
Abstract
Faced with the burden of increasing resistance to antifungals in many fungal pathogens and the constant emergence of new drug-resistant strains, it is essential to assess the importance of various resistance mechanisms. Fungi have relatively plastic genomes and can tolerate genomic copy number variation (CNV) caused by aneuploidy and gene amplification or deletion. In many cases, these genomic changes lead to adaptation to stressful conditions, including those caused by antifungal drugs. Here, we specifically examine the contribution of CNVs to antifungal resistance. We undertook a thorough literature search, collecting reports of antifungal resistance caused by a CNV, and classifying the examples of CNV-conferred resistance into four main mechanisms. We find that in human fungal pathogens, there is little evidence that gene copy number plays a major role in the emergence of antifungal resistance compared to other types of mutations. We discuss why we might be underestimating their importance and new approaches being used to study them.
Similar content being viewed by others

How can fungi resist antifungal drugs?
Whether naturally occurring or intentionally designed, antimicrobials play a vital role in controlling bacteria, fungi, parasites, and viruses, helping to mitigate the effects of infectious diseases in both animals and plants. Adaptation to antimicrobials in bacteria occurs largely through novel mutations in genes encoding drug targets and through horizontal transfer of resistant genes and alleles1. However, horizontal gene transfer is not an appreciable driver of resistance in pathogenic fungi2. This limits the potential for the spread of resistance mutations but simultaneously makes the identification and tracking of resistance mutations more difficult, as many different resistance mutations can occur in a large array of genes. Already, resistance to the four clinically used classes of antifungal drugs has been found in clinical strains, and in strains isolated in the wild for some species2. Fungal genomes undergo many sorts of mutations but these can be broadly separated into two groupings. Firstly, small-scale changes, including missense, nonsense and indel mutations, which we will collectively refer to as point mutations. Secondly, copy number variations (CNVs) that lead to gene dosage changes3.
The term CNV encompasses a wide range of genomic changes that can affect genomic regions of differing size. Segmental duplications or gene loss can include as little as a single gene or less, though they can also affect wider regions including many genes at once. Isochromosomes involve the duplication of an entire chromosome arm. Aneuploidies are when an entire chromosome is present in more or less copies than in a wild-type strain4,5. In all cases, the main effect is the change in gene dosage, and so we consider all these cases together under the umbrella term CNV, distinguishing when necessary. Aneuploidies differ from accessory chromosomes, as the former is a change in the copy number of an already existing chromosome while the latter is a novel chromosome found in certain strains only6. The major change that follows a modification of gene dosage through CNV is the modulation of mRNA levels and protein expression7,8.
Both point mutations and CNVs have been reported to confer resistance in a large diversity of human fungal pathogens, but their relative importance is not clear3. Assessing the relative weight of CNVs and point mutations is vital because detecting them requires different approaches, and the mechanisms through which they confer resistance could differ. For instance, it has been suggested that CNVs could be more likely to cause cross-resistance than individual point mutations9,10,11,12, because they can increase the abundance of drug targets, which impacts all drugs that share this target. Alternative treatment strategies for resistance strains harboring resistance-causing CNVs should therefore be considered, if this is indeed the case.
CNVs can also coexist with point mutations, and they could theoretically work synergistically with point mutations to increase drug resistance. CNVs can provide additional gene copies in which point mutations can occur, increasing the chances of a resistant point mutation appearing. CNVs could also help temper the effects of point mutations, by providing a wild-type copy of a particular gene to maintain fitness, in cases where a resistant allele has a high cost to the cell. Gene copy number has been shown to influence which specific point mutations will occur, as diploid and tetraploid strains developed different resistance-conferring mutations in the same gene13. However, in the literature, no simultaneous detection of point mutations and CNVs that acted together has been reported, though one azole resistant Aspergillus fumigatus was found to have a duplication of a mutated allele of CYP51A14.
Resistance mutations operate under two main genetic patterns: loss of function (LOF) or gain of function (GOF). In the case of LOF mutations, the mutated protein-coding gene is not expressed or is rendered non-functional. This leads to resistance by directly removing the drug target, or through affecting biological processes that indirectly lead to resistance. For example, in Candida lusitaniae15, either the loss of the FCY1 gene or the appearance of mutations that impair its protein activity is sufficient to cause resistance to flucytosine. LOF mutations in genes that are not the drug targets can also lead to resistance, for example, by stopping the import of the drug16, or by affecting the metabolism of critical cellular components such as ergosterol for the polyene class of drugs17. In principle, CNVs causing gene loss could confer resistance, but we find that CNVs contribute to resistance mainly through the gain of gene copies. One reason could be that CNVs usually encompass large genomic regions, and the loss of such areas could also remove essential genes, making mutants unviable. In addition, in diploid species, the loss of a single copy of genes whose LOF leads to resistance could be insufficient to confer a growth advantage in the presence of antifungals18. Another possibility is that the basal rate of loss of genomic segments is lower than the rate of gains, making LOF through CNVs rarer in general19. In contrast to LOF, mutations causing GOF modify the function or increase expression of a protein. GOF can lead to resistance through modification of the drug binding site, for example, by reducing the affinity of echinocandin drugs for their targets, the FKS proteins20. In the case of CNVs, GOF occurs through changes in gene expression, which can have effects on the drug target as well as on cell metabolism. However, CNVs are not the only method through which gene expression modulation can confer resistance. For example, overexpression of the efflux pumps Cdr1 and Cdr2 caused by a GOF amino acid substitution in the transcription factor Tac1 leads to azole resistance in Candida albicans21.
