
Abstract
Trade-offs among traits determine microbial community dynamics and affect soil carbon-cycling feedback to climate change. Here, we determined soil prokaryotic traits based on a novel marker gene-based workflow using cross-continental temperature and aridity gradients. Genome streamlining and high 16S rRNA gene copy numbers per genome (RRN) conferred high maximal growth rates, possibly by allowing for smaller cells with higher surface to volume ratio. Small genomes and high maximal growth rates were found under high precipitation seasonality and in barren soils. Large genomes and low maximal growth rates were found in forests, characterized by high water availability and by abundant and complex organic resources. Our findings suggest that large genomes confer versatility to cope with resource fluctuations and moderate climatic fluctuations while extreme climatic fluctuations and scarcity of resources promote genome streamlining. Seasonal fluctuations in water availability were associated with the ability to form spores and with rapid resuscitation, promoted by high RRN. Moreover, Prokaryotes were less dispersal limited compared to Fungi, presumably due to their smaller size, but within Prokaryotes, small taxa were not more ubiquitous. Our trait-based framework highlights that particularly changes in precipitation patterns and vegetation type will cause changes in microbial processes under future climate.

Similar content being viewed by others


Introduction
Soil microorganisms are key players in carbon (C) and nitrogen (N) cycling and mediate feedback to climate change through changes in their activity and community dynamics1. Trade-offs among microbial traits determine the outcome of competitive dynamics and consequently C- and nutrient cycling at the ecosystem level2,3. Ecological concepts have been employed to describe life history strategies in microbial communities, such as the copiotroph-oligotroph framework4, the “competitor”-“stress tolerator”-“ruderal” (CSR) framework5,6 and its microbial adaptation, the “yield”-“acquisition”-“stress resistance” (YAS) framework2.
Important traits reflecting prokaryotic life histories include growth rate, cell size, genome size, 16S rRNA gene copy numbers per genome (RRN) and traits enabling survival under harsh conditions, such as spore formation and tolerance to high salinity. High growth rates enhance fitness and are promoted by high RRN, enabling rapid ribosome production7,8. Diverse and complex organic resources and/or fluctuating environments require versatility and therefore a high number of genes and a large genome9,10,11,12. Due to the costs of genome maintenance and gene expression, large genomes are associated with resource- and water-rich environments13,14,15 and with an aerobic lifestyle16. A trade-off between genome size and growth rates has been suggested because replication of large genomes requires more N and phosphorus, which thus is not available for biosynthesis17. However, this is not unambiguously supported by empirical data7,18,19,20. Moreover, growth yield changes with growth rate both within single species and between taxa, but both decreasing and increasing relationships have been reported8,21. Lipson22 reconciled these contrasting relationships by suggesting a hump-shaped relationship where growth yield increases with rate in the lower range of growth rates when growth rates are limited by nutrients, low substrate quality, and physiological stress. Conversely, a rate-yield trade-off was suggested to manifest in the higher range of growth rates in resource-rich environments. A high-yield strategy was proposed as a cooperative strategy among clonal populations isolated in spatially structured environments to economize resources, while in spatially mixed environments, the presence of competitors would favor taxa that maximize growth rate even at the expense of yield22,23,24.
Cell size is another key trait that affects multiple fitness-relevant properties, including resource uptake, dispersal, attachment to surfaces and susceptibility to predation (reviewed by Young25). A cell needs to fit its genome and biosynthetic machinery, suggesting that cell size scales with metabolic complexity and genome size26,27,28. Moreover, large cells may store greater amounts of resources to outlast periods of scarcity29. Because the surface quadratically scales with the radius while the volume scales with its cube, processes related to the surface, such as nutrient uptake and cell wall synthesis, are enhanced in smaller cells30,31,32. It remains a matter of debate, however, how metabolic rates and growth rates scale with cell size in microorganisms20,26,27,33,34,35,36,37,38,39. Due to its importance for organic matter turnover, cell size has been implemented in microbial trait-based biogeochemical models40.
Empirical evidence for trade-offs in microbial life history strategies mainly comes from cultured microorganisms7,8,16,20, owing to the difficulty of measuring traits in the environment. Genomic traits, such as genome size and RRN, have repeatedly been studied in environmental microbial communities13,14,28,41. Yet, microbial strategies can only be fully understood from the combination of multiple traits and therefore, the ecological strategies of environmental microorganisms remain elusive. Recent metagenomic studies inferred life history strategies based on functional categories from genomic databases as well as from genes and genomic traits associated with CSR and YAS strategies15,42. For instance, Piton et al.15 revealed genome size and thus metabolic complexity as well as investment into nutrient recycling versus environmental responsiveness as major dimensions of trait variation in soils, which was driven by precipitation patterns and pH. However, the findings were based on broad gene categories that do not necessarily capture ecologically relevant and interpretable traits or on very specific genes from CSR and YAS frameworks. Thus, beyond these theory-based frameworks, the trait combinations and trade-offs enabling microorganisms to thrive in a particular environment remain poorly understood. In addition, in these previous studies, trait-based strategies have been inferred from community weighted trait means (CWM), calculated as abundance-weighted average of traits/genes across community members15, or from metagenome-assembled genomes (MAGs)42. Because CWMs do not necessarily represent combinations of traits occurring in the same individual, using CWMs does not allow for assessing trade-offs. The MAG-approach, conversely, allows to infer trade-offs, but a limitation is the small number of MAGs obtained43. Hence, a comprehensive assessment of trade-offs within individual microorganisms beyond a limited number of the most abundant taxa remains lacking.
Besides environmental selection, the biogeographical distribution of prokaryotic and fungal communities is affected by dispersal limitation. Consequently, the turnover of community composition depends on the geographical distance44. In ecology, dispersal is often considered to be a stochastic process45, but the probability of an organism to disperse and colonize a remote site depends on its size and capacity to survive transport46,47.
Distance-decay relationships have mostly been assessed within the same continent and within a range of <5000 km48 and depend on the taxonomic domain (Archaea, Bacteria, Microeukaryotes), the spatial scale and the habitat type49. Due to their small size, microorganisms are easily dispersed50,51,52,53 and biodiversity at the local scale shows considerable overlap with biodiversity at the global scale, particularly for Prokaryotes54. Consequently, environmental variables typically explain more variation than geographical distance49.
Here, we use a cross-continental dataset comprising multiple temperature and precipitation gradients to investigate soil microbial community assembly and adaptation to different climatic and edaphic conditions. We assess the contribution of dispersal limitation and environmental filtering to the formation of soil prokaryotic and fungal communities and identify their environmental drivers as well as traits affecting dispersal. We furthermore assess environmental selection on prokaryotic traits and identify within-organism trade-offs using a novel approach to infer prokaryotic genomic and physiological traits from marker gene sequences43. Finally, we elucidate prokaryotic strategies based on multiple traits enabling occupation of a particular niche. Thereby, we include previously unconsidered traits, such as cell size and reveal ecological strategies beyond established frameworks, such as the CSR and YAS frameworks. We hypothesize that (i) Fungi are more limited by dispersal than Prokaryotes, (ii) small Prokaryotes are more easily dispersed and therefore found in a wider range of samples compared to large Prokaryotes, and (iii) harsh climatic conditions and scarcity of resources select for slow growing Prokaryotes with small genomes.
Results
Influence of geographical distance and environmental selection on prokaryotic and fungal community structures
Our dataset included temperature gradients related to latitude (Greenlandic and European gradients), altitude (Alpine gradient) and aridity gradients (Spanish and South African gradients; Fig. S1, https://doi.org/10.6084/m9.figshare.30010576. Bray-Curtis dissimilarities between fungal community structures increased faster than between prokaryotic community structures (decay constant b = 0.00040 and 0.00022 for Fungi and Prokaryotes, respectively) and approached saturation with geographical distance faster than for prokaryotic communities. Thus, 95% dissimilarity was reached at 929 and 5186 km for Fungi and Prokaryotes, respectively (Fig. 1A). Fungal community structures also changed faster with environmental distance than bacterial community structures, but the difference in decay rate was smaller than for geographical distances (b = 8.0 and 6.2 for Fungi and Prokaryotes, respectively; Fig. S2A). Conversely, weighted UniFrac distances increased faster for Prokaryotes than for Fungi (b = 9.6 × 10-6 and 3.8 × 10-6 for Prokaryotes and Fungi, respectively; Fig. S2B).

