
Abstract
A pressure-induced insulator-to-metal transition (IMT) has recently been discovered in the nodal-line ferrimagnet Mn3Si2Te6. The electronic phase transition is accompanied by anomalies in the magnetic ordering temperature and the anomalous Hall conductivity, which peak at or near the critical pressure of the IMT. We perform density functional theory (DFT) calculations as a function of pressure to establish the connection between the IMT and the magnetic anomalies in Mn3Si2Te6. We extract Heisenberg Hamiltonians as a function of pressure based on our DFT calculations. Our classical Monte Carlo simulations for these Hamiltonians yield ordering temperatures and magnetic ordering patterns, in agreement with the experimental data. Although we can accurately explain the evolution of magnetism with pressure, it seems that the anomalous Hall conductivity in Mn3Si2Te6 can only be accounted for by extrinsic contributions or moderate electron doping of the samples in the experiment.

Similar content being viewed by others
Introduction
Magnetic materials with non-trivial band topology have recently emerged as a platform for novel transport phenomena with potential spintronic applications1,2,3,4,5. In particular, nodal-line semiconductors and semimetals exhibit a particularly large anomalous Hall effect6,7,8 and angular magnetoresistance9,10,11,12,13.
Among them, Mn3Si2Te6 stands out as a ferrimagnetic nodal-line semiconductor that exhibits record-high colossal magnetoresistance (CMR)14 and angular magnetoresistance (AMR)12. These phenomena have been linked to chiral orbital currents15 and highlight the material as a candidate system where magnetism, topology, and electronic correlations are strongly intertwined.
At ambient pressure, Mn3Si2Te6 crystallizes in a trigonal structure (space group \({{\rm{P}}}\overline{3}1{{\rm{c}}}\))16 and orders ferrimagnetically below TC ≈ 78 K17,18,19. Previous studies suggest that its magnetic and electronic properties are closely coupled, with signatures of a field-driven insulator-to-metal transition20 and current-driven effects on magnetism21. Single-crystal neutron diffraction shows a slight non-collinearity of magnetic moments, which points to a delicate frustration of magnetic couplings22.
The magnetic anisotropy at ambient pressure has been estimated as ~0.7 meV/Mn23,24, which translates to ~1 K after dividing out the spin factor S2 = 25/4. The spin waves of Mn3Si2Te6 at ambient pressure have been mapped with inelastic neutron scattering. A fit of the spectrum with isotropic interactions plus single-ion anisotropy also yields ~1 K25. One study also finds a Dzyaloshinskii–Moriya interaction23.
Application of pressure reveals even richer physics. At Pc = 15.4 GPa, Mn3Si2Te6 undergoes a structural transition to a monoclinic phase, concurrent with an insulator-to-metal transition26. A recent study places the critical pressure at a slightly lower pressure of 10–12 GPa27. From ambient to critical pressure, the ferrimagnetic ordering temperature increases almost linearly to nearly room temperature, before decreasing again at higher pressures, resulting in a dome-shaped evolution of TC26. After the transition to the monoclinic crystal structure, a pronounced anomalous Hall effect emerges, which peaks near 17 GPa26. These correlated anomalies strongly suggest that changes in the electronic structure under compression are intimately linked to the evolution of magnetism.
Here, we investigate the microscopic origin of these pressure-induced anomalies. Using density functional theory (DFT) calculations combined with classical Monte Carlo simulations, we extract exchange couplings, ordering temperatures and anisotropies across the structural transition, as well as the intrinsic anomalous Hall conductivity. Our results provide a comprehensive picture of how pressure tunes the interplay between electronic and magnetic degrees of freedom in Mn3Si2Te6.
Results
For pressures up to P = 15.4 GPa in the trigonal space group \(P\bar{3}1c\), we determine the crystal structures by DFT structure relaxation applied to the internal coordinates in combination with experimental lattice parameters. We proceed this way because in the case of strongly correlated materials like Mn3Si2Te6, structure prediction under hydrostatic pressure is not always reliable. We use a GGA+U functional for a proper description of strong electronic interactions of the 3d electrons of the Mn2+ ions. We fix the value of the Hund’s rule coupling at JH = 0.76 eV28. The relaxed structural parameters are not strongly dependent on the precise value of the on-site interaction U; for this reason, we use a generic value of U = 5 eV, which is between U = 4.5 eV describing MnSc2S429 and U = 5.2 eV that works well for Na3Mn(CO3)2Cl30. For the crystal structures, we use the lattice parameters determined experimentally in ref. 26. We interpolate the lattice parameters in order to obtain a regular and dense mesh of pressure values.
For the trigonal structures, we obtain the internal positions by DFT+U structure relaxation in the ferrimagnetic state that is known to be the ground state. Note, however, that while the structures differ significantly between relaxation in non-magnetic and magnetic states, the magnetic order (ferromagnetic or ferrimagnetic) does not play a significant role. We compared the structures we obtained at P = 0 GPa by relaxing internal coordinates with GGA+U = 5 eV in ferromagnetic and ferrimagnetic states. We find that Mn-Te distances differ by less than 0.006 Å, Mn-Te-Mn angles by less than 0.01 degrees and the total energy by less than 0.4 meV per formula unit. This indicates that the choice of magnetic order during relaxation does not have a significant influence. More details on the structures are given in the Supplementary Information.
Preparation of the monoclinic high-pressure structures is more involved. We again interpolate the lattice parameters of the candidate C2c structures between P = 15.4 GPa and 25 GPa that were experimentally determined to be the best candidate for the high-pressure space group26. However, we found that DFT relaxation of internal parameters took the structure too far away from the single fully determined crystal structure at P = 22.3 GPa. This happens independent of the exchange-correlation functional and is comparable to the situation in CrGeTe3, where we similarly found that the experimental crystal structure cannot be precisely reproduced by any DFT functional31. For this reason, we keep the internal structure parameters constant at the values determined experimentally for P = 22.3 GPa.
