
Abstract
Nitrous oxide (N2O) is the third most important greenhouse gas and originates primarily from natural and engineered microbiomes. Effective emission mitigations are currently hindered by the largely unresolved ecophysiological controls of coexisting N2O-converting metabolisms in complex communities. To address this, we used biological wastewater treatment as a model ecosystem and combined long-term metagenome-resolved metaproteomics with ex situ kinetic and full-scale operational characterization over nearly 2 years. By leveraging the evidence independently obtained at multiple ecophysiological levels, from individual genetic potential to actual metabolism and emergent community phenotype, the cascade of environmental and operational triggers driving seasonal N2O emissions has ultimately been resolved. We identified nitrifier denitrification as the dominant N2O-producing pathway and dissolved O2 as the prime operational parameter, paving the way to the design and fostering of robust emission control strategies. This work exemplifies the untapped potential of multi-meta-omics in the mechanistic understanding and ecological engineering of microbiomes towards reducing anthropogenic impacts and advancing sustainable biotechnological developments.
Similar content being viewed by others


Main
The yearly anthropogenic emissions of nitrous oxide (N2O), currently the third most important greenhouse gas, are projected to increase by 50% in the coming 50 years if no mitigation strategies are employed1. N2O is mainly produced by microbial communities in natural, managed and engineered ecosystems2. Yet, the mechanisms governing biological N2O emissions in these ecosystems remain largely unknown. The main challenge lies in the coexistence of nitrogen-converting guilds in complex microbiomes, each emitting N2O under a range of complementary conditions that alternate or overlap in most ecosystems (for example, alternating oxic–anoxic conditions in wastewater treatment plants3 and sea sediments4, and substrate concentration gradients in oceans5, soils6 and wastewater treatment biofilms7). In general, high ammonium (NH4+) and oxygen (O2) concentrations stimulate N2O production through the oxidation of hydroxylamine (NH2OH) by ammonia-oxidizing bacteria (AOB), while high nitrite (NO2−) and low O2 concentrations enhance the nitrifier denitrification pathway8,9 (Fig. 1a). High NO2− and O2 concentrations result in N2O accumulation as a result of imbalanced denitrification by heterotrophic denitrifying bacteria (DEN)8,10 (Fig. 1a). High concentrations of reactive intermediates (NO2−, NH2OH and nitric oxide (NO)) and metals (for example, Fe and Mn) can lead to abiotic N2O formation (previously reviewed in ref. 11), particularly in ammonia-oxidizing archaea (AOA)-dominated marine environments12,13,14. Yet, in soils and conventional wastewater treatment plants (WWTPs), abiotic rates have been shown to be minor compared to biological N2O production by AOB and DEN15,16,17,18. Seemingly ubiquitous is the strong seasonality of N2O emissions in many natural and managed environments, such as oceans19,20, soils21,22,23, lakes24,25 and rivers26, and engineered systems, such as WWTPs27,28,29,30,31,32,33,34 (summarized in Supplementary Table 1). This indicates that seasonally impacted macroscopic factors directly influence biological N2O turnover. Yet, studying the interactions between environmental conditions, complex microbiome dynamics and N2O emissions and capturing the underlying ecological principles is inherently challenging. To this end, we used biological wastewater treatment as a more tractable model ecosystem as the N2O seasonality is well represented, while other variables (for example, aeration and biomass concentration) are controlled or extensively monitored35.

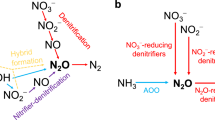
a, Nitrogen conversions in the biological nitrogen removal process and the corresponding enzyme complexes. AOB aerobically oxidize NH4+ to NH2OH with ammonia monooxygenase (AMO), NH2OH to NO with hydroxylamine oxidoreductase (HAO) and NO to NO2− with a yet unknown enzyme. AOB can biologically produce N2O through the oxidation of NH2OH with cytochrome P460 (Cyt P460) or through the reduction of NO, produced by NH2OH oxidation or nitrifier denitrification (NO2− reduction with nitrite reductase (NIR)), with nitric oxide reductase (NOR; dotted arrows). NOB aerobically oxidize NO2− to nitrate (NO3−) with nitrite oxidoreductase (NXR) and encode NIR, but its activity and function remain to be resolved66,67,68,69,70. Normally under anoxic conditions, DEN reduce NO3− to NO2− with membrane-bound or periplasmic nitrate reductase (NAR and NAP), NO2− to NO with NIR, NO to N2O with NOR and N2O to N2 with nitrous oxide reductase (NOS). Some DEN perform only some of the steps of the denitrification pathway, while others perform the entire pathway. b, Overview of the methodological approach adopted in this study for the 18-month characterization of a full-scale WWTP to resolve the microbial mechanisms underlying seasonal N2O emissions. Sludge samples were used for metagenomics (6 samples), metaproteomics (12 samples) and ex situ activity tests at 20 °C (29 samples). Predicted proteins in the metagenomics data were used as protein database (DB) in the metaproteomics analysis. The activity tests were carried out by following the decrease in nitrogen substrates concentrations (Csubstrate) over time. Created in BioRender. Roothans, N. (2025) https://BioRender.com/q43b584.
Most WWTPs emit the majority of their yearly N2O during a winter or spring peak lasting 3 to 4 months, with simultaneous NO2− accumulation27,31,32,33,34,36 (Supplementary Table 1). Similarly, higher N2O emissions during colder seasons are widely reported for oceans20, soils22,23 and lakes24. Low or increasing temperatures have been hypothesized as the underlying causes of these seasonal N2O emissions, but a clear correlation is often missing20,23,24,28,29,37,38. The immediate effect of diverse environmental and process parameters on the N2O production rates of AOB and DEN largely explain the short-term N2O dynamics in WWTPs3,39 and natural environments5,6,40,41, but fail to describe the widely observed seasonality. Emblematic is the reported higher production of N2O by AOB at high temperatures42, while most seasonal emissions occur in winter. Broadly applied correlation analyses between N2O and environmental and operational parameters have proved insufficient to explain the seasonal emissions in WWTPs28,34,43, oceans19,20, soils21,22,23 and freshwater systems24,25,26. Despite the evident central role of microbes in N2O conversions, most studies do not take the potential seasonal dynamics of the microbiome’s metabolism into account, probably overlooking key mechanisms linking environmental triggers and emissions. A delay between triggers, metabolic adaptations and emergent phenotype is expected in slow-growing natural and WWTP communities38. Only a few studies have investigated microbial dynamics during seasonal peaks in nitrogen oxides in WWTPs, with seemingly contradictory results. Seasonal NO2− and N2O accumulation events have been attributed to decreased nitrite-oxidizing bacteria (NOB) 16S ribosomal RNA gene abundances29,33 and an increased difference between AOB and NOB activity27,32, while in other instances, no seasonal fluctuations were observed in the nitrifying community44. So far, the operational and metabolic mechanisms controlling seasonal N2O emissions remain largely unknown, hindering effective mitigation.
We combined long-term metagenomic-resolved metaproteomic analyses with ex situ kinetic and full-scale process characterizations to address the mechanistic gap in seasonal N2O emissions. The cascade of environmental and operational triggers underlying N2O emissions is resolved by leveraging the evidence obtained at multiple ecophysiological levels, from individual genetic potential to actual metabolism and emergent community phenotype. We identified nitrifier denitrification as the prime N2O-producing pathway and dissolved O2 as the central operational parameter to minimize emissions. This work exemplifies the yet-to-be-realized potential of multi-meta-omics approaches to inform ecologically driven microbiome management and engineering, ultimately reducing anthropogenic emissions and advancing sustainable biotechnological developments.
Signature metabolite accumulation profiles
The ecophysiological response of N2O-emitting complex microbial communities to seasonal environmental and operational dynamics was studied using the Amsterdam-West WWTP as the model ecosystem (Fig. 1a,b). The monitoring and sampling period lasted 18 months and covered two highly comparable N2O emission peaks (Fig. 2). The peaks occurred during periods with low water temperatures, namely February–May 2021 and November 2021–March 2022, and were preceded by sequential accumulations of NH4+, O2 and NO2− (Fig. 2, Supplementary Fig. 2 and Supplementary Data 1). Central to the plant operation is the control of the dissolved O2 (DO) concentration as a function of the residual NH4+ concentration in the aerated compartment. To counteract the temperature-induced nitrification rate reduction and consequent increase in NH4+ concentration, the weekly average DO concentration increased from 1 up to almost 3 mg O2 l−1 (Fig. 2). In spite of this, O2 remained the rate-limiting substrate for nitrification during low-temperature periods with high N2O emissions, as evidenced by a lower O2/NH4+ ratio in the aerated compartment compared to warmer periods with low N2O emissions (Supplementary Fig. 3). Following the increase in DO, the average NO2− concentration in the pooled effluent rapidly increased up to 1.1 mg N l−1. Finally, N2O started to accumulate, reaching maximum daily rates of 110 kg N d−1 (first peak) and 101 kg N d−1 (second peak; Fig. 2 and Supplementary Fig. 2). The delay between the maximum DO concentration and the maximum N2O emission rate ranged between 6 and 7 weeks for both peaks (Fig. 2). This timeframe aligns with the imposed average sludge retention time of 11–15 days, indicating that seasonal N2O emissions are driven by changes in microbial composition and/or protein expression, rather than solely by shifts in microbial activity. Statistically, NO2− strongly correlated with the O2 concentration (Pearson correlation coefficient of 0.8), and N2O with NO2− (correlation coefficient 0.7), while it only weakly correlated with the other parameters, including the temperature (Supplementary Fig. 4 and Supplementary Table 2).

Weekly average parameters measured at the Amsterdam-West WWTP from October 2020 to July 2022 (from back to front, light green to dark blue): concentrations of NH4+ and dissolved O2 in the nitrification compartment (left axis), pooled effluent NO2− concentration and N2O emission rates measured in the off-gas from all reactor compartments (right axis). The water temperature inside the reactor is also shown (circles, right axis). All metabolites were measured in a single biological nutrient removal lane of the WWTP, except the effluent NO2− (seven lanes pooled together). Occasional sharp NH4+ peaks were caused by outliers on rainy days (Supplementary Fig. 2). The scheme above the plot shows the sampling time points for metagenomic (DNA), metaproteomic (protein) and ex situ activity (bioreactor) tests. Timeline icons created in BioRender. Roothans, N. (2025) https://BioRender.com/q43b584.
