
Abstract
The expansion of ocean oxygenation during the Paleoproterozoic Lomagundi carbon isotope excursion remains highly debated. Here, we present iron isotope data for the coeval marine Francevillian Group strata. The data scatter from heavy to light iron isotope composition, with predominantly minimal isotope fractionation. The latter reflects complete oxidation and quantitative removal of dissolved iron from seawater, followed by complete reduction of oxidized iron in the sediments. Conversely, heavy and light iron isotope compositions are linked to equilibrium and kinetic isotopic effects, respectively during diagenesis. We propose that the oxygenation of the Paleoproterozoic oceans, at least below the shelf break, was responsible for the complete oxidation and quantitative removal of iron at depth, leaving behind a depleted marine iron reservoir during the Lomagundi Event. Oxidizing conditions to significantly build up a pool of oxyanions stable at much higher redox potential, such as nitrate, were only reached in the photic zone.
Similar content being viewed by others

Introduction
Early in Earth’s history, the atmosphere-hydrosphere system lacked sufficient oxygen to sustain stable aerobic ecosystems, starkly contrasting with conditions in modern-day surface environments. In so far as the timing and tempo of Earth’s surface oxygenation remain highly debated, a wide range of biogeochemical proxies based on Fe, Mn, C, S, P, N, Cr, Mo, V, Se, and U, have been employed to constrain its evolution better1,2,3,4,5,6,7,8,9,10,11. While the Archean Eon was predominantly anoxic, whiffs of atmospheric oxygenation and episodic development of shallow-marine oxygen oases are reported to have kicked off, before the Great Oxidation Event (GOE), as early as during the Mesoarchean1,2,4,5,6,7,11,12,13,14.
Transitioning to the Paleoproterozoic Era, the first permanent rise in atmospheric oxygen levels occurred between ca. 2.43 and 2.06 Ga known as the GOE1,3,8,11,15. This significant milestone in Earth’s history heralded a transformative shift in atmospheric composition, shaping the trajectory from an anaerobic to a more aerobic-dominated biosphere. The emergence and variability of complex life on Earth are intricately linked to fluctuations in atmospheric oxygen levels throughout Earth history8,11,16. Gaining insight into the extent of oxygenation of the ocean system during the Early Proterozoic holds significant promise for elucidating the emergence and proliferation of oxygen-dependent organisms, including eukaryotes. During the initial phase of the GOE, oxygen levels remained relatively low, estimated at around 10−6 of present atmospheric levels (PAL)1,3,11,17,18,19. The identification of increased oxygen concentrations above 10−5 PAL during the Paleoproterozoic is characterized by the disappearance of mass-independent fractionation of sulfur isotopes and redox-sensitive detrital minerals (e.g., detrital uraninite and pyrite) in the sedimentary record, alongside the emergence of red beds1,3,11,16. The later stage of the GOE, particularly between ~2.22 and 2.06 Ga, is marked by the Lomagundi Event (LE), the Earth’s longest-lived and most pronounced positive carbon isotope excursion recorded in marine carbonates1,20. During the LE, an overshoot of atmospheric oxygen levels and expansion of oceanic oxygenation are proposed, although models show that atmospheric oxygen levels remained below 1–10% PAL1,3,8,11,21,22,23,24.
The Paleoproterozoic era’s surge in oxygen levels has garnered significant attention, with efforts aimed at precisely quantifying the magnitude and extent of oxygenation in the atmosphere-ocean system yielding diverse and, sometimes, conflicting views. The prevailing view about ocean oxygenation suggests a scenario of a pronounced redox-stratified ocean, with a redoxcline fluctuating within shelf environments, throughout the GOE and its aftermaths8,11,25,26,27,28,29. This persisted until the subsequent increase in Earth’s surface oxygenation, approaching a modern atmosphere-ocean redox landscape during the Neoproterozoic era25,30,31. It is conjectured that during the GOE, only marine environments above the storm wave base were oxygenated, with minimum oxygen concentrations ranging from 0.4 to 1 µM28,32. At the same time, seawater below this bathymetric threshold remained anoxic28,29,32. This led to extensive upwelling of Fe(II) and Mn(II) dissolved in deep anoxic waters followed by their oxidation and co-precipitation in continental shelf environments during the early stage of the GOE33,34.
Conversely, an alternative view posits that enhanced oxygenation of the atmosphere-hydrosphere system during the LE allowed continental shelf water columns to become fully oxygenated with nitrate and selenate-replete conditions21. Others propose near-modern Earth’s surface oxygenation during the LE35 and shortly after its end36. More recently, a study on thallium (Tl) isotopes identified dynamic atmosphere-ocean oxygenation characterized by an episode of widespread precipitation of Mn-oxides on an oxygenated seafloor between ca. 2.3 and 2.2 Ga37. However, how the ocean redox landscape evolved following initial extensive seafloor oxygenation during the LE—a period of long-term enhanced atmospheric oxygen level during the late stage of the GOE—remains poorly constrained.
Oxygen (O2) serves as an energy source for living organisms and holds ecological significance for the proliferation and expansion of aerobic life forms30,31,38,39. Consequently, it is imperative to ascertain the extent of atmospheric oxygen concentrations on Earth’s surface during its initial rise, especially in marine environments where life and ecosystems are proposed to have already been more complex than usually thought during the GOE38,39. The conflicting views outlined above underscore the ongoing debate regarding the extent of Earth’s surface oxygenation and its role in facilitating the emergence and expansion of complex aerobic ecosystems. In this study, Fe isotope analysis (δ56Fe values expressed in ‰, relative to IRMM-014 standard) is used on one of the best-preserved sedimentary successions, the Francevillian Group (Supplementary Fig. S1, Fig. 1), deposited during the LE21,38,39,40,41,42,43,44,45,46,47. The Francevillian Group is particularly noteworthy for fossilized structures of putative multicellular organisms preserved within continental shelf deposits38,39. The main objective of this study is to provide novel insights into the Paleoproterozoic ocean redox landscape during the LE which is closely linked to the scale and geographic distribution of marine habitats conducive to developing more complex aerobic ecosystems. It also aims to shed new light on the interplay between environmental redox conditions and evolutionary dynamics during this critical period in Earth’s history.
Geological setting
The Francevillian Group, containing unmetamorphosed black shales rich in expandable clay minerals38,42, dates back to approximately 2.1Ga43,45,48. It crops out in the southeast region of Gabon and encompasses around 44,000 km2 within the expansive Francevillian basin (Supplementary Fig. S1)38,39,41,42,43,44,45,46,47. The basin is subdivided into four distinct sub-basins, including Booué (Plateau des Abeilles), Lastoursville, Franceville, and Okondja (Supplementary Fig. S1).