Prolonged antifungal treatment favors the evolution of resistant strains, including in clinical settings22. While resistance-conferring point mutations and CNVs can appear rapidly, the time it takes to fix under such selection pressure depends on factors such as the drug dosage and effective population size23. The relative prevalence of point mutations and CNVs will critically determine the relative contribution of these two genetic changes to the evolution of resistance. Given a similar selective pressure coming from antifungal treatment, resistance is more likely to occur first through the mechanism with a higher mutation rate, if fungal population sizes are limited24. Mechanisms of CNV formation have not been thoroughly explored in many fungal species, but certain mechanisms are conserved across the tree of life25. Non-allelic homologous recombination can cause either deletion or duplications through recombination of sequences in disparate parts of the genome, while non-homologous end-joining can affect sequences that are not homologous across the genome25. Additionally, CNVs can be caused by replication-based mechanisms, such as fork stalling and template switching26. In particular, C. albicans can form characteristic CNVs through a mechanism involving a dicentric chromosome intermediate followed by breakage-fusion-bridge cycles27. The drug regimen fungi are exposed to could also affect whether resistance-conferring CNVs arise. One experimental evolution assay found that C. albicans was more likely to develop CNVs causing resistance when exposed to drug concentrations near the minimum concentration that affect the cells, than when exposed to higher concentrations28. The authors suggest that a small number of cell divisions could still occur in these conditions, allowing CNVs to appear through replication-mediated mechanisms. Modeling has also been used to examine this question. One model shows that the time necessary for a resistant trisomy to fix in a population will depend not only on the aneuploidy and mutation rates, but also on the fitness advantage bestowed by an aneuploidy, which can change based on the antifungal concentration29. Another model showed that resistant aneuploid strains were more likely to be outcompeted by resistant euploid strains as the number of generations increased30. Both in vivo and in vitro C. albicans populations are generally genotypically heterogeneous and clonal interference plays an integral role in the evolution of antifungal resistance over many generations31. Competition between resistant clones carrying point mutations and those carrying CNVs should therefore favor the least costly adaptation. In larger populations, where selection is more efficient, this should be even more pronounced30.
The fitness cost of CNVs can vary widely, depending on the size and the genes affected32. For example, duplications of different chromosomes in C. albicans have different costs33. The fitness costs of CNVs are thought to come mainly from the cost of increased protein production, and the overabundance and imbalance of proteins creating hypo-osmotic stress-like effects in the cell34,35. In addition, studies have proposed a link between the number of genes affected, and the fitness cost to the cell29,36,37. While many CNVs may be tolerated when providing antifungal resistance, they could be purged from the population when the antifungals are removed, such as growth in laboratory conditions27,38. However, studies in Saccharomyces cerevisiae also show that certain aneuploid cells are able to limit the fitness costs of aneuploidies by controlling gene expression, known as dosage compensation or attenuation, including through the translational regulator Ssd139,40.
Mutation rates in fungal pathogens have not been studied in detail. Nonetheless, studies in S. cerevisiae have shown that rates for single nucleotide mutations are around 10−10/base/generation, while rates for both large duplications and whole chromosome aneuploidies are closer to 10−4-10−5/generation, though aneuploidy rates differ across chromosomes41,42. We can thus conclude that the appearance of CNVs is relatively common, and their occurrence is not a limiting factor in the prevalence of antifungal resistance through CNVs. Interestingly, antifungal drugs themselves have been demonstrated to trigger changes in cell division that increase the rate of aneuploidy. For example, under exposure to the azole drug fluconazole, C. albicans and other Candida species cells often undergo improper bud separation, leading to the formation of aneuploid cells43. Of the fungal pathogens studied, C. albicans stands out for its ability to form CNVs. This ability depends on many factors, most notably that its genome is enriched in long repeat sequences, which lead to increased recombination-based CNV formation44. For other species, no in-depth work has revealed a particular propensity for CNVs, and it is not clear if different clades of fungi have different rates of CNV formation.
How do CNVs confer antifungal resistance?
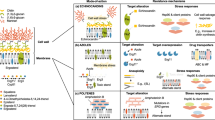
There are multiple potential mechanisms through which CNVs could confer resistance to antifungal drugs. Changes in expression brought on by CNVs (or other types of mutations) can affect many cellular processes, which can affect the organism’s interactions with different drugs. By performing a review of the literature on CNVs (See Supplementary Table 1), we grouped mechanisms into four general classes (Fig. 1). The most direct resistance mechanism through CNVs is overexpression of the drug target (Fig. 1a i). Resistance could also occur through CNV-mediated overexpression of efflux pumps, which reduces the intracellular concentration of the drug (Fig. 1a ii). An additional mechanism is changes to the metabolism of non-protein drug targets, such as ergosterol (Fig. 1a iii). Finally, CNVs could either activate or inhibit cellular stress response pathways through modified expression of regulators of these pathways (Fig. 1a iv). We tried to quantify the relative proportions of each of these mechanisms across fungal pathogens for which we could find reports in the literature. In total, we found 38 articles in the literature concerning antifungal resistance which is thought to be conferred by a CNV or an aneuploidy (Supplementary Table 1). In total, we find only 21 articles concerning clinical strains, and in total, these articles contain only 30 distinct observations of CNV-mediated antifungal resistance. A further 32 observations come from laboratory strains and 4 from environmental strains (Fig. 1b). We found that the majority of reports unsurprisingly concern C. albicans, the species most commonly involved in human infections and the most studied pathogen in terms of clinical isolates and experimental evolution. In clinical strains, while overexpression of the target protein and overactivation of efflux pumps are the most commonly reported resistance mechanisms, it is worth noting that the same genes are frequently implicated within their respective mechanisms, while other mechanisms with fewer reports are caused by different genes. We give a few detailed examples below.

a Illustration of four mechanisms through which CNVs can lead to antifungal resistance. i) CNVs leading to overexpression of a drug target protein can confer resistance. In cases where drug concentration is low, a high enough concentration of the drug target can lead to enough copies of the protein that are free of drug binding to allow cell survival. ii) CNV-mediated overexpression of small molecule efflux pumps could confer resistance to certain antifungal drugs. Different pumps can significantly reduce the intracellular concentration of certain molecules, including certain antifungal drugs. iii) Changes to cell metabolism brought on by CNVs can confer resistance. Changes in the copy number of metabolic enzymes (light purple) can modify the metabolic flux in the cell and lead to resistance in various pathways. In particular, CNVs affecting enzymes involved in cell membrane and cell wall synthesis can change the composition of these structures, and thus modulate the effect of antifungal drugs. iv) Stress response mechanisms and proteins, including chaperones, play an important role in the antifungal drug response101,102,103,104, and so CNVs that affect the activity of stress response pathways could have a large impact on fungi’s ability to adapt to antifungal drugs. b Count of the mechanisms through which CNVs confer antifungal resistance. The bar plot indicates the number of reported examples of each mechanism in the literature (as described in panel a), for each species.“Other” indicates proposed mechanisms that do not neatly fit into one of the categories, and “Unknown” indicates that no potential mechanism was reported. The pie chart titled “Strain origin” indicates whether the CNV was found in a clinical, a laboratory, or an environmental strain. The pie chart titled “Clinical only” represents the mechanisms of resistance found for only the strains of clinical origin. All data used in making panel b are available in Supplementary Table 1.