In fact, the crystal structure of Mn3Si2Te6 is similar to van der Waals chalcogenides: layers of MnSiTe3 are self-intercalated with Mn atoms, leading to alternating honeycomb and triangular Mn layers16,26. At the critical pressure of Pc = 15.4 GPa, the monoclinic phase transition splits the tellurium positions into three inequivalent sites and the MnSiTe3 layers slide slightly with respect to each other26.
Our DFT calculations with ferrimagnetic spin configuration reproduce the experimentally observed IMT of Mn3Si2Te6 at a pressure of Pc = 15.4 GPa. For lower pressures, the system is a band insulator. For higher pressures, the system becomes metallic. The Berry curvature in the metallic phase shows a complex momentum structure with positive and negative contributions of high absolute value (see Fig. 1a, c, g, i). In the metallic phase, both the electronic band structure (see Fig. 1b, d, h, j) and the corresponding momentum-space structures of the Berry curvature (see Fig. 1e, f, k, l) evolve gradually as a function of pressure. Band structures for pressures below 15.4 GPa are shown in Supplementary Note 1 with Supplementary Figs. 4–6. In these, we observe an insulating ferrimagnetic ground state with spins oriented in the ab plane. For spins oriented along the c axis, the IMT occurs below the structural phase transition, reminiscent of the field-driven IMT in Mn3Si2Te620. This shows how both pressure and magnetic field can be used to manipulate the electronic structure of this material.

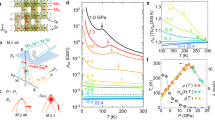
a, c Show the total Berry curvature Ωx and Ωz (in units of squared Bohr radii \({a}_{0}^{2}\)) and b, d show the electronic band structure with orbital weights on a high-symmetry path through the Brillouin zone for spin quantization axis parallel to the a direction at a pressure of 16 and 23 GPa, respectively. e, f Show cuts of the total Berry curvature Ωz in the kx-ky plane at kz = 0 for the same pressures and the same orientation of spin quantization axis. The colour scale is cut off at a value of ±60. The grey shaded area represents the Fermi surface. g–l Show the electronic structure and total Berry curvature for spin quantization axis parallel to the c direction and all other parameters equal to (a–f).
For the anomalous Hall conductivity (AHC), we perform full-relativistic DFT calculations including spin-orbit coupling (SOC). The AHC arises from the Berry curvature as the integral over large positive and negative contributions, i.e. we can expect the AHC to be very sensitive to details of the electronic structure. Although changes in the AHC as a function of pressure are gradual (see Fig. 2), we observe sign changes in some components of the conductivity tensor, while others are relatively stable. The xz component is zero within numerical accuracy, regardless of the choice of quantization axis (which corresponds to the direction of an applied magnetic field in experiment). In fact, the xz component is exactly zero due to the presence of antiunitary symmetries \(C{2}^{{\prime} }(y)\) and \({m}^{{\prime} }(y)\). The xy and yz components show qualitative changes when the spin quantization axis is rotated from the easy axis (along a, see Fig. 2a) to the hard axis (along c, see Fig. 2b). The pressure dependence of these components results from changes in details of the electronic band structure due to the increased bandwidth and spin-orbit coupling when changing the direction of the spin quantization axis. The anisotropy of AHC in Mn3Si2Te6 follows from the layered crystal structure, which gives rise to an anisotropic electronic band structure, and the presence of strong SOC.

a, b Show the intrinsic contribution to the anomalous Hall conductivity in the xy, xz and yz planes as a function of pressure calculated from relativistic DFT calculations (i.e. including spin-orbital coupling) in the ferrimagnetic state with spin quantization axis parallel to the a direction (easy axis) and the c direction (hard axis), respectively. c Shows the calculated AHC in the xy plane with shifted chemical potential μ, which simulates charge doping of Mn3Si2Te6, in comparison to the experimental data from ref. 26.
Normally, the AHC is measured in large magnetic field. Therefore, which orientation of the spin quantization axis in DFT is appropriate for comparison to experiment depends on the spin-flop field of Mn3Si2Te6, which we determine together with other magnetic properties of this material. In addition, we have calculated the AHC with spins parallel to the c axis under a shift of the chemical potential to simulate charge doping of the Mn3Si2Te6 samples. In Fig. 2c we show the xy component of the AHC for moderate electron doping in comparison to theoretical values without shift and the experimental results. Electron doping strongly alters the results and a moderate doping level of μ = 0.16 eV yields a dome-like AHC similar to experiment. To scan the range of achievable AHC, we performed our calculations at fixed chemical potential μ, which (due to pressure-induced changes in the electronic structure) is not the same as a fixed doping level. Comparison to future experimental results with a precisely known doping level will have to take this into account.
Next, we establish the Heisenberg Hamiltonian parameters for Mn3Si2Te6 as a function of pressure. We base our calculations on experimental crystal structures as described above. In order to determine the exchange interactions, we now apply the DFT energy mapping technique. This involves DFT calculations for a large number of spin configurations in low-symmetry structures of Mn3Si2Te6 and fitting the total energies with the Heisenberg Hamiltonian
where Si are spin operators, and we do not double count bonds. This approach regularly yields excellent results in magnetic insulators32,33 and it has also been useful for understanding the magnetism in semiconducting and metallic CrGeTe3 under pressure31,34. In fact, the latter material shares many similarities with Mn3Si2Te6, for example, the structural elements of the Ge2Te6 or Si2Te6 units and the observation of an anomalous Hall effect under pressure26,35. Examples for alternative approaches to magnetic Hamiltonian parameters are the four-state method36, which we do not employ here because we rely on the safety net provided by our statistical approach, and the force theorem method as implemented, for example, in the tb2j package37; we applied the latter to ferrimagnetic Mn3Si2Te6 but did not arrive at a meaningful result.