The Francevillian Group comprises five lithostratigraphic formations ranging from the Francevillian A (FA) at the base to FE at the top (Supplementary Fig. S1)43. The FA is mainly represented by terrestrial siliciclastic deposits (sandstone and conglomerate). The FB-FE formations are marine deposits, including siliciclastic (black shale and sandstone) and chemical (carbonate and chert) sediments, with depositional environments fluctuating between the inner and outer shelf21,43,46. This study focuses on black shales and carbonates from the FB to FD formations (Figs. 1, 2, Supplementary Fig. S1). These sedimentary rocks record the peak (from FB to lower FC formations) and end (upper FC and FD formations) of the LE21,38,41,42,44,46,47. Through sedimentological analysis and various geochemical indicators, such as high manganese oxidation rates, a minor amount of iron enrichment in upwelling zones43,44, and elevated V/Al ratios46,47, it is evident that the Francevillian basin maintained a strong connection with the open ocean during the deposition of the FB and FC formations.

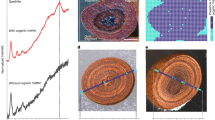
A Photomicrograph of black shale showing authigenic diagenetic Fe-bearing minerals, including pyrite (Py), authigenic clay (Aut. clay), and dolomite (Dol). B Backscattered electron (BSE) image of black shale showing Fe-dolomite (Fe-Dol) and pyrite (Py) as the main diagenetic Fe host minerals. C BSE image of black shale showing Fe mainly hosted in diagenetic clay minerals (Fe-Chl) together with diagenetic Fe-rich and Fe-poor dolomite (Fe-Dol and Dol., respectively). BSE images of the non-fossil related pyrite nodules (D) and pyrite beds (E and F) showing preserved inclusions of sulfate. G–I BSE images of pyritized Francevillian fossilized structure showing pyritization on organic-rich structures (in the core, H and in the edge, I) preserved in the black shale. Pyritization of the non-fossil related nodule only involves organic matter in the host sediment (high S/C ratios and equilibrium Fe isotope fractionation), while pyritization of fossilized structures involves both organic matter in the bodies of the structures and that of the host sediment (low S/C ratios and kinetic Fe isotope fractionation). Qtz Quartz, Fe-Chl iron-rich chlorite, ill illite, H. shale host shale.
The deposition of the Francevillian Group occurred during the LE, dated between 2.22 to 2.06 billion years ago38,39,40,41,42, 44,46,47. The most reliable ages of deposition of the Francevillian Group, although not very precise, include the age of the N’Goutou complex that marks the transition between the FA and FB formations, at ca. 2.19 Ga49, and the age of the FD Formation at ca. 2.08 and 2.07 Ga48,50. This range of ages has been recently confirmed by Re-Os dates of the FB-FD formations between ca. 2.15 and 2.05 Ga51. The samples analyzed in this study are from the FB to the FC formations, therefore, range in age from ca. 2.15 to 2.07 Ga.
Results
Iron isotopes were investigated on bulk samples from a range of lithologies, including marine carbonates, thin sedimentary pyrite beds, early diagenetic pyrite and carbonate nodules, as well as pyritized fossilized structures that were further compared with data obtained from bulk black shales and in situ analyses on early diagenetic pyrite nodules (Figs. 1–3; Table 1 and Supplementary Table S1). The selected samples are characterized by very limited to no post-depositional alteration (Fig. 1) in sedimentary facies that did not experience metamorphism, including at both a regional and local scale, as demonstrated in previous works21,39,42. Furthermore, most of the Fe is characterized by highly reactive components mainly hosted by authigenic mineral phases rather than detrital components38,42,44,47. Therefore, detrital mineral phases have minimal to no impact on the Fe composition presented in this study with the exemption of black shales, especially those with low FeHR/FeT ratios (<0.22; Supplementary Table S2), because of their siliciclastic origin mainly dominated by detrital components.

These include direct precipitation in seawater and precipitation in the sediments below the water column.
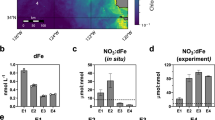
Analyses for bulk marine carbonates, early diagenetic carbonate and pyrite nodules combined with black shales yield δ56Fe values ranging from −0.46‰ to 1.03‰ (Figs. 2, 3; Table 1). The primary carbonate δ56Fe values average 0.19‰ with 11 out of 13 analyzed samples in the range of 0 ± 0.5‰. The diagenetic pyrites nodules average 0.44‰ with 3 out of the 5 analyzed samples in the range of 0 ± 0.5‰ whereas two diagenetic carbonate concretions yield δ56Fe values of −0.14 and −0.09‰. These ranges overlap with the bulk shale δ56Fe values at 0.22‰, on average, with 21 out of the 30 analyzed samples in the range of 0 ± 0.5‰. By contrast, analyses of the bulk pyritized Francevillian fossils and microbial mats yield δ56Fe values exclusively <−0.5‰ (Figs. 2, 3; Table 1).

The shaded gray band represents the boundary of the near-to-zero δ56Fe values of 0 ± 0.5‰.
In situ analysis of the early diagenetic pyritized fossilized structures and non-fossil pyritized nodules yield δ56Fe values ranging from −3.14‰ to 1.03‰ (averaging −1.15‰ with 45 out of 58 <−0.5‰), and from −1.39‰ to 1.71‰ (averaging −0.12‰ with 6 out of 44 > 0.5‰), respectively (Fig. 3; Supplementary Table S1).
Discussion
Origin of Fe isotope fractionation in the Francevillian basin
Upwelled submarine hydrothermal fluids with δ56Fe values near 0‰ ( − 0.5 to +0.1)52,53,54,55,56,57 and continental weathering of silicate rocks, having δ56Fe values of 0 ± 0.3‰55,56 represent the most important input sources of Fe to the oceans. By contrast, the main output channels for Fe leaving the oceans are oxidation of Fe(II) and precipitation of Fe(III) as oxides, reduction of Fe(III) to Fe(II) and precipitation through sulfidation and/or pyritization. Therefore, the fractionation pathways affecting this output of oceanic Fe pool, mostly involving biotic and abiotic oxidation-reduction processes, drives the isotopic signatures (δ56Fe values) recorded in marine sediments. The main mechanisms driving Fe(II) oxidation in seawaters include O2 produced through oxygenic photosynthesis, anoxygenic photosynthesis (e.g., photoferrotrophy), and photo-oxidation52,53,54,56,58,59,60. Fe(II) could either be completely or partially oxidized, which may depend on the marine oxygen concentration. Complete oxidation and quantitative Fe removal from seawater would cause minimal Fe isotope fractionation resulting in Fe(III)-bearing sediments with near-to-zero δ56Fe values overlapping with primary Fe inputs. In partial oxidation, sediments would record positive δ56Fe values of up to 2‰52,53,54,58,59,60. Moreover, Fe(III) may be partially or completely reduced or sulfidized either in the water column or during diagenesis in the sediments54,61,62,63. Partial reduction of Fe (oxyhydr)oxides, could result in a range of negative and positive δ56Fe values depending on the initial signature of the oxidized Fe or the process involved in its reduction or precipitation, with the complete reduction and sulfidation being characterized by minimal Fe isotope fractionation54. Partial reduction of Fe (oxyhydr)oxides can occur either through abiotic processes, yielding δ56Fe values ranging from −1 to −3.1‰54,64,65, or via dissimilatory iron reduction (DIR), resulting in δ56Fe values as low as −2.9‰ as observed in ancient and modern marine Fe deposits5,6,12,54,66,67,68. In contrast, more recent studies reveal that the sulfidation of Fe(III) will result in large Fe isotope fractionation with δ56Fe values between −3.5 and +1.4‰ depending on the degree of FeS precipitation and equilibration, i.e., kinetic and equilibrium isotope effects61,62,63, 69.