CNVs can significantly contribute to antifungal resistance by increasing the gene copies encoding the antifungal drug targets. A prominent example is the formation of an isochromosome 5 L (i(5 L)), in azole-resistant clinical isolates45. This chromosomal alteration results in the amplification of ERG11, which encodes lanosterol 14α-demethylase, the target of azole antifungals46. In Cryptococcus neoformans, the disomy of chromosome 1 leads to an increase in the copy number of ERG11, also providing increased resistance47. Resistance to azoles due to an increase in copy number of ERG11 or its homologs has also been observed to be a common mechanism in other species such as Candida parapsilosis, Candida tropicalis and Malassezia pachydermatis48,49,50. Interestingly, overexpression of the drug target protein through CNV has been reported to confer resistance only in the case of azoles, even though echinocandins also work by binding to their respective target proteins3. There are a few reasons why such incongruence has been observed. Amplification of the echinocandin target genes (FKS) by CNV may not be sufficient to confer resistance, for instance, if changes in gene dosage do not lead to a change in protein abundance51. Higher rates of CNV may be better tolerated in ERG11 compared to FKS1 due to the lower fitness costs of ERG11 amplification, and other co-amplified genes. This would prevent strains containing CNVs affecting FKS genes from increasing in frequency even under selection for resistance. Indeed, the fitness costs of CNVs also vary across the genome, especially between chromosomes, with one study demonstrating that trisomy of chromosome 1 (harboring FKS1) leads to fitness loss in the absence of the drug while trisomy of chromosome 5 (harboring ERG11) showed nearly the same fitness as the euploid parent strain in C. albicans33. Furthermore, experiments overexpressing ERG11 in S. cerevisiae and C. albicans suggest that there is only a slight fitness cost in standard laboratory conditions, although this has not been thoroughly investigated52. Fitness costs can also differ widely between growth conditions, as in C. neoformans, underexpression of FKS1 had no fitness cost in in vitro growth in laboratory conditions, while both under- and overexpression of FKS1 in a murine model lead to reduced growth compared to a control strain53.
CNVs can also lead to the amplification of genes that encode major efflux pumps and can often involve multiple genes that are nearby on the same chromosome, such as CDR1 and CDR2 in C. albicans and C. parapsilosis27,54. The previously mentioned disomy of chromosome 1 in C. neoformans also leads to an increase in copy number of AFR1, a major transporter of azoles47. Such an increase in expression of efflux pumps can also arise through overexpression of their transcription factor as seen in the case of (i(5 L)) in C. albicans where there is an increase in copy number of TAC1, a transcription factor regulating CDR1 and CDR246. A study also reported that CDR1 and CDR2 lie in the region close to MRR1 and the amplification of MRR1 gene was accompanied by the increase in gene copies of these genes as well27. MRR1 is a transcription factor of the multidrug efflux pump MDR1, so increase in its copy number can also lead to increased expression of Mdr1, resulting in multidrug resistance in C. albicans55.
CNVs can alter fungal metabolism by changing the copy number of genes involved in key metabolic pathways by rerouting the metabolic processes to evade the effects of antifungal drugs or alternatively, to make the fungi more susceptible to these drugs. The trisomy of chromosome 1 in clinical isolates of C. albicans has been linked to resitance to aureobasidin A, through the resulting higher expression of PDR16 and AUR138. These two genes contribute to antifungal resistance by altering membrane composition and enhancing efflux pump activity, respectively38. In fluconazole resistant Candida auris clinical isolates, the aneuploidy of chromosome 5 leads to an increase in copy number of NCP1, ERG9, ERG13, and ZCF22 and consequently the transcription upregulation of two genes related to ergosterol biosynthesis, ERG11 and ERG1, situated on other chromosomes56.
CNVs can also amplify genes involved in the stress response systems that help survive adverse conditions, including exposure to antifungal agents. Stress response genes such as HSP70, CGR1, ERO1, TPK1, ASR1, and PBS2 in C. albicans have been observed within amplified regions of the genomes of isolates resistant to fluconazole27. Deletion of PKC or genes expressing proteins involved in the calcineurin pathways have also been shown to impact the ability of C. albicans to tolerate both tunicamycin and caspofungin11.
Beyond these primary mechanisms, CNVs can contribute to antifungal resistance through a variety of other, less studied mechanisms such as accumulation of intracellular fluorinated nucleotides, elevation of intracellular Ca2+ levels and disruption of iron homeostasis, indirectly impacting the cell’s susceptibility to antifungal agents57,58,59. In other cases, the mechanisms remain unknown. Indeed, despite clear evidence that certain CNVs are associated with resistance, the specific genes and pathways involved are not always identifiable, as CNVs and especially aneuploidies can affect a large number of genes concurrently. This aspect requires more investigation.
There are also cases where CNVs may not confer resistance, however, they can enable growth at a reduced rate at inhibitory drug concentrations, a phenomenon known as drug tolerance60. An in vitro study of C. albicans has associated the trisomy of at least one of chromosomes 3, 6, and R with azole tolerance, though a causal link has not been demonstrated61. Other studies on C. albicans’ response to fluconazole revealed that tolerant and resistant strains developed different genomic alterations depending on the drug concentration they were exposed to. Tolerance was acquired when exposed to higher concentrations of the drug and predominantly exhibited ChrR aneuploidy. Resistance was acquired upon exposure to low concentration of the drug and showed a distinctive absence of chromosome R aneuploidy28,62. Additionally, different genes present on the same aneuploid chromosome can confer tolerance to different molecules. For instance, exposure to tunicamycin favors the development of chromosome 2 trisomy, which induces cross-tolerance to caspofungin and hydroxyurea with certain genes being specifically responsible for resistance to each drug, while not playing a role in resistance to the other two11. Aneuploidy can also lead to susceptibility to other drugs as seen in the case of trisomy of chromosome 7 in C. albicans which renders affected cells susceptible to undecylenic acid63.