A Distance decay for prokaryotic (left) and fungal community structures (right) based on Bray-Curtis dissimilarities. Curves were fitted with an exponential model, f(x) denotes the fitted decay function 1 - ae-bx. Fill and edge color indicate the gradients from which samples were compared. Dashed vertical lines indicate the distance where community structures between sites reach 95% dissimilarity according to the model. B Ordinations of prokaryotic (left) and fungal (right) community structures constrained with environmental parameters based on Bray-Curtis dissimilarities of relative abundances. Non-colinear environmental variables were selected stepwise with adjusted R2 as additional stopping criterium. Numbers in brackets indicate the variation explained by each axis. Vectors indicate loadings of environmental parameters on ordination axes. Dashed arrows and light text indicate non-significant land cover classes. AL = Alpine, EU = Europe, GL = Greenland, SA = South Africa, SP = Spain, BIO15 = precipitation seasonality, MAP = mean annual precipitation, Tsoil = in situ soil temperature at the time of sampling.
We tested the contribution of dispersal limitation (i.e., geographical distance) and environmental selection to the assembly of microbial community structures. For both prokaryotic and fungal community structures, the greatest part of constrained variation was explained by environmental parameters alone (Table 1). While the total explained variation was higher for Prokaryotes than for Fungi (53.1 and 44.5%, respectively), the relative importance of environmental variables and geographical distances to the constrained ordination was similar for both groups. For both Prokaryotes and Fungi, the most important parameters structuring communities were bioclimatic variable 15 (BIO15; precipitation seasonality, measured as the coefficient of variation of monthly precipitation), in situ soil temperature at the time of sampling (Tsoil), land cover, soil pH and C:N (Fig. 1B, Tables S1, S2). Prokaryotic communities were, in addition, influenced by mean annual precipitation (MAP) and soil silt content, while fungal communities were influenced by the amount of plant litter. pH had a stronger influence on prokaryotic than on fungal communities. Interestingly, in contrast to prokaryotic communities, fungal communities showed distinct clusters associated with deciduous broadleaf and mixed forests versus evergreen needleleaf forests. For both Prokaryotes and Fungi, South African samples were clearly distinct from all the European and the Greenlandic samples, which was mostly driven by Tsoil and precipitation seasonality.
We hypothesized that smaller prokaryotic cells would be more easily dispersed and therefore found in a wider range of sites. However, we only found a weak positive correlation between cell size (diameter and length) and the number of samples, as well as between cell size and the maximum geographical distance an ASV occurred in (all Spearman’s ρ < 0.1; Table 2). A larger genome should confer versatility, allowing microorganisms to colonize a wider range of environments9,12. Nevertheless, we only found a weak positive correlation between genome size and sample number, maximum geographical distance as well as maximum difference in environmental conditions (Spearman’s ρ = 0.065, 0.063 and 0.062, respectively; Table 2).
Environmental selection on prokaryotic traits
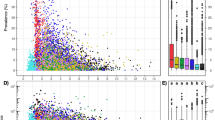
We identified environmental drivers of multiple prokaryotic traits (cell diameter and length, minimal doubling time, genome size, RRN, optimum pH, optimum temperature, salinity preference, oxygen preference, motility and sporulation [endospores, exospores and myxospores]). An analogous method to classify fungal traits is currently not available; hence, we focused on Prokaryotes. The trait classification method uses a bootstrap procedure to assess the confidence of the trait classifications based on which we determined if an ASV was considered classified using stringent quality criteria43. Thus, we were able to classify 49 to 93% of the sequences and 6965 to 17491 out of 20892 ASVs for these traits with high confidence, but with considerable variation across samples (Fig. S3). Optimum pH showed the lowest fraction of classified sequences (49 ± 16%) while sporulation showed the highest (93 ± 4.8%). We combined two types of models (random forest model [RF] and stepwise generalized linear model [GLM]) to link CWMs (abundance-weighted average of traits across community members) with environmental drivers. Figure S4 shows the distribution of the data for the relationships between predictors and CWMs. In a previous, method-focused study with the same dataset, we used this approach to identify environmental drivers of genome size43. Here, we expand on our previous study identifying key predictors of CWMs for multiple traits, which were parameters associated with water availability (BIO15, soil water-holding capacity [WHC], water activity [aw], MAP), Tsoil, vegetation properties (land cover, litter content), texture (silt content) and pH (Fig. 2, Fig. S5). The two models mostly agreed on the selection of the most important predictors but showed some differences in the ranking and the relationship between predictors and CWMs. For the stepwise GLM, the five most important predictors were part of the model for most cross validation runs with stepwise selection, confirming their robustness (Fig. 2).

The five most important predictors are shown for each trait. Small panels next to each bar indicate the response of the trait to each predictor. Larger response plots for land cover are shown in Fig. S5. Black areas of rectangles show the fraction of 200 repeated split sampling cross validation runs where a variable was selected in the stepwise GLM indicating the robustness of the variables to different subsets of the data. Variable importance was determined as % increase in mean squared errors when permuting the variable of interest (mean of 100 permutations). aw = water activity, BIO5 = maximum temperature warmest month, BIO15 = precipitation seasonality, LC = land cover, MAP = mean annual precipitation, SOM = soil organic matter, TC = total carbon, TOC = total organic carbon, Tsoil = in situ soil temperature at the time of sampling, WHC = water holding capacity.
BIO15 (high BIO15 values indicate unevenly distributed precipitation throughout the year) was positively related to sporulation, salinity preference, optimum pH, oxygen preference, RRN, motility, cell length as well as optimum temperature and negatively related to minimal doubling time, cell diameter and genome size. Barren soils were associated with low sporulation, low minimal doubling time, high optimum pH, small genomes, small cell diameter, high optimum temperature and preference for oxic environments. Litter content was positively related with minimal doubling time, cell diameter and genome size and negatively related with sporulation, salinity preference and optimum pH. Notably, pH and SOM were less important predictors than climatic conditions and vegetation related properties (Fig. 2). Some land cover classes were related to BIO15, where evergreen needleleaf, mixed and deciduous broadleaf forests encompassed low BIO15 values, while woody savannas encompassed high BIO15 values (Fig. S6). Thus, precipitation seasonality may also mediate effects on soil prokaryotic traits via vegetation properties.
We then cross validated the models’ predictive accuracy. For the RF, Pearson correlations between observed and predicted CWMs ranged from 0.45 to 0.83 and for the stepwise GLM from 0.52 to 0.79. (Table 3). Cell length and salinity preference showed the lowest accuracy for the RF and the GLM, respectively, while oxygen preference showed the highest accuracy for both models. A weighted ensemble performed slightly better than the individual models for cell diameter, minimal doubling time, RRN and salinity preference (Table 3).
Prokaryotic strategies to adapt to their environment
To pinpoint ecological strategies and identify trade-offs, we evaluated co-occurrence patterns among traits across ASVs. We found significant positive correlations (Padj. <0.05 and Spearman’s ρ ≥ 0.2) between optimum pH, optimum temperature and motility, as well as between sporulation, cell length and RRN (Fig. 3). Genome size was positively related with oxygen preference, cell diameter and minimal doubling time, all of which were negatively related with optimum temperature and pH, except oxygen preference with optimum pH. Further, RRN was negatively associated with cell diameter and minimal doubling time and positively associated with motility and sporulation. In addition, cell length was positively correlated with oxygen preference and motility, and negatively correlated with minimal doubling time, salinity preference, and optimum temperature.

Significant correlations (Padj. <0.05) with ρ ≥ 0.2 are shown (n = 20 892). ASV = amplicon sequence variant. Unclassified ASVs were omitted for each pairwise comparison. P-values were adjusted for multiple comparisons using the Benjamini-Hochberg procedure. The circle size indicates the strength of the correlation.