It is not clear how microbial sulfate reduction (MSR) affects the Fe isotope composition of sedimentary sulfides, although it has been suggested that the consumption of sulfate and Fe concurrently may induce large Fe isotope fractionation60. For example, in shallow sedimentary environments where DIR is favored by the presence of ferric oxide or hydroxide minerals and MSR is less constrained by sulfate availability, δ56Fe values tend to be low (up to −3.5‰)54,60. Conversely, deeper sedimentary deposits where DIR is less prevalent and sulfate limitation occurs tend to exhibit positive δ56Fe values (up to 1.5‰) when the initial δ56Fe values are near-to-zero60. However, observation in the modern environments shows no significant Fe isotope fractionation associated with MSR70.
It has been proposed that partial Fe(II) oxidation in Archean oceans caused the removal of heavy Fe isotopes, resulting in near-surface seawater becoming enriched in the light isotopes as recorded in shallow marine sediments53. By contrast, the Paleoproterozoic oceans were described to have been characterized by extensive Fe sulfide precipitation from seawater at the expense of Fe(oxyhydr)oxides, which resulted in the record of positive δ⁵⁶Fe values in marine sediments53. However, this view has since been challenged by experimental work on modern environments and studies on Archean and Proterozoic sulfides63,71,72. According to these studies, positive δ56Fe values in sulfides can be explained by equilibrium isotope effect— when the precipitation rate is slower relative to the isotope exchange between the more soluble pyrite precursor and the porewater Fe(II)—while negative δ56Fe values can be induced by kinetic isotope effect— when precipitation is faster relative to isotope exchange61,62,63, often in early-stage nucleation, in the presence of high organic carbon concentration63. Although highly negative δ56Fe values in Archean sedimentary pyrites are instead attributed to a partial reduction of Fe (oxyhydr)oxides, pyrite precipitation, kinetic isotope effect during precipitation from dissolved Fe(II) pool, or a combination of all processes72. Studies comparing pyrite-depleted shallow- and deep-marine Mesoarchean and Neoarchean ferruginous shales demonstrated a strong positive correlation between δ56Fe values and water depth—highly negative δ56Fe values in shallow-marine ferruginous shales and positive δ56Fe values in deep-water ferruginous shales5,12.—which is in favor of the pioneering study on Archean sedimentary sulfides53. Moreover, positive δ56Fe values in Paleoproterozoic pyrites are thought to reflect partial Fe(II) oxidation in the water column followed by sulfidation in the sediments72. Regardless of the exact pathways that controlled Fe isotope fractionation in Precambrian oceans before ca. 1.7 Ga, most of these studies are in agreement with non-quantitative Fe precipitation in late Archean oceans, often characterized by oxygen oases, and in a redox stratified Paleoproterozoic oceans that are significantly enriched in sulfate—in agreement with the recently proposed nonequilibrium Fe cycle in the Precambrian oceans73. In other words, the deep Paleoproterozoic Ocean is considered to have remained completely anoxic, with respect to Fe biogeochemical cycle, at least below upwelling zones.
The Francevillian Group’s marine carbonate and thin pyrite beds, diagenetic carbonates and (non-fossil) pyrite nodules exhibit near-to-zero δ56Fe values (Figs. 2, 3, Table 1). The same applies to black shale samples (Fig. 3, Table 1). These near-to-zero δ56Fe values could reflect 1) Fe isotope signature of detrital input, or 2) Fe isotope composition carried by authigenic minerals and induced by complete oxidation of dissolved Fe(II) followed by quantitative removal and complete reduction of Fe(III) in the sediments. Detrital components can be ruled out in the chemical precipitates as they are mainly characterized by authigenic mineral phases. By contrast, the near-to-zero δ56Fe values measured in black shales, for which Fe speciation data yield low FeHR/FeT ratios <0.22, i.e., FB1a, FB1b and FB2b Francevillian units (see Supplementary Table S2)38,42,44,46,47, may be influenced by detrital Fe components. It is also apparent that low FeHR/FeT < 0.22 points to exhausted dissolved Fe(II) in the well-oxygenated water column during the deposition of these units. However, Francevillian Group black shales in sedimentary units including FB1b, FB1c, FB2b, upper FC and lower FD, yield high FeHR/FeT ratios >0.38 (see Supplementary Table S2) and near-to-zero δ56Fe values are dominated by authigenic Fe precipitation. Among these sedimentary levels, the FB1c and upper FC units are even associated with high authigenic Mn enrichment (Mn oxidation in the water column and precipitation in the sediments)43,44,46 which further support authigenic, rather than detrital, Fe isotope composition. The near-to-zero δ56Fe values found in these black shales with high FeHR/FeT > 0.3844,46,47 demonstrate that even when dissolved Fe(II) episodically increased in seawater, as a result of enhanced submarine volcanic activity44, it went through complete oxidation in the water column and complete reduction in the sediments which is consistent with the near-to-zero Fe isotope records. The preservation of sulfate inclusions in the pyrite nodules (Fig. 1) points to Fe(II) scarcity as a result of complete, rather than partial, Fe (oxyhydr)oxide reduction in sediments underneath a water column depleted in Fe(II) which caused limited Fe(III) flux to the sediments. Therefore, near-to-zero δ56Fe values predominantly recorded in the Francevillian Group can be attributed to the authigenic signature imparted from the isotope fractionation between the input Fe(II) pool and output Fe(III) to the sediments in Paleoproterozoic marine environments rather than Fe isotope composition being controlled by detrital materials in black shales. These near-to-zero δ56Fe values are thus interpreted to typically reflect complete Fe(II) oxidation in seawater, followed by complete reduction of Fe (oxyhydr)oxide in the sediments, akin to previously described modern-like Fe biogeochemical cycle which is proposed to have kicked off only from ca. 1.7 Ga53.
Moreover, positive δ56Fe values measured in some bulk non-fossil related pyrites combined with their concretion origin strongly strengthens the notion that Fe(III) reduction and pyritization occurred within the sediments, rather than in the water column, before lithification as extensively demonstrated in previous studies38,39,44,74. This is further supported by the stability of nitrate and selenate in the entire Francevillian seawater column21. Based on the presence of highly negative δ34S values, it was proposed that MSR controlled this diagenetic pyritization38,39,74. However, as discussed above, the influence of MSR on Fe isotope composition remains unclear (e.g., refs. 60,70). In the case of the Francevillian basin, the stability of nitrate and selenate in the entire Francevillian seawater column21 at least supports the notion that Fe(III) and sulfate reduction mainly occurred in the deeper parts of the organic-rich sediments.