Maximum nitrogen metabolite conversion rates
To quantify seasonal changes in the microbiome metabolic potential, we estimated every second week the maximum oxidation and reduction rates of the main nitrification (that is, NH4+ and NO2−) and denitrification (that is, NO3−, NO2− and N2O) intermediates, respectively. The maximum NH4+ oxidation rate almost always exceeded the NO2− oxidation rate, with the difference being the highest at the seasonal full-scale metabolite accumulation peaks (Supplementary Fig. 5 and Supplementary Data 1). No clear seasonality emerged in the NO3−, NO2− and N2O maximum reduction rates and the N2O reduction capacity was 1.4- to 2.1-fold higher than all other nitrifying and denitrifying rates (Supplementary Fig. 5). The average N2O accumulation rates remained negligible or constant during NO3− and NO2− reduction tests, respectively, throughout the seasons. In turn, the available data on N2O accumulation rates during NH4+ and NO2− oxidation tests show a higher degree of fluctuation (Supplementary Fig. 6a–d and Supplementary Data 1). The average NO2− accumulation rates were sixfold higher during NH4+ oxidation than during NO3− reduction tests (Supplementary Fig. 6e).
Genome-resolved taxonomic diversity
The WWTP metagenome was sequenced at six time points to follow the dynamics in microbial composition and functional potential, and to serve as a database for metaproteomic analysis (Fig. 1b). Combined short-read (two samples, average 147 million reads per sample) and long-read DNA sequencing (five samples, one of which was also sequenced with short-reads, average 4.3 million reads per sample) resulted in 143 gigabase (Gb) data after quality filtering and trimming. A total of 349 high-quality metagenome-assembled genomes (HQ MAGs, ≥90% completeness and ≤5% contamination; Fig. 3 and Supplementary Data 1) were obtained. The 89 MAGs generated from the five long-read samples were de-replicated with the HQ MAGs from Singleton et al.45 at 95% average nucleotide identity of open reading frames to increase the genome-resolved read coverage. Of the final 349 HQ MAGs, 44 were unique to our dataset, 268 were unique to the dataset of Singleton et al.45 and 37 overlapped both datasets (Supplementary Fig. 7). Overall, the HQ MAGs covered 31 phyla and 272 different genera, and included two archaeal species (only bacterial MAGs are represented in Fig. 3). The full 16S rRNA gene was identified in 347 (99.4%) MAGs. The relative abundance of the individual MAGs showed no marked seasonal trend and little variation over the six time points (Supplementary Fig. 8 and Supplementary Data 1). We therefore discuss the average of their relative abundance among all samples. The two most abundant MAGs belonged to the Candidatus Microthrix (4.0%) and Nitrospira (2.7%) genera (Fig. 3). All other MAGs had an average relative abundance of less than 1%. The majority of the non-nitrifying MAGs contained at least one denitrification gene (DEN, 304; Fig. 3 and Supplementary Data 1). Furthermore, 51 MAGs had the genetic potential to perform dissimilatory nitrite reduction to ammonia (containing the nrfAH genes), 46 of which also had at least one denitrification gene (Supplementary Fig. 15 and Supplementary Data 1). Seven MAGs harboured the amoABC genes (AOB) and eight harboured the nxrAB genes (NOB), and most of these also had at least one denitrification gene, mainly nir and nor encoding the NO2− and NO reductases, respectively (Supplementary Fig. 15 and Supplementary Data 1). Complete ammonia-oxidizing (comammox), AOA and anaerobic ammonia-oxidizing (anammox) MAGs were not found in the metagenomes.

From the inner to the outer circle: circular phylogenetic tree with the identification of the key activated sludge genera Nitrosomonas, Nitrospira, Ca. Accumulibacter and Ca. Microthrix; identification of AOB (containing amoABC genes, dark blue), NOB (containing nxrAB genes, light blue), denitrifying organisms (DEN, non-AOB and non-NOB MAGs harbouring at least one denitrification gene, yellow) and other organisms (white); some of the AOB and NOB MAGs also contained one or more denitrification genes (Supplementary Data 4); average relative DNA abundance of each MAG in the community; average relative protein abundance of each MAG in the community; identification of the six most abundant phyla. The two archaeal MAGs are not represented.
Metaproteomic-based functional profile
The dynamics of protein expression of the entire microbial community across twelve samples was assessed by shotgun metaproteomics. We used protein expression as a proxy for active metabolisms and to estimate the protein-based relative abundance of each MAG. In total, 3,868 unique protein groups were detected, and 1,884 had at least two unique peptides (accounting for 44 ± 1% of the total mass-normalized spectral counts); 1,105 of the identified proteins (accounting for 68 ± 1% of the two unique peptides filtered normalized spectral counts) uniquely matched a single protein predicted in the metagenome (including all MAGs and unbinned sequences). The remaining 779 proteins (accounting for 32 ± 1% of the two unique peptides filtered normalized spectral counts) matched multiple highly similar proteins and could not be linked to a single MAG, yet could be functionally and taxonomically annotated at the genus level. Of the 349 HQ MAGs, proteins from 143 MAGs (101 genera) were detected (Supplementary Data 1). The HQ MAGs covered 39 ± 1% of the total protein pool, more than the 28 ± 4% coverage of the total community DNA (Fig. 4a). On average, the relative abundance of key activated sludge taxa (for example, Ca. Microthrix, Candidatus Accumulibacter, Nitrosomonas and Nitrospira) differed up to 20-fold between the metagenomic and metaproteomic approaches (Supplementary Fig. 13). For example, the AOB/NOB ratio was 0.1 in the metagenome and 3.6 in the metaproteome (see Supplementary Section 6 for a discussion). Taxonomically, the diversity was greatest within the DEN guild (proteins from 124 MAGs were detected) with no clear dominant MAG (Fig. 4b). Owing to this high diversity, many DEN organisms were present in too low abundance to be recovered as MAGs, even at the already high sequencing depth employed here (20–25 Gb per sample). Consequently, DNA sequences from many DEN remained in the unbinned portion of the metagenomes, resulting in the majority of the detected denitrification enzymes, namely nitrate, nitrite and nitrous oxide reductases, being assigned to the unbinned fraction (Supplementary Fig. 16). Proteins from all seven AOB and four NOB MAGs were detected in the metaproteome. The AOB consisted entirely of Nitrosomonas MAGs and were dominated by one MAG (Fig. 4b). The NOB were dominated by a Nitrospira and a Chloroflexota MAG belonging to the Promineofilaceae family (Fig. 4b), but the alpha- and beta-subunits of the nitrite oxidoreductase (NxrA and NxrB) were only expressed by Nitrospira and Candidatus Nitrotoga (Supplementary Fig. 16). Almost all detected nitrifying enzymes belonged entirely to the recovered MAGs, highlighting the nearly full coverage of the active nitrifying community by the MAGs (Supplementary Fig. 16). Throughout the monitoring period, the relative proteomic abundance of DEN barely fluctuated, while the AOB and NOB guilds fluctuated similarly over time (Fig. 4c). The maximum guild-specific fold change in the proteome was 1.1 for DEN, 1.8 for AOB and 2.5 for NOB. Overall, there was no major shift in the MAG-based composition of each guild at both the DNA and protein level (Supplementary Figs. 9–11), and there were no strong correlations between protein-level taxa abundance and WWTP performance (Supplementary Table 5).

a, Average relative abundance of denitrifying bacteria (DEN, non-AOB and non-NOB MAGs containing at least one denitrification gene, yellow), NOB (containing nxrAB genes, light blue), AOB (containing amoABC genes, dark blue), other MAGs (dark grey) and unbinned sequences (light grey) in the total metagenome (DNA) and metaproteome (protein) of the activated sludge. Some of the AOB and NOB MAGs also contained one or more denitrification genes (Supplementary Data 4). The bars represent the mean and the error bars represent the standard deviation for 6 (DNA) and 12 (protein) activated sludge samples taken at different time points throughout 18 months. b, MAG-based composition of the DEN, NOB and AOB guilds. The most abundant genera in the DEN (Ca. Accumulibacter and Candidatus Competibacter), NOB (unidentified Promineofilaceae genus, Ca. Nitrotoga and Nitrospira) and AOB (Nitrosomonas) guilds are highlighted. c, Temporal fluctuations in the relative protein abundance of the DEN, NOB and AOB guilds. The error bars represent standard deviations between technical duplicates and are all smaller than the symbols.
Unbalanced nitrification drives seasonal nitrite accumulation
The net accumulation and potential emission of any nitrogen intermediate is a result of the imbalance between its production and consumption rates. Nitrite, a central metabolite exchanged between AOB, NOB and DEN (Fig. 1a), always accumulated before the N2O peaks (Fig. 2). To understand the NO2− flux balance dynamics, we focused on the DNA, expressed proteins and ex situ activity ratios of NO2−-producing and -consuming guilds. At all levels (genomic, proteomic and kinetic), the DEN guild did not display notable seasonal dynamics (Figs. 4c and Supplementary Figs. 9 and 20). In contrast, the (im)balance between AOB (NO2− producer) and NOB (NO2− consumer) fluctuated during the monitored period. The ratio between the total abundances of AOB and NOB, both at the DNA and protein level, was up to threefold higher during periods of high effluent NO2− concentrations compared with the rest of the year (Fig. 5a,b). At the individual protein level, including MAG and unbinned proteins, the ratios between the expression of the key NH3-consuming enzyme (represented by the beta-subunit of the ammonia monooxygenase, AmoB) and NO2−-producing enzyme (represented by the hydroxylamine oxidoreductase, Hao) of AOB relative to the catalytic subunit of the NO2− oxidoreductase of NOB (NxrA) were also higher at high effluent NO2− concentrations (Fig. 5c and Supplementary Data 1). The ratio between the maximum NH4+ and NO2− oxidation activities was consistently higher during high NO2− concentration periods (Fig. 5d).