CNVs can be studied in two ways: either by creating artificial CNVs and measuring their impact on antifungal resistance, or by studying strains that have been found to resist antifungal drugs, some of which harbor CNVs. Both these methods have their respective limitations. Artificially recreated CNVs are most often precise deletions or duplications of genes52,64,65, which do not always reflect the situation in natural CNVs. The latter can vary in size, and thus be spread across multiple genes and open reading frames, or even entire chromosomes in the case of aneuploidies25. Furthermore, most artificial examples of CNVs are gene deletions, which were not initially constructed in an effort to study CNVs, but rather to determine the function of the gene by measuring the null mutant phenotype57,64,65,66,67,68. We found that most examples of CNV-conferring resistance in clinical or experimental evolution strains are increases in copy number rather than deletions (Supplementary Table 1), and most gene deletions found to confer antifungal resistance have not been observed to occur naturally64,65,66. In the case of naturally occurring CNVs, the limitation lies in clearly showing the cause of resistance. In many instances, especially in cases of aneuploidies, a large number of genes are implicated, and it is often unclear which gene or genes are conferring resistance, alone or jointly. CNVs can also coexist with point mutations that are present either inside or outside the CNV, which can make it even harder to pinpoint the exact cause of resistance. One method that can shed some light on the cause of resistance is to measure the expression of genes present within the CNV to detect any differential expression that could be conferring resistance69. Experimental evolution can also be used to identify recurrent CNVs that repeatedly affect the same genes when exposed to the same drug, further cementing the role of the CNV in resistance28,61, with the caveat that repeated instances of the same CNV could also be caused by an increased appearance rate. In cases where large regions or entire chromosomes are affected, follow-up studies on genes of interest are often necessary to confirm which genes in particular are conferring resistance10.
How prevalent are CNVs in antifungal resistance?
Now that we have examined the number of reports of antifungal resistance conferred by CNVs and presented examples of mechanisms of resistance through CNVs, we can compare CNVs to point mutations. So, what is the relative contribution of CNVs to resistance? Even though CNVs have a high mutation rate and can have profound physiological consequences, they have a limited contribution globally to the emergence of resistance in clinical strains, at least in terms of the number of observations. As mentioned above, we find only 30 observations of clinical strains of antifungal-resistant fungi in which resistance was thought to be conferred by a CNV. Reports compiling resistance conferred by point mutations find many more observations than our search of the literature concerning CNVs. For instance, a recent compilation of antifungal resistance examples catalogs over 5200 point mutations that confer resistance to antifungals70. This far outweighs the few examples of CNV-mediated resistance we could find in the literature. The pattern for point mutations being more frequently reported than CNVs holds across species and drugs, though some genes, such as CYP51, are particularly enriched in resistance-conferring point mutations70. Given, as discussed above, the rate at which CNVs are generated41,42, and their strong impact on gene expression, one could ask why CNVs are not more frequently found in antifungal-resistant clinical isolates. Two possibilities emerge: either point mutations are truly more widespread in antifungal resistance, or CNVs are massively underreported.
One possibility explaining the lack of reports of CNV-mediated resistance is that CNVs may be common, but transient. In particular, a model of transient aneuploidies in antifungal resistance has been proposed71. Under this model, certain cells exposed to antifungal stress develop copy number increases of specific chromosomes, and certain aneuploidies confer antifungal resistance. In these aneuploid chromosomes, mutations continue to accumulate, and eventually resistance-conferring point mutations appear. The additional copies of the chromosomes not harboring the resistance-conferring point mutation are subsequently lost while the point mutation continues to confer antifungal resistance to the now euploid cell. There are many interesting aspects to this model. First, increasing the copy number of genes involved in antifungal resistance increases the probability of a mutation occurring in a resistance-associated gene. Yeast cells with aneuploidies have also been found to have higher mutation rates than their wild-type counterparts, further increasing the odds of a resistance-conferring mutation appearing72,73. The eventual loss of additional chromosomes is also corroborated by experimental data, as aneuploid chromosomes are known to have a fitness cost to the cell, so purging non-essential additional chromosomes is beneficial33. However, there are some limitations to this model as well. Despite an initial report of transient aneuploidy allowing adaptation to heat stress in S. cerevisiae24, recent reanalysis of the experimental data has shown that most of the adapted strains did not have an aneuploid ancestor30. Despite the many discussions of this hypothetical model23,29,36,71,74,75, we have not found reports of direct observations of this phenomenon in pathogenic fungi, either in experimental evolution or clinical infections. One report found transient aneuploidies in sequential sampling of the same infections, which developed resistance to azoles, but the disappearance of these aneuploidies was not directly tied to the appearance of point mutations, and it is not clear if the aneuploidies were the target of point mutations, or if other mutants outcompeted and replaced the aneuploid strains76. Additionally, theoretical analysis of this model suggests that adaptation through transient aneuploidy would only be favored in small populations, whereas in larger populations adaptation would most likely occur directly in the initial euploid cells without an intermediate aneuploidy state30. Still, if this model is even partially accurate, many cases in which antifungal resistance was initially conferred by an aneuploidy but then replaced by a point mutation would not be detected as CNV-conferred resistance in the clinical isolate, leading to underinflated estimates of the importance of CNVs in the initial stages of resistance acquisition.
Another possibility that could explain why CNVs are rarely reported is that they are lost during isolation and culturing steps that precede phenotypic testing and sequencing. CNVs conferring antifungal resistance increase in frequency in response to antifungal-induced stress. However, if this stress is removed, CNVs often have a fitness cost, as mentioned above. Basic evolutionary principles suggest that any culturing steps, for example plating a clinical sample on a petri dish to isolate individual colonies, could favor strains that have reverted to a non-CNV state if the antifungal stress is not constantly maintained at an appropriate level. Indeed, there have been many observations of loss of antifungal resistance and reversions of CNVs when culturing strains on non-selective media5,10,27,38,77.