To constrain which mechanisms drove positive δ56Fe values, of up to 0.98‰ in non-fossil diagenetic pyrites, in situ Fe isotopes have been measured for both fossil and non-fossil-related pyrites as both pyrite formations were controlled by MSR in the same sediment layers (Fig. 2)21,38,39,44,74. The majority of analytical spots for non-fossil pyrite nodules tend toward heavy and near-to-zero Fe isotope composition, consistent with what is observed with bulk analyses, with 30 (out of 44) positive and near-to-zero δ56Fe values, and 14 (out of 44) negative values (Supplementary Table S1; Fig. 3). However, in situ analysis of pyrite concretions of fossilized Francevillian biota and their bulk analyses yield negative δ56Fe values as low as –3.14 and –0.979‰, respectively (Figs. 1, 3; Table 1 and Supplementary Table S1). This large Fe isotope fractionation, from highly negative to highly positive δ56Fe values, can be explained by 1) DIR (e.g., refs. 54,60) and 2) kinetic vs. equilibrium Fe isotope effects between soluble pyrite precursors, such as mackinawite, and porewater Fe(II) (e.g., refs. 61,62,63,69,72). For the highly negative δ56Fe values measured on pyritized fossilized structures, DIR and kinetic Fe isotope effect appear as the most plausible explanations while equilibrium Fe isotope effect can explain the highly positive δ56Fe values recorded in non-fossil pyrite nodules. Pyritization of the organic matter of Francevillian biota likely caused their low S/C ratios and ultimately the faster pyrite nucleation, relative to isotope exchange, responsible for the incorporation of the light Fe isotope—the kinetic Fe isotope effects63. The expression of this kinetic Fe isotope effect would have been amplified by the contribution of light Fe isotopes from DIR60. It is thus possible that the combination of kinetic and DIR Fe isotope effects favored the incorporation of light Fe isotopes in fossilized structures and would have left the remaining pore water sediments, with higher S/C ratios, enriched in the heavy Fe isotopes that were incorporated in the slowly growing non-fossil related pyrites—the equilibrium Fe isotope effects63. Following observations on both groups of early diagenetic pyrite nodules analyzed in this study, it also clearly appears that MSR, involved in both pyritization processes, has no obvious influence on Fe isotope fractionation on sedimentary pyrites.
Moreover, the heterogeneity in the in situ δ56Fe values observed in the pyritized Francevillian biota, with the occurrence of some near-to-zero and positive δ56Fe values, could be attributed to the continuous growth of pyrite later during diagenesis. Such a process would incorporate the remaining heavier Fe isotopes left in the porewater sediments and/or the diagenetic release of poorly reactive Fe from the dissolution of unstable detrital minerals such as biotite and chlorite from the host black shales. This is not unexpected given that in situ δ34S values previously measured in these fossilized biota are also heterogeneous38,74. Indeed, the δ34S values of pyrite are predominantly highly negative as a result of MSR in sediments, with few of them being near-to-zero to highly positive which reflects late diagenetic pyrite growth38,74. Alternatively, it cannot be excluded that negative δ56Fe values recorded in pyritized fossils reflect the preferential uptake of light Fe isotopes by the putative large multicellular organisms in Francevillian marine environments as previously observed for Zn isotopes39. However, it remains unclear what exact metabolism could drive such Fe isotope fractionation. Regardless of any evidence of biological uptake, the low δ56Fe values found in fossilized structures represent at least another line of evidence for the biogenicity of the Francevillian biota. Nevertheless, the presence of lower δ56Fe values in the in situ analyses of non-fossil pyrite likely reflects the incorporation of lighter isotopes during the initial stages of pyrite formation.
In summary, while both complete abiotic and biotic (microbially-induced) reduction of Fe (oxyhydr)oxides during diagenesis in marine sediments, following complete Fe(II) oxidation in the water column, seem to affect pyritized fossil structures and non-fossil pyrite nodules, it appears that different S/C ratios are the main driver of Fe isotope fractionation in the studied Paleoproterozoic pyrites in marine sediments. During diagenesis, kinetic isotope fractionation mainly drives pyritized fossil structures towards highly negative δ56Fe values as a result of the low S/C ratios caused by the higher organic carbon content of the Francevillian biota. In the non-fossil pyrite nodules, highly positive δ56Fe values are driven by equilibrium isotope fractionation in the deeper parts of the sediments as a result of high S/C ratios. Such Fe isotope records of sedimentary Fe sulfides reshape our understanding on Precambrian Fe biogeochemical cycle and its associated isotopic effects. This was made possible because the whole Fe(II) reservoir was already exhausted, as a consequence of complete Fe(II) oxidation and quantitative removal, in the Paleoproterozoic oceans during the LE (Fig. 4). This prevented any significant Fe isotopic effects to be expressed during the changes of Fe redox states between the oceanic Fe input and output channels (Figs. 4, 5).

The data show that Fe(II) was significantly oxidized and removed from seawater in mildly oxic deep oceans while dissolved Mn(II) was still transported to well-oxygenated shelf environments where it was quantitatively oxidized and removed from seawater.

The data show that large Fe isotope fractionation in pyrite is mainly driven by diagenetic processes involving both equilibrium and kinetic effects, and that the previously defined late Proterozoic to modern stage III (53; now stage III-2) was already reached at least for the duration of the ca. 2.22-2.06 Ga LE (stage III-1). The previously described stage II (53) remains restricted during the early part of the GOE when Mn and Fe were still co-precipitating significantly in shelf environments. The shaded gray band represents the boundary of the near-to-zero δ56Fe values of 0 ± 0.5‰. (The plotted literature data are presented in the Supplementary Data 1).
Implication for oceanic redox landscape and ecosystem structure during the Lomagundi event (LE)
The Paleoproterozoic oceans are traditionally thought to have remained strongly redox-stratified, with mainly anoxic ferruginous conditions in offshore environments, at depth below the inner shelf, i.e. bathymetry above storm wave base, with expanding euxinic conditions during and after the GOE15,75,76,77,78. These conditions might have favored the reduction and instability of oxyanions that are sensitive at higher redox thresholds, i.e. nitrate, and very critical to sustain the proliferation and ecological prominence of more complex aerobic life forms such as eukaryotes27,28,29,79,80,81,82,83. However, an emerging view suggests episodic development of seafloor oxygenation, involving the stability of Mn oxides during the early part of the GOE at ca. 2.3 Ga37. Additionally, others suggest an expansion of the ocean oxygenation to depth below the shelf-break with both inner and outer continental shelf being fully oxidized with nitrate- and selenate-replete conditions during the ca. 2.22-2.06 Ga LE21,40. Moreover, findings from Fe speciation studies coupled with N and Se isotope data already considered the possibility that deeper marine regions of the Paleoproterozoic oceans might have been oxygenated during the LE21,47, although the extent of such deep ocean oxygenation as well as the trajectory of seafloor redox conditions remains poorly constrained. Our Fe isotope data, pointing to extensive Paleoproterozoic ocean oxygenation that was significantly depleted in dissolved Fe(II), support this view. Such extensive ocean oxygenation likely expanded down to bathymetry significantly below the shelf break in the open oceans (Fig. 4). This is particularly supported by the lack of Fe deposits associated with the LE-aged Francevillian Mn-ore deposits, unlike the early-GOE Fe-rich Mn-ore deposits of the giant Kalahari manganese field33,34. Iron was still precipitated alongside Mn in continental shelf environments when the deeper part of the oceans were still predominantly anoxic during the early stage of the GOE, before ca. 2.3 Ga, while Mn was oxidized and precipitated alone in shelf environments during the late part of the GOE corresponding to the ca. 2.22-2.06 Ga LE.