a,b, Ratios between the total relative abundance of DNA (a) and protein (b) of AOB and NOB (circles). The symbols represent the mean and the error bars represent the standard deviations of technical duplicates independently analysed by liquid chromatography–tandem mass spectrometry (LC–MS/MS); some error bars are smaller than the symbols. c, Ratios between the relative abundance of NO2−-producing and -consuming enzymes of AOB and NOB, respectively: AmoBAOB/NxrANOB (diamonds) and HaoAOB/NxrANOB (circles). The enzyme abundances include proteins belonging to the MAGs and unbinned fraction. The symbols in b,c represent the mean and the error bars represent the standard deviations of technical duplicates independently analysed by LC–MS/MS; some error bars are smaller than the symbols. The corresponding enzyme conversions are represented on the right. d, Ratio between the maximum ex situ NH4+ and NO2− oxidation rates (rmax) measured at 20 °C (circles). The data in a–d are overlaid on the weekly average NO2− concentration in the effluent (seven parallel lanes pooled together, grey shading, right axis). The correlation coefficients between the WWTP parameters and microbial ratios represented here and their statistical significance are reported in Supplementary Table 7.
Overexpressed nitrifier denitrification during N2O accumulation
In analogy to nitrite, we used ratios between the relative abundance of enzymes directly or indirectly producing and consuming N2O as a proxy for the N2O flux balance. The total enzyme abundances include MAG and unbinned protein abundances (Supplementary Data 1). The seasonally accumulated NO2− can be reduced to N2O by both AOB and DEN by sequentially using Cu-type (NirK) or cd1-type (NirS) NO2− reductase and the NO reductase (Fig. 1a). Here, NirK and NirS were exclusively expressed by nitrifiers and DEN, respectively (Supplementary Fig. 16). Four Nitrosomonas (AOB) and one Nitrospira MAG (NOB) accounted for most of the NirK expression (75% and 17%, respectively, Supplementary Fig. 16). Within the nitrifying community, the relative abundance of NirK over the key AOB enzymes AmoB and Hao was highest during periods of high NO2− and N2O accumulation (Fig. 6a). The ratio of the total relative abundance of NirK over the competing NO2−-oxidizing NxrA (NOB) and NO2−-reducing NirS (DEN) followed a similar trend (Fig. 6b). NosZ is the only known N2O-reducing enzyme and the ratio NirK/NosZ clearly reflected the seasonal dynamics, being higher during seasonal peaks (Fig. 6c). Similarly, yet to a considerably lower extent, the ratios between the NH2OH-producing AmoB and -consuming Hao and CytP460 (Supplementary Fig. 19) and the ratio NirS/NosZ (Supplementary Fig. 20c) also displayed some seasonality. The protein extraction protocol employed here did not allow for the quantification of membrane-bound proteins, such as the NO reductases46, which therefore have not been included in the discussion. All microbial ratios (Figs. 5 and 6) negatively correlate with temperature and overall positively correlate with NH4+, DO, NO2− and N2O emissions (Supplementary Table 7).

a, Ratios between the total relative abundance of NO2−-consuming NirK and the key AOB enzymes AmoB (triangles) and Hao (circles). b, Ratios between the total relative abundance of NO2−-consuming NirK and the NO2−-competing enzymes NirS (DEN, triangles) and NxrA (NOB, circles). c, NirK in N2O balance. Ratio between the total relative abundance of NirK (producing the N2O precursor NO) and the only known enzymatic N2O-sink N2O reductase (NosZ). The enzyme abundances include proteins belonging to the MAGs and unbinned fraction. The symbols represent the mean and the error bars represent the standard deviations of technical duplicates independently analysed by LC–MS/MS; some error bars are smaller than the symbols. All enzymatic conversions are schematically represented on the right. NirK is expressed by both AOB and NOB, but the activity and function of the enzyme in NOB are yet unknown. The data in a–c are overlaid on weekly average NO2− concentration in the effluent of the WWTP (seven parallel lanes pooled together, grey shading, right axis) and N2O emission rates measured in the off-gas from all the reactor compartments in one lane at the WWTP (grey line, right axis). The correlation coefficients between the WWTP parameters and the microbial ratios represented here and their statistical significance are reported in Supplementary Table 7.
Discussion
We postulate that the seasonal accumulation of NO2− and subsequent emissions of the potent greenhouse gas N2O at a full-scale WWTP are related to fluctuations in the balance of key nitrogen-converting populations, rather than their individual abundance or activity. No major changes in the DNA and protein composition, nor notable correlations with plant performance, were observed throughout 18 months of operation. This is consistent with previous metagenomic and 16S rRNA gene amplicon sequencing reports in WWTPs47,48,49,50. The microbiome was dominated by a taxonomically diverse DEN community (74% of the binned community proteome), in line with most genomic and transcriptional analyses of conventional WWTPs29,51,52. While the high DEN abundance may have masked fluctuations at guild level, the absence of major changes at the activity and individual protein level further supports the DEN stability. Instead, the DNA and protein abundances of the nitrifying community, dominated by one AOB and two NOB MAGs, fluctuated over time, yet not consistently with the observed accumulation dynamics of nitrogen oxides. This aligns with most studies reporting limited to no correlation between AOB and NOB 16S rRNA gene abundances and seasonal nitrification failures44, or AOB and NOB conversion rates and N2O production32. Only a few studies observed a correlation between increased N2O emissions and increased relative AOB abundances (16S)53, AOB ex situ activities54 or decreased NOB abundances (16S)29,33. Yet evidence remains sparse and seemingly conflicting, ultimately hindering mechanistic generalizations. This lack of general consensus resides in the fundamental dependency between metabolite dynamics and the trade-off between their production and consumption rates (that is, the balance between the producing and consuming guilds) rather than their individual magnitudes.
Against a relatively stable DEN community, featuring a fairly constant nitrite production and reduction potential, we identified the imbalance between AOB (NO2− producer) and NOB (NO2− consumer) as the primary cause for seasonal nitrite accumulation. During the nitrite peaks preceding the N2O ones, a higher ratio of AOB/NOB was observed at genomic, proteomic and kinetic levels. So far, only Bae et al.32 have quantitatively linked N2O emissions with increased AOB/NOB ex situ activity ratios in an otherwise stable nitrifying community based on 16S rRNA gene sequencing. Gruber et al.33 observed stable AOB but lower NOB and filamentous bacteria 16S rRNA gene abundances during winter N2O emissions and hypothesized a selective NOB washout due to compromised floc integrity. Here, the fluctuations in sludge settleability (representing floc integrity) and DNA and protein abundances of Ca. Microthrix (filamentous bacteria) did not follow the full-scale metabolite profiles, nor the NOB abundance or AOB/NOB ratio (Supplementary Figs. 2 and 12 and Supplementary Table 2). The known higher sensitivity of NOB to toxic free ammonia and nitrous acid compared to AOB55,56,57 has also been suggested as a potential cause of nitrite accumulation56. However, in our case, the estimated concentrations of free ammonia (0.03 mg N l−1) and nitrous acid (0.001 mg N l−1) were far below the NOB toxicity thresholds (Supplementary Tables 7 and 8)55,56,57,58. Instead, we argue that the unbalanced AOB/NOB ratio results from a cascade of separate environmental and operational perturbations differentially impacting their respective growth rates (Fig. 7). The decrease in temperature reduces the growth rates of both AOB and NOB and may alone promote the selective washout of the slower growing NOB (as estimated in this work and consistent with literature values; Supplementary Table 10 and Supplementary Fig. 22). In addition, reduced AOB growth rates lead to the accumulation of ammonium, with the operationally undesired deterioration of effluent quality. In response, most WWTPs increase the operational DO set point to promote nitrification. The increased availability of ammonium selectively favours AOB, while, in principle, the increase in DO positively impacts the growth rate of both AOB and NOB. However, the reported lower apparent affinity of AOB for O2 in activated sludge59,60,61,62 is likely to favour AOB over NOB, further enhancing the initial differential temperature impact on their growth rates. Ultimately, nitrite accumulation is the result of the progressive relative enrichment of AOB over NOB. To test our hypothesis, we developed and implemented a mathematical model based on the experimentally estimated kinetic parameters and literature-derived stoichiometric parameters (Supplementary Tables 10–13). The model comprised all three known metabolic pathways leading to N2O formation and reproduced all observed seasonal metabolite peaks induced by decreasing temperatures and the consequent increase in ammonium and operational DO. The simulations also captured the progressive increase in the relative biomass of AOB over NOB (Supplementary Fig. 24). These results strongly indicate that the sequential seasonal peaks in nitrogen oxides are the result of a cascade of distinguishable events, where temperature is the initial trigger, but not the sole direct cause, as commonly hypothesized. The absence of a single parameter correlating with nitrite and the subsequent N2O emissions probably explains the difficulties in identifying direct correlations in past studies28,34,43. Importantly, the DO concentration emerged as the central operational parameter to act upon, and we posit that the AOB/NOB imbalance may be largely prevented by anticipating in time, that is, before measurable NH4+ accumulation, the operational O2 increase.

A decrease in temperature causes lower growth rates (μ) of AOB and NOB, promoting ammonium accumulation and a selective washout of the slower growing NOB. The resulting increased ammonium concentration stimulates the growth of AOB and induces the process control to increase the operational concentration of DO. The increased O2 concentration increases the growth rates of both AOB and NOB, but may selectively benefit AOB due to its lower apparent affinity for O2. The resulting increased AOB/NOB ratio leads to the accumulation of nitrite and consequent stimulation of nitrifier denitrification by AOB, as observed in the overexpression of the Cu-type nitrite reductase (NirK). The increases in concentrations of ammonium, nitrite and N2O are a result of changes in the microbial community metabolism, while the increase in O2 concentration is the only manually controlled parameter in the cascade.