Cluster analysis revealed ten major prokaryotic strategies characterized by different combinations of traits (Fig. 4) of which Clusters 3, 10 and 6 were the most abundant (Fig. S7). Cluster 10 was particularly found in South African grasslands and woody savannas with high precipitation seasonality and harbored cells with small diameter and intermediate length, relatively small genomes, extremely high RRN and short minimal doubling time, motility, ability to form spores and high salinity preference (Fig. 4, Fig. S8). Clusters 4, 6 and 7 were associated with different types of forests and low pH. Cluster 3 occurred in vegetated soils with high clay content and was characterized by short, non-motile cells unable to form spores with low minimal doubling time, pH optimum and salinity preference. Clusters 4 and 7 were also associated with high clay content and both encompassed large cells with high minimal doubling time, high salinity preference and low optimum pH (Fig. 4, Fig. S8). In contrast to Cluster 7, Cluster 4 had a very high RRN and high oxygen preference. Cluster 6 correlated positively with aw and showed motility and low oxygen preference as well as small cells and genomes with short minimal doubling times. Clusters 1, 2, 5 were associated with barren soils and/or open shrublands with coarse texture and high pH. Clusters 1, 2 and 5 had in common small genomes, low RRN, relatively small cells and inability to form spores. Cluster 1 was motile and had a high salinity preference and a higher minimal doubling time compared to Clusters 2 and 5. Cluster 5 displayed a particularly high temperature optimum. Cluster 9 was positively associated with open shrublands and precipitation seasonality and showed very distinct features and was characterized by spore formation, motility, extremely long cells and a large genome. Cluster 8 was not associated with a particular environment and was characterized by large genome, short minimal doubling time, spore formation, high oxygen preference and high optimum pH.

A Heatmap of trait values for each cluster. Trait values were scaled and centered. Darker colors indicate higher values. For oxygen preference, higher values indicate a higher preference for presence of oxygen. For salinity preference, higher values indicate more halophilic preferences. B Constrained ordination of community structures based on Bray-Curtis dissimilarities of relative abundances of clusters. Points represent community structures based on clusters. Vectors represent loadings of environmental variables on the ordination axes, numbers in squares indicate loadings for clusters. Non-colinear environmental variables were selected stepwise with adjusted R2 as additional stopping criterium. Dashed gray arrows and text indicate non-significant land cover classes. Numbers in brackets indicate variation explained by each axis. C Spearman correlations between abundances of trait clusters and environmental variables (n = 70). Significant correlations (Padj. <0.05) with ρ ≥ 0.2 are shown. Only variables with at least one significant correlation are shown. P-values were adjusted for multiple comparisons using the Benjamini-Hochberg procedure. The landcover categories Forests, Barren / shrublands and Savannas / Grasslands represent combined variables for the land cover classes indicated in the figure legend. aw = water activity, BIO15 = precipitation seasonality.
Some associations between trait clusters and environmental variables were also found within individual gradients, while others manifested only between gradients (i.e., in the full dataset) or showed different relationships in different gradients and compared to the full dataset (Fig. S9). For instance, positive associations with pH for Clusters 2 and 5 and a negative association for Cluster 7 were found in the Spanish and Alpine gradients as well as in the full dataset. Conversely, relationships with BIO15 within gradients mostly differed from patterns in the global dataset. This is because a strong contrast in BIO15 was found between the South African gradient and the other gradients, while the range of BIO15 within each gradient was relatively small (Fig. S1).
Discussion
Overlapping prokaryotic and fungal communities across the globe55,56 suggest that dispersal rates are high relative to diversification rates for both groups. In line with this, prokaryotic community structures displayed <0.95 dissimilarity up to >5000 km distance and biogeographical patterns were more driven by selection than by dispersal limitation. Conversely, fungal communities displayed >0.95 dissimilarity at <1000 km distance, with an abrupt increase in dissimilarity between samples from different continents. This suggests that Fungi are more dispersal-limited than Prokaryotes, particularly across oceans. However, for both groups, a similar fraction of variation was explained by geographical distance alone. The higher fraction of unexplained variation for Fungi may be explained by greater dispersal limitation as community dissimilarity becomes independent of geographical distance when reaching complete dissimilarity. In addition, unmeasured environmental variables that affect Fungi more strongly than Prokaryotes could explain the higher fraction of unexplained variation. For instance, plant diversity has been shown to be an important driver of fungal diversity57 which was not considered in our study. Fungal communities also exhibited stronger distance decay with environmental distances than prokaryotic communities, but the difference was smaller than that observed with geographical distances. Thus, in addition to being more dispersal-limited, fungal communities may be more sensitive to environmental filtering compared to prokaryotic communities. In contrast to prokaryotic communities, fungal communities differed strongly between evergreen needleleaf forests compared to deciduous and mixed forests. These differences may be driven by plant-fungus interactions, such as mycorrhizal symbiosis58 and by different litter quantity and quality as Fungi are primary decomposers of lignocellulose59. In contrast to taxonomic turnover based on Bray-Curtis distances, prokaryotic communities exhibited stronger phylogenetic turnover than fungal communities based on weighted Unifrac distances. This could be explained by a faster pace of evolution relative to dispersal limitation in Prokaryotes compared to Fungi, which could be a consequence of shorter generation times and thus higher mutation rates60.
In a simulation study, the range of aerial dispersal for microorganisms with sizes between 9 and 60 µm decreased with size51, which may explain differences in dispersal capacities between Prokaryotes and Fungi in our study. For Prokaryotes, however, we found a weak positive relationship between ubiquity and size. Wilkinson et al.51 showed that microorganisms <9 µm were passively dispersed across global distances, indicating that most Prokaryotes theoretically could traverse the globe by aerial dispersal. In the size range of Prokaryotes, smaller particles would be subject to a longer aerial time61, which may negatively affect survival and thus (viable) dispersal capacities, explaining the lack of relationship between size and ubiquity. Alternatively, dispersal followed by successful colonization of a wide range of environments may be facilitated by large, versatile genomes, which require larger cells26,28, overriding the effect of increased aerial dispersal of small cells.
We established several universal trait trade-offs across all ASVs in the dataset, e.g., the positive relationship between cell diameter, genome size and minimal doubling time. Larger genomes confer versatility and more complex metabolic networks62 but require larger cells to fit the genome and the metabolic machinery26. As energy-generating processes are located in the membrane, the decreasing surface-to-volume ratio with increasing size limits the energy available per nucleotide and may slow down growth rates31. Accordingly, we and others16 found that genome size was correlated with high oxygen preference, suggesting that aerobic metabolism is needed to provide sufficient energy to express and replicate a large genome. RRN was negatively correlated with minimal doubling time7,8,20 substantiating their growth-promoting role through enhanced protein synthesis in soil Prokaryotes. Notably, RRN correlated with the ability to form spores, indicating that high rates of protein synthesis and rapid growth are particularly relevant during spore resuscitation, as they enable quick cellular restructuring and metabolic changes, thereby maximizing the benefit from periods of favorable conditions. It is noteworthy that although we found universal relationships among traits, in accordance with Westoby et al.20, correlations were relatively weak, highlighting the importance of more complex relationships than bivariate trade-offs, such as environment-dependent relationships and trade-offs among multiple traits37,63.
We revealed precipitation seasonality and land cover as key drivers of prokaryotic traits at the community level, in agreement with a metagenome-based study15. Figure 5 and Table 4 summarize trait-environment relationships. High precipitation seasonality favored traits that suggest drought resistance, such as high salinity preference. Importantly, increased spore formation together with short minimal doubling times, small genomes and high RRN under high precipitation seasonality indicate that alternations between drought and abundant water select for outlasting stressful periods in a resting stage and maximizing growth rates under favorable conditions. Community-level traits under high precipitation seasonality corresponded to high abundances of Cluster 10, which united the above-named properties. These trait combinations may become more dominant in the future, as more variable precipitation is predicted in many regions of the world64.