In the DFT energy mapping approach, there are two parameters that affect the overall energy scale of the exchange couplings, the on-site interaction U and the Hund’s rule coupling JH. The latter is expected to have little material dependence, and we fix it to JH = 0.76 eV as suggested in ref. 28. For the on-site Coulomb interaction U, we exploit experimental observations at ambient pressure and then fix U, assuming that it is not strongly pressure dependent. The experimental Curie–Weiss temperature TCW = − 277 K and the ferrimagnetic ordering temperature TC = 78 K at ambient pressure17 are well reproduced in our theoretical calculations if we choose U = 4.2 eV for this entire study (see Fig. 3a). With the fixed parameters U = 4.2 eV and JH = 0.76 eV, we now calculate the Heisenberg Hamiltonian parameters; they are shown in Fig. 3b, d, e, f. Table 1 identifies the most important bonds and specifies how they evolve across the structural phase transition.

a DFT calculated exchange parameters for Mn3Si2Te6 at ambient pressure as a function of the on-site Coulomb repulsion U. The dashed line denotes the value of on-site Coulomb repulsion U = 4.2 eV, which we choose for the remainder of our study. For this interaction strength, we obtain ΘCW = − 247 K as the mean-field Curie–Weiss temperature and TC = 65 K as the ordering temperature in classical Monte Carlo, which both agree well with experimental values. b Relevant exchange paths in Mn3Si2Te6, shown for the high symmetry, trigonal \(P\bar{3}1c\) space group. c Charge gap of the ferrimagnetic ground state and average charge gap of all magnetic configurations included in the DFT energy mapping as a function of pressure. In the high-pressure phase, the charge gap is zero. d Relevant exchange paths in Mn3Si2Te6, shown for the low symmetry, monoclinic C2/c space group. e Antiferromagnetic intra-trimer (nearest neighbour) exchange coupling J1 as a function of pressure. f Next-nearest neighbour and longer range exchange couplings between Mn atoms as a function of pressure. The inset shows the ground state spin structure. The interaction parameters are U = 4.2 eV and JH = 0.76 eV.
We find that in the trigonal low-pressure region, Hamiltonians are dominated by antiferromagnetic J1 and J3. J1 mediates the exchange between two Mn1 and one Mn2 in the nearest-neighbour Mn trimer; in this sense, the network defined by J1 is zero-dimensional, and below the ordering temperature for J1, the trimers order in up-down-up or down-up-down states and the effective moment of the system is reduced to one third. The existence of a large intra-trimer coupling J1 is consistent with experiments that show signs of short-range magnetic order well above the ordering temperature TC17,22,38. Mn trimers form a triangular lattice in the ab plane, and triangle centres define the position to which triangular lattices above and below are shifted. As a consequence, J3 couples the Mn trimers into a buckled honeycomb lattice in the ab plane. However, as this occurs above and below each triangular lattice of trimers, J3 provides a fully three-dimensional connectivity of the lattice. It couples Mn1 and Mn2 sites, and therefore the Neel state formed by unfrustrated J3 corresponds to a ferromagnetic order of the trimers with effective spin of \({S}_{{{\rm{eff}}}}=\frac{5}{2}\). The resulting ferrimagnetic state is an antiparallel arrangement of Mn1 and Mn2 sublattices, which occur in a ratio 2:1 (see inset of Fig. 3f). Therefore, the nearly linear increase of J3 in the insulating phase up to P = 15.4 GPa indicates that we can expect a similar linear increase for the ferrimagnetic ordering temperature of the material. Note that our calculations do not support a substantial J2, which we find to be 3% of J1 rather than the 18% obtained in ref. 17.
We confirm this expectation by classical Monte Carlo (cMC) simulation of the full Hamiltonians shown in Fig. 3. In Fig. 5, we show that the calculated TC from our Hamiltonians can explain the TC increase seen in experimental resistivity and susceptibility data from ref. 26 very well (for details see Supplementary Note 2 with Supplementary Figs. 7–12). In our calculations, the pressure-induced insulator to metal transition in the ferrimagnetic ground state (with spins oriented along the a axis) occurs between 14 and 16 GPa (see Fig. 3c). With spins quantized along the c axis (hard axis), the IMT occurs somewhat earlier, indicating the field-tunability of this material. Between 14 and 16 GPa, we observe a clear change in the pressure evolution of the exchange interactions: J1 levels off, and most of the subleading couplings begin to decrease or even turn negative (ferromagnetic). The phase transition from trigonal to monoclinic space group leads to significant changes in the exchange couplings.