The last metabolite to accumulate along the reconstructed ecophysiology cascade is N2O. Relative increases in nitrite concentrations are well known to lead to N2O emissions through both nitrifier and heterotrophic denitrification3, yet the dominant pathway underlying seasonal N2O emissions remains unclear31,34,54. Abiotic conversions can reasonably be neglected in conventional WWTPs such as the one analysed here8,18 as the reactive precursor nitrogen species (NH2OH, NO and NO2−) are known to accumulate primarily at lower pH values in systems treating higher-strength wastewaters, such as digester supernatant, and partial nitritation reactors8,18,63. We used the nitrite reductases NirK and NirS as proxies for N2O production and their genome-resolved taxonomy to differentiate between nitrifier and heterotrophic denitrification. Considering the rapid turnover of NO (ref. 8), the use of Nir allows the challenges in detecting the membrane-bound hydrophobic NO reductase in metaproteomic analyses to be overcome64,65. Unbalanced heterotrophic denitrification is unlikely to be the main N2O-producing pathway during seasonal emissions owing to the relatively constant ratio between NirS and NosZ, both exclusively expressed by DEN, and their rates. The marginal contribution of denitrification to N2O emissions under the WWTP conditions is further supported by the developed mathematical model, where denitrification was always a net consumer of N2O (Supplementary Fig. 24). The nitrite reductase NirK was exclusively expressed by nitrifiers, primarily by AOB, so it was used as a proxy for nitrifier denitrification. NOB Nitrospira contributed to about one-fifth of the total detected NirK, but its activity and function remain to be experimentally verified (hitherto measured activities are low)66,67,68,69,70. A marked increase in the ratios of NirK over other AOB enzymes (AmoB and Hao) and the competing NO2−-consuming enzymes (NxrA from NOB and NirS from DEN) was observed during the seasonal peaks in nitrogen oxides. The higher expression of NirK could have been induced by the seasonally increased ammonia, nitrite and/or NO concentrations65,71,72,73 and may suggest an increased relative nitrite flux towards nitrifier denitrification rather than nitrite oxidation or heterotrophic nitrite reduction. Emissions also coincided with periods in which O2 was identified as the metabolically limiting substrate for AOB (that is, lower O2/NH4+ ratios compared to the rest of the year), probably forcing AOB to resort to nitrifier denitrification as an additional electron sink74,75. The potential existence of an alternative nitrite reductase warrants caution on the use of NirK as the sole proxy for nitrite reduction in AOB. Sustained nitrite reduction and N2O production by nirK-deficient Nitrosomonas europaea mutant cells has been documented in batch experiments76,77,78,79. Yet, the hypothesized alternative nitrite reduction mechanism is suggested to be stimulated solely in the absence of NirK76. Moreover, another study showed that nirK-deficient N. europaea did not reduce nitrite in a chemostat and produced N2O abiotically from hydroxylamine80. The universal existence and physiological role of an alternative nitrite reductase in AOB therefore remains to be confirmed. Importantly, consistent across all studies is the centrality of NirK for efficient NH3 oxidation and AOB growth76,77,80. NirK can quickly regenerate the electron carriers needed for Hao, avoiding nitrosative stress caused by NH2OH (and NO) accumulation during high NH3 turnover65,76,77,81. In line with this, the observed slight imbalance between hydroxylamine-producing AmoB and hydroxylamine-consuming Hao and cytochrome P460 in the metaproteome suggests that hydroxylamine accumulated as a result of the kinetic O2 limitation74, further supporting an electron imbalance in the AOB metabolism and the increased need for NirK as an electron sink. Optimized membrane metaproteomics could fill the gap in the nitrifier denitrification pathway by clarifying whether the Nor dynamics aligns with NirK82,83. So far, only one report has suggested a correlation between N2O emissions in WWTPs and nirK gene transcript abundance, quantified by quantitative PCR with reverse transcription84. Yet, the nirK transcripts were not taxonomically classified and were assumed to belong entirely to heterotrophic denitrifiers84. All other studies discussing seasonal N2O emissions in WWTPs infer the main N2O-producing pathways based on metabolite profiles and a general consensus is still lacking28,30,31,34,54 (Supplementary Table 1). Most studies identified nitrifier or heterotrophic denitrification as the main N2O-producing pathway in WWTPs using isotopic signatures85,86,87, but seasonal dynamics were not captured. Even more importantly, the isotopic signatures of the produced N2O (from natural abundance or 15N/18O tracers) largely overlap when nitrite is the starting substrate, as the biochemical pathways of nitrifier and heterotrophic denitrification are identical and rely on the same enzymes. This challenges the possibility of univocally distinguishing the two pathways in ecosystems where they potentially co-occur (reviewed previously in refs. 9,88,89). Instead, by integrating metagenomic-guided metaproteomics with kinetic analyses and full-scale operational data, we have provided independent evidence on multiple ecophysiological levels, further supported by mathematical modelling, identifying nitrifier denitrification as the prime N2O-producing pathway during seasonal emissions. More broadly, our results demonstrate the untapped potential of multi-meta-omics integration into biotechnological developments to reduce anthropogenic impacts by resolving the complexity and advancing the engineering of microbiomes.
Methods
WWTP operation
The Amsterdam-West WWTP has the daily capacity to treat 200,000 m3 municipal wastewater under dry weather conditions (1 million population equivalents). After fine screening and primary sedimentation, carbon, phosphorus and nitrogen are biologically removed in a modified University of Cape Town configuration in seven independent parallel cylindric plug-flow activated sludge tanks (Supplementary Fig. 1). Nutrient removal occurs in four compartments: anaerobic (biological phosphorus removal), anoxic (denitrification), facultative (aerated when additional nitrification capacity is required) and aerobic (nitrification; Supplementary Fig. 1). The setpoint for the dissolved O2 concentration in the aerobic and facultative zones is set as a function of the measured NH4+ concentration in the aerated compartment. The average sludge retention time is 11–15 days and is controlled to maintain average total suspended solids of 4.2 g l−1. N2O was measured in the combined gas exhaust of all compartments (anaerobic + anoxic + facultative + aerobic) of a single lane using an Rosemount X-STREAM gas analyser (Emerson). NH4+, NO3− and N2O were measured in a single biological nutrient removal lane of the WWTP, and NO2− was measured in the pooled effluent of seven lanes.
Ex situ batch activity tests with full-scale activated sludge
The maximum nitrification and denitrification activities of the activated sludge were measured every 2 weeks between January 2021 and May 2022. For consistency, the sludge sampling, handling and storage, as well as the activity tests, were always performed in the same manner. Samples were collected from the aerated compartment of the monitored full-scale activated sludge reactor and stored in 2-l glass bottles in a fridge for a maximum of 4 h. The sludge was transported under cold conditions (never reaching a temperature above 10 °C) and immediately placed in a 3-l jacketed glass bioreactor with a 2 l working volume (Applikon). The sludge was made anoxic by sparging with N2 for 1 h at 0.5 l min−1 (after which the bioreactor was sealed) and then incubated overnight with 50 mg N l−1 NaNO3 to consume the internal carbon storage. During overnight storage and the subsequent activity tests, the sludge was stirred at 750 r.p.m. by two six-blade turbines, the temperature was maintained at 20 ± 1 °C using a cryostat bath (Lauda), and the pH was automatically maintained at 7.0 ± 0.1 with 1 M HCl and 1 M NaOH using two peristaltic pumps (Watson Marlow) controlled by an in-Control process controller (Applikon). The pH and dissolved oxygen were continuously monitored with probes (Applikon AppliSens). Influent gas flows were controlled by mass-flow controllers (Brooks). After overnight incubation with NO3−, the sludge was activated by adding a spike of NaNO3 (5 mg N l−1) and a mixture of organic carbon (acetate, pyruvate and glucose, 37.5 mg chemical oxygen demand (COD) per litre each). The batch activity tests were performed sequentially on the same day in the following order: N2O, NO2− and NO3− reduction (denitrification) and NH4+ and NO2− oxidation (nitrification; Supplementary Table 3). The depletion of the previous nitrogen compound was ensured before each test. Substrates were added to the bioreactor via a syringe and needle through a rubber septum, marking the start of the batches. The progress of the batch tests was monitored with NO2− and NO3− MQuant colourimetric test strips (Merck).
Nitrogen compounds were added at 12 mg N l−1, in the form of N2O (sparging with 1.5% N2O + 98.5% N2 at 0.5 l min−1 for 15–20 min), NaNO2 (1.2 ml), NaNO3 (1.2 ml) and NH4HCO3 (1.2 ml) from concentrated stocks. The proportion of bicarbonate to nitrogen was kept the same for the two nitrification batches by adding 0.9 mM NaHCO3 to the NO2− oxidation batch. Organic carbon compounds were added at the start of the denitrification batches (75 mg COD l−1 each, at least twofold higher than stoichiometrically needed) from anoxic concentrated stock solutions: sodium acetate (C2H3NaO2, 3 ml), sodium pyruvate (C3H3NaO3, 3 ml) and glucose (C6H12O6, 3 ml). The concentration of pyruvate was fourfold lower in the batch tests from January until mid-August 2021, but this had no effect on the measured activities. Before each denitrification test, anoxic conditions were ensured by sparging with N2 at 0.5 l min−1 for 20 min, after which the reactor was sealed off from the exterior. The transition from anoxic to oxic conditions was achieved by sparging with air at 0.5 l min−1 for at least 1 h. During each nitrification test, oxic conditions (>70% air saturation) were ensured by continuously sparging with air at 0.5 l min−1. When necessary, foam formation was reduced with a few drops of sixfold diluted antifoam C 391 emulsion (Merck Life Science NV). For supernatant analysis, samples were taken every 3, 5, 10 and 15 min (depending on the length of the batches) and immediately filtered through a 0.45 µm polyvinylidene fluoride (PVDF) Millex syringe filter (Merck) and placed on ice. The samples were stored at 4 °C until analysis the following day.
Analytical methods
The concentrations of NH4+, NO2− and NO3− in the filtered supernatant were spectrophotometrically measured on the day following the batch tests using the Gallery Discrete Analyzer (Thermo Fisher Scientific) or cuvette test kits (LCK339, LCK342 and LCK304, Hach Lange). When measuring NO3− with the cuvette test kits, the samples were diluted 1:1 with 20 g l−1 sulfamic acid to remove interference from NO2−. The volatile suspended solids concentration (ash content subtracted from the dried biomass), measured in triplicate, was taken as a proxy for the biomass concentration. Immediately upon arrival, 3 × 25 ml of sludge was centrifuged at 4,000 g for 20 min, the pellet was resuspended in 15 ml MilliQ water, dried at 105 °C (24 h) and burned at 550 °C (2 h). The concentrations of O2, CO2 and N2O in the condenser-dried reactor off-gas were monitored by a Rosemount NGA 2000 off-gas analyser (Emerson). The dissolved N2O concentrations were monitored and recorded every minute with a standard N2O-R microsensor (customized concentration range 0.4–2 mM, Unisense) and a PA2000 picoammeter (Unisense). The dissolved N2O concentrations were calculated using the average of calibrations performed 1–2 days before every batch series.