Corners of the triangle represent environments. The vertical double arrow specifies an additional axis of trait variation within the high resource environment, possibly related to a trade-off between maximizing growth and stress response traits. Clusters were placed close to the environment with which they were associated. Low resource environments promote genome streamlining, high maximal growth rate and either extremely small cells or very long (filamentous) cells allowing for efficient nutrient uptake. Conversely, environments with high amounts of resources as well as complex resources favor large, versatile genomes associated with slow growth. Seasonal fluctuations drive spore formation with rapid resuscitation and growth during favorable periods through high RRN. Within the high resource environments, clusters separate by stress response traits, such as halophilic preference, which are associated with larger cells and genomes as well as higher minimal doubling time. For Clusters 8 and 9 association with dominant environments was less clear and they displayed trait features found in different environments. aw = water activity, RRN = 16S rRNA gene copy numbers per genome.
In contrast to seasonally fluctuating environments, barren soils and open shrublands represent continuously harsh environments due to low and rapidly fluctuating water availability, enhanced exposure to UV radiation, and scarcity of organic C and nutrients, the availability of which is further limited by the slow transport across disconnected water films. Trait profiles in these environments (Clusters 1, 2 and 5) shared with Cluster 10 a small cell diameter, short minimal doubling time and small genomes, but were unable to form spores. For these clusters, small cells and genomes may represent a mechanism to sustain constitutive growth under low resource availability and to compensate for energy dedicated to elevated maintenance costs under extreme environmental conditions28,65. Moreover, small cells may be an adaptation to prevent lysis due to changes in osmotic pressures, as tensile forces are smaller in smaller cells66. Properties of Clusters 1, 2 and 5 are consistent with CWMs found in barren soils. It should be noted that the doubling times in this study are potential doubling times, which are unlikely to occur under natural conditions most of the time, due to energy and/or nutrient limitations. Accordingly, based on stable isotope probing, smaller genomes have been associated with higher growth rates in glucose-amended soils where energy limitation was alleviated, but not in unamended soils19.
Our findings highlight that the amplitude and duration of favorable versus unfavorable conditions, as well as their predictability, affect microbial strategies. For instance, fluctuations in microclimatic conditions in barren soils are likely too rapid and unpredictable for microorganisms to survive in a resting stage. In contrast, seasonal fluctuations, which include benign conditions of sufficient length, promote spore formation and resuscitation. Similarly, in phytoplankton, the timing and magnitude of resource fluctuations determined if continuously growing resource storage specialists or species with high nutrient uptake that maximize growth under resource pulses were the better competitors29,67.
We found slow-growing cells and large genomes (clusters 3, 4, 6 and 7) in resource-rich environments with high litter input and with diverse and recalcitrant SOM from woody vegetation, such as forest soils. We propose that these traits confer high metabolic versatility, enabling Prokaryotes to degrade diverse C sources obtained from plant litter or microbial necromass, in line with large genomes found in environments with fluctuating and complex resources9. Interestingly, despite a high minimal doubling time, Cluster 4 exhibited relatively high RRN. We suggest that multiple RRN support the expression of complex metabolic and regulatory networks from a large genome. Hence, the same trait can serve different functions depending on the environmental context. The energy-demanding lifestyle of Cluster 4 is supported by its high oxygen preference16. High salinity preference and extremely low optimum pH in Cluster 4, moreover, suggest the presence of mechanisms to deal with osmotic imbalances that require additional energy. Conversely, Cluster 6 exhibited traits to thrive under low oxygen conditions and its abundance correlated with high aw, which may cause anoxic conditions. A lower energy yield from anoxic metabolism may explain smaller genomes and cells compared to Clusters 4 and 7. Lastly, Cluster 3 exhibited relatively large genomes and cells, but rather short minimal doubling time while traits related to thriving under adverse conditions, such as sporulation and high salinity preference were absent. An explanation for higher maximal growth rates relative to the genome size for Cluster 3 compared to Clusters 4 and 7 could be a lower investment into stress response and maintenance. Moreover, the broader association of Cluster 3 with vegetated soils in general but not forest soils in particular points towards the usage of diverse but less recalcitrant C sources and may explain the higher maximal growth rates compared to Clusters 4 and 7. While some cluster abundance-environment relationships were consistent across gradients and the full dataset, many relationships were specific to individual gradients or manifested only in the global dataset, contrasting different gradients. These results indicate that the range of values considered for each environmental factor, as well as the combination with other factors, affects its importance as a driver of microbial trait variation in each study area and highlight the difficulty in generalizing trait environment relationships across studies.
Previous studies reported ambiguous relationships between genome size and growth rates7,18,19,20. Beier et al.68 found a positive relationship for genomes <4 Mbp and a negative relationship for genomes >5 Mbp and suggested different environment-dependent mechanisms for these two ranges. In agreement with our study, the authors found a trade-off between genome size and growth rate in soils, while they found no relationship in aquatic and digestive tract metagenomes68. In contrast to our study, Leff et al.69 found decreasing genome sizes and a tendency for increasing growth rates under nutrient addition. We predominantly found large genomes in environments with high litter input, high vegetation biomass and woody vegetation. Although resources in these environments are abundant on average, availability is expected to fluctuate depending on plant inputs and water availability. Thus, we suggest that a versatile strategy enabling growth on various substrates, including complex ones such as lignocellulosic material, is most successful in these environments. In an environment with more stable and less complex resources, conversely, we would expect a strategy that maximizes growth rates at the expense of versatility to dominate. Experimental nutrient addition as in Leff et al.69 may represent such constant resource supply where streamlining is not penalized and higher growth rates are promoted. In our study, however, genome streamlining was driven by fluctuations in climatic conditions and periods of severe resource limitations, underscoring the importance of environmental context in understanding ecological strategies in microorganisms. In scarce environments, a growth rate-yield trade-off has been suggested to promote slow growth with high efficiency8,70 and thus, it seems surprising to find high maximal growth rates in such environments as barren soils. However, the rate-yield trade-off may manifest only under resource-rich conditions and high growth rates22. Thus, the potentially fast-growing taxa in barren soils will not be subjected to a rate-yield trade-off when realized growth rates are limited by resource supply and physiological stress rather than their inherent maximum. Moreover, maximizing growth yield at the cost of low growth rates is only a viable strategy under limited competition23,24. In soil environments, rather than being inherently linked with growth rates, we expect growth yield to vary with the capacity to withstand stress and respond to environmental change.
Large genomes have been associated with versatility to thrive in a wide range of conditions varying across space and time as well as with high substrate complexity9,10,11,12,71. In contrast to previous studies11,12, genome size correlated poorly with the range of environments a taxon could inhabit. One possible explanation is a trade-off in cell size owing to dispersal capacity as outlined above. In addition, the geographical scale and the habitats differed from these previous studies and our study encompassed a large fraction of desert-like environments favoring small genomes, which tended to be located at each end of the geographical range (Greenland and South Africa), blurring genome size-ubiquity relationships.
Compared to metagenome-based trait inference, the amplicon-based approach used here has both strengths and weaknesses. Amplicon-based trait inference depends on the phylogenetic conservation of traits and assumes that closely related taxa share similar traits. In a previous study, we confirmed this relationship by cross-validating trait predictions across a range of phylogenetic distances between query and reference sequences43. This setup allowed us to evaluate realistic scenarios for environmental sequences with no closely related reference sequence available. A strength of our method is that it allows us to quantify the uncertainty of trait predictions and thus the relationship between phylogenetic relatedness and trait conservation for environmental sequences by bootstrapping. Trait conservation may vary across different microbial phyla, resulting in lower accuracies of trait predictions and a higher fraction of unclassified sequences for phyla with less conserved traits. From a microbial point of view, soils are highly diverse environments72, and the availability of trait information varies across taxonomic groups. Hence, groups with fewer reference sequences are expected to encompass a higher fraction of unclassified environmental sequences. It should be noted that similar limitations apply to the annotation of functional genes from metagenomes, which assumes that genes with high sequence identity have the same function, where conservation of genes may vary across taxonomic groups and where the availability of reference genomes is not equal across taxonomic groups. Both approaches will greatly benefit from increasing the coverage of genome databases, particularly for currently underrepresented taxa. While it is relatively straightforward to estimate the uncertainty of amplicon-based trait predictions, it remains difficult to evaluate the uncertainty of trait predictions from environmental metagenomes that encompass multiple, complex bioinformatic processes, such as assembly, gene annotation and binning, each of which could introduce errors.