The dominant coupling J1 that is responsible for the S = 5/2 trimers remains constant at around 100 K up to the highest pressure P = 25 GPa. The second neighbour coupling J2, which defines a honeycomb network for Mn1, splits into three distinct couplings; while J2 was near zero in the trigonal structure, possibly due to a compensation between ferromagnetic and antiferromagnetic contributions to the exchange, the three couplings \({J}_{2}^{{\prime} }\), \({J}_{2}^{{\prime\prime} }\) and \({J}_{2}^{{\prime\prime} {\prime} }\) in the monoclinic structure take on substantial ferromagnetic values. The coupling J3, which defines a buckled honeycomb network connecting Mn1 and Mn2, also splits into three couplings at the transition to monoclinic symmetry. They remain mostly antiferromagnetic but are significantly smaller than J3 before the transition. The average of \({J}_{3}^{{\prime} }\), \({J}_{3}^{{\prime\prime} }\) and \({J}_{3}^{{\prime\prime} {\prime} }\) shows an almost linear decrease. The coupling J4a connects second nearest neighbours in the Mn1 honeycomb network while J4b form a Mn2 triangular lattice. Both are small in \(P\bar{3}1c\) but become significant and ferromagnetic in C2c. The coupling J5 is the second nearest neighbour in the Mn trimer at twice the length of J1. Small and antiferromagnetic in trigonal symmetry (slightly destabilizing the up-down-up order in the trimer), it becomes increasingly ferromagnetic in monoclinic symmetry, where it stabilizes the up-down-up trimer order. Across the pressure range, the sub-leading couplings introduce some frustration into the system, which agrees well with the findings in ref. 22.
To determine the anisotropies from DFT, we calculate the total energy in ferri- and ferromagnetic spin configuration as a function of the spin quantization axis. We then perform an additional energy mapping, considering that the isotropic part of the Hamiltonian Hiso (Eq. (1)) is independent of the quantization axis. This allows us to resolve the anisotropy terms in
where \({J}_{ij}^{yy}\) and \({J}_{ij}^{zz}\) are nearest neighbour exchange anisotropies and \({K}_{i}^{y}\) and \({K}_{i}^{z}\) are single-ion anisotropies. In the trigonal phase we have \({J}_{ij}^{yy}=0\) and \({K}_{i}^{y}=0\) due to the crystal symmetry.
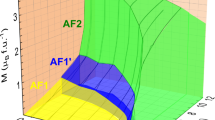
The DFT energies, evaluated as a function of pressure, magnetic ordering pattern (FM or ferrimagnetic), and spin quantization axis (see Fig. 4a, b), clearly indicate that ferrimagnetic ordering is more stable than FM, and that the ab plane constitutes the easy plane in trigonal Mn3Si2Te6. In the monoclinic phase, the energies of the a and b directions split (see Fig. 4b). Alignment parallel to the b axis yields an energy slightly above the a axis. The energy distance with respect to the c axis increases, so that c clearly remains the hard axis. The ferromagnetic state, which is much higher in energy than the ferrimagnetic state, shows a similar trend (see Fig. 4a), although the b axis is slightly lower in energy than the a axis. In order to ensure that this finding is not dependent on our choice of interaction strength of U = 4.2 eV, we performed a sample calculation at U = 5 eV for Mn3Si2Te6 at P = 20 GPa. We find exactly the same hierarchy of energies, and the energy differences differ only insignificantly from those shown in Fig. 4a, b.

All data points were calculated with U = 4.2 eV. Labels C2/c and \(P\bar{3}1c\) denote the crystal symmetries on both sides of the phase transition. a Shows the raw energy differences per Mn atom (also divided by the square of the spin S = 5/2) for the ferromagnetic state with three possible spin quantization axes and as a function of pressure. b Shows the raw energy differences per Mn atom (also divided by the square of the spin S = 5/2) for the ferrimagnetic ground state and compares these energy differences to the experimentally determined c axis spin-flop fields from ref. 26. c Shows the anisotropic single-ion (Ky and Kz) and nearest-neighbour exchange (\({J}_{1}^{yy}\) and \({J}_{1}^{zz}\)) parameters as a function of pressure determined from raw energy differences.
The anisotropy energies can be converted into a magnetic field, which is necessary to rotate spins from the easy plane or axis to the hard axis. For this, we need to consider the rotation angle between the crystal axes, which differs from 90° in the monoclinic phase. These estimates agree very well with the observed spin-flop (coercive) fields in ref. 26 (see right axis in Fig. 4b), which is rather puzzling given that ref. 26 claims a c easy axis in the monoclinic phase. It also seems that some experimental spin-flop fields assigned to pressures above Pc are similar to our calculations in the trigonal phase. This could indicate some uncertainty in the experimentally applied pressures or a sample-dependent critical pressure. We return to the issue of magnetic anisotropy in the discussion section.
As expected from the energies, in the trigonal phase we find positive exchange and single-ion anisotropies for the z direction, while the anisotropies for the y direction are basically zero, except for small convergence errors in the DFT energies (see Fig. 4c). This agrees well with polarized neutron powder diffraction experiments at ambient pressure38. Since nearest neighbour spins in the ferrimagnetic trimer of the ground state are antiparallel, the energy difference between ab plane and c axis is mostly due to the single-ion anisotropy Kz, while the exchange anisotropy Jzz actually decreases the anisotropy energy.
This qualitative picture for the parameters associated with the c axis persists into the monoclinic phase: the exchange anisotropy Jzz remains almost constant, while the single-ion anisotropy Kz increases slightly (see Fig. 4c). The parameters associated with the b axis become non-zero: while the positive single-ion anisotropy Ky is quite small, the anisotropy energy for the b axis is generated mostly by the negative exchange anisotropy Jyy.
Discussion
Our results for the electronic structure of Mn3Si2Te6 reproduce the experimentally observed IMT at the structural phase transition. Interestingly, the average charge gap of the magnetic configurations in our study decreases smoothly as a function of pressure and finally vanishes at the structural phase transition (see Fig. 3c). This result seems to agree with the smooth decrease of longitudinal resistivity as a function of pressure observed in experiment26.