Activity test calculations
The maximum NO2− and NO3− reduction and NH4+ and NO2− oxidation rates were obtained by linear regression of the substrate concentration profiles over time. The slopes were determined from the line through at least four linear concentration data points. The maximum N2O reduction rate was calculated using Spyder IDE v5.1.5 using Python v3.9.12 and the NumPy v1.21.5 (ref. 90), SciPy v1.7.3 (ref. 91) and Pandas v1.4.2 (ref. 92) packages, taking into account the gas–liquid transfer between the reactor broth and headspace throughout the batch test (Supplementary Section 13). A system of ordinary differential equations, representing the liquid and headspace mass balances, was defined to describe the gas–liquid transfer over time:
where \(c_{{{\rm{N}}}_{2}{\rm{O,liq}}}\) and \(c_{{{\rm{N}}}_{2}{\rm{O,gas}}}\) are the N2O concentrations in the liquid and headspace, \(r_{{{\rm{N}}}_{2}{\rm{O}}}\) is the unknown N2O consumption rate, kLa is the experimentally determined volumetric mass transfer coefficient (5 h−1), \(K_{{{\rm{H,N}}}_{2}{\rm{O}}}\) is the Henry coefficient (27.05 mM atm−1), R is the ideal gas constant (8.206 × 10−5 l atm mmol K−1), T is the temperature, p is the pressure, and Vliq and Vgas are the broth and headspace volumes, respectively. The rates were obtained by fitting the model to the experimental data, that is, by minimizing the sum of the squared errors between the experimentally measured and calculated (equations (1) and (2)) N2O concentrations (see the code in Supplementary Section 13).
DNA extraction, library preparation and sequencing
Samples of 2 ml were taken immediately after cold transport of the sludge and centrifuged at 16,200 g for 5 min at 4 °C to separate the biomass from the supernatant. The biomass pellets were stored at −80 °C until DNA extraction. The DNA of the 12 November 2020, 9 June and 16 December 2021, and 11 May 2022 samples was extracted with the DNeasy PowerSoil Pro Kit (47014, Qiagen). The manufacturer’s instructions were followed, with the exception of these steps: ~50 mg biomass was resuspended in the CD1 solution by vortexing before transferring to the PowerBead tube, 3 × 40 s bead-beating (Beadbeater-24, Biospec) was alternated with 2 min incubation on ice and tubes were gently inverted instead of vortexed to prevent DNA shearing45. The DNA of the 20 January and 3 March 2021 samples (1/3 pellet) was extracted with the DNeasy UltraClean Microbial Kit (12224, Qiagen) following the manufacturer’s instructions. The DNA concentration and quality were assessed with a Qubit 4 fluorometer (Thermo Fisher Scientific) and a BioTek Synergy HTX multimode microplate reader (Agilent), respectively.
The samples of 12 November 2020 (np1), 9 June (np2) and 16 December 2021 (np3), and 11 May 2022 (np4) were prepared for long-read sequencing using the Ligation Sequencing Kit V14 (SQK-LSK114, Oxford Nanopore Technologies), the NEBNext Companion Module for Oxford Nanopore Technologies ligation sequencing (E7672, New England BioLabs) and ultrapure BSA (50 mg ml−1; AM2616, Thermo Fisher Scientific). The incubations in the Hula mixer were replaced with slow manual inversions, all resuspensions were performed by flicking the tube and the last room temperature incubation step was performed at 37 °C to improve the recovery of long DNA fragments. Four MinION R10.4 flow cells (Oxford Nanopore Technologies), one for each sample, were used to sequence on a MinION for 89–96 h in accurate mode (260 bases per second (bps)), yielding 21–29 Gb per sample. The sample of 20 January 2021 (np1.5) was prepared with a Ligation Sequencing Kit V12 and sequenced on a GridION with MinION R9.4 flow cells (Oxford Nanopore Technologies), generating 11.2 Gb. Short-read sequencing was also performed on the samples of 20 January (il1) and 3 March 2021 (il2) on an Illumina NovaSeq 6000 platform (Novogene), resulting in over 20 Gb (per sample) of 150 bp paired-end reads with a 350 bp insert.
Processing of metagenomic data and MAG recovery
After sequencing, the DNA data were processed to obtain MAGs. The final set of MAGs was obtained from the five nanopore-sequenced samples (np1–np4 and np1.5). The Illumina reads (il1 and il2) were used solely for differential coverage binning and to estimate the relative abundance of each MAG on the respective dates. The raw long reads were basecalled in super-accurate mode with the dna_r10.4.1_450bps_sup.cfg configuration file and --do_read_splitting option using Guppy v6.4.2 (np1–np4) or with dna_r9.4.1_450bps_sup.cfg using Guppy v5.0.7 (np1.5; Oxford Nanopore Technologies). The duplex reads of np1–np4 were filtered using pairs_from_summary and filter_pairs from Duplex tools v0.2.19 (Oxford Nanopore Technologies). The duplex reads were basecalled using the duplex basecaller of Guppy and merged with the remaining simplex reads using SeqKit v2.3.0 (ref. 93). The reads were filtered, trimmed and inspected with NanoFilt v2.8.0 (options -q 10 -l 200; ref. 94), Porechop v0.2.4 (https://github.com/rrwick/Porechop) and NanoPlot v1.41.0 (ref. 94). The Illumina reads were filtered and trimmed using Trimmomatic v0.39 (ref. 95) with the options LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:35 HEADCROP:5. The kmer algorithm of Nonpareil v3.401 (ref. 96) estimated a diversity coverage of 69.9% (il1) and 71.3% (il2) for the trimmed Illumina reads.
The long reads were individually assembled and pairwise co-assembled (np1–np2, np2–np3 and np3–np4) with Flye v2.9.1 (ref. 97) in --meta mode. The reads were mapped on the assembly with Minimap2 v2.24 (ref. 98). The individual assemblies were polished once with Racon v1.4.3 (https://github.com/isovic/racon) and twice with Medaka v1.5.0 (https://github.com/nanoporetech/medaka). The reads from all samples were mapped to each assembly using Minimap2, the alignments were converted from Sequence Alignment/Map (SAM) to Binary Alignments/Map (BAM) and sorted with SAMtools v1.10 (ref. 99), and the contig coverage in each sample was calculated with jgi_summarize_bam_contig_depths (ref. 100). The differential coverages were used for the automatic binning of each assembly with MetaBAT2 v2.15 (ref. 100), MaxBin2 v2.2.7 (ref. 101) and CONCOCT v1.1.0 (ref. 102), setting the minimum contig length at 2,000 bp. The outputs were combined into an optimized set of non-redundant bins with DAS Tool v1.1.3 (ref. 103), which used Prodigal v2.6.3 (ref. 104) and DIAMOND v2.0.8 (ref. 105). The bins obtained from all assemblies (np1, np1.5, np2, np3, np4, np1–np2, np2–np3 and np3–np4) were de-replicated with the 1,083 HQ MAGs from Singleton et al.45 at 95% average nucleotide identity of open reading frames using dRep v3.2.2 (ref. 106) with the options -comp 70 -con 10 -sa 0.95 --S_algorithm gANI.
Bin completeness and contamination were assessed with the lineage_wf workflow of CheckM v1.1.3 (ref. 107). The relative abundance of the bins in each sample (np1, np2, np3, np4, il1 and il2) was determined with CoverM v0.6.1 (https://github.com/wwood/CoverM) using the options --methods relative_abundance mean --min-read-percent-identity 95 --min-read-aligned-percentage 50. Bins with a completeness <90%, contamination >5% or with zero abundance in all samples were discarded, resulting in a non-redundant set of 349 HQ MAGs. The HQ MAGs were taxonomically classified using the classify_wf mode of GTDB-Tk v2.3.0 (ref. 108) and GTDB release 207 (gtdbtk_r207_v2_data.tar.gz; ref. 109). The presence of 16S rRNA genes was verified with barrnap v0.9 (https://github.com/tseemann/barrnap). A bacterial phylogenetic tree was made with FastTree v2.1.11 (ref. 110) using the multiple sequence alignment generated with the identify and align modes of GTDB-Tk, adjusted with the TreeTools v1.10.0 (ref. 111) package in RStudio v22.0.3 (ref. 112) with R v4.2.2 (ref. 113) and visualized with iTol v6.8.2 (ref. 114).
Gene prediction and functional annotation
Genes were predicted in all assemblies using Prodigal v2.6.3 (ref. 104) with the -p meta option. The gene sequences were concatenated and duplicates were removed using grep and rmdup from SeqKit v2.3.1 (ref. 93), resulting in a unique set of genes covering all metagenomic samples. The predicted genes were functionally annotated with the annotate pipeline of EnrichM v0.6.5 (https://github.com/geronimp/enrichM) using DIAMOND v2.0.8 (ref. 105), HMMER v3.2.1 (http://hmmer.org/) and the EnrichM v10 database (https://data.ace.uq.edu.au/public/enrichm/), including a KEGG orthology (KO)-annotated UniRef100 2018_11 (ref. 115) DIAMOND database and HMM libraries of the KEGG 88.2 (ref. 116), Pfam 32.0 (ref. 117) and TIGRFAMs 15.0 (ref. 118) databases. In general, the genes of interest from the nitrogen cycle were identified by their KO identifier (Supplementary Table 4). Cytochrome P460 was identified through its Pfam identifier PF16694. KO identifier-sharing genes (nar/nxr, amo/pmo, norB/norZ and nosZI/nosZII) were distinguished through phylogenetic analyses, BLAST or multiple sequence alignments (Supplementary Section 4). Data processing was performed using RStudio v22.0.3 (ref. 112) with R v4.2.2 (ref. 113) and the plyr v1.8.8 (ref. 119), tidyverse v2.0.0 (ref. 120), readxl v1.4.2 (ref. 121), data.table v1.15.0 (ref. 122), aplot v0.2.2 (ref. 123) and reshape2 v1.4.4 (ref. 124) packages.