Another factor that may influence our estimation of the relative impact of CNVs in antifungal resistance is our ability to detect them. Until recently, region-specific methods such as quantitative PCR and Southern blots were used to measure the gene copy numbers of individual loci5,78. Array Comparative Genome Hybridization (aCGH) was also used to determine the copy number of a large number of loci, by measuring relative DNA binding to a microarray5,79. Electrophoresis based methods, such as Contour-clamped Homogeneous Electric Field (CHEF), allow separation of very large DNA fragments by their size, and consequently were used for detection of CNV-affected chromosomes80. However, these methods have generally been replaced by more powerful sequencing-based approaches, which work by comparing read depths for different loci along chromosomes or the entire genome using sliding windows45,81. In particular, long-read sequencing technologies are well suited for detection of CNVs, as they can detect segmental duplications and their beginning and endpoints more accurately82. Various computational tools and pipelines have been developed to aid in the analysis of sequencing data, which are designed to detect both CNVs and point mutations from fungal genome sequences45,81,83. Sequencing-based approaches allow the analysis of entire genomes and can be scaled up relatively quickly and cost-effectively to analyze multiple isolates, both from clinical or laboratory origins. Thus, our ability to detect CNVs should no longer limit our evaluation of the prevalence of CNV-mediated resistance if resistant isolates are sequenced.
Going forward, increased surveillance of fungal CNVs in health settings using sequencing-based approaches, ideally directly in clinical isolates without intermediate culturing, could help determine the true prevalence and impact of CNVs in antifungal resistance. Indeed, even with the current best technologies, detection will remain rare if CNVs are lost after lab culturing. Molecular diagnostic kits in current use can identify a wide range of fungi, and in some cases detect nucleotide changes known to confer resistance, but no commercial method currently detects CNVs in any fungus84. Consequently, clinical detection of CNVs remains rare, although this situation could be remedied by the development of standard methods and technologies that can detect CNVs in fungi, similarly to current methods used to detect CNVs in tumor DNA85. An additional factor that could help develop the analysis of CNVs is the use of pangenomes as references instead of conventional reference strains. Pangenomes reveal a broader spectrum of structural variations, including CNVs, that are often missed by single reference genomes. This is due to the inclusion of accessory genes and chromosomal rearrangements that contribute to genomic plasticity and adaptive evolution in pathogens86,87. Such an approach ensures a more exhaustive coverage of genes that may be present or absent in an isolate, leading to detection of CNVs that would not have been detected previously88,89. We have not found reports of resistance mutations specifically found using this approach but given that the accessory genomes of plant pathogens do contain pathogenesis-related function90, accessory genes need to be better investigated in human pathogens as well.
Beyond detecting CNVs, it is critical that we can validate their role in resistance. These large-scale molecular changes can occur alongside other mutations and genomic changes, or in strains that are quite distant to laboratory reference strains, so it is challenging to isolate or precisely quantify their contribution to resistance. In addition, CNVs often impact multiple genes and it is not always clear which gene or genes are under selection for increased resistance. However, new techniques can be used to measure the impact of CNVs. Gene editing techniques can be used to insert additional copies of genes into fungal genomes91, and telomere-mediated truncation methods have been used to artificially create aneuploidies and other CNVs92,93. Furthermore, CRISPRa (activation) and CRIPSRi (inhibition) systems have been developed to respectively drive the activation or inhibition of gene transcription in C. albicans and Nakaseomyces glabratus (formerly known as Candida glabrata)94,95,96,97, thus recreating the over or under-expression phenotypes that can result from CNVs. In laboratory evolution experiments, better measurements of gene copy numbers and their rate of appearance through whole genome sequencing and novel methods will also help in assessing both the prevalence and fitness consequences of CNVs. One promising method uses reporter genes inserted adjacent to genes of interest so that the signal of the reporter gene can be tied to the copy number of the gene of interest at the single-cell level98,99,100. This technique could help select genes particularly prone to CNVs for subsequent surveillance in clinical settings.
Although changes in gene copy number through CNVs develop relatively frequently in pathogenic fungi, they seem to be less frequently identified as sources of antifungal resistance than point mutations70, but the small number of reports concerning CNVs makes this comparison difficult. To completely settle this question, antifungal-resistant laboratory strains and especially clinical strains should undergo whole genome sequencing with limited intermediate culturing steps, to detect both point mutations and CNVs, to properly establish the relative contribution of each mechanism.
Data availability
The data is provided as a supplementary table.
References
Munita, J. M. & Arias, C. A. Mechanisms of antibiotic resistance. in Virulence Mechanisms of Bacterial Pathogens 481–511 (ASM Press, Washington, DC, USA, 2016).
Fisher, M. C. et al. Tackling the emerging threat of antifungal resistance to human health. Nat. Rev. Microbiol. 20, 557–571 (2022).
Robbins, N., Caplan, T. & Cowen, L. E. Molecular evolution of antifungal drug resistance. Annu. Rev. Microbiol. 71, 753–775 (2017).
Pös, O. et al. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomed. J. 44, 548–559 (2021).
Selmecki, A., Forche, A. & Berman, J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313, 367–370 (2006).
Witte, T. E., Villeneuve, N., Boddy, C. N. & Overy, D. P. Accessory chromosome-acquired secondary metabolism in plant pathogenic fungi: The evolution of biotrophs into host-specific pathogens. Front. Microbiol. 12, 664276 (2021).
Steenwyk, J. L. & Rokas, A. Copy number variation in fungi and its implications for wine yeast genetic diversity and adaptation. Front. Microbiol. 9, 288 (2018).
Tucker, C. et al. Transcriptional Regulation on Aneuploid Chromosomes in Divers Candida albicans Mutants. Sci. Rep. 8, 1630 (2018).
Cools, H. J., Hawkins, N. J. & Fraaije, B. A. Constraints on the evolution of azole resistance in plant pathogenic fungi. Plant Pathol 62, 36–42 (2013).
Yang, F. et al. Aneuploidy enables cross-adaptation to unrelated drugs. Mol. Biol. Evol. 36, 1768–1782 (2019).
Yang, F. et al. Tunicamycin potentiates antifungal drug tolerance via aneuploidy in Candida albicans. MBio 12, e0227221 (2021).
Yang, F. et al. Adaptation to fluconazole via aneuploidy enables cross-adaptation to Amphotericin B and Flucytosine in Cryptococcus neoformans. Microbiol. Spectr. 9, e0072321 (2021).
Avramovska, O., Smith, A. C., Rego, E. & Hickman, M. A. Tetraploidy accelerates adaptation under drug selection in a fungal pathogen. Front. Fungal Biol. 3, 984377 (2022).