Although our calculations faithfully describe the experimentally known electronic and magnetic properties of Mn3Si2Te6 on both sides of the structural phase transition, we cannot fully explain the experimentally observed dome in the anomalous Hall conductivity as a function of pressure (see Fig. 2) upon entering the monoclinic metallic phase26. Since the AHE is measured in large magnetic field far above the spin-flop field of ~1 T (see Fig. 4b), we can assume the spins to be oriented along the hard axis (c axis). Therefore, the experimental AHC should be compared to Fig. 2b or potentially Fig. 2c. DFT predicts a large intrinsic contribution with negative sign to the xy component of the AHC, while experiment shows small positive values, which decrease between about 16 and 21 GPa26. Based on our findings in Fig. 2b it seems possible that the experimentally observed behaviour could be the result of partial cancellation between the large negative intrinsic contribution and a (hypothetical) positive extrinsic contribution of about the same size. A similar qualitative mismatch has been observed for the closely related van der Waals ferromagnet CrGeTe3, where discrepancies have been attributed to extrinsic effects35,39,40. Alternatively, the experimentally observed AHC could be the consequence of electron doping, e.g. due to Te deficiency, as shown in Fig. 2c. A shift of the chemical potential to μ = 0.16 eV produces a dome-shaped AHC remarkably similar to experiment. Eventually, the actual high-pressure structure may differ from the C2/c candidate employed in our calculations. This issue merits further investigation.
Our results for the isotropic exchange couplings of Mn3Si2Te6 show a rather simple picture of relatively unfrustrated antiferromagnetic exchange in the trigonal high-symmetry structure. In the metallic monoclinic low-symmetry structure, many exchange paths contribute with opposite signs (see Fig. 3f), which potentially leads to cancellation of contributions and a de-stabilization of the ferrimagnetic ground state as well as a lower ordering temperature. In this sense, the decreasing ordering temperature (see Fig. 5) cannot be attributed to the decrease or increase of a single exchange coupling. It is rather the product of qualitatively different exchange Hamiltonians in the trigonal vs. monoclinic structures. Nevertheless, our classical Monte Carlo simulations for the ordering temperature (see Fig. 5) and the ground state faithfully reproduce the experimentally observed behaviour, also in the low-symmetry phase whose structure is not yet resolved with absolute certainty. This indicates that the C2/c structure is a good candidate for the low-symmetry phase, where the complex cancellation of ferromagnetic and antiferromagnetic exchange paths seems to be the root cause of the decreased ordering temperature.

Theoretical values are shown in blue, while experimental data from ref. 26 are shown in yellow, orange and green symbols for comparison. The theoretical prediction is based on the isotropic Hamiltonian determined from the DFT energy mapping, which we use to simulate magnetic configurations in classical Monte Carlo.
Our estimates of the exchange interactions and single-ion anisotropies are consistent with multiple studies at ambient pressure23,24,25, as well as with experimentally observed spin-flop (coercive) fields under pressure26. Our calculations indicate that Mn3Si2Te6 in the low-pressure high-symmetry phase exhibits an ab easy plane and a hard axis along the c direction (see Fig. 4b). Upon entering the monoclinic phase, the degeneracy between the a and b directions is lifted: the a direction becomes the easy axis, while the b direction lies only slightly higher in energy. The c direction remains the hard axis.
To the best of our knowledge, ref. 26 reports a c easy axis based on an extrapolation of the magnetic anisotropy energy in the trigonal phase and two low-field (0.2 T) magnetization measurements along the c axis. In contrast, our calculations unambiguously identify the a direction as the easy axis in the monoclinic phase and show no indication of a decreasing magnetic anisotropy energy. Consequently, our results are in clear contradiction with the c easy axis proposed in ref. 26.
The behaviour of individual anisotropy parameters is rather complex and non-monotonic as a function of pressure (see Fig. 4c). Nevertheless, the resulting spin-flop fields (see Fig. 4b) quantitatively agree with experimental data from ref. 26. With a spin-flop field of ~1T and the strong effect of the orientation of the spin quantization axis on electronic structure, anomalous Hall effect and magnetic properties, comparisons between experiment and theory need to carefully consider the size of magnetic field applied in experiment and its implications for the spin quantization axis in DFT.
In summary, our combined DFT and cMC analysis provides a coherent microscopic picture of the pressure-induced behaviour in Mn3Si2Te6. The evolution of isotropic and anisotropic exchange couplings across the structural phase transition successfully reproduces the experimentally observed variation of the ferrimagnetic ordering temperature and the spin-flop field. The calculated electronic structure captures the pressure-driven insulator-metal transition, demonstrating an intimate connection between electronic band structure and magnetic properties of Mn3Si2Te6. However, the experimentally observed dome in the anomalous Hall conductivity cannot be accounted for by intrinsic Berry curvature effects alone, suggesting additional extrinsic contributions or charge doping of experimental samples. Mn3Si2Te6 emerges as a model platform for exploring how pressure couples lattice and spin degrees of freedom with band structure topology in van der Waals magnets.
Methods
Density functional theory-based energy mapping
We perform density functional theory (DFT) calculations within the full potential local orbital (FPLO) method41 and using the generalized gradient approximation (GGA)42 for the exchange-correlation functional. We account for the strong interactions on Mn2+ ions using a DFT+U correction43. We use collinear DFT to determine the Heisenberg Hamiltonian parameters, the single ion anisotropy energies and selected anisotropic exchange interactions. The experimental crystal structures under pressure were imported from ref. 26 and interpolated smoothly using splines (for details see Supplementary Note 1 with Supplementary Figs. 1–3).
The DFT-based energy mapping approach requires accurate DFT energies for a large set of distinct spin configurations, which are then used to fit the exchange parameters between the spin-5/2 manganese atoms in Eq. (1) by matching the classical energies of the Heisenberg Hamiltonian with the calculated DFT energies using the method of ordinary least squares (OLS).