A main disadvantage of the amplicon-based trait inference approach compared to metagenome-based trait inference is that it may not resolve trait variation between closely related taxa. We accounted for this problem by assessing the confidence of the trait classification with the bootstrap procedure, thus excluding unreliable trait classifications due to limited resolution which would result in low bootstrap confidence. On the other hand, we were able to classify traits for > 5000 ASVs, which is much higher than the number of MAGs typically obtained from metagenomic datasets, and we were able to include several traits, such as cell size that are not available from metagenomes. Moreover, the same trait classification approach could be applied to full-length 16S rRNA gene sequences extracted from metagenomes to improve resolution as well as to partial 16S rRNA gene sequences, given that full-length genes often cannot be recovered from metagenomes. Overall, our results align well with metagenomic studies (e.g., ref. 15), and although we acknowledge that amplicon-based trait classification, as any method, may suffer from some biases, in some aspects it can outperform metagenomics, for instance by recovering trait information for rare taxa with great ecological importance, such as nitrifying bacteria and archaea.
Conclusion
Our study provides novel insights into microbial traits shaping ecological strategies and their interactions with the environment. By directly predicting a unique combination of traits for environmental microorganisms, we extend our knowledge beyond studies in isolates16,20, studies focusing on single traits, such as genome size14 as well as studies using broad gene categories as a surrogate for traits or focusing on specific ecological frameworks15. For instance, our data indicates that both cell width and length constitute adaptations to specific environments and are linked with key fitness traits, such as maximal growth rates and metabolic complexity, through trade-offs. Importantly, our study revealed distinct strategies to adapt to seasonally dry environments with intermittent periods of rainfall and to barren soils, constituting constantly harsh environments, with precipitation seasonality and land cover as key drivers of these strategies. These results underscore that the frequency, magnitude and predictability of disturbances, such as drought events under climate change should be considered to evaluate soil microbial resistance and resilience and thus ecosystem functioning. Besides increasing temperatures, climate change imposes changes in precipitation patterns and vegetation cover, such as treeline shifts and desertification64,73,74. Our findings highlight that these global change components will lead to profound changes in key prokaryotic traits including maximal growth rates and genome size with possible feedback on global biogeochemical cycles.
Material and methods
Site description and sampling
In 2021, we sampled 70 sites along five temperature or aridity gradients following standardized protocols, which cover a wide range of climatic conditions, land cover types and soil properties. The sites include latitudinal gradients across Greenland (5 sites; GL) and Europe (13 sites; EU), an altitudinal gradient across Switzerland (13 sites; denoted as Alpine gradient, AL) as well as aridity gradients across Spain (12 sites; SP) and South Africa (28 sites; SA; https://doi.org/10.6084/m9.figshare.30010576). Across all gradients, mean annual temperatures (MAT) ranged from −18 °C to 22 °C and mean annual precipitation (MAP) from 45 to 1635 mm (Fig. S1). Land cover classes included “Barren” (5 sites), “Grasslands” (15 sites), “Open Shrublands” (5 sites), “Closed shrublands” (1 site), “Savannas” (9 sites), “Woody Savannas” (6 sites), “Evergreen Broadleaf Forests” (3 sites), “Deciduous Broadleaf Forests” (11 sites), “Mixed Forests” (4 sites) and “Evergreen Needleleaf Forests” (11 sites). Soil organic matter (SOM) content ranged from 0.34 to 59.1% of dry weight and pH from 2.6 to 8.1. Within an area of 20 × 20 m, we took five replicate soil cores with height and diameter of 10 cm. We sieved the soils through a 4-mm mesh and stored a subsample for DNA extraction at −20 °C75,76.
Climatic, soil and vegetation properties
We determined pH in a soil extract with 0.01 M CaCl2 (extractant - soil ratio 2:1 v/w) with a pH meter. Total organic C (TOC) content was measured after HCl-fumigation using an elemental analyzer77. We quantified soil and litter total C (TC) and nitrogen (TN) with an elemental analyzer (NC-2500; CE Instruments, Wigan, United Kingdom) after drying at 60 °C and grinding the material. Soil organic matter (SOM) was measured by loss-on-ignition, where we combusted the samples at 450 °C for 4 h78. We used the hydrometer method according to Gee and Bauder79 to determine soil texture (sand, silt and clay content). Water activity (aw), which represents the microbe available water80, was measured with an aw-meter (LabSwift-aw; Novasina AG, Lachen, Switzerland). Further environmental variables were obtained from georeferenced databases as described previously43. We extracted the bioclimatic variables BIO1 (mean annual temperature), BIO5 (maximum temperature warmest month), BIO7 (temperature, annual range; maximum temperature of warmest month minus minimum temperature of coldest month), BIO12 (mean annual precipitation) and BIO15 (precipitation seasonality; ratio of the standard deviation of the monthly total precipitation to the mean of the monthly total precipitation) from the worldclim database81 at 30 arc-seconds resolution. Further, land cover classifications following the International Geosphere-Biosphere Program classification were extracted using the MODIS product MCD12Q1_LC182 for the year 2020 at 500 m resolution and revised manually with the help of photos from the actual sites. Water holding capacity was obtained from the ISRIC-WISE30sec data set83 at a resolution of 30 arc-seconds.
DNA extraction, amplicon sequencing and bioinformatics
We used the DNeasy PowerSoil Pro Kit (Qiagen, Hilden, Germany) to isolate DNA from 250 mg of soil according to the manufacturer’s instructions and determined DNA concentrations using PicoGreen (Molecular Probes, Eugene, OR, USA). We amplified the V3-V4 region of the prokaryotic16S rRNA gene and fungal ITS2 region with primers and PCR conditions as described previously84. Amplicon barcoding using the Fluidigm Access Array technology (Fluidigm) and paired end sequencing on the Illumina MiSeq v3 platform (Illumina Inc., San Diego, CA, USA) were conducted at the Genome Quebec Innovation Center (Montreal, Canada). Raw sequences were quality filtered and denoised using DADA285 implemented in Qiime286. We removed primers with cutadapt87 with default settings and then quality filtered and denoised sequences with DADA2 (--p-trunc-len-f 270, --p-trunc-len-r 220, --p-max-ee 5 for 16S rRNA gene fragment amplicons and --p-trunc-len-f 270, --p-trunc-len-r 230, --p-max-ee 4 for ITS2 amplicons). Prokaryotic and fungal ASVs were classified against the SILVA v13888 and UNITE v989 databases, respectively, using the scikit-learn multinomial naive Bayes classifier in qiime290,91 (feature-classifier classify-sklearn) with default parameters. Contaminant sequences were removed using the isContaminant function (method = “prevalence”) in the R package decontam92. We obtained 3,920,603 high-quality prokaryotic sequences (11,234 ± 3577 per sample) and 5,309,207 fungal sequences (15,213 ± 3741 per sample) that formed 20,892 (147 ± 46 per sample) and 19 066 ASVs (117 ± 49 per sample), respectively. Rarefaction curves reached a plateau for all samples, confirming sufficient sequencing depth (Fig. S10). Rarefaction curves were calculated with the function rarecurve from the R package vegan93. For Prokaryotes, 0.96% of the ASVs (1.17% of the reads) were of archaeal origin, while the remaining ASVs were of bacterial origin. The most abundant bacterial phyla were Proteobacteria, Firmicutes and Planctomycetota and the most abundant archaeal phyla were Crenarchaeaota, Euryarchaeota and Nanoarchaeota. For Fungi, the most abundant phyla were Ascomycota, Basidiomycota and Mortierellomycota. Raw sequences were deposited in the NCBI Sequence Read Archive under the accession numbers PRJNA1073882 for 16S rRNA gene fragments and PRJNA1161578 for fungal ITS2 fragments.
Data analysis
All plots and statistical analyses were produced in R version 4.3.394. All code and data are available on figshare (https://doi.org/10.6084/m9.figshare.30010576). All analyses were conducted using the mean of five replicates.