During the LE, significant amounts of dissolved Mn(II) were transported by upwelling Fe(II)-depleted waters from weakly oxidized deep oceans to well-oxygenated shelf environments where it was quantitatively oxidized and removed from seawater (Fig. 4). Fe(II) was quantitatively oxidized and removed from seawaters and/or was readily oxidized near hydrothermal vents under mildly oxygenated deep oceanic conditions (Fig. 4) which has been proposed, for Precambrian oceans younger than ca. 1.7 Ga (stage III Fig. 5)53,58,59. This may explain the scarcity of BIFs in sedimentary records during the LE53,58,59 indicating depleted Fe(II) reservoir in the Paleoproterozoic oceans and that the Mesoproterozoic ( < 1.7 Ga) oceanic Fe biogeochemical cycle53—was already established during the LE and lasted for at least ca. 200 million years (Fig. 5). In so far as Paleoproterozoic records of deep marine sediments that formed at the bottom of ocean basins (i.e., deeper than 300 m) are hard to find, and considering the observation of Fe-lean Mn deposits, the data allow only to propose that the LE-aged Paleoproterozoic oceans were characterized by an expanded aerobic iron biogeochemical cycle that completely oxidized dissolved Fe(II) and quantitatively removed it from seawater. However, extensive oxidation of Fe(II) may have been an important O2 buffer that contributed to maintaining low redox thresholds (e.g., low Eh) below the shelf break and ultimately prevented deeper oceanic environments from reaching oxidizing conditions that require the stability of Mn (oxyhydr)oxides and other elements of similar redox thresholds, i.e., nitrate and selenate. These higher oxidizing conditions were thus only reached near the ocean surface, as a result of enhanced oxygenic photosynthesis during the LE, but not at depth below upwelling zones (Fig. 4)21. This ocean redox landscape likely restricted a more global expansion of key limiting nutrients such as nitrate which ultimately prevented the ecological prominence of more complex aerobic life forms and ecosystems.
Conclusion
The disparity in δ56Fe values within LE-aged marine sediments offers valuable insights into the Paleoproterozoic ocean’s redox landscape with implications for biological processes. The prevalence of near-to-zero δ56Fe values in primary marine carbonate, diagenetic carbonate, ferruginous black shales and diagenetic pyrite point to extensive aerobic Fe cycle associated with complete oxidation of marine Fe(II) pool. This left the Paleoproterozoic oceans depleted in dissolved Fe(II). The distribution of negative and positive δ56Fe values in pyrites is attributed to the variability of S/C ratios during diagenesis in the sediments rather than processes in the water column. These Fe isotope signatures reflect a swift removal of dissolved Fe(II) at depth, thus allowing precipitation of Fe(oxyhydr)oxides well below upwelling zones. These conditions imply that the oxygenation level of seawater reached the redox threshold of some elements such as iron with lower redox potential, i.e., Eh <0 at circumneutral pH, to allow its complete to nearly complete removal from seawater at oceanic scale. By contrast, extensive oxidation of Fe(II) acted as an important O2 buffer which prevented the water column below the shelf break from reaching the redox thresholds of elements with higher redox potential, i.e., Eh > 0 and closer to O2 at circumneutral pH, such as Mn oxides, nitrate and selenate that became only stable in continental shelf environments or photic zone’s water column where the rate of oxygenic photosynthesis was extremely high during the LE. This likely prevented the spread of more complex aerobic ecosystems that only remained restricted in well-oxygenated continental shelf marine habitats.
Methods
Bulk Fe stable isotope analysis
Stable Fe isotope data for bulk samples reported in this study were performed on the ThermoFisher Scientific NeptunePlus multicollector inductively coupled plasma mass spectrometer (MC-ICP-MS) housed at the Isotope Geochemistry facilities of the University of Tuebingen using measuring protocols previously published5,84,85. The analysis of carbonate and sulfide minerals was performed on microdrilled and powdered samples while black shales were exclusively powdered. In summary, an aliquot corresponding to 15 μg Fe was taken from a stock solution of the digested sample and mixed with a 57Fe-58Fe double spike to yield 1:1 sample-to-spike Fe ratio. Fe purification included a one-step anion exchange chromatography86. Measurements of samples on MC-ICP-MS include an introduction of samples as solutions of 2.5 μg g−1 Fe in 0.3 M HNO3 via a dual-glass spray chamber and a PFA pneumatic nebulizer. The instrument was operated in static mode to simultaneously detect all Fe isotopes and the 52Cr+ and 60Ni+ monitors to correct for isobaric interferences of 54Cr+ on 54Fe+ and 58Ni+ on 58Fe+.
40Ar12C+ on 52Cr+, 40Ar14N+ on 54Fe+, 40Ar16O+ on 56Fe+, 40Ar17O+ on 57Fe+, and 40Ar18O+ on 58Fe+ polyatomic interferences were discriminated by enabling a medium-resolution mode with a resolving power M/ΔM of 6000–800087. Individual Fe isotope measurements comprised 90 to 120 cycles with an integration time of 4.2 s. Background signals were corrected by subtracting on-peak-zero measurements on the pure 0.3 M HNO3 carrier solution before and after each sample and standard measurement. Double-spike deconvolution was carried out based on a method by Compston and Oversby88, assuming an exponential mass fractionation law. The data are reported in δ-notation relative to the standard IRMM-014 according to the following formula, thereby multiplication by a factor of 1000 expresses the result in per mille
Over the course of this study IRMM-014 yielded a δ56/54Fe value of 0.000 ± 0.033‰ (2 SD, n = 20) matching the long-term reproducibility of 0.000 ± 0.036‰ (2 SD, n = 328) of the Isotope Geochemistry laboratory of Tuebingen since 2015. The in-house standards Han-Fe and Tueb-Fe gave δ56/54Fe values of 0.289 ± 0.040‰ (2 SD, n = 9) and −0.372 ± 0.022‰ (2 SD, n = 7), respectively, being in excellent agreement with previous studies5,84,85 and the long-term reproducibility of 0.289 ± 0.038‰ (2 SD, n = 133) and −0.377 ± 0.041‰ (2 SD, n = 113), respectively. Furthermore, analytical accuracy was determined by measuring USGS reference materials OU-6 (shale) and 782-1 (dolostone), which were run together with processed samples, and yielded a δ56/54Fe of 0.071 ± 0.010‰ (2 SD; n = 4, four digests) and −0.923 ± 0.016‰ (2 SD, n = 3, three digests). Procedural blanks were <2.7 ng, contributing less than 0.02% to the processed amount of sample Fe and thus considered to be negligible.