In the trigonal \(P\bar{3}1c\) space group, we use 2 × 1 × 1 supercells with 12 distinct Mn2+ ions. Calculating 30 spin configuruations, we can resolve 14 exchange interactions up to a distance of about three times the nearest neighbour Mn-Mn distance. In the monoclinic C2c space group, we ensure low statistical error bars on the exchange interactions by calculating 100 spin configurations in order to resolve 16 exchange couplings.
For the monoclinic phase, we observe that parameter estimates for the isotropic Heisenberg couplings can be sensitive to small deviations in the DFT energies. Therefore, we minimize the mean squared error plus a regularization parameter α times an L1 penalty term, which enforces sparsity of the resulting parameter set44. We find the optimal regularization parameter α = 0.04 from the onset of the mean squared error as a function of α45. For more details, see Supplementary Note 3, which includes Supplementary Figs. 13–17.
Single-ion and exchange anisotropies were extracted from DFT by rotating the spin quantization axis and calculating total energies in both ferri- and ferromagnetic spin configuration. Using six DFT energies (from the two spin configurations multiplied by the three choices for the spin quantization axis), we can solve the corresponding system of linear equations for the anisotropies.
In the trigonal phase, we know that the a and b directions are equivalent. Therefore, we have \({J}_{1}^{yy}=0\) and Ky = 0. The classical energies of the Hamiltonian with anisotropic terms (Eq. (2)) in each spin configuration and direction of spin quantization axis are therefore given by:
Due to the crystal symmetry, the energies in a and b direction should be the same. In DFT estimates for the energies on the left-hand side, small deviations may occur due to imperfect convergence.
In the high-pressure monoclinic phase, the a and b directions are not equivalent any more. Therefore, finite anisotropy parameters in the y direction occur. The system of equations in the monoclinic phase reads:
We use Eq. (4) in both the trigonal and monoclinic phase and solve for the unknown parameters on the right-hand side using ordinary least squares. This also allows us to check the quality of our DFT energies, since perfect convergence should provide zero anisotropies in the trigonal phase for \({J}_{1}^{yy}\) and Ky. Figure 4c shows that we recover the zero values for \({J}_{1}^{yy}\) and Ky almost perfectly.
From the known DFT energies in the ferrimagnetic state \({E}_{M\parallel \{a,b,c\}}^{{{\rm{ferri}}}}\), we can also estimate the spin-flop field, i.e. the magnetic field strength needed to rotate the spins in the direction of the magnetic field. The energy of a spin with S = 5/2 in a magnetic field is Hfield = − gμBB ⋅ S. With \({{\bf{S}}}=\frac{5}{2}{{\bf{u}}}\), B = Bv and g = 2, we can write Hfield = − 5μBB v ⋅ u. The energy difference between two spin orientations u and w in a fixed magnetic field is therefore ΔEfield = − 5μBB v ⋅ (u − w).
Let us now assume that w is the low-energy axis in the zero field. That means a spin-flop will occur from the w direction to the u direction if B is parallel to u and sufficiently large to overcome the energy difference between the two spin orientations in zero field:
We now assume v∥u, where u is the high-energy direction in zero field. With \({{\bf{v}}}\cdot {{\bf{w}}}=\cos (\beta )\), we can solve for the magnetic field required for the spin-flop:
The experimental values in Fig. 4b are extracted from Fig. S3b of ref. 26 by taking the magnetic field necessary for full ferrimagnetic spin polarization along the c axis (~0.55 T at 14.1 GPa) and from Fig. 2b and Fig. S11c of ref. 26 by taking the field along the c axis necessary for saturation of the anomalous Hall conductivity σxy. These are ~0.48 T at 16.2 GPa, ~0.64 T at 17.5 GPa, ~0.48 T at 18.4 GPa, and ~0.88 T at 21.0 GPa for Fig. 2b of ref. 26 as well as ~0.45 T at 7.7 GPa and ~0.9 T at 16.8 GPa for Fig. S11c of ref. 26.
Classical Monte Carlo
We performed classical Monte Carlo (cMC) simulations using the Metropolis-Hastings algorithm with local updates for Mn3Si2Te6 as a function of pressure. We use a supercell of size 8 × 8 × 8 with 3072 Mn sites in total. For each simulation we perform 5000 lattice sweeps for the warmup. Subsequently, we perform 3000 measurements with 20 lattice sweeps for each. Each cMC run is averaged over the measurements of 512 parallel threads. Therefore, we perform 3072 ⋅ (5000 + 3000 ⋅ 20) ⋅ 512 ≈ 1011 Metropolis steps for each cMC result.
The exchange couplings illustrated in Fig. 3 were used to calculate and plot the specific heat, magnetic susceptibility and ordering temperatures. At ambient pressure, we find a peak in specific heat at 65 K (see Supplementary Information for information on crystal structures, additional classical Monte Carlo results and regularization). Upon investigating the equilibrium spin configurations from our cMC data, we constructed polar plots of the spins in the ensemble. This allowed us to confirm that the ground state is ferrimagnetic, and the peak in specific heat corresponds to the ferrimagnetic transition temperature TC (see Supplementary Information for information on crystal structures, additional classical Monte Carlo results and regularization). We observe a nearly linear increase in TC as a function of pressure in the trigonal phase. In the monoclinic phase, we anticipate a consequent nearly linear decrease in TC based on experimental observations, which is indeed reflected in our Monte Carlo calculations in this phase. Hence, we successfully reproduced the dome-shaped behaviour in TC as a function of pressure through our TC simulations.