Bader, O. et al. cyp51A-Based mechanisms of Aspergillus fumigatus azole drug resistance present in clinical samples from Germany. Antimicrob. Agents Chemother. 57, 3513–3517 (2013).
Papon, N. et al. Molecular mechanism of flucytosine resistance in Candida lusitaniae: contribution of the FCY2, FCY1, and FUR1 genes to 5-fluorouracil and fluconazole cross-resistance. Antimicrob. Agents Chemother. 51, 369–371 (2007).
Noël, T. et al. Flucytosine-fluconazole cross-resistance in purine-cytosine permease-deficient Candida lusitaniae clinical isolates: indirect evidence of a fluconazole uptake transporter. Antimicrob. Agents Chemother. 47, 1275–1284 (2003).
Young, L. Y., Hull, C. M. & Heitman, J. Disruption of ergosterol biosynthesis confers resistance to amphotericin B in Candida lusitaniae. Antimicrob. Agents Chemother. 47, 2717–2724 (2003).
Luna-Tapia, A. et al. Loss of Upc2p-inducible ERG3 transcription is sufficient to confer niche-specific azole resistance without compromising Candida albicans pathogenicity. MBio 9, e00225-18 (2018).
Lynch, M. et al. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl. Acad. Sci. USA. 105, 9272–9277 (2008).
Perlin, D. S. Echinocandin Resistance in Candida. Clin. Infect. Dis. 61, S612–S617 (2015).
Coste, A. et al. A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics 172, 2139–2156 (2006).
Rabaan, A. A. et al. Potential strategies to control the risk of antifungal resistance in humans: A comprehensive review. Antibiotics (Basel) 12, 608 (2023).
Vande Zande, P., Zhou, X. & Selmecki, A. The Dynamic Fungal Genome: Polyploidy, Aneuploidy and Copy Number Variation in Response to Stress. Annu. Rev. Microbiol. 77, 341–361 (2023).
Yona, A. H. et al. Chromosomal duplication is a transient evolutionary solution to stress. Proc. Natl. Acad. Sci. USA. 109, 21010–21015 (2012).
Hastings, P. J., Lupski, J. R., Rosenberg, S. M. & Ira, G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 10, 551–564 (2009).
Hull, R. M., Cruz, C., Jack, C. V. & Houseley, J. Environmental change drives accelerated adaptation through stimulated copy number variation. PLoS Biol 15, e2001333 (2017).
Todd, R. T. & Selmecki, A. Expandable and reversible copy number amplification drives rapid adaptation to antifungal drugs. Elife 9, e58349 (2020).
Todd, R. T. et al. Antifungal Drug Concentration Impacts the Spectrum of Adaptive Mutations in Candida albicans. Mol. Biol. Evol. 40, msad009 (2023).
Pompei, S. & Cosentino Lagomarsino, M. A fitness trade-off explains the early fate of yeast aneuploids with chromosome gains. Proc. Natl. Acad. Sci. USA. 120, e2211687120 (2023).
Kohanovski, I. et al. Aneuploidy Can Be an Evolutionary Diversion on the Path to Adaptation. Mol. Biol. Evol. 41, msae052 (2024).
Huang, M. & Kao, K. C. Population dynamics and the evolution of antifungal drug resistance in Candida albicans. FEMS Microbiol. Lett. 333, 85–93 (2012).
Natesuntorn, W. et al. Genome-wide construction of a series of designed segmental aneuploids in Saccharomyces cerevisiae. Sci. Rep. 5, 12510 (2015).
Yang, F. et al. The fitness costs and benefits of trisomy of each Candida albicans chromosome. Genetics 218, iyab056 (2021).
Tsai, H.-J. et al. Hypo-osmotic-like stress underlies general cellular defects of aneuploidy. Nature 570, 117–121 (2019).
Torres, E. M. et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317, 916–924 (2007).
Gilchrist, C. & Stelkens, R. Aneuploidy in yeast: Segregation error or adaptation mechanism? Yeast 36, 525–539 (2019).
Scopel, E. F. C., Hose, J., Bensasson, D. & Gasch, A. P. Genetic variation in aneuploidy prevalence and tolerance across Saccharomyces cerevisiae lineages. Genetics 217, iyab015 (2021).
Zheng, L. et al. Chromosome 1 trisomy confers resistance to aureobasidin A in Candida albicans. Front. Microbiol. 14, 1128160 (2023).
Gasch, A. P. et al. Further support for aneuploidy tolerance in wild yeast and effects of dosage compensation on gene copy-number evolution. Elife 5, e14409 (2016).
Hose, J. et al. The genetic basis of aneuploidy tolerance in wild yeast. Elife 9, (2020).
Zhu, Y. O., Siegal, M. L., Hall, D. W. & Petrov, D. A. Precise estimates of mutation rate and spectrum in yeast. Proc. Natl. Acad. Sci. USA. 111, E2310–E2318 (2014).
Sharp, N. P., Sandell, L., James, C. G. & Otto, S. P. The genome-wide rate and spectrum of spontaneous mutations differ between haploid and diploid yeast. Proc. Natl. Acad. Sci. USA. 115, E5046–E5055 (2018).
Harrison, B. D. et al. A tetraploid intermediate precedes aneuploid formation in yeasts exposed to fluconazole. PLoS Biol 12, e1001815 (2014).
Todd, R. T., Wikoff, T. D., Forche, A. & Selmecki, A. Genome plasticity in Candida albicans is driven by long repeat sequences. Elife 8, e45954 (2019).
Todd, R. T. & Selmecki, A. Copy Number Variation and Allele Ratio Analysis in Candida albicans Using Whole Genome Sequencing Data. Methods Mol. Biol. 2658, 105–125 (2023).
Selmecki, A., Gerami-Nejad, M., Paulson, C., Forche, A. & Berman, J. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol. Microbiol. 68, 624–641 (2008).
Sionov, E., Lee, H., Chang, Y. C. & Kwon-Chung, K. J. Cryptococcus neoformans Overcomes Stress of Azole Drugs by Formation of Disomy in Specific Multiple Chromosomes. PLoS Pathog 6, e1000848 (2010).
