wdry) was recorded after allowing the sample to dry in an 80 °C oven overnight. All measurements were repeated 3 times and averaged.
Hydroxide conductivity measurement
The in-plane hydroxide conductivity of membranes (40 μm, 2 inch × 0.3 inch) was measured using the a.c. impedance on a four-electrode cell with platinum electrodes. The measurement cell was submersed in de-aerated deionized water in an N2-purged glovebox. Impedance measurements were carried out at open circuit over the frequency range 1–100 kHz, with a Solartron SI 1260 frequency response analyzer and a Solartron SI 1289 potentiostat.
Tensile measurement
Tensile strength of the membranes in bicarbonate form (2 cm × 0.5 cm, 30 µm thick) were measured using a TA Instruments Q800 DMA at ambient conditions with a 10% min−1 strain rate, 0.1% initial strain, and 0.001 N preload.
Contact angle testing
The hydrophilicity of ionomer films in their dry state was assessed with contact angle measurements. A bead (of controlled size) of deionized water was dropped onto the surface of an ionomer film. It was let to sit for ~10 s because the image of its contact angle was taken. The value of the angle was determined with software within the instrument which calculated the inner, right-side angle of the water bead on the surface.
Dynamic vapor sorption
Water vapor absorption of ionomer films was determined with a DVS analyzer. Ionomer films were cut into 8 mm diameter circles with a punch tool. One cut-out was then placed into a humidity chamber of the DVS which could accurately control humidity from 0 to 80% RH, although allowed up to 100% RH. The test began at 0% RH and moved onto 10% RH when the mass of the sample was stable (dm/dt = 0.003) or 3600 s passed. The RH increased in increments of 10% until 100% RH was reached. All testing was performed at 80 °C.
Data availability
The data that support the findings of this study are available on request from the corresponding author.
References
Amir Mehtab et al. Hydrogen energy as sustainable energy resource for carbon-neutrality realization. ACS Sustain. Resour. Manag. 1, 604–620 (2024).
Cetinkaya, E., Dincer, I. & Naterer, G. F. Life cycle assessment of various hydrogen production methods. Int. J. Hydrog. Energy 37, 2071–2080 (2012).
Hydrogen Strategy. Enabling a low-carbon economy. https://www.energy.gov/sites/prod/files/2020/07/f76/USDOE_FE_Hydrogen_Strategy_July2020.pdf (2020).
Kolb et al. J. Renewable hydrogen imports for the German energy transition – a comparative life cycle assessment. J. Clean. Prod. 373, 133289–133289 (2022).
Seyedehhoma Ghavam, Vahdati, M., Wilson, G. & Styring, P. Sustainable ammonia production processes. Front. Energy Res. 9, 580808 (2021).
Singh, J. K. & Rout, A. K. Advances in green steel making technology - a review. Am. J. Mater. Eng. Technol. 6, 8–13 (2018).
Abbasi, R. et al. A roadmap to low-cost hydrogen with hydroxide exchange membrane electrolyzers. Adv. Mater. 31, 1805876 (2019).
Turner, J. A. Sustainable hydrogen production. Science 305, 972–974 (2004).
Bartlett, J. & Krupnick, A. Decarbonized hydrogen in the US power and industrial sectors: identifying and incentivizing opportunities to lower emissions. Resources for the Future, https://www.rff.org/publications/reports/decarbonizing-hydrogen-us-power-and-industrial-sectors/ (2020).
Du, N. et al. Anion-exchange membrane water electrolyzers. Chem. Rev. 122, 11830–11895 (2022).
Park, S. et al. Low-cost and high-performance anion-exchange membrane water electrolysis stack using non-noble metal-based materials. ACS Appl. Energy Mater. 6, 8738–8748 (2023).
Choi, W.-S. et al. Three-dimensional honeycomb-like Cu0.81Co2.19O4 nanosheet arrays supported by Ni foam and their high efficiency as oxygen evolution electrodes. ACS Appl. Mater. Interfaces 10, 38663–38668 (2018).
Proch, S. et al. Coated stainless steel as bipolar plate material for anion exchange membrane fuel cells (AEMFCs). Int. J. Hydrog. Energy 45, 1313–1324 (2019).