Anomalous Hall conductivity
We perform full-relativistic DFT electronic structure calculations, i.e. including spin-orbit coupling (SOC), based on which we estimate the intrinsic component of the anomalous Hall effect. The anomalous Hall conductivity σxy is defined as the integral of the total Berry curvature \({\Omega }_{z}(\vec{k})\) over the entire Brillouin zone (BZ)46:
The total Berry curvature \({\Omega }_{z}(\vec{k})\), calculated using Wannier interpolation within FPLO47,48, is defined as the sum over all bands n of the band-resolved Berry curvature \({\Omega }_{n,z}(\vec{k})\) weighted by the respective occupation number \({f}_{n}(\vec{k})\)46:
As in our previous study for the similar van der Waals (vdW) ferromagnet CrGeTe335, we calculated the integral over the Brillouin zone in Eq. (7) using the vegas adaptive Monte Carlo algorithm49,50.
For each calculation of the anomalous Hall conductivity σxy we performed ten independent Monte Carlo runs with 106 evaluations of the integrand for training the adaptive part of the algorithm and subsequent 106 evaluations for the actual calculation of the conductivity. The ten independent runs allow us to estimate the standard deviation of the obtained results, i.e. the Monte Carlo uncertainty.
Data availability
The data and scripts for all figures within this study are deposited at Zenodo51 under the following URL: https://doi.org/10.5281/zenodo.18376083.
Code availability
The DFT code used in this study is available from https://www.fplo.de. The code for the cMC calculations of this study is available from the corresponding author upon request.
References
Manna, K., Sun, Y., Muechler, L., Kübler, J. & Felser, C. Heusler, Weyl and Berry. Nat. Rev. Mater. 3, 244 (2018).
Tokura, Y., Yasuda, K. & Tsukazaki, A. Magnetic topological insulators. Nat. Rev. Phys. 1, 126 (2019).
Nagaosa, N., Morimoto, T. & Tokura, Y. Transport, magnetic and optical properties of Weyl materials. Nat. Rev. Mater. 5, 621 (2020).
Nakatsuji, S. & Arita, R. Topological magnets: Functions based on Berry phase and multipoles. Annu. Rev. Condens. Matter Phys. 13, 119 (2022).
Bernevig, B. A., Felser, C. & Beidenkopf, H. Progress and prospects in magnetic topological materials. Nature 603, 41 (2022).
Liu, E. et al. Giant anomalous Hall effect in a ferromagnetic kagome-lattice semimetal. Nat. Phys. 14, 1125 (2018).
Wang, Q. et al. Large intrinsic anomalous Hall effect in half-metallic ferromagnet Co3Sn2S2 with magnetic Weyl fermions. Nat. Commun. 9, 3681 (2018).
Kim, K. et al. Large anomalous Hall current induced by topological nodal lines in a ferromagnetic van der Waals semimetal. Nat. Mater. 17, 794 (2018).
Ali, M. N. et al. Butterfly magnetoresistance, quasi-2D Dirac Fermi surface and topological phase transition in ZrSiS. Sci. Adv. 2, e1601742 (2016).
Suzuki, T. et al. Singular angular magnetoresistance in a magnetic nodal semimetal. Science 365, 377 (2019).
Hassinger, E. & Meng, T. Magnetic modification of electrical resistance. Science 365, 324 (2019).
Seo, J. et al. Colossal angular magnetoresistance in ferrimagnetic nodal-line semiconductors. Nature 599, 576 (2021).
Das, A. & Mukhopadhyay, S. Tuning the chiral orbital currents in a colossal magnetoresistive nodal-line ferrimagnet. Phys. Rev. B 111, 174419 (2025).
Ni, Y. et al. Colossal magnetoresistance via avoiding fully polarized magnetization in the ferrimagnetic insulator Mn3Si2Te6. Phys. Rev. B 103, L161105 (2021).
Zhang, Y. et al. Control of chiral orbital currents in a colossal magnetoresistance material. Nature 611, 467 (2022).
Vincent, H., Leroux, D., Bijaoui, D., Rimet, R. & Schlenker, C. Crystal structure of Mn3Si2Te6. J. Solid State Chem. 63, 349 (1986).
May, A. F. et al. Magnetic order and interactions in ferrimagnetic Mn3Si2Te6. Phys. Rev. B 95, 174440 (2017).
Liu, Y. & Petrovic, C. Critical behavior and magnetocaloric effect in Mn3Si2Te6. Phys. Rev. B 98, 064423 (2018).
May, A. F., Cao, H. & Calder, S. Magnetic properties of ferrimagnetic Mn3Si2Te6. J. Magn. Magn. Mater. 511, 166936 (2020).
Gu, Y. et al. Unconventional insulator-to-metal phase transition in Mn3Si2Te6. Nat. Commun. 15, 8104 (2024).
Zhang, Z. et al. Variation of magnetic properties with current in ferrimagnetic semiconductor Mn3Si2Te6. AIP Adv. 14, 035238 (2024).
Ye, F. et al. Magnetic structure and spin fluctuations in the colossal magnetoresistance ferrimagnet Mn3Si2Te6. Phys. Rev. B 106, L180402 (2022).
Bigi, C. et al. Covalency, correlations, and interlayer interactions governing the magnetic and electronic structure of Mn3Si2Te6. Phys. Rev. B 108, 054419 (2023).
Zhang, Y., Lin, L.-F., Moreo, A. & Dagotto, E. Electronic structure, magnetic properties, spin orientation, and doping effect in Mn3Si2Te6. Phys. Rev. B 107, 054430 (2023).
Sala, G. et al. Ferrimagnetic spin waves in honeycomb and triangular layers of Mn3Si2Te6. Phys. Rev. B 105, 214405 (2022).