Protein extraction
Biomass samples were taken and stored as detailed in the DNA extraction, library preparation and sequencing section. Proteins were extracted from 12 samples, as described previously125. Briefly, ~60 mg of biomass pellet was homogenized in three vortex–ice incubation cycles using glass beads (150–212 µm, Sigma Aldrich), 50 mM TEAB buffer, 1% (w/w) sodium deoxycholate and bacterial protein extraction reagent (B-PER, Thermo Scientific). Proteins in the supernatant were precipitated with 1:4 trichloroacetic acid/supernatant (Sigma Aldrich). The pellet was washed and disrupted twice with acetone and redissolved in a 200 mM ammonium bicarbonate solution with 6 M urea (Sigma Aldrich). Human serum albumin (0.1 µg, Sigma Aldrich) was added to all samples to control the digestion efficiency. The mixture was then reduced with 10 mM dithiothreitol (Sigma Aldrich) at 37 °C for 60 min and alkylated with 20 mM iodoacetamide (Sigma Aldrich) in the dark for 30 min. Samples were diluted with 100 mM ammonium bicarbonate to obtain a urea concentration of less than 1 M. Protein was digested overnight at 37 °C and 300 r.p.m. with 1.5 µg sequencing-grade trypsin (Promega). Next, 0.5 pmol Pierce Peptide Retention Time Calibration Mixture (Thermo Scientific) was added to all samples to control the chromatographic performance. Solid-phase extraction was performed with an Oasis HLB 96-well µElution Plate (2 mg sorbent per well, 30 µm, Waters) and a vacuum pump. The columns were conditioned with MeOH, equilibrated with water twice, loaded with peptide samples, washed with two rounds of 5% MeOH and sequentially eluted with 2% formic acid in 80% MeOH and 1 mM ammonium bicarbonate in 80% MeOH. The samples were dried in a Concentrator plus centrifuge (Eppendorf) at 45 °C and stored at −20 °C until analysis.
Shotgun metaproteomics
Peptide samples were dissolved in 20 µl of 3% acetonitrile and 0.01% trifluoroacetic acid, incubated at room temperature for 30 min and vortexed thoroughly. The protein concentration was measured at a wavelength of 280 nm with a NanoDrop ND-1000 spectrophotometer (Thermo Scientific), diluting samples to a concentration of 0.5 mg ml−1. Shotgun metaproteomics was performed as described previously125 with a randomized sample order. Briefly, ~0.5 µg protein digest was analysed using a nano-liquid chromatography using an EASY nano-LC 1200 unit equipped with an Acclaim PepMap RSLC RP C18 separation column (50 μm × 150 mm, 2 μm, catalogue number 164568) and a QE plus Orbitrap mass spectrometer (Thermo Fisher Scientific). The flow rate was maintained at 350 nl min−1 over a linear gradient from 5% to 25% solvent B over 90 min, from 25% to 55% solvent B over 60 min, followed by back equilibration to the starting conditions (solvent A was a 0.1% formic acid solution in water and solvent B consisted of 80% acetonitrile in water and 0.1% formic acid). The Orbitrap mass spectrometer was operated in data dependent acquisition mode acquiring peptide signals in the m/z range of 385–1,250 at 70,000 resolution in full MS mode with a maximum ion injection time of 75 ms and an automatic gain control (AGC) target of 3 × 106. The top 10 precursors were selected for MS/MS analysis and subjected to fragmentation using higher-energy collisional dissociation at a normalized collision energy of 28. MS/MS scans were acquired at 17,500 resolution with an AGC target of 2 × 105 and injection time of 75 ms and 1.2 m/z isolation width. The protein reference sequence database was generated through whole metagenome sequencing of the microbial samples, which included all MAGs and unique unbinned sequences from all samples. The raw MS data from each sample were analysed against this database using PEAKS Studio X (Bioinformatics Solutions) in a two-round database search process. The initial round was conducted without considering variable modifications and missed cleavages. Subsequently, the focused database was further searched, allowing for a 20 ppm parent ion and a 0.02 m/z fragment ion mass error tolerance, up to three missed cleavages, and iodoacetamide as a fixed modification, with methionine oxidation and asparagine (N)/glutamine (Q) deamidation as variable modifications.
Metaproteomic data analysis
Peptide spectrum matches were filtered against 5% false discovery rates and protein identifications with ≥2 unique peptide sequences were considered meaningful. Human serum albumin, added as an internal process control, was filtered out. Proteins were grouped according to their unique protein group identification. Peptide spectral counts were divided by their molar mass for normalization and technical duplicates were averaged. The relative abundance of each protein in a certain sample was determined by dividing the respective normalized spectral counts by the sum of the normalized spectral counts of all proteins detected in that sample. The total relative abundance of each MAG in the metaproteome was calculated by summing the relative abundance of all proteins belonging to that MAG. The same process was performed to calculate the total relative abundance of functionally identical proteins. Some functionally identical proteins belonging to different MAGs from the same genus could not be distinguished because of their high similarity. Therefore, these proteins were grouped by their functional annotation and genus for the data analysis. Proteins that simultaneously matched unbinned sequences and one or more MAG from a certain genus were classified as belonging to that genus. The catalytic subunits of the nitrogen-converting enzymes of interest were used as representative of that protein during data analysis, with the exception of AMO. The catalytic alpha-subunit (AmoA) is located in the cell membrane126 and is thus hydrophobic, so was not well detected in the proteomic analysis (Supplementary Fig. 16). The beta-subunit (AmoB), only partially in the membrane, was detected in much higher amounts so here it was used as a proxy for AMO. The results were similar for AmoA and AmoB, suggesting that both subunits are reliable for proteomic analysis (Supplementary Fig. 18). Data processing was performed using RStudio v22.0.3 (ref. 112) with R v4.2.2 (ref. 113), plyr v1.8.8 (ref. 119), tidyverse v2.0.0 (ref. 120), readxl v1.4.2 (ref. 121), data.table v1.15.0 (ref. 122), aplot v0.2.2 (ref. 123), reshape2 v1.4.4 (ref. 124) and matrixStats v1.2.0 (ref. 127) packages.
Data availability
The raw DNA reads have been deposited in the NCBI Sequence Read Archive and the 54 high-quality MAGs in Genbank under BioProject identifier PRJNA1082082. The raw mass spectrometry proteomics data acquired in this project have been deposited in the ProteomeXchange Consortium database under the dataset identifier PXD051095. The processed proteomics data, as well as the functional annotations of all hypothetical coding sequences predicted from the DNA, have been deposited in the 4TU.ResearchData repository128. The operational data from the WWTP during the monitoring period, the concentration profiles from all nitrogen species during the activity measurements, the accession numbers, quality and abundance of the HQ MAGs, and the gene presence and protein abundance in the MAGs and the unbinned sequences are presented in Supplementary Data 1.
Code availability
The Python codes used to calculate the maximum N2O rates and simulate the biological nitrogen removal process are provided in the Supplementary Information.
References
IPCC Climate Change 2014: Synthesis Report (eds Core Writing Team, Pachauri, R. K. & Meyer L. A.) (IPCC, 2014).
Tian, H. et al. A comprehensive quantification of global nitrous oxide sources and sinks. Nature 586, 248–256 (2020).
Vasilaki, V., Massara, T. M., Stanchev, P., Fatone, F. & Katsou, E. A decade of nitrous oxide (N2O) monitoring in full-scale wastewater treatment processes: a critical review. Water Res. 161, 392–412 (2019).
Marchant, H. K. et al. Denitrifying community in coastal sediments performs aerobic and anaerobic respiration simultaneously. ISME J. 11, 1799–1812 (2017).
Freing, A., Wallace, D. W. R. & Bange, H. W. Global oceanic production of nitrous oxide. Philos. Trans. R. Soc. B 367, 1245–1255 (2012).
Butterbach-Bahl, K., Baggs, E. M., Dannenmann, M., Kiese, R. & Zechmeister-Boltenstern, S. Nitrous oxide emissions from soils: how well do we understand the processes and their controls? Philos. Trans. R. Soc. B 368, 20130122 (2013).
Sabba, F., Picioreanu, C., Boltz, J. P. & Nerenberg, R. Predicting N2O emissions from nitrifying and denitrifying biofilms: a modeling study. Water Sci. Technol. 75, 530–538 (2017).
Schreiber, F., Wunderlin, P., Udert, K. M. & Wells, G. F. Nitric oxide and nitrous oxide turnover in natural and engineered microbial communities: biological pathways, chemical reactions, and novel technologies. Front. Microbiol. 3, 372 (2012).
Wrage-Mönnig, N. et al. The role of nitrifier denitrification in the production of nitrous oxide revisited. Soil Biol. Biochem. 123, A3–A16 (2018).
Morley, N., Baggs, E. M., Dörsch, P. & Bakken, L. Production of NO, N2O and N2 by extracted soil bacteria, regulation by NO2− and O2 concentrations. FEMS Microbiol. Ecol. 65, 102–112 (2008).
Zhu-Barker, X., Cavazos, A. R., Ostrom, N. E., Horwath, W. R. & Glass, J. B. The importance of abiotic reactions for nitrous oxide production. Biogeochemistry 126, 251–267 (2015).
Stein, L. Y. et al. Comment on “A critical review on nitrous oxide production by ammonia-oxidizing archaea” by Lan Wu, Xueming Chen, Wei Wei, Yiwen Liu, Dongbo Wang, and Bing-Jie Ni. Environ. Sci. Technol. 55, 797–798 (2021).
Stieglmeier, M. et al. Aerobic nitrous oxide production through N-nitrosating hybrid formation in ammonia-oxidizing archaea. ISME J. 8, 1135–1146 (2014).
Santoro, A. E., Buchwald, C., McIlvin, M. R. & Casciotti, K. L. Isotopic signature of N2O produced by marine ammonia-oxidizing archaea. Science 333, 1282–1285 (2011).
Hink, L., Gubry-Rangin, C., Nicol, G. W. & Prosser, J. I. The consequences of niche and physiological differentiation of archaeal and bacterial ammonia oxidisers for nitrous oxide emissions. ISME J. 12, 1084–1093 (2018).
Hink, L., Nicol, G. W. & Prosser, J. I. Archaea produce lower yields of N2O than bacteria during aerobic ammonia oxidation in soil. Environ. Microbiol. 19, 4829–4837 (2017).