Mayyas, A. T., Ruth, M. F., Pivovar, B. S., Bender, G. & Wipke, K. B. Manufacturing cost analysis for proton exchange membrane water electrolyzers. OSTI OAI (U.S. Department of Energy Office of Scientific and Technical Information), https://doi.org/10.2172/1557965 (2019).
Quaino, P., Juarez, F. & Santos, E. & Wolfgang Schmickler. Volcano plots in hydrogen electrocatalysis – uses and abuses. Beilstein J. Nanotechnol. 5, 846–854 (2014).
Kim, M., Lee, D., Qi, M. & Kim, J. Techno-economic analysis of anion exchange membrane electrolysis process for green hydrogen production under uncertainty. Energy Convers. Manag. 302, 118134–118134 (2024).
BCC Publishing. Global Market for PEM Electrolyzer. BCC Research LLC, https://www.bccresearch.com/market-research/fuel-cell-and-battery-technologies/global-pem-electrolyzer-and-mea-component-market.html?srsltid=AfmBOooJWacUHu2KroMNny9nk_qhAHTVfrWSQvUqn1Jt3O0vU1TBK1qP (2017).
Mauger, S. A. et al. Development of high-performance roll-to-roll-coated gas-diffusion-electrode-based fuel cells. J. Power Sources 506, 230039–230039 (2021).
Park, J., Kang, Z., Bender, G., Ulsh, M. & Mauger, S. A. Roll-to-roll production of catalyst coated membranes for low-temperature electrolyzers. J. Power Sources 479, 228819–228819 (2020).
Mauger, S. Roll to roll (R2R) manufacturing of electrolysis electrodes for low cost hydrogen production. Nrel.gov, https://www.nrel.gov/docs/fy22osti/83194.pdf (2024).
Hydrogen and Fuel Cell Technologies Office. Energy.gov, https://www.energy.gov/eere/fuelcells/articles/us-department-energy-announces-8-million-projects-advance-electrolyzer-and (2024).
Wang, J. et al. Poly(aryl piperidinium) membranes and ionomers for hydroxide exchange membrane fuel cells. Nat. Energy 4, 392–398 (2019).
Olsson, J. S., Pham, T. H. & Jannasch, P. Poly(arylene piperidinium) hydroxide ion exchange membranes: synthesis, alkaline stability, and conductivity. Adv. Funct. Mater. 28, https://doi.org/10.1002/Adfm.201702758 (2018).
Panchenko, O. et al. Non-destructive in-operando investigation of catalyst layer degradation for water electrolyzers using synchrotron radiography. Mater. Today Energy 16, 100394 (2020).
Li, D. et al. Highly quaternized polystyrene ionomers for high performance anion exchange membrane water electrolysers. Nat. Energy 5, 378–385 (2020).
Zheng, Y. et al. Water uptake study of anion exchange membranes. Macromolecules 51, 3264–3278 (2018).
Cao, H. et al. Interaction regulation between ionomer binder and catalyst: active triple-phase boundary and high performance catalyst layer for anion exchange membrane fuel cells. Adv. Sci. 8, 2101744 (2021).
Liu, X. J., Shi, Y. F., Kalbassi, M. A., Underwood, R. & Liu, Y. S. Water vapor adsorption isotherm expressions based on capillary condensation. Sep. Purif. Technol. 116, 95–100 (2013).
Vogt, B. D., Soles, C. L., Lee, H. J., Lin, E. K. & Wu, W. L. Moisture absorption into ultrathin hydrophilic polymer films on different substrate surfaces. Polymer 46, 1635–1642 (2005).
Luo, X., Rojas-Carbonell, S. & Yan, Y. & Ahmet Kusoglu. Structure-transport relationships of poly(aryl piperidinium) anion-exchange membranes: Effect of anions and hydration. J. Membr. Sci. 598, 117680–117680 (2019).
Yang, H. & Wu, N. Ionic conductivity and ion transport mechanisms of solid-state lithium-ion battery electrolytes: a review. Energy Sci. Eng. 10, 1643–1671 (2022).
Hyun, J. et al. Impact of the binding ability of anion exchange ionomer on the initial performance degradation of anion exchange membrane water electrolyzers. Chem. Eng. J. 469, 143919–143919 (2023).
Zhou, H. et al. Experimental investigation of degradation mechanism in proton exchange membrane water electrolyzer under prolonged and severe bubble accumulation condition. Chem. Eng. J. 491, 152202–152202 (2024).