Community structures across space and environments
To assess relationships between microbial community structure and geographical distance we calculated distance matrices for prokaryotic and fungal community structures based on Bray-Curtis dissimilarities and weighted UniFrac distances of relative abundances as well as a distance matrix of geographical distances based on great circle distances between the coordinates. We fitted an exponential relationship (Eq. 1) for Bray-Curtis dissimilarities or weighted UniFrac distances as a function of geographical distances using the function decay.model in the package betapart95.
a denotes the community dissimilarity when x = 0 and b denotes the decay rate constant where greater b indicates faster increase in dissimilarity. To assess the influence of environmental filtering on microbial communities, we calculated a constrained ordination of microbial community structures with stepwise selection of environmental variables in forward direction with adjusted R2 as an additional stopping criterion using the function ordiR2step in the R package vegan93. Only non-colinear variables with variance inflation factor <10 were included in this analysis96. To assess the influence of geographical distance on microbial community structures, we calculated principal coordinates of geographical distances based on a neighborhood matrix (function pcnm in vegan), which we used as independent variables in a constrained ordination of microbial community structures. Subsequently, we used variation partitioning to assess the contribution of environmental variables and geographical distance in structuring prokaryotic and fungal community structures (function varpart in vegan). We moreover tested if the environmental variables significantly explained variation in microbial community structures after conditioning for geographical distance and vice versa using the adonis2 function in vegan. In addition, we fitted curves for the decay of Bray-Curtis dissimilarities with environmental distances, which were determined as Gower distances of non-colinear environmental variables.
Classification of prokaryotic traits
We used the trait sequence database ampliconTraits43,97 and the SINAPS algorithm98 to classify genomic and physiological traits for prokaryotic ASVs. ampliconTraits combines a trait database99 with sequences from the SILVA small ribosomal subunit database88. Moreover, we determined the sequence identity with the best hit in the database and considered ASVs with a bootstrap value > 70 and sequence identity with the top hit >80% as classified. To enable classification of continuous traits, ampliconTraits bins them into intervals and offers versions with different numbers of intervals across the range of values for each trait, the choice of which affects the resolution but also the fraction of classified ASVs. Intervals were set according to Donhauser et al.43 to balance between resolution and a sufficiently high fraction of classified sequences: cell diameter: 20; cell length: 30; minimal doubling time: 30; genome size: 10; optimum pH: 20; optimum temperature: 20; 16S rRNA gene copy numbers: exact numbers. To assess the influence of environmental drivers on prokaryotic traits, we calculated CWMs (abundance weighted average of trait values across all classified ASVs in a sample), based on the mean of the interval for continuous traits. The levels of the traits oxygen preference and salinity preference were ranked as follows and then treated as continuous variables: oxygen preference: obligate_anaerobic = 1, anaerobic = 2, facultative = 3, microaerophilic = 4, aerobic = 5, obligate aerobic = 6; salinity preference: non-halophilic = 1; stenohaline and halotolerant = 2, moderate-halophilic = 3, halophilic = 4, extreme-halophilic = 5. We summarized different forms of motility as motile and transformed motility in a binary variable with motile = 1 and non-motile = 0. Similarly, spore-forming was recoded as 1 and non-spore-forming as 0.
Modeling trait-environment relationships
We used the set of 16 non-colinear variables as predictors of prokaryotic CWMs, which we then modeled with RFs (function randomForest from the package randomForest100) and stepwise GLMs with the Akaike information criterion (AIC) as stopping criterion and selection in both directions (function stepAIC from the package MASS101). The distribution of the response variables were determined with the function fitdist from the package fitdistrplus102 based on the AIC and diagnostic plots. Variable importance was evaluated for the RF model as part of the randomForest function and for the GLM with the function VarImp.glm in the MicEnvMod package43,103 and response plots were generated using the function respMono in MicEnvMod. Model accuracy was assessed for both model types by repeated split sampling with the function crossVal in MicEnvMod with default parameters based on Pearson correlations between observed and predicted values. Stability of parameter selection across datasets for the stepwise GLM was assessed with the function crossVal.step (MicEnvMod) as the fraction of cross-validation runs where the variable appeared in the model. Accuracy of an ensemble model between RF and stepwise GLM, weighted by the Pearson correlation coefficients of the models, was assessed with the function crossVal.ensemble (MicEnvMod). To reduce overfitting with the RF model, in addition to the model with all predictors, we also created a model using the same numbers of the most important predictors as defined by the stepwise GLM. For the ensemble model, we used the RF that performed the best (RF with all parameters or RF with most important parameters), i.e. the model with only the most important parameters for all traits except RRN.
Identifying ASV level trait trade-offs and strategies
To identify trait trade-offs at the ASV levels, we calculated a correlation matrix of Spearman correlations for the ASVs in the dataset using the function rcorr from the Hmisc package104, which we plotted with the corrplot function from the corrplot package105. P-values adjusted for multiple comparisons were calculated using the p.adjust function using the Benjamini-Hochberg method. We conducted a cluster analysis for the trait combinations across ASVs, including complete cases, i.e., ASVs where all traits were classified, using a bootstrap cutoff of 50 and a sequence of identity with the top hit >70% for classification. This allowed us to include 24.7% of the ASVs. We used the function pam in the R package cluster106 based on a Gower distance matrix enabling the use of mixed data types. Oxygen preference and salinity preference were implemented as ordinal variables, motility and sporulation as binary variables and all other variables as numeric. We compared clustering with 2 to 100 clusters to determine the optimal number of clusters based on silhouette widths. 10 clusters had a silhouette width of 0.52 which was the highest value for cluster numbers between 2 and 20. 50 and 100 clusters had higher silhouette widths than 10 clusters (0.63 and 0.71) but would be difficult to interpret. Therefore, we chose to represent a clustering analysis with 10 clusters using heatmaps based on scaled and centered cluster means. Analogously to the analyses for ASV abundances, we performed a constrained ordination of cluster abundances, producing ordination plots with both environmental variables and species scores overlayed as vectors. Moreover, we calculated Spearman correlations between trait cluster abundances using the function rcorr from the Hmisc package104. To this end, the levels of land cover were converted to binary variables using the function model.matrix. We also created combined groups for land cover classes that showed similar relationships with trait cluster abundances, e.g., different types of forests. P-values adjusted for multiple comparisons were calculated using the p.adjust function using the Benjamini-Hochberg method.
Relationships between prokaryotic traits and ubiquity
To evaluate whether the ubiquity of prokaryotes was linked with traits that either facilitate dispersal (cell size) or confer versatility (genome size), we calculated Spearman correlations between the trait values of each ASV and the number of sites, the maximum geographical distance, and the maximum difference in environmental conditions that each ASV inhabited. Difference in environmental conditions was calculated as Gower distances between sites based on environmental variables, including non-colinear variables as in previous analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Sequences were deposited in the NCBI Sequence Read Archive under the accession numbers PRJNA1073882 for 16S rRNA gene fragments and PRJNA1161578 for fungal ITS2. All other data is available on figshare: https://doi.org/10.6084/m9.figshare.30010576.
Code availability
Code for statistical analyses is available on figshare: https://doi.org/10.6084/m9.figshare.30010576.
References
Walker, T. W. N. et al. Microbial temperature sensitivity and biomass change explain soil carbon loss with warming. Nat. Clim. Change 8, 885–889 (2018).
Malik, A. A. et al. Defining trait-based microbial strategies with consequences for soil carbon cycling under climate change. ISME J https://doi.org/10.1038/s41396-019-0510-0 (2019).
Wang, B. & Allison, S. D. Climate-Driven Legacies in Simulated Microbial Communities Alter Litter Decomposition Rates. Front. Ecol. Evol. 10, (2022).
Poindexter, J. S. Oligotrophy. In Advances in Microbial Ecology (ed. Alexander, M.) 63–89 (Springer US, Boston, MA, 1981). https://doi.org/10.1007/978-1-4615-8306-6_2.
Grime, J. P. Evidence for the existence of three primary strategies in plants and its relevance to ecological and evolutionary theory. Am. Nat. 111, 1169–1194 (1977).
Ho, A., Di Lonardo, D. P. & Bodelier, P. L. E. Revisiting life strategy concepts in environmental microbial ecology. FEMS Microbiol. Ecol. 93, fix006 (2017).
Vieira-Silva, S. & Rocha, E. P. C. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet. 6, e1000808 (2010).
Roller, B. R. K., Stoddard, S. F. & Schmidt, T. M. Exploiting rRNA operon copy number to investigate bacterial reproductive strategies. Nat. Microbiol. 1, 16160 (2016).