In situ Fe stable isotope analysis
Iron isotopic compositions were measured using a Cameca ims 1280 HR2 at SwissSIMS (University of Lausanne), following the method described by Decraene et al.89. Thick sections were polished and pressed into 1-inch indium mounts within a Balmat pyrite standard (δ56Fe = −1.46 ± 0.01‰)90.
Briefly, a 10 kV, 3 nA Gaussian 16O- primary beam was focused into a 5 μm spot. 52Cr+, 54Fe+ and 56Fe+ secondary ions were collected simultaneously by electron multipliers (L2 for 52Cr+) and offset Faraday cups (C for 54Fe+, H1 for 56Fe+). The field aperture was set to 2000µm and the energy slit to 50 eV. A mass resolving power of 7000 was obtained by the combination of an entrance slit width of 61 µm and the slit 3 of the multicollection. This mass resolving power allows to resolve the interference of 53CrH on 54Fe. 52Cr+ secondary ion is measured to monitor the 54Cr+ isobaric interference on the 54Fe+90,91. A 90 s pre-sputtering time was applied to remove surface contamination, followed by secondary ion beam centering and detector background measurements. Data acquisition was then performed for 300 sec. The typical count rate for 56Fe+ on Balmat standard was 3.3 E+7 counts per second (cps) and the mean reproducibility was ±0.40‰ (2 SD). The data reduction procedure is described in detail in Pasquier et al.90. After Cr and drift correction, the instrumental mass fractionation was determined on Balmat standard, measured every 10–15 analyses and applied to correct the data.
Data availability
All the data acquired and used in this study is presented in the main manuscript text and the supplementary file.
References
Bekker, A. The Great Oxidation Event. In: Encyclopedia of Astrobiology (eds, Gargaud, M. et al.) (Springer, Berlin, Heidelberg) (2022).
Wang, X. et al. Uranium isotope evidence for Mesoarchean biological oxygen production in shallow marine and continental settings. Earth Planet. Sci. Lett. 551, 116583 (2020).
Bekker, A. et al. Dating the Rise of atmospheric oxygen. Nature 427, 117–120 (2004).
Ossa Ossa, F. O. et al. Limited oxygen production in the Mesoarchean ocean. Proc. Natl. Acad. Sci. USA 116, 6647–6652 (2019).
Ossa Ossa, F. et al. Aerobic iron and manganese cycling in a redox-stratified Mesoarchean epicontinental sea. Earth Planet. Sci. Lett. 500, 28–40 (2018).
Eickmann, B. et al. Isotopic evidence for oxygenated Mesoarchaean shallow oceans. Nat. Geosci. 11, 133–138 (2018).
Planavsky, N. J. et al. Evidence for oxygenic photosynthesis half a billion years before the Great Oxidation Event. Nat. Geosci. 7, 283–286 (2014).
Lyons, T. W., Reinhard, C. T. & Planavsky, N. J. The rise of oxygen in Earth’s early ocean and atmosphere. Nature 506, 307–315 (2014).
Farquhar, J. et al. Isotopic evidence for Mesoarchaean anoxia and changing atmospheric sulphur chemistry. Nature 449, 706–709 (2007).
Ohmoto, H., Watanabe, Y., Ikemi, H., Poulson, S. R. & Taylor, B. E. Sulphur isotope evidence for an oxic Archaean atmosphere. Nature 442, 908–911 (2006).
Holland, H. D. The oxygenation of the atmosphere and oceans. Philos. Trans. R. Soc. B Biol. Sci. 361, 903–915 (2006).
Kurzweil, F., Wille, M., Gantert, N., Beukes, N. J. & Schoenberg, R. Manganese oxide shuttling in pre-GOE oceans – evidence from molybdenum and iron isotopes. Earth Planet. Sci. Lett. 452, 69–78 (2016).
Anbar, A. D. et al. A whiff of oxygen before the great oxidation event? Science 317, 1903–1906 (2007).
Eroglu, S. et al. Open ocean vs. continentally-derived iron cycles along the neoarchean campbellrand-malmani carbonate platform, South Africa. Am. J. Sci. 318, 367–408 (2018).
Poulton, S. W. et al. A 200-million-year delay in permanent atmospheric oxygenation. Nature 592, 232–236 (2021).
Lyons, T. W., Fike, D. A. & Zerkle, A. Emerging biogeochemical views of Earth’s ancient microbial worlds. Elements 11, 415–421 (2015).
Warke, M. R. et al. The great oxidation event preceded a paleoproterozoic ‘snowball earth’. Proc. Natl. Acad. Sci. USA 117, 13314–13320 (2020).
Catling, D. C. & Zahnle, K. J. The Archean atmosphere. Sci. Adv. 6, eaax1420 (2020).
Farquhar, J., Bao, H. & Thiemens, M. Atmospheric Influence of Earth’s Earliest Sulfur Cycle. Science 289, 756–758 (2000).
Karhu, J. A. & Holland, H. D. Carbon isotopes and the rise of atmospheric oxygen. Geology 24, 867–870 (1996).
Ossa Ossa, F. et al. Moderate levels of oxygenation during the late stage of Earth’s Great Oxidation Event. Earth Planet. Sci. Lett. 594, 117716 (2022).
Colwyn, D. A. et al. A paleosol record of the evolution of Cr redox cycling and evidence for an increase in atmospheric oxygen during the Neoproterozoic. Geobiology 17, 579–593 (2019).
Kanzaki, Y. & Murakami, T. Estimates of atmospheric O2 in the Paleoproterozoic from paleosols. Geochim. Cosmochim. Acta 174, 263–290 (2016).
Bekker, A. & Holland, H. D. Oxygen overshoot and recovery during the early Paleoproterozoic. Earth Planet. Sci. Lett. 317–318, 295–304 (2012).
Alcott, L. J., Mills, B. J. W. & Poulton, S. W. Stepwise Earth oxygenation is an inherent property of global biogeochemical cycling. Science 366, 1333–1337 (2019).
Asael, D., Rouxel, O., Poulton, S. W., Lyons, T. W. & Bekker, A. Molybdenum record from black shales indicates oscillating atmospheric oxygen levels in the early paleoproterozoic. Am. J. Sci. 318, 275–299 (2018).
Cheng, C. et al. Nitrogen isotope evidence for stepwise oxygenation of the ocean during the Great Oxidation Event. Geochim. Cosmochim. Acta 261, 224–247 (2019).
Kipp, M. A., Stüeken, E. E., Bekker, A. & Buick, R. Selenium isotopes record extensive marine suboxia during the Great Oxidation Event. Proc. Natl. Acad. Sci. USA 114, 875–880 (2017).
Stüeken, E. E., Kipp, M. A., Koehler, M. C. & Buick, R. The evolution of Earth’s biogeochemical nitrogen cycle. Earth Sci. Rev. 160, 220–239 (2016).
Canfield, D. E., Poulton, S. W. & Narbonne, G. M. Late-Neoproterozoic deep-ocean oxygenation and the rise of animal life. Science 315, 92–95 (2007).