Susilo, R. A. et al. High-temperature concomitant metal-insulator and spin-reorientation transitions in a compressed nodal-line ferrimagnet Mn3Si2Te6. Nat. Commun. 15, 3998 (2024).
Hou, J. et al. Pressure-driven semiconductor-to-metal, magnetic, and structural transitions in the ferrimagnetic semiconductor Mn3Si2Te6. Chin. Phys. Lett. 42, 057403 (2025).
Mizokawa, T. & Fujimori, A. Electronic structure and orbital ordering in perovskite-type 3d transition-metal oxides studied by Hartree-Fock band-structure calculations. Phys. Rev. B 54, 5368 (1996).
Iqbal, Y., Müller, T., Jeschke, H. O., Thomale, R. & Reuther, J. Stability of the spiral spin liquid in MnSc2S4. Phys. Rev. B 98, 064427 (2018).
Harald O. Jeschke, private communication.
Xu, H.-X., Shimizu, M., Guterding, D., Otsuki, J. & Jeschke, H. O. Pressure evolution of electronic structure and magnetism in the layered van der Waals ferromagnet CrGeTe3. Phys. Rev. B 108, 125142 (2023).
Gonzalez, M. G. et al. Dynamics of K2Ni2(SO4)3 governed by proximity to a 3D spin liquid model. Nat. Commun. 15, 7191 (2024).
Jaubert, L. D. C., Iqbal, Y. & Jeschke, H. O. Spin-peierls transition in the frustrated spinels ZnCr2O4 and MgCr2O4. Phys. Rev. Lett. 134, 086702 (2025).
Ebad-Allah, J. et al. Near room-temperature ferromagnetism from double exchange in the van der Waals material CrGeTe3: Evidence from optical conductivity under pressure. Phys. Rev. B 111, L140402 (2025).
Scharf, G. et al. Pressure tuning of intrinsic and extrinsic sources to the anomalous Hall effect in CrGeTe3. Phys. Rev. Res. 7, 013127 (2025).
Xiang, H. J., Kan, E. J., Wei, S.-H., Whangbo, M.-H. & Gong, X. G. Predicting the spin-lattice order of frustrated systems from first principles. Phys. Rev. B 84, 224429 (2011).
He, X., Helbig, N., Verstraete, M. J. & Bousquet, E. TB2J: A python package for computing magnetic interaction parameters. Comput. Phys. Commun. 264, 107938 (2021).
Baral, R., May, A. F. & Calder, S. Short-range magnetic order and planar anisotropy in the topological ferrimagnet Mn3Si2Te6. Phys. Rev. B 112, 024405 (2025).
Scharf, G. et al. Light-induced ferromagnetism above the Curie temperature in CrGeTe3. Phys. Rev. B 111, L100408 (2025).
Zhang, S., Harii, K., Yokouchi, T., Okayasu, S. & Shiomi, Y. Amorphous ferromagnetic metal in van der Waals materials. Adv. Electron. Mater. 10, 2300609 (2024).
Koepernik, K. & Eschrig, H. Full-potential nonorthogonal local-orbital minimum-basis band-structure scheme. Phys. Rev. B 59, 1743 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Liechtenstein, A. I., Anisimov, V. I. & Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 52, R5467 (1995).
Tibshirani, R. Regression shrinkage and selection via the Lasso. J. R. Statist. Soc. B 58, 267 (1996).
Hastie, T., Tibshirani, R. & Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction, 2nd ed. (Springer, New York, 2009) https://doi.org/10.1007/978-0-387-84858-7.
Wang, X., Yates, J. R., Souza, I. & Vanderbilt, D. Ab initio calculation of the anomalous Hall conductivity by Wannier interpolation. Phys. Rev. B 74, 195118 (2006).
Eschrig, H. & Koepernik, K. Tight-binding models for the iron-based superconductors. Phys. Rev. B 80, 104503 (2009).
Koepernik, K., Janson, O., Sun, Y. & van den Brink, J. Symmetry-conserving maximally projected Wannier functions. Phys. Rev. B 107, 235135 (2023).
Lepage, G. P. A new algorithm for adaptive multidimensional integration. J. Comput. Phys. 27, 192 (1978).
Lepage, G. P. Adaptive multidimensional integration: VEGAS enhanced. J. Comput. Phys. 439, 110386 (2021).
Venkatasubramanian, V., Shimizu, M., Guterding, D. & Jeschke, H. O. Data accompanying: Origin of pressure-induced anomalies in the nodal-line ferrimagnet Mn3Si2Te6, https://doi.org/10.5281/zenodo.18376083 (2026).
Acknowledgements
We are grateful to Resta A. Susilo for help with the experimental crystal structures. H.O.J. acknowledges support through JSPS KAKENHI Grants No. 24H01668 and No. 25K08460. M.S. is supported by ISHIZUE 2025 of Kyoto University. Part of the computation in this work has been done using the facilities of the Supercomputer Center, the Institute for Solid State Physics, the University of Tokyo. The authors gratefully acknowledge the computing time provided on the supercomputers Lise and Emmy at NHR@ZIB and NHR@Göttingen as part of the German NHR infrastructure.
Author information
Authors and Affiliations
Contributions
The project was conceived by D.G. and H.O.J. M.S. developed the classical Monte Carlo code. V.V., M.S., D.G. and H.O.J. performed the calculations. V.V., D.G. and H.O.J. analysed and discussed the results and drafted the manuscript. All authors revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks Shiyu Zhu and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Venkatasubramanian, V., Shimizu, M., Guterding, D. et al. Origin of pressure-induced anomalies in the nodal-line ferrimagnet Mn3Si2Te6. Commun Mater 7, 94 (2026). https://doi.org/10.1038/s43246-026-01132-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43246-026-01132-x
