
Abstract
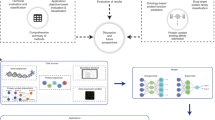
Human essential proteins (HEPs) are indispensable for individual viability and development. However, experimental methods to identify HEPs are often costly, time consuming and labor intensive. In addition, existing computational methods predict HEPs only at the cell line level, but HEPs vary across living human, cell line and animal models. Here we develop a sequence-based deep learning model, Protein Importance Calculator (PIC), by fine-tuning a pretrained protein language model. PIC not only substantially outperforms existing methods for predicting HEPs but also provides comprehensive prediction results across three levels: human, cell line and mouse. Furthermore, we define the protein essential score, derived from PIC, to quantify human protein essentiality and validate its effectiveness by a series of biological analyses. We also demonstrate the biomedical value of the protein essential score by identifying potential prognostic biomarkers for breast cancer and quantifying the essentiality of 617,462 human microproteins.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Data availability
Protein essentiality data were collected from gnomAD (https://gnomad.broadinstitute.org/), Project Score (https://www.sanger.ac.uk/tool/project-score-database/) and OGEE (https://v3.ogee.info/#/home) databases to train PIC-human, PIC-cell and PIC-mouse, respectively. PPI network node degree is accessible through the STRING (https://cn.string-db.org/) database. Gene expression values in normal tissue and cancer tissue are accessible through the Human Protein Atlas (https://www.proteinatlas.org/) database. Phylop and phastCons are accessible through the UCSC genome browser (https://genome.ucsc.edu/). The numbers of protein-related diseases are accessible through the DisGeNet (https://www.disgenet.com/) database. The transcriptome data and clinical information for patients with breast cancer can be collected from the TCGA (https://www.cancer.gov/ccg/research/genome-sequencing/tcga) database. Additional experimental validation cohort data were obtained from the GEO (https://www.ncbi.nlm.nih.gov/geo/) database including GSE58644, GSE25066, GSE86166, GSE96058, GSE199633 and GSE202203.The protein sequences of microproteins are accessible through the smORFunction (http://www.cuilab.cn/smorfunction) website. The data used in this study are available on Zenodo at https://doi.org/10.5281/zenodo.13994480 (ref. 39). Source data are provided with this paper.
Code availability
The PIC web server is available at http://www.cuilab.cn/pic. The PIC source code is available on GitHub at https://github.com/KangBoming/PIC and Zenodo at https://doi.org/10.5281/zenodo.13994480 (ref. 39).
References
Bartha, I., di Iulio, J., Venter, J. C. & Telenti, A. Human gene essentiality. Nat. Rev. Genet. 19, 51–62 (2018).
Ji, X., Rajpal, D. K. & Freudenberg, J. M. The essentiality of drug targets: an analysis of current literature and genomic databases. Drug Discov. Today 24, 544–550 (2019).
Aromolaran, O., Aromolaran, D., Isewon, I. & Oyelade, J. Machine learning approach to gene essentiality prediction: a review. Brief. Bioinf. 22, bbab128 (2021).
Joy, M. P., Brock, A., Ingber, D. E. & Huang, S. High-betweenness proteins in the yeast protein interaction network. J. Biomed. Biotechnol. 2005, 96–103 (2005).
Wuchty, S. & Stadler, P. F. Centers of complex networks. J. Theor. Biol. 223, 45–53 (2003).
Hahn, M. W. & Kern, A. D. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Mol. Biol. Evol. 22, 803–806 (2005).
Li, G. et al. Predicting essential proteins based on subcellular localization, orthology and PPI networks. BMC Bioinf. 17, 279 (2016).
Guo, F. B. et al. Accurate prediction of human essential genes using only nucleotide composition and association information. Bioinformatics 33, 1758–1764 (2017).
Hasan, M. A. & Lonardi, S. DeeplyEssential: a deep neural network for predicting essential genes in microbes. BMC Bioinf. 21, 367 (2020).
Zhang, X., Xiao, W. & Xiao, W. DeepHE: accurately predicting human essential genes based on deep learning. PLoS Comput. Biol. 16, e1008229 (2020).
Zeng, M. et al. A deep learning framework for identifying essential proteins by integrating multiple types of biological information. IEEE/ACM Trans. Comput. Biol. Bioinf. 18, 296–305 (2021).
Li, Y., Zeng, M., Wu, Y., Li, Y. & Li, M. Accurate prediction of human essential proteins using ensemble deep learning. IEEE/ACM Trans. Comput. Biol. Bioinf. 19, 3263–3271 (2022).
Li, Y., Zeng, M., Zhang, F., Wu, F. X. & Li, M. DeepCellEss: cell line-specific essential protein prediction with attention-based interpretable deep learning. Bioinformatics 39, btac779 (2023).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Elnaggar, A. et al. ProtTrans: toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127 (2022).
Thumuluri, V., Almagro Armenteros, J. J., Johansen, A. R., Nielsen, H. & Winther, O. DeepLoc 2.0: multi-label subcellular localization prediction using protein language models. Nucleic Acids Res. 50, W228–W234 (2022).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
Hou, X., Wang, Y., Bu, D., Wang, Y. & Sun, S. EMNGly: predicting N-linked glycosylation sites using the language models for feature extraction. Bioinformatics 39, btad650 (2023).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Eppig, J. T. Mouse Genome Informatics (MGI) Resource: genetic, genomic, and biological knowledgebase for the laboratory mouse. ILAR J. 58, 17–41 (2017).