Lenton, T. M., Boyle, R. A., Poulton, S. W., Shields-Zhou, G. A. & Butterfield, N. J. Co-evolution of eukaryotes and ocean oxygenation in the Neoproterozoic era. Nat. Geosci. 7, 257–265 (2014).
Hardisty, D. S. et al. An iodine record of Paleoproterozoic surface ocean oxygenation. Geology 42, 619–622 (2014).
Gutzmer, J. & Beukes, N. J. Fault-Controlled Metasomatic Alteration of Early Proterozoic Sedimentary Manganese Ores in the Kalahari Manganese Field, South Africa. Econ. Geol. 90, 823–844 (1995).
Tsikos, H. & Moore, J. M. The Kalahari manganese field: An enigmatic association of iron and manganese. South Afr. J. Geol. 101, 287–290 (1998).
Blättler, C. L. et al. Two-billion-year-old evaporites capture Earth’s great oxidation. Science 360, 320–323 (2018).
Mänd, K. et al. Palaeoproterozoic oxygenated oceans following the Lomagundi–Jatuli Event. Nat. Geosci. 13, 302–306 (2020).
Ostrander, C. M. et al. Onset of coupled atmosphere–ocean oxygenation 2.3 billion years ago. Nature 631, 335–340 (2024).
El Albani, A. et al. Large colonial organisms with coordinated growth in oxygenated environments 2.1 Gyr ago. Nature 466, 100–104 (2010).
Ossa Ossa, F. et al. Zinc enrichment and isotopic fractionation in a marine habitat of the c. 2.1 Ga Francevillian Group: A signature of zinc utilization by eukaryotes? Earth Planet. Sci. Lett. 611, 118147 (2023).
Ossa Ossa, F., König, S., Hofmann, A., Bekker, A. & Schoenberg, R. Increase in ecosystem complexity aided organic carbon sequestration during the c. 2. 22-2. 06 Ga Lomagundi carbon isotope excursion, https://doi.org/10.5194/egusphere-egu21-10828.
Bekker, A. et al. The Paleoproterozoic Francevillian succession of Gabon and the Lomagundi-Jatuli event. Geology 49, e528 (2021).
Ossa Ossa, F. et al. Exceptional preservation of expandable clay minerals in the ca. 2.1Ga black shales of the Francevillian basin, Gabon and its implication for atmospheric oxygen accumulation. Chem. Geol. 362, 181–192 (2013).
Gauthier-Lafaye, F. & Weber, F. Natural nuclear fission reactors: Time constraints for occurrence, and their relation to uranium and manganese deposits and to the evolution of the atmosphere. Precambrian Res. 120, 81–100 (2003).
Ossa Ossa, F. et al. Two-step deoxygenation at the end of the Paleoproterozoic Lomagundi Event. Earth Planet. Sci. Lett. 486, 70–83 (2018).
Bros, R., Stille, P., Gauthier-Lafaye, F., Weber, F. & Clauer, N. SmNd isotopic dating of Proterozoic clay material: An example from the Francevillian sedimentary series, Gabon. Earth Planet. Sci. Lett. 113, 207–218 (1992).
Ossa Ossa, F. et al. Limited expression of the Paleoproterozoic Oklo natural nuclear reactor phenomenon in the aftermath of a widespread deoxygenation event ~2.11–2.06 billion years ago. Chem. Geol. 578, 120315 (2021).
Canfield, D. E. et al. Oxygen dynamics in the aftermath of the Great Oxidation of Earth’s atmosphere. Proc. Natl. Acad. Sci. USA 110, 16736–16741 (2013).
Horie, k., Hidaka, H. & Gauthier-Lafaye, F. U-Pb geochronology and geochemistry of zircon from the Franceville series at Bidoudouma, Gabon. In: The 15th Annual Goldschmidt Conference (Goldschmidt, 2005).
Sawaki, Y. et al. Chronological constraints on the Paleoproterozoic Francevillian Group in Gabon. Geosci. Front. 8, 397–407 (2017).
Thiéblemont, D. et al. Transition from alkaline to calc-alkaline volcanism during evolution of the Paleoproterozoic Francevillian basin of eastern Gabon (Western Central Africa). J. Afr. Earth Sci. 99, 215–227 (2014).
ghoubou, N. et al. The first application of Re–Os dating on Paleoproterozoic Francevillian sediments (Gabon). Comptes Rendus Géosci. 356, 57–66 (2024).
Beard, B. L. et al. Application of Fe isotopes to tracing the geochemical and biological cycling of Fe. Chem. Geol. 195, 87–117 (2003).
Rouxel, O. J., Bekker, A. & Edwards, K. J. Iron isotope constraints on the Archean and Paleoproterozoic ocean redox state. Science 307, 1088–1092 (2005).
Johnson, C. M., Beard, B. L., Klein, C., Beukes, N. J. & Roden, E. E. Iron isotopes constrain biologic and abiologic processes in banded iron formation genesis. Geochim. Cosmochim. Acta 72, 151–169 (2008).
Fantle, M. S. & DePaolo, D. J. Iron isotopic fractionation during continental weathering. Earth Planet. Sci. Lett. 228, 547–562 (2004).
Beard, B. L. et al. Iron isotope biosignatures. Science 285, 1889–1891 (1999).
Dauphas, N. et al. Clues from Fe isotope variations on the origin of early Archean BIFs from Greenland. Science 306, 2077–2080 (2004).
Bekker, A. et al. Iron formation: The sedimentary product of a complex interplay among mantle, tectonic, oceanic, and biospheric processes. Econ. Geol. 105, 467–508 (2010).
Konhauser, K. O. et al. Iron formations: A global record of Neoarchaean to Palaeoproterozoic environmental history. Earth Sci. Rev. 172, 140–177 (2017).
Johnson, C. M. & Beard, B. L. Fe isotopes: An emerging technique for understanding modern and ancient biogeochemical cycles. GSA Today 16, 4–10 (2006).
Guilbaud, R., Butler, I. B., Ellam, R. M., Rickard, D. & Oldroyd, A. Experimental determination of the equilibrium Fe isotope fractionation between Feaq2+ and FeSm (mackinawite) at 25 and 2 °C. Geochim. Cosmochim. Acta 75, 2721–2734 (2011).
Rolison, J. M. et al. Iron isotope fractionation during pyrite formation in a sulfidic Precambrian ocean analogue. Earth Planet. Sci. Lett. 488, 1–13 (2018).
Mansor, M. & Fantle, M. S. A novel framework for interpreting pyrite-based Fe isotope records of the past. Geochim. Cosmochim. Acta 253, 39–62 (2019).
Wiederhold, J. G. et al. Iron isotope fractionation during proton-promoted, ligand-controlled, and reductive dissolution of goethite. Environ. Sci. Technol. 40, 3787–3793 (2006).
McAnena, A., Severmann, S., Guilbaud, R. & Poulton, S. W. Iron isotope fractionation during sulfide-promoted reductive dissolution of iron (oxyhydr)oxide minerals. Geochim. Cosmochim. Acta 369, 17–34 (2024).