Kozlowski, J. A., Stieglmeier, M., Schleper, C., Klotz, M. G. & Stein, L. Y. Pathways and key intermediates required for obligate aerobic ammonia-dependent chemolithotrophy in bacteria and Thaumarchaeota. ISME J. 10, 1836–1845 (2016).
Su, Q., Domingo-Félez, C., Jensen, M. M. & Smets, B. F. Abiotic nitrous oxide (N2O) production is strongly pH dependent, but contributes little to overall N2O emissions in biological nitrogen removal systems. Environ. Sci. Technol. 53, 3508–3516 (2019).
Yang, S. et al. Global reconstruction reduces the uncertainty of oceanic nitrous oxide emissions and reveals a vigorous seasonal cycle. Proc. Natl Acad. Sci. USA 117, 11954–11960 (2020).
Wan, X. S., Lin, H., Ward, B. B., Kao, S. J. & Dai, M. Significant seasonal N2O dynamics revealed by multi-year observations in the Northern South China Sea. Glob. Biogeochem. Cycles 36, e2022GB007333 (2022).
Bremner, J. M., Robbins, S. G. & Blackmer, A. M. Seasonal variability in emission of nitrous oxide from soil. Geophys. Res. Lett. 7, 641–644 (1980).
Wagner-Riddle, C. et al. Globally important nitrous oxide emissions from croplands induced by freeze–thaw cycles. Nat. Geosci. 10, 279–283 (2017).
Wang, X. et al. Hot moment of N2O emissions in seasonally frozen peatlands. ISME J. 17, 792–802 (2023).
Kortelainen, P. et al. Lakes as nitrous oxide sources in the boreal landscape. Glob. Chang. Biol. 26, 1432–1445 (2020).
Liang, X. et al. Nitrification regulates the spatiotemporal variability of N2O emissions in a eutrophic lake. Environ. Sci. Technol. 56, 17430–17442 (2022).
Beaulieu, J. J., Shuster, W. D. & Rebholz, J. A. Nitrous oxide emissions from a large, impounded river: the Ohio River. Environ. Sci. Technol. 44, 7527–7533 (2010).
Brotto, A. C. et al. Factors controlling nitrous oxide emissions from a full-scale activated sludge system in the tropics. Environ. Sci. Pollut. Res. 22, 11840–11849 (2015).
Chen, X. et al. Assessment of full-scale N2O emission characteristics and testing of control concepts in an activated sludge wastewater treatment plant with alternating aerobic and anoxic phases. Environ. Sci. Technol. 53, 12485–12494 (2019).
Valk, L. et al. Exploring the microbial influence on seasonal nitrous oxide concentration in a full-scale wastewater treatment plant using metagenome assembled genomes. Water Res. 219, 118563 (2022).
Sieranen, M. et al. Seasonality of nitrous oxide emissions at six full-scale wastewater treatment plants. Water Sci. Technol. 89, 603–612 (2023).
Daelman, M. R. J., van Voorthuizen, E. M., van Dongen, U. G. J. M., Volcke, E. I. P. & van Loosdrecht, M. C. M. Seasonal and diurnal variability of N2O emissions from a full-scale municipal wastewater treatment plant. Sci. Total Environ. 536, 1–11 (2015).
Bae, W. et al. Temporal triggers of N2O emissions during cyclical and seasonal variations of a full-scale sequencing batch reactor treating municipal wastewater. Sci. Total Environ. 797, 149093 (2021).
Gruber, W. et al. Linking seasonal N2O emissions and nitrification failures to microbial dynamics in a SBR wastewater treatment plant. Water Res. X 11, 100098 (2021).
Gruber, W. et al. Estimation of countrywide N2O emissions from wastewater treatment in Switzerland using long-term monitoring data. Water Res. X 13, 100122 (2021).
Daims, H., Taylor, M. W. & Wagner, M. Wastewater treatment: a model system for microbial ecology. Trends Biotechnol. 24, 483–489 (2006).
Gruber, W. et al. N2O emission in full-scale wastewater treatment: proposing a refined monitoring strategy. Sci. Total Environ. 699, 134157 (2020).
Kosonen, H. et al. Nitrous oxide production at a fully covered wastewater treatment plant: results of a long-term online monitoring campaign. Environ. Sci. Technol. 50, 5547–5554 (2016).
Daelman, M. R. J., Van Voorthuizen, E. M., Van Dongen, L. G. J. M., Volcke, E. I. P. & Van Loosdrecht, M. C. M. Methane and nitrous oxide emissions from municipal wastewater treatment—results from a long-term study. Water Sci. Technol. 67, 2350–2355 (2013).
Chen, H., Zeng, L., Wang, D., Zhou, Y. & Yang, X. Recent advances in nitrous oxide production and mitigation in wastewater treatment. Water Res. 184, 116168 (2020).
Baggs, E. M. Soil microbial sources of nitrous oxide: recent advances in knowledge, emerging challenges and future direction. Curr. Opin. Environ. Sustain. 3, 321–327 (2011).
Thomson, A. J., Giannopoulos, G., Pretty, J., Baggs, E. M. & Richardson, D. J. Biological sources and sinks of nitrous oxide and strategies to mitigate emissions. Philos. Trans. R. Soc. B 367, 1157–1168 (2012).
Pijuan, M. & Zhao, Y. in Quantification and Modelling of Fugitive Greenhouse Gas Emissions from Urban Water Systems (eds Ye, L. et al.) Ch. 2 (IWA, 2022).
Vasilaki, V., Volcke, E. I. P., Nandi, A. K., van Loosdrecht, M. C. M. & Katsou, E. Relating N2O emissions during biological nitrogen removal with operating conditions using multivariate statistical techniques. Water Res. 140, 387–402 (2018).
Johnston, J., LaPara, T. & Behrens, S. Composition and dynamics of the activated sludge microbiome during seasonal nitrification failure. Sci. Rep. 9, 4565 (2019).
Singleton, C. M. et al. Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing. Nat. Commun. 12, 2009 (2021).
Shiro, Y. Structure and function of bacterial nitric oxide reductases: nitric oxide reductase, anaerobic enzymes. Biochim. Biophys. Acta Bioenerg. 1817, 1907–1913 (2012).
Wang, Y. et al. Successional dynamics and alternative stable states in a saline activated sludge microbial community over 9 years. Microbiome 9, 199 (2021).
Ju, F., Guo, F., Ye, L., Xia, Y. & Zhang, T. Metagenomic analysis on seasonal microbial variations of activated sludge from a full-scale wastewater treatment plant over 4 years. Environ. Microbiol. Rep. 6, 80–89 (2014).
Saunders, A. M., Albertsen, M., Vollertsen, J. & Nielsen, P. H. The activated sludge ecosystem contains a core community of abundant organisms. ISME J. 10, 11–20 (2016).
Herold, M. et al. Integration of time-series meta-omics data reveals how microbial ecosystems respond to disturbance. Nat. Commun. 11, 5281 (2020).
Yu, K. & Zhang, T. Metagenomic and metatranscriptomic analysis of microbial community structure and gene expression of activated sludge. PLoS ONE 7, e38183 (2012).
Lu, H., Chandran, K. & Stensel, D. Microbial ecology of denitrification in biological wastewater treatment. Water Res. 64, 237–254 (2014).
Castellano-Hinojosa, A., Maza-Márquez, P., Melero-Rubio, Y., González-López, J. & Rodelas, B. Linking nitrous oxide emissions to population dynamics of nitrifying and denitrifying prokaryotes in four full-scale wastewater treatment plants. Chemosphere 200, 57–66 (2018).
Song, M. J. et al. Identification of primary effecters of N2O emissions from full-scale biological nitrogen removal systems using random forest approach. Water Res. 184, 116144 (2020).
Duan, H. et al. Improving wastewater management using free nitrous acid (FNA). Water Res. 171, (2020).
Zhou, Y., Oehmen, A., Lim, M., Vadivelu, V. & Ng, W. J. The role of nitrite and free nitrous acid (FNA) in wastewater treatment plants. Water Res. 45, 4672–4682 (2011).
Park, S. & Bae, W. Modeling kinetics of ammonium oxidation and nitrite oxidation under simultaneous inhibition by free ammonia and free nitrous acid. Process Biochem. 44, 631–640 (2009).
Anthonisen, A. C., Srinath, E. G., Loehr, R. C. & Prakasam, T. B. S. Inhibition of nitrification and nitrous acid compounds. J. Water Pollut. Control Fed. 48, 835–852 (1976).
Cao, Y. et al. The influence of dissolved oxygen on partial nitritation/anammox performance and microbial community of the 200,000 m3/d activated sludge process at the Changi water reclamation plant (2011 to 2016). Water Sci. Technol. 78, 634–643 (2018).
Law, Y. et al. High dissolved oxygen selection against Nitrospira sublineage I in full-scale activated sludge. Environ. Sci. Technol. 53, 8157–8166 (2019).
Picioreanu, C., Pérez, J. & van Loosdrecht, M. C. M. Impact of cell cluster size on apparent half-saturation coefficients for oxygen in nitrifying sludge and biofilms. Water Res. 106, 371–382 (2016).
Regmi, P. et al. Control of aeration, aerobic SRT and COD input for mainstream nitritation/denitritation. Water Res. 57, 162–171 (2014).
Terada, A. et al. Hybrid nitrous oxide Pproduction from a partial nitrifying bioreactor: hydroxylamine interactions with nitrite. Environ. Sci. Technol. 51, 2748–2756 (2017).
Olaya-Abril, A. et al. Exploring the denitrification proteome of Paracoccus denitrificans PD1222. Front. Microbiol. 9, 1137 (2018).
Zorz, J. K., Kozlowski, J. A., Stein, L. Y., Strous, M. & Kleiner, M. Comparative proteomics of three species of ammonia-oxidizing bacteria. Front. Microbiol. 9, 938 (2018).
Mundinger, A. B., Lawson, C. E., Jetten, M. S. M., Koch, H. & Lücker, S. Cultivation and transcriptional analysis of a canonical Nitrospira under stable growth conditions. Front. Microbiol. 10, 1325 (2019).
Daims, H., Lücker, S. & Wagner, M. A new perspective on microbes formerly known as nitrite-oxidizing bacteria. Trends Microbiol. 24, 699–712 (2016).