Dwane, L. et al. Project Score database: a resource for investigating cancer cell dependencies and prioritizing therapeutic targets. Nucleic Acids Res. 49, D1365–D1372 (2021).
Breiman, L. Bagging predictors. Mach. Learn. 24, 123–140 (1996).
Chen, H. et al. New insights on human essential genes based on integrated analysis and the construction of the HEGIAP web-based platform. Brief. Bioinf. 21, 1397–1410 (2020).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Wang, J. D., Xu, J. Q., Long, Z. J. & Weng, J. Y. Disruption of mitochondrial oxidative phosphorylation by chidamide eradicates leukemic cells in AML. Clin. Transl. Oncol. 25, 1805–1820 (2023).
Liu, L. et al. High metabolic dependence on oxidative phosphorylation drives sensitivity to metformin treatment in MLL/AF9 acute myeloid leukemia. Cancers 14, 486 (2022).
UniProt Consortium. UniProt: the Universal Protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Jabbarzadeh Kaboli, P. et al. Unlocking c-MET: a comprehensive journey into targeted therapies for breast cancer. Cancer Lett. 588, 216780 (2024).
Zheng, L. et al. A potential tumor marker: chaperonin containing TCP‑1 controls the development of malignant tumors (Review). Int. J. Oncol. 63, 106 (2023).
Cai, M., Li, H., Chen, R. & Zhou, X. MRPL13 promotes tumor cell proliferation, migration and EMT process in breast cancer through the PI3K–AKT–mTOR pathway. Cancer Manag. Res. 13, 2009–2024 (2021).
Zhao, Y. et al. Deubiquitinase PSMD7 regulates cell fate and is associated with disease progression in breast cancer. Am. J. Transl. Res. 12, 5433–5448 (2020).
Vishnubalaji, R. & Alajez, N. M. Single-cell transcriptome analysis revealed heterogeneity and identified novel therapeutic targets for breast cancer subtypes. Cells 12, 1182 (2023).
Gui, Z., Liu, P., Zhang, D. & Wang, W. Clinical implications and immune implications features of TARS1 in breast cancer. Front. Oncol. 13, 1207867 (2023).
Song, S. et al. CHMP4A stimulates CD8+ T-lymphocyte infiltration and inhibits breast tumor growth via the LSD1/IFNβ axis. Cancer Sci. 114, 3162–3175 (2023).
Ji, X., Cui, C. & Cui, Q. smORFunction: a tool for predicting functions of small open reading frames and microproteins. BMC Bioinf. 21, 455 (2020).
Polycarpou-Schwarz, M. et al. The cancer-associated microprotein CASIMO1 controls cell proliferation and interacts with squalene epoxidase modulating lipid droplet formation. Oncogene 37, 4750–4768 (2018).
Makarewich, C. A. et al. MOXI is a mitochondrial micropeptide that enhances fatty acid β-oxidation. Cell Rep. 23, 3701–3709 (2018).
Bhatta, A. et al. A mitochondrial micropeptide is required for activation of the Nlrp3 inflammasome. J. Immunol. 204, 428–437 (2020).
Kang, B., Fan, R., Cui, C. & Cui, Q. Comprehensive prediction and analysis of human protein essentiality based on a pre-trained protein large language model(v1.0). Zenodo https://doi.org/10.5281/zenodo.13994480 (2024).
Martin, F. J. et al. Ensembl 2023. Nucleic Acids Res. 51, D933–D941 (2023).
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (62025102, 32301239 and 81921001) and the Scientific and Technological Research Project of Xinjiang Production and Construction Corps (2023AB057).
Author information
Authors and Affiliations
Contributions
Q.C. presented the original idea. B.K. and R.F. designed the study. B.K. performed the study. B.K., R.F., C.C. and Q.C. wrote or edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Computational Science thanks Min Li and Stefano Lonardi for their contribution to the peer review of this work. Primary Handling Editor: Jie Pan, in collaboration with the Nature Computational Science team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Notes 1–3, Figs. 1–8 and Tables 1–14 and descriptions of Supplementary Data 1–8.
Supplementary Data (download ZIP )
Supplementary Data 1–6 and 8.
Source data
Source Data Fig. 2 (download XLSX )
The source data for Fig. 2.
Source Data Fig. 3 (download XLSX )
The source data for Fig. 3.
Source Data Fig. 4 (download XLSX )
The source data for Fig. 4.
Source Data Fig. 5 (download XLSX )
The source data for Fig. 5.
Source Data Fig. 6 (download XLSX )
The source data for Fig. 6.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kang, B., Fan, R., Cui, C. et al. Comprehensive prediction and analysis of human protein essentiality based on a pretrained large language model. Nat Comput Sci 5, 196–206 (2025). https://doi.org/10.1038/s43588-024-00733-1
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s43588-024-00733-1
This article is cited by
-
Accurate prediction of gene deletion phenotypes with Flux Cone Learning
Nature Communications (2025)
-
Protein-DNA Binding Sites Prediction via Integrating Pretrained Large Language Models and Contrastive Learning
Interdisciplinary Sciences: Computational Life Sciences (2025)