Konstantinidis, K. T. & Tiedje, J. M. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. USA 101, 3160–3165 (2004).
Guieysse, B. & Wuertz, S. Metabolically versatile large-genome prokaryotes. Curr. Opin. Biotechnol. 23, 467–473 (2012).
Cobo-Simón, M. & Tamames, J. Relating genomic characteristics to environmental preferences and ubiquity in different microbial taxa. BMC Genomics 18, 499 (2017).
Lear, G. et al. Following Rapoport’s Rule: the geographic range and genome size of bacterial taxa decline at warmer latitudes. Environ. Microbiol. 19, 3152–3162 (2017).
Gravuer, K. & Eskelinen, A. Nutrient and rainfall additions shift phylogenetically estimated traits of soil microbial communities. Front. Microbiol. 8, 01271 (2017).
Liu, H. et al. Warmer and drier ecosystems select for smaller bacterial genomes in global soils. iMeta 2, e70 (2023).
Piton, G. et al. Life history strategies of soil bacterial communities across global terrestrial biomes. Nat. Microbiol. 8, 2093–2102 (2023).
Nielsen, D. A. et al. Aerobic bacteria and archaea tend to have larger and more versatile genomes. Oikos 130, 501–511 (2021).
Hessen, D. O., Jeyasingh, P. D., Neiman, M. & Weider, L. J. Genome streamlining and the elemental costs of growth. Trends Ecol. Evol. 25, 75–80 (2010).
Lee, M.-C. & Marx, C. J. Repeated, Selection-Driven Genome Reduction of Accessory Genes in Experimental Populations. PLOS Genet 8, e1002651 (2012).
Li, J. et al. Predictive genomic traits for bacterial growth in culture versus actual growth in soil. ISME J. 13, 2162–2172 (2019).
Westoby, M. et al. Cell size, genome size, and maximum growth rate are near-independent dimensions of ecological variation across bacteria and archaea. Ecol. Evol. 11, 3956–3976 (2021).
Pirt, S. J. The maintenance energy of bacteria in growing cultures. Proc. R. Soc. Lond. B Biol. Sci. 163, 224–231 (1965).
Lipson, D. A. The complex relationship between microbial growth rate and yield and its implications for ecosystem processes. Front. Microbiol. 6, 615 (2015).
Kreft, J.-U. Biofilms promote altruism. Microbiology 150, 2751–2760 (2004).
Freilich, S. et al. Metabolic-network-driven analysis of bacterial ecological strategies. Genome Biol. 10, 1–8 (2009).
Young, K. D. The Selective Value of Bacterial Shape. Microbiol. Mol. Biol. Rev. 70, 660–703 (2006).
DeLong, J. P., Okie, J. G., Moses, M. E., Sibly, R. M. & Brown, J. H. Shifts in metabolic scaling, production, and efficiency across major evolutionary transitions of life. Proc. Natl. Acad. Sci. 107, 12941–12945 (2010).
Kempes, C. P., Wang, L., Amend, J. P., Doyle, J. & Hoehler, T. Evolutionary tradeoffs in cellular composition across diverse bacteria. ISME J. 10, 2145–2157 (2016).
Sorensen, J. W., Dunivin, T. K., Tobin, T. C. & Shade, A. Ecological selection for small microbial genomes along a temperate-to-thermal soil gradient. Nat. Microbiol. 4, 55–61 (2019).
Grover, J. P. Resource storage and competition with spatial and temporal variation in resource availability. Am. Nat. 178, E124–E148 (2011).
Schulz, H. N. & Jørgensen, B. B. Big Bacteria. Annu. Rev. Microbiol. 55, 105–137 (2001).
Lane, N. & Martin, W. The energetics of genome complexity. Nature 467, 929–934 (2010).
Trickovic, B. & Lynch, M. Resource allocation to cell envelopes and the scaling of bacterial growth rate. Phys. Biol. 22, 046002 (2025).
Bååth, E. Thymidine and leucine incorporation in soil bacteria with different cell size. Microb. Ecol. 27, 267–278 (1994).
Jiang, H. & Sun, S. X. Morphology, growth, and size limit of bacterial cells. Phys. Rev. Lett. 105, 028101 (2010).
Kempes, C. P., Dutkiewicz, S. & Follows, M. J. Growth, metabolic partitioning, and the size of microorganisms. Proc. Natl. Acad. Sci. 109, 495–500 (2012).
García, F. C. et al. The allometry of the smallest: superlinear scaling of microbial metabolic rates in the Atlantic Ocean. ISME J. 10, 1029–1036 (2016).
Gounand, I. et al. Size evolution in microorganisms masks trade-offs predicted by the growth rate hypothesis. Proc. R. Soc. B Biol. Sci. 283, 20162272 (2016).
Gallet, R. et al. The evolution of bacterial cell size: the internal diffusion-constraint hypothesis. ISME J. 11, 1559–1568 (2017).
Lynch, M., Trickovic, B. & Kempes, C. P. Evolutionary scaling of maximum growth rate with organism size. Sci. Rep. 12, 22586 (2022).
Kaiser, C., Franklin, O., Dieckmann, U. & Richter, A. Microbial community dynamics alleviate stoichiometric constraints during litter decay. Ecol. Lett. 17, 680–690 (2014).
Donhauser, J., Niklaus, P. A., Rousk, J., Larose, C. & Frey, B. Temperatures beyond the community optimum promote the dominance of heat-adapted, fast growing and stress resistant bacteria in alpine soils. Soil Biol. Biochem. 148, 107873 (2020).
Karaoz, U. & Brodie, E. L. MicroTrait: a toolset for a trait-based representation of microbial genomes. Front. Bioinforma. 2, 918853 (2022).
Donhauser, J. et al. Modelling soil prokaryotic traits across environments with the trait sequence database ampliconTraits and the R package MicEnvMod. Ecol. Inform. 83, 102817 (2024).
Martiny, J. B. H. et al. Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4, 102–112 (2006).
Vellend, M. Conceptual Synthesis in Community Ecology. Q. Rev. Biol. 85, 183–206 (2010).
Jenkins, D. G. et al. Does size matter for dispersal distance? Glob. Ecol. Biogeogr. 16, 415–425 (2007).
Locey, K. J. et al. Dormancy dampens the microbial distance–decay relationship. Philos. Trans. R. Soc. B Biol. Sci. 375, 20190243 (2020).
Clark, D. R., Underwood, G. J. C., McGenity, T. J. & Dumbrell, A. J. What drives study-dependent differences in distance–decay relationships of microbial communities? Glob. Ecol. Biogeogr. 30, 811–825 (2021).
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C. & Martiny, J. B. H. Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 10, 497–506 (2012).
Wilkinson, D. M. What is the upper size limit for cosmopolitan distribution in free-living microorganisms? J. Biogeogr. 28, 285–291 (2001).
Wilkinson, D. M., Koumoutsaris, S., Mitchell, E. A. D. & Bey, I. Modelling the effect of size on the aerial dispersal of microorganisms. J. Biogeogr. 39, 89–97 (2012).
Favet, J. et al. Microbial hitchhikers on intercontinental dust: catching a lift in Chad. ISME J. 7, 850–867 (2013).
Archer, S. D. J. et al. Contribution of soil bacteria to the atmosphere across biomes. Sci. Total Environ. 871, 162137 (2023).
Ramirez, K. S. et al. Biogeographic patterns in below-ground diversity in New York City’s Central Park are similar to those observed globally. Proc. R. Soc. B Biol. Sci. 281, 20141988 (2014).
Thompson, L. R. et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463 (2017).
Větrovský, T. et al. A meta-analysis of global fungal distribution reveals climate-driven patterns. Nat. Commun. 10, 5142 (2019).
Chen, Y.-L. et al. Plant diversity represents the prevalent determinant of soil fungal community structure across temperate grasslands in northern China. Soil Biol. Biochem. 110, 12–21 (2017).
Davison, J. et al. Plant functional groups associate with distinct arbuscular mycorrhizal fungal communities. N. Phytol. 226, 1117–1128 (2020).