Ning, Y., Dai, R., Zhang, L., Xu, Y. & Xiao, M. Copy number variants of ERG11: mechanism of azole resistance in Candida parapsilosis. Lancet Microbe 5, e10 (2024).
Fan, X. et al. Author Correction: Tandem gene duplications contributed to high-level azole resistance in a rapidly expanding Candida tropicalis population. Nat. Commun. 15, 587 (2024).
Kim, M. et al. Genomic Tandem Quadruplication is Associated with Ketoconazole Resistance in Malassezia pachydermatis. J. Microbiol. Biotechnol. 28, 1937–1945 (2018).
Ascencio, D. et al. Expression attenuation as a mechanism of robustness against gene duplication. Proc. Natl. Acad. Sci. USA. 118, (2021).
Fiori, A. & Van Dijck, P. Potent synergistic effect of doxycycline with fluconazole against Candida albicans is mediated by interference with iron homeostasis. Antimicrob. Agents Chemother. 56, 3785–3796 (2012).
Beattie, S. R., Jezewski, A. J., Ristow, L. C., Wellington, M. & Krysan, D. J. FKS1 is required for Cryptococcus neoformans fitness in vivo: Application of copper-regulated gene expression to mouse models of cryptococcosis. mSphere 7, e0016322 (2022).
Bergin, S. et al. Analysis of clinical Candida parapsilosis isolates reveals copy number variation in key fluconazole resistance genes. Antimicrob. Agents Chemother. 68, e0161923 (2024).
Morschhäuser, J. et al. The transcription factor Mrr1p controls expression of the MDR1 efflux pump and mediates multidrug resistance in Candida albicans. PLoS Pathog 3, e164 (2007).
Bing, J. et al. Experimental Evolution Identifies Adaptive Aneuploidy as a Mechanism of Fluconazole Resistance in Candida auris. Antimicrob. Agents Chemother. 65, e01466–20 (2020).
Gabriel, F. et al. Deletion of the uracil permease gene confers cross-resistance to 5-fluorouracil and azoles in Candida lusitaniae and highlights antagonistic interaction between fluorinated nucleotides and fluconazole. Antimicrob. Agents Chemother. 58, 4476–4485 (2014).
Oyama, M. et al. Deletion of the Golgi Ca2+-ATPase PMR1 gene potentiates antifungal effects of dodecanol that depend on intracellular Ca2+ accumulation in budding yeast. FEMS Yeast Res. 20, foaa003 (2020).
Chen, M., Zhong, G., Wang, S., Chen, P. & Li, L. Deletion of cox7c Results in Pan-Azole Resistance in Aspergillus fumigatus. Antimicrob. Agents Chemother. 66, e0015122 (2022).
Berman, J. & Krysan, D. J. Drug resistance and tolerance in fungi. https://www.nature.com/articles/s41579-019-0322-2 (2020) https://doi.org/10.1038/s41579-019-0322-2.
Kukurudz, R. J. et al. Acquisition of cross-azole tolerance and aneuploidy in Candida albicans strains evolved to posaconazole. G3 12, jkac156 (2022).
Yang, F. et al. Antifungal Tolerance and Resistance Emerge at Distinct Drug Concentrations and Rely upon Different Aneuploid Chromosomes. MBio 14, e0022723 (2023).
Ma, Q., Ola, M., Iracane, E. & Butler, G. Susceptibility to Medium-Chain Fatty Acids Is Associated with Trisomy of Chromosome 7 in Candida albicans. mSphere 4, msphere.00402-19 (2019).
Li, W.-J. et al. FLO8 deletion leads to azole resistance by upregulating CDR1 and CDR2 in Candida albicans. Res. Microbiol. 170, 272–279 (2019).
Yu, S.-J., Chang, Y.-L. & Chen, Y.-L. Deletion of ADA2 Increases Antifungal Drug Susceptibility and Virulence in Candida glabrata. Antimicrob. Agents Chemother. 62, aac.01924-17 (2018).
Bhattacharya, S., Esquivel, B. D. & White, T. C. Overexpression or Deletion of Ergosterol Biosynthesis Genes Alters Doubling Time, Response to Stress Agents, and Drug Susceptibility in Saccharomyces cerevisiae. MBio 9, mbio.01291-18 (2018).
Rybak, J. M. et al. Abrogation of Triazole Resistance upon Deletion of CDR1 in a Clinical Isolate of Candida auris. Antimicrob. Agents Chemother. 63, aac.00057-19 (2019).
Chin, C. et al. Deletion of AIF1 but not of YCA1/MCA1 protects Saccharomyces cerevisiae and Candida albicans cells from caspofungin-induced programmed cell death. Microb. Cell 1, 58–63 (2014).
Bergin, S. A. et al. Systematic Analysis of Copy Number Variations in the Pathogenic Yeast Candida parapsilosis Identifies a Gene Amplification in RTA3 That is Associated with Drug Resistance. MBio 13, e0177722 (2022).
Bédard, C. et al. FungAMR: A comprehensive portrait of antimicrobial resistance mutations in fungi. bioRxiv 2024.10.07.617009 https://doi.org/10.1101/2024.10.07.617009 (2024).
Bennett, R. J., Forche, A. & Berman, J. Rapid mechanisms for generating genome diversity: whole ploidy shifts, aneuploidy, and loss of heterozygosity. Cold Spring Harb. Perspect. Med. 4, a019604–a019604 (2014).
Kaya, A. et al. Molecular signatures of aneuploidy-driven adaptive evolution. Nat. Commun. 11, 588 (2020).
Sheltzer, J. M. et al. Aneuploidy drives genomic instability in yeast. Science 333, 1026–1030 (2011).
Beekman, C. N. & Ene, I. V. Short-term evolution strategies for host adaptation and drug escape in human fungal pathogens. PLoS Pathog 16, e1008519 (2020).
Berman, J. Ploidy plasticity: a rapid and reversible strategy for adaptation to stress. FEMS Yeast Res 16, fow020 (2016).
Ford, C. B. et al. The evolution of drug resistance in clinical isolates of Candida albicans. Elife 4, e00662 (2015).
Barda, O. et al. Aneuploidy Formation in the Filamentous Fungus Aspergillus flavus in Response to Azole Stress. Microbiol Spectr 11, e0433922 (2023).