Albut, G. et al. Modern weathering in outcrop samples versus ancient paleoredox information in drill core samples from a Mesoarchaean marine oxygen oasis in Pongola. Geochim. Cosmochim. Acta 265, 330–353 (2019).
Moeller, K. et al. Comparison of iron isotope variations in modern and Ordovician siliceous Fe oxyhydroxide deposits. Geochim. Cosmochim. Acta 126, 422–440 (2014).
Crosby, H. A., Roden, E. E., Johnson, C. M. & Beard, B. L. The mechanisms of iron isotope fractionation produced during dissimilatory Fe(III) reduction by Shewanella putrefaciens and Geobacter sulfurreducens. Geobiology 5, 169–189 (2007).
Heard, A. W. & Dauphas, N. Constraints on the coevolution of oxic and sulfidic ocean iron sinks from archean-paleoproterozoic iron isotope records. Geology 48, 358–362 (2020).
Severmann, S., Johnson, C. M., Beard, B. L. & McManus, J. The effect of early diagenesis on the Fe isotope compositions of porewaters and authigenic minerals in continental margin sediments. Geochim. Cosmochim. Acta 70, 2006–2022 (2006).
Heard, A. W. et al. Triple iron isotope constraints on the role of ocean iron sinks in early atmospheric oxygenation. Science 370, 4–7 (2020).
Dupeyron, J., Decraene, M. N., Marin-Carbonne, J. & Busigny, V. Formation pathways of Precambrian sedimentary pyrite: Insights from in situ Fe isotopes. Earth Planet. Sci. Lett. 609, 118070 (2023).
Dauphas, N. et al. Past and Present Dynamics of the Iron Biogeochemical Cycle. Treatise on Geochemistry, 3rd ed. https://doi.org/10.1016/B978-0-323-99762-1.00059-0 (2023).
El Albani, A. et al. The 2.1 Ga old Francevillian biota: Biogenicity, taphonomy and biodiversity. PLoS One 9, e99438 (2014).
Poulton, S. W. & Canfeld, D. E. Ferruginous conditions: A dominant feature of the ocean through Earth’s history. Elements 7, 107–112 (2011).
Sperling, E. A. et al. Statistical analysis of iron geochemical data suggests limited late Proterozoic oxygenation. Nature 523, 451–454 (2015).
Poulton, S. W., Fralick, P. W. & Canfield, D. E. Spatial variability in oceanic redox structure 1.8 billion years ago. Nat. Geosci. 3, 486–490 (2010).
Planavsky, N. J. et al. Widespread iron-rich conditions in the mid-Proterozoic ocean. Nature 477, 448–451 (2011).
Stüeken, E. E. et al. The evolution of the global selenium cycle: Secular trends in Se isotopes and abundances. Geochim. Cosmochim. Acta 162, 109–125 (2015).
Mitchell, K., Mansoor, S. Z., Mason, P. R. D., Johnson, T. M. & Van Cappellen, P. Geological evolution of the marine selenium cycle: Insights from the bulk shale δ82/76Se record and isotope mass balance modeling. Earth Planet. Sci. Lett. 441, 178–187 (2016).
Kipp, M. A., Stüeken, E. E., Yun, M., Bekker, A. & Buick, R. Pervasive aerobic nitrogen cycling in the surface ocean across the Paleoproterozoic Era. Earth Planet. Sci. Lett. 500, 117–126 (2018).
Fennel, K., Follows, M. & Falkowski, P. G. The co-evolution of the nitrogen, carbon and oxygen cycles in the proterozoic ocean. Am. J. Sci. 305, 526–545 (2005).
Anbar, A. D. & Knoll, A. H. Proterozoic ocean chemistry and evolution: A bioinorganic bridge? Science 297, 1137–1142 (2002).
Wagner, L. J. et al. Coupled stable chromium and iron isotopic fractionation tracing magmatic mineral crystallization in Archean komatiite-tholeiite suites. Chem. Geol. 756, 120121, (2021).
Swanner, E. D. et al. Fractionation of Fe isotopes during Fe(II) oxidation by a marine photoferrotroph is controlled by the formation of organic Fe-complexes and colloidal Fe fractions. Geochim. Cosmochim. Acta 165, 44–61 (2015).
Schoenberg, R., Zink, S., Staubwasser, M. & von Blanckenburg, F. The stable Cr isotope inventory of solid Earth reservoirs determined by double spike MC-ICP-MS. Chem. Geol. 249, 294–306 (2008).
Weyer, S. & Ionov, D. A. Partial melting and melt percolation in the mantle: The message from Fe isotopes. Earth Planet. Sci. Lett. 259, 119–133 (2007).
Compston, W. & Oversby, V. M. Lead Isotopic Analysis Using a Double Spike. J. Geophys Res. 74, 4338–4348 (1969).
Decraene, M. N. et al. High-spatial-resolution measurements of iron isotopes in pyrites by secondary ion mass spectrometry using the new Hyperion-II radio-frequency plasma source. Rapid Commun. Mass Spectrom. 35, 0–2 (2021).
Pasquier, V. et al. SIMS Iron Isotope Measurements of the Balmat Pyrite Reference Material: A Non-Unique δ56Fe Signature. Geostand. Geoanalytical Res. 48, 423–431 (2024).
Decraene, M. N. et al. Intense biogeochemical iron cycling revealed in Neoarchean micropyrites from stromatolites. Geochim. Cosmochim. Acta 312, 299–320 (2021).
Acknowledgements
This study was funded by Khalifa University of Science and Technology (KUST grant FSU-2023-020/8474000494). FOO, IK and RS acknowledge financial support from the University of Tübingen and the German Research Foundation (DFG Grant SCHO1071/11-1). AH and FOO acknowledge support from National Research Foundation of South Africa (NRF Grant 75892). The SwissSIMS is part of the Center of Advanced Surface Analyses, a research consortium between EPFL and UNIL. Sample permission was not applicable in this study.
Author information
Authors and Affiliations
Contributions
The work formed part of the PhD research of A.A.A. that was supervised by F.O.O and A.A. The study was conceptualized by F.O.O. and R.S. Fieldwork and sample collection were completed by F.O.O., A.H., R.S. and A.A.A.; F.O.O., R.S., I.C.K. and J.M-C. conducted Fe isotope analyses. Sample evaluation and petrographic data were acquired by A.A.A, F.O.O and J.M-C. The paper was written by A.A.A. and F.O.O. with significant editorial input from all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth & Environment thanks Eva Stüeken and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Carolina Ortiz Guerrero. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ajagunjeun, A.A., Ossa Ossa, F., Kleinhanns, I.C. et al. Expansion of the aerobic iron biogeochemical cycle during the Paleoproterozoic Lomagundi event. Commun Earth Environ 6, 113 (2025). https://doi.org/10.1038/s43247-025-02106-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43247-025-02106-6
This article is cited by
-
Pyrite morphology and sulfur isotopes refine taphonomic models for the 2.1 Ga Francevillian biota, Gabon
Scientific Reports (2025)