Voříšková, J. & Baldrian, P. Fungal community on decomposing leaf litter undergoes rapid successional changes. ISME J. 7, 477–486 (2013).
Bromham, L. Why do species vary in their rate of molecular evolution? Biol. Lett. 5, 401–404 (2009).
Ginoux, P. et al. Sources and distributions of dust aerosols simulated with the GOCART model. J. Geophys. Res. Atmospheres 106, 20255–20273 (2001).
Parter, M., Kashtan, N. & Alon, U. Environmental variability and modularity of bacterial metabolic networks. BMC Evol. Biol. 7, 169 (2007).
Edwards, K. F., Klausmeier, C. A. & Litchman, E. Evidence for a three-way trade-off between nitrogen and phosphorus competitive abilities and cell size in phytoplankton. Ecology 92, 2085–2095 (2011).
IPCC. Summary for Policymakers. In Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change 3–32 (Cambridge University Press, Cambridge, United Kingdom and New York, NY, US, 2021). https://doi.org/10.1017/9781009157896.001.
Sabath, N., Ferrada, E., Barve, A. & Wagner, A. Growth temperature and genome size in bacteria are negatively correlated, suggesting genomic streamlining during thermal adaptation. Genome Biol. Evol. 5, 966–977 (2013).
National Research Council (US) Steering Group for the Workshop on Size Limits of Very Small Microorganisms. Size Limits of Very Small Microorganisms: Proceedings of a Workshop. (National Academies Press, 1999).
Grover, J. P. Resource competition in a variable environment: phytoplankton growing according to the variable-internal-stores model. Am. Nat. 138, 811–835 (1991).
Beier, S., Werner, J., Bouvier, T., Mouquet, N. & Violle, C. Trait-trait relationships and tradeoffs vary with genome size in prokaryotes. Front. Microbiol. 13, 985216 (2022).
Leff, J. W. et al. Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc. Natl. Acad. Sci. USA 112, 10967–10972 (2015).
Roller, B. R. K. & Schmidt, T. M. The physiology and ecological implications of efficient growth. ISME J. 9, 1481 (2015).
Choudoir, M. J., Barberán, A., Menninger, H. L., Dunn, R. R. & Fierer, N. Variation in range size and dispersal capabilities of microbial taxa. Ecology 99, 322–334 (2018).
Walters, K. E. & Martiny, J. B. H. Alpha-, beta-, and gamma-diversity of bacteria varies across habitats. PLoS ONE 15, e0233872 (2020).
Steinbauer, M. J. et al. Accelerated increase in plant species richness on mountain summits is linked to warming. Nature 556, 231–234 (2018).
Mirzabaev, A. et al. Chapter 3: Desertification. In Climate Change and Land: an IPCC special report on climate change, desertification, land degradation, sustainable land management, food security, and greenhouse gas fluxes in terrestrial ecosystems (Ali Mohammed, 2019).
Han, X. et al. Soil organic matter properties drive microbial enzyme activities and greenhouse gas fluxes along an elevational gradient. Geoderma 449, 116993 (2024).
Doménech-Pascual, A. et al. Soil functions are shaped by aridity through soil properties and the microbial community structure. Appl. Soil Ecol. 213, 106313 (2025).
Walthert, L. et al. Determination of organic and inorganic carbon, δ13C, and nitrogen in soils containing carbonates after acid fumigation with HCl. J. Plant Nutr. Soil Sci. 173, 207–216 (2010).
Davies, B. E. Loss-on-ignition as an estimate of soil organic matter. Soil Sci. Soc. Am. J. 38, 150–151 (1974).
Gee, G. W. & Bauder, J. W. Particle-size analysis. In Methods of soil analysis 383–411 (American Society of Agronomy, Madison, 1986). https://doi.org/10.2136/sssabookser5.1.2ed.c15.
Daniel, R. M., Finney, J. L., Stoneham, M. & Grant, W. D. Life at low water activity. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 359, 1249–1267 (2004).
Fick, S. E. & Hijmans, R. J. WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 37, 4302–4315 (2017).
Friedl, M. A. et al. MODIS Collection 5 global land cover: Algorithm refinements and characterization of new datasets. Remote Sens. Environ. 114, 168–182 (2010).
Batjes, N. H. Harmonized soil property values for broad-scale modelling (WISE30sec) with estimates of global soil carbon stocks. Geoderma 269, 61–68 (2016).
Frey, B. et al. Microbial diversity in European alpine permafrost and active layers. FEMS Microbiol. Ecol. 92, fiw018 (2016).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Martin, M. CUTADAPT removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–D596 (2013).
Abarenkov, K. et al. The UNITE database for molecular identification of fungi – recent updates and future perspectives. N. Phytol. 186, 281–285 (2010).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 1–17 (2018).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Davis, N. M., Proctor, D.iM., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 1–14 (2018).
Oksanen, J. et al. Vegan: Community Ecology Package. (2022).
R Core Team. R: A Language and Environment for Statistical Computing. (R Foundation for Statistical Computing, 2022).
Baselga, A. et al. Betapart: Partitioning Beta Diversity into Turnover and Nestedness Components. (Betapart, 2023).
Thuiller, W., Guisan, A. & Zimmermann, N. E. Habitat Suitability and Distribution Models: 6 Environmental Predictors: Issues. In Habitat Suitability and Distribution Models (Cambridge University Press, 2019).
Donhauser, J. ampliconTraits. GitHub repository (2023).
Edgar, R. C.SINAPS: Prediction of microbial traits from marker gene sequences. bioRxiv 124156 https://doi.org/10.1101/124156 (2017).
Madin, J. S. et al. A synthesis of bacterial and archaeal phenotypic trait data. Sci. Data 7, 170 (2020).
Liaw, A. & Wiener, M. Classification and Regression by randomForest. R. N. 2, 18–22 (2022).
Venables, W. N. & Ripley, B. D. Modern Applied Statistics with S. (Springer, New York, 2002).
Delignette-Muller, M. L. & Dutang, C. fitdistrplus: An R Package for Fitting Distributions. J. Stat. Softw. 64, 1–34 (2015).
Donhauser, J. MicEnvMod: Modelling microbial community - environment relationships. Ecol. Informat. 83, 102817 (2024).
Harrell, F. E. Hmisc: Harrell Miscellaneous. (2024).
Wei, T. & Simko, V. R Package ‘Corrplot’: Visualization of a Correlation Matrix. (2021).
Maechler, M., Rousseeuw, P., Struyf, A., Hubert, M. & Hornik, K. Cluster: Cluster Analysis Basics and Extensions. (2022).
Acknowledgements
We acknowledge Pallas core facilities at the University of Copenhagen and the Danish National Life Science Supercomputing Center Computerome for providing access to high-performance computing facilities. Further, we acknowledge the contribution of scientists at the McGill University and Génome Québec Innovation Center, Montréal, Canada, for paired-end Illumina MiSeq sequencing. Funding: This work was supported by the Innovation Fund Denmark [grant number BiodivClim-76 GRADCATCH], the Swiss National Science Foundation [grant number SNF 31BD30_193667], the Spanish State Research Agency [grant number PCI2020-120702-2/AEI/10.13039/501100011033] and the Department of Science and Innovation of the Republic of South Africa [grant number GRADCATCH], through the 2019-2020 BiodivERsA joint call for research proposals, under the BiodivClim ERA-Net COFUND program. Moreover, The Danish National Research Foundation supported activities within the Center for Volatile Interactions [VOLT, DNRF168].
Author information
Authors and Affiliations
Contributions
J.D., A.P., A.F., A.R., and J.-B.R. conceived the study. J.D., A.P., A.F., A.R., X.H., A.D.-P., J.C.-R., K.J., and G.S. performed the experimental work. J.D. analyzed the data and wrote the original manuscript. All authors critically reviewed the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declares no competing interests.
Peer review
Peer review information
Communications Earth and Environment thanks Ulas Karaoz, Roland Marmeisse and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Somaparna Ghosh [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Donhauser, J., Han, X., Doménech-Pascual, A. et al. Cross-continental soil prokaryotic traits driven by precipitation regime and land cover. Commun Earth Environ 7, 15 (2026). https://doi.org/10.1038/s43247-025-03028-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43247-025-03028-z