Li, X. et al. Trisomy of chromosome R confers resistance to triazoles in Candida albicans. Med. Mycol. 53, 302–309 (2015).
Theisen, A. Microarray-based Comparative Genomic Hybridization (aCGH). Nature Education 1, 45 (2008).
Perepnikhatka, V. et al. Specific chromosome alterations in fluconazole-resistant mutants of Candida albicans. J. Bacteriol. 181, 4041–4049 (1999).
Abbey, D. A. et al. YMAP: a pipeline for visualization of copy number variation and loss of heterozygosity in eukaryotic pathogens. Genome Med 6, 100 (2014).
Vollger, M. R. et al. Long-read sequence and assembly of segmental duplications. Nat. Methods 16, 88–94 (2019).
Schikora-Tamarit, M. À. & Gabaldón, T. PerSVade: personalized structural variant detection in any species of interest. Genome Biol. 23, 175 (2022).
Garcia-Effron, G. Molecular Markers of Antifungal Resistance: Potential Uses in Routine Practice and Future Perspectives. J. Fungi. (Basel) 7, 197 (2021).
Jung, H.-S., Lefferts, J. A. & Tsongalis, G. J. Utilization of the oncoscan microarray assay in cancer diagnostics. Appl. Canc. Res. 37, 1–8 (2017).
Plissonneau, C., Hartmann, F. E. & Croll, D. Pangenome analyses of the wheat pathogen Zymoseptoria tritici reveal the structural basis of a highly plastic eukaryotic genome. BMC Biol 16, 5 (2018).
Croll, D., Zala, M. & McDonald, B. A. Breakage-fusion-bridge cycles and large insertions contribute to the rapid evolution of accessory chromosomes in a fungal pathogen. PLoS Genet 9, e1003567 (2013).
Dunn, B., Richter, C., Kvitek, D. J., Pugh, T. & Sherlock, G. Analysis of the Saccharomyces cerevisiae pan-genome reveals a pool of copy number variants distributed in diverse yeast strains from differing industrial environments. Genome Res. 22, 908–924 (2012).
Tralamazza, S. M., Gluck-Thaler, E., Feurtey, A. & Croll, D. Copy number variation introduced by a massive mobile element facilitates global thermal adaptation in a fungal wheat pathogen. Nat. Commun. 15, 5728 (2024).
Badet, T., Oggenfuss, U., Abraham, L., McDonald, B. A. & Croll, D. A 19-isolate reference-quality global pangenome for the fungal wheat pathogen Zymoseptoria tritici. BMC Biol 18, 12 (2020).
Uthayakumar, D., Sharma, J., Wensing, L. & Shapiro, R. S. CRISPR-Based Genetic Manipulation of Candida Species: Historical Perspectives and Current Approaches. Front Genome Ed 2, 606281 (2020).
Uddin, W., Dhabalia, D., Prakash, S. M. U. & Kabir, M. A. Systematic truncations of chromosome 4 and their responses to antifungals in Candida albicans. J. Genet. Eng. Biotechnol. 19, 92 (2021).
Tada, S. et al. Telomere-mediated chromosomal truncation in Aspergillus oryzae. J. Biosci. Bioeng. 119, 43–46 (2015).
Gervais, N. C. et al. Development and applications of a CRISPR activation system for facile genetic overexpression in Candida albicans. G3 13, jkac301 (2023).
Wensing, L. et al. A CRISPR Interference Platform for Efficient Genetic Repression in Candida albicans. mSphere 4, e00002-19 (2019).
Billerbeck, S., Prins, R. C. & Marquardt, M. A Modular Cloning Toolkit Including CRISPRi for the Engineering of the Human Fungal Pathogen and Biotechnology Host Candida glabrata. ACS Synth. Biol. https://doi.org/10.1021/acssynbio.2c00560 (2023).
Maroc, L., Shaker, H. & Shapiro, R. S. Functional genetic characterization of stress tolerance and biofilm formation in Nakaseomyces glabrata via a novel CRISPR activation system. bioRxiv 2023.12.07.569977 https://doi.org/10.1101/2023.12.07.569977 (2023).
Lauer, S. et al. Single-cell copy number variant detection reveals the dynamics and diversity of adaptation. PLoS Biol 16, e3000069 (2018).
Lauer, S. & Gresham, D. An evolving view of copy number variants. Curr. Genet. 65, 1287–1295 (2019).
Zhou, X. et al. Single-cell detection of copy number changes reveals dynamic mechanisms of adaptation to antifungals in Candida albicans. Nat. Microbiol. 9, 2923–2938 (2024).
LaFayette, S. L. et al. PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog 6, e1001069 (2010).
Lamoth, F., Alexander, B. D., Juvvadi, P. R. & Steinbach, W. J. Antifungal activity of compounds targeting the Hsp90-calcineurin pathway against various mould species. J. Antimicrob. Chemother. 70, 1408–1411 (2015).
Lamoth, F., Juvvadi, P. R., Gehrke, C. & Steinbach, W. J. In vitro activity of calcineurin and heat shock protein 90 Inhibitors against Aspergillus fumigatus azole- and echinocandin-resistant strains. Antimicrob. Agents Chemother. 57, 1035–1039 (2013).
Singh, S. D. et al. Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog 5, e1000532 (2009).
Acknowledgements
The authors would like to thank Romain Durand and Camille Bédard for their comments on an earlier version of the manuscript.
Author information
Authors and Affiliations
Contributions
A.J. curated the literature; D.F.J. and A.J. analyzed the data; A.J. and D.F.J. wrote the manuscript with contributions from A.G. and C.R.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jay, A., Jordan, D.F., Gerstein, A. et al. The role of gene copy number variation in antimicrobial resistance in human fungal pathogens. npj Antimicrob Resist 3, 1 (2025). https://doi.org/10.1038/s44259-024-00072-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44259-024-00072-1
This article is cited by
-
Collectively charting the landscape of antifungal and fungicide resistance
Nature Microbiology (2025)
-
Drivers of antimicrobial resistance in pig production systems of Uganda
Communications Earth & Environment (2025)
-
Frontiers in superbug management: innovating approaches to combat antimicrobial resistance
Archives of Microbiology (2025)
-
FungAMR: a comprehensive database for investigating fungal mutations associated with antimicrobial resistance
Nature Microbiology (2025)