Veld, P. J.; Rutledge, G. C. Temperature-dependent elasticity of a semicrystalline interphase composed of freely rotating chains. Macromolecules 36, 7358–7365 (2003).
Thompson, A. P. et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, Meso, and Continuum Scales. Comput. Phys. Commun. 271, 108171 (2022).
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996).
Kaminski, G. & Jorgensen, W. L. Performance of the AMBER94, MMFF94, and OPLS-AA force fields. Modeling Org. Liq. J. Phys. Chem. 100, 18010–18013 (1996).
Dahlgren, M. K., Schyman, P., Tirado-Rives, J. & Jorgensen, W. L. Characterization of biaryl torsional energetics and its treatment in OPLS all-atom force fields. J. Chem. Inf. Model. 53, 1191–1199 (2013).
Rizzo, R. C. & Jorgensen, W. L. OPLS all-atom model for amines: resolution of the amine hydration problem. J. Am. Chem. Soc. 121, 4827–4836 (1999).
Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271 (1987).
Bonthuis, D. J., Mamatkulov, S. I. & Netz, R. R. Optimization of classical nonpolarizable force fields for OH- and H3O+. J. Chem. Phys. 144, 104503 (2016).
Clary, J. M., Wang, L., Yan, Y., Frischknecht, A. L. & Vigil-Fowler, D. Effect of stoichiometry and hydration level on water domain size and transport in poly (aryl piperidinium) alkaline anion-exchange membranes. J. Membr. Sci. 717, 123517 (2025).
Ryckaert, J.-P., Ciccotti, G. & Berendsen, H. J. C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Abbott, L. J., Hart, K. E. & Colina, C. M. Polymatic: a generalized simulated polymerization algorithm for amorphous polymers. Theor. Chem. Acc. 132, 1334 (2013).
Abbott, L. J. & Frischknecht, A. L. Nanoscale structure and morphology of sulfonated polyphenylenes via atomistic simulations. Macromolecules 50, 1184–1192 (2017).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). 27–28.
Sarkisov, L., Bueno-Perez, R., Sutharson, M. & Fairen-Jimenez, D. Materials Informatics with PoreBlazer v4.0 and the CSD MOF Database. Chem. Mater. 32, 9849–9867 (2020).
Xiao, J. et al. Water-fed hydroxide exchange membrane electrolyzer enabled by a fluoride-incorporated nickel–iron oxyhydroxide oxygen evolution electrode. ACS Catal. 11, 264–270 (2020).
Acknowledgements
This material is based upon work supported by the US Department of Defense under Award No. W911NF-23-2-0028. We acknowledge financial support from the US DOE Office of Energy Efficiency and Renewable Energy, Hydrogen and Fuel Cell Technologies Office, under the ElectroCat Consortium, DOE technology managers McKenzie Hubert and William Gibbons, and DOE program managers David Peterson and Dimitrios Papageorgopolous. With gratitude to Chevy. This work was authored by the National Renewable Energy Laboratory, operated by the Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. The research was performed using computational resources sponsored by the Department of Energy’s Office of Energy Efficiency and Renewable Energy and located at the National Renewable Energy Laboratory.
Author information
Authors and Affiliations
Contributions
A.P. created the MEAs, performed the electrolyzer testing (polarization curves and EIS) for the PAP-TP-85 and PAP-TP-100 samples, completed water uptake testing, SEM with EDS, dynamic mechanical analysis, dynamic vapor sorption analysis, processed & analyzed the data, and wrote all but the modeling section of the paper. J.C. performed the computational tasks and contributed to the modeling section of the results and methods. T.W. completed the hydroxide conductivity testing. A.O. assembled MEAs and performed electrolyzer testing for the PAP-TP-85-MQN tests. Y.Y., L.W., and D.V. supervised the research.
Corresponding author
Ethics declarations
Competing interests
Y.Y. is a founder and CEO of Versogen, where all the ionomer and membrane materials were sourced from.
Peer review
Peer review information
Communications Materials thanks Haitao Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Haotian Wang and Jet-Sing Lee. [A peer review file is available.]
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Polsky, A., Clary, J., Oliveira, A.M. et al. Impact of processing humidity on ionomer film structure and performance in hydroxide exchange membrane electrolyzers. Commun Mater 6, 171 (2025). https://doi.org/10.1038/s43246-025-00900-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43246-025-00900-5