Starkenburg, S. R. et al. Genome sequence of the chemolithoautotrophic nitrite-oxidizing bacterium Nitrobacter winogradskyi Nb-255. Appl. Environ. Microbiol. 72, 2050–2063 (2006).
Koch, H. et al. Expanded metabolic versatility of ubiquitous nitrite-oxidizing bacteria from the genus Nitrospira. Proc. Natl Acad. Sci. USA 112, 11371–11376 (2015).
Lücker, S. et al. A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc. Natl Acad. Sci. USA 107, 13479–13484 (2010).
Beaumont, H. J. E., Lens, S. I., Reijnders, W. N. M., Westerhoff, H. V. & Van Spanning, R. J. M. Expression of nitrite reductase in Nitrosomonas europaea involves NsrR, a novel nitrite-sensitive transcription repressor. Mol. Microbiol. 54, 148–158 (2004).
Yu, R. & Chandran, K. Strategies of Nitrosomonas europaea 19718 to counter low dissolved oxygen and high nitrite concentrations. BMC Microbiol. 10, 70 (2010).
Cua, L. S. & Stein, L. Y. Effects of nitrite on ammonia-oxidizing activity and gene regulation in three ammonia-oxidizing bacteria. FEMS Microbiol. Lett. 319, 169–175 (2011).
Soler-Jofra, A., Schmidtchen, L., Olmo, L., van Loosdrecht, M. C. M. & Pérez, J. Short and long term continuous hydroxylamine feeding in a granular sludge partial nitritation reactor. Water Res. 209, 117945 (2022).
Perez-Garcia, O., Villas-Boas, S. G., Swift, S., Chandran, K. & Singhal, N. Clarifying the regulation of NO/N2O production in Nitrosomonas europaea during anoxic–oxic transition via flux balance analysis of a metabolic network model. Water Res. 60, 267–277 (2014).
Cantera, J. J. L. & Stein, L. Y. Role of nitrite reductase in the ammonia-oxidizing pathway of Nitrosomonas europaea. Arch. Microbiol. 188, 349–354 (2007).
Kozlowski, J. A., Price, J. & Stein, L. Y. Revision of N2O-producing pathways in the ammonia-oxidizing bacterium Nitrosomonas europaea ATCC 19718. Appl. Environ. Microbiol. 80, 4930–4935 (2014).
Kozlowski, J. A., Kits, K. D. & Stein, L. Y. Comparison of nitrogen oxide metabolism among diverse ammonia-oxidizing bacteria. Front. Microbiol. 7, 1090 (2016).
Beaumont, H. J. E. et al. Nitrite reductase of Nitrosomonas europaea is not essential for production of gaseous nitrogen oxides and confers tolerance to nitrite. J. Bacteriol. 184, 2557–2560 (2002).
Schmidt, I., van Spanning, R. J. M. & Jetten, M. S. M. Denitrification and ammonia oxidation by Nitrosomonas europaea wild-type, and NirK- and NorB-deficient mutants. Microbiology 150, 4107–4114 (2004).
Hink, L. et al. Kinetics of NH3-oxidation, NO-turnover, N2O-production and electron flow during oxygen depletion in model bacterial and archaeal ammonia oxidisers. Environ. Microbiol. 19, 4882–4896 (2017).
Morris, R. M. et al. Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction. ISME J. 4, 673–685 (2010).
Wilmes, P., Heintz-Buschart, A. & Bond, P. L. A decade of metaproteomics: where we stand and what the future holds. Proteomics 15, 3409–3417 (2015).
Vieira, A., Galinha, C. F., Oehmen, A. & Carvalho, G. The link between nitrous oxide emissions, microbial community profile and function from three full-scale WWTPs. Sci. Total Environ. 651, 2460–2472 (2019).
Gruber, W. et al. Tracing N2O formation in full-scale wastewater treatment with natural abundance isotopes indicates control by organic substrate and process settings. Water Res. X 15, 100130 (2022).
Toyoda, S. et al. Isotopomer analysis of production and consumption mechanisms of N2O and CH4 in an advanced wastewater treatment system. Environ. Sci. Technol. 45, 917–922 (2011).
Wunderlin, P. et al. Isotope signatures of N2O in a mixed microbial population system: constraints on N2O producing pathways in wastewater treatment. Environ. Sci. Technol. 47, 1339–1348 (2013).
Duan, H., Ye, L., Erler, D., Ni, B. & Yuan, Z. Quantifying nitrous oxide production pathways in wastewater treatment systems using isotope technology—a critical review. Water Res. 122, 96–113 (2017).
Bakken, L. R. & Frostegård, Å. Sources and sinks for N2O, can microbiologist help to mitigate N2O emissions? Environ. Microbiol. 19, 4801–4805 (2017).
Harris et al. Array programming with NumPy. Nature 585, 357–362 (2020).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
McKinney, W. pandas: a foundational Python library for data analysis and statistics. Python High Perform. Sci. Comput. 1–9 (2011).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 11, e0163962 (2016).
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669 (2018).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Rodriguez-R, L. M. & Konstantinidis, K. T. Nonpareil: a redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 30, 629–635 (2014).
Kolmogorov, M. et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 17, 1103–1110 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Wu, Y. W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Sieber, C. M. K. et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843 (2018).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2014).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316 (2022).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, 785–794 (2022).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Smith, M. TreeTools: create, modify and analyse phylogenetic trees. Comprehensive R Archive Network https://doi.org/10.5281/zenodo.3522725 (2019).
RStudio Team. RStudio: Integrated development environment for R (v22.0.3). https://www.rstudio.com/ (2021).
R Core Team. R: A language and environment for statistical computing (v4.2.2). https://www.r-project.org/ (2022).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Suzek, B. E., Wang, Y., Huang, H., McGarvey, P. B. & Wu, C. H. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Mistry, J. et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 49, 412–419 (2021).
Haft, D. H. et al. TIGRFAMs and genome properties in 2013. Nucleic Acids Res. 41, 387–395 (2013).
Wickham, H. The split-apply-combine strategy for data analysis. J. Stat. Softw. 40, 1–29 (2011).
Wickham, H. et al. Welcome to the Tidyverse. J. Open Source Softw. 4, 1686 (2019).
Wickham, H. & Bryan, J. readxl: Read Excel files (v1.4.2). https://cran.r-project.org/package=readxl (2023).
Barrett, T. et al. data.table: Extension of ‘data.frame’ (v1.15.0). https://rdatatable.gitlab.io/data.table (2024).
Yu, G. aplot for decorating a plot with associated information. Github https://github.com/YuLab-SMU/aplot (2023).
Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 21, 1–20 (2007).
Kleikamp, H. B. C. et al. Metaproteomics, metagenomics and 16S rRNA sequencing provide different perspectives on the aerobic granular sludge microbiome. Water Res. 246, 120700 (2023).
Musiani, F., Broll, V., Evangelisti, E. & Ciurli, S. The model structure of the copper‑dependent ammonia monooxygenase. J. Biol. Inorg. Chem. 25, 995–1007 (2020).
Bengtsson, H. et al. matrixStats: Functions that apply to rows and columns of matrices (and to vectors). Github https://github.com/HenrikBengtsson/matrixStats (2023).
Roothans, N. et al. Data underlying the PhD thesis: Ecophysiology of N2O-emitting microbial communities. 4TU.ResearchData https://doi.org/10.4121/e70ef94f-95f2-4270-ad8c-b52761ed3ab2.v2 (2025).
Acknowledgements
We thank D. Geerts (TU Delft) for valuable support with the bioreactor, C. de Ram and D. Heikens (TU Delft) for help with the protein extraction protocol, Waternet for providing the activated sludge (H. Schijfsma, A. Veltman and A. Azé for sampling), M. Albertsen and team at Aalborg University for sequencing one of the samples with Oxford Nanopore R9.4 during the Hands-on metagenomics using Oxford Nanopore DNA sequencing course, D. Sorokin (TU Delft) for valuable feedback and A. Deeke (Waterschap de Dommel), C. Uijterlinde (STOWA), I. Pistorius, R. Kras, F. de Heer and M. Ramaker (Waterschap Aa en Maas), M. Hoekstra (HHNK), M. Ronteltap (Hoogheemraadschap van Delfland), the Dutch Community of Practice for N2O, A. Joss (Eawag) and W. Gruber (upwater AG) for insightful discussions. This work was financed by Stichting Toegepast Onderzoek Waterbeheer (STOWA; JG191217009/732.750/CU, M.L.), Hoogheemraadschap Hollands Noorderkwartier (HHNK; 20.0787440, M.L.) and Waterschap de Dommel (Z62737/U131154, M.L.). M.L. was partially supported by a Veni grant from the Dutch Research Council (NWO; project number VI.Veni.192.252).
Author information
Authors and Affiliations
Contributions
N.R., M.C.M.v.L. and M.L. conceptualized the study. N.R., M.P., M.v.D., C.H.M., M.C.M.v.L. and M.L. designed the experiments. N.R., M.v.D. and C.H.M. performed the maximum activity measurements. M.Z. collected and pre-processed the WWTP data. N.R. performed the nanopore sequencing, implemented the bioinformatics pipeline and performed metagenomic analysis with input from T.A. N.R. performed the protein extraction with input from M.P. M.P. performed the shotgun metaproteomics. N.R. wrote the Python and RStudio scripts for data analysis and N.R., M.v.D. and C.H.M. analysed the data with inputs from M.Z., M.C.M.v.L. and M.L. N.R. wrote the draft paper and created the visuals with strong inputs from M.L. and contributions from all co-authors. All authors reviewed and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Water thanks Barth Smets and Lisa Stein for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information (download PDF )
Supplementary Methods, Discussion, Code, Figs. 1–25 and Tables 1–16.
Supplementary Data 1 (download XLSX )
WWTP operational data, concentration profiles during activity measurements, accession numbers, quality and abundance of the MAGs, and the gene presence and protein abundance in the MAGs and unbinned sequences.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Roothans, N., Pabst, M., van Diemen, M. et al. Long-term multi-meta-omics resolves the ecophysiological controls of seasonal N2O emissions during wastewater treatment. Nat Water 3, 590–604 (2025). https://doi.org/10.1038/s44221-025-00430-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44221-025-00430-x
