
Abstract
Bacterial infections remain a major global health issue, with antimicrobial resistance (AMR) worsening the crisis. However, treatment failure can occur even when bacteria show antibiotic susceptibility in diagnostic tests. We explore factors such as phenotypic resilience, bacterial lifestyles such as biofilms, and differences between laboratory tests and real infection sites, highlighting the need for improved platforms to better predict treatment outcomes, and reviewing emerging technologies aimed at improving susceptibility testing.
Similar content being viewed by others


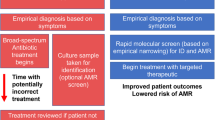
Introduction and scope of review
Bacterial infections remain a significant source of morbidity and mortality despite advances in modern medicine. A recent report estimated their role in 7.7 million (13.6%) deaths worldwide regardless of antimicrobial resistance status—the second highest cause of mortality after ischaemic heart disease1. In parallel, antimicrobial resistance (AMR) is a growing crisis associated with 1.27 million deaths globally2. While AMR has received a significant amount of attention, treatment failure unrelated to bacterial AMR genes is far less understood.
The first step in microbiological diagnosis is identifying the causative organism(s), followed by determining susceptibility to antibiotics to guide prescribing. However, such in vitro susceptibility does not always translate to successful bacterial clearance or treatment outcome in the patient. We will discuss how treatment failure in infection with a susceptible microorganism is likely multifactorial, due to phenotypic resilience behaviours (e.g. tolerance and persistence) but also bacterial lifestyles such as biofilms; host factors; and environmental differences in diagnostic culture media compared with the in vivo infection site. Finally, we will examine emerging infection models that may better predict treatment outcomes, and offer future perspectives.
The return of infection despite “appropriate” antibiotic treatment: terms of reference
Treatment failure despite antibiotic treatment represents a significant clinical challenge. As there are various related terms in the literature, we will first clarify the nomenclature we employ in this review (Fig. 1). We use the term treatment failure when referring to the inability of a drug to permanently clear the infection, no matter what the cause of failure. If measurable infection returns after treatment, this may be due to de novo infection of a new organism (reinfection), which is not a type of treatment failure. In the treatment failure category, one can describe recrudescence (outright failure of a drug to clear an infection during treatment). Alternatively, if the treatment initially succeeds but then the original agent returns, we deem this recurrence, which can be relapse (reactivation of the original infection from dormancy or sequestration in the body at some point after treatment) or the acquisition of classical antimicrobial resistance3, herein defined as an acquired bacterial genetic trait (e.g. an AMR gene such as beta-lactamase). This review will not deal with reinfection with a new strain, antibiotic resistance nor intrinsic resistance (e.g. membrane impermeability) that will never allow a drug to be efficacious in that particular bacterial class, e.g. cephalosporins against Enterococci4. We will also not discuss scenarios where the organism identified in the diagnostic laboratory (e.g. within a mixed growth) was not the organism causing the symptoms, and therefore treatment failed because the wrong bacteria was targeted by antibiotics. Instead, we will be discussing short- or long-term treatment failure in infections caused by bacteria that were predicted to be susceptible to a particular antimicrobial.

Scheme presented is for an otherwise healthy individual (e.g. not immunosuppressed) with an acute infection.
It is challenging to distinguish between true recurrence involving the same bacterial strain, and reinfection with a new strain of the same species with the same antibiogram (defined as the profile of susceptibility of a strain to an array of different drugs). To definitively determine the cause of a persisting infection, longitudinal sampling coupled with genotyping (preferably whole-genome sequencing) would be required, which is logistically challenging, resource-intensive in clinical settings, and rarely performed5,6,7,8. Hence, as a caveat, sometimes relapse and recrudescence are attributed as such without definitive proof. Therefore, this limitation should be kept in mind when we discuss the evidence below.
Another important point is that distinct terms are used within disease specialities which might differ. For instance, in the Helicobacter pylori infection community, recrudescence is used interchangeably with relapse, while re-infection is defined as infection with a new H. pylori strain after successful eradication. If infection returns after initially successful clearance within one year, it is known as recrudescence, while return after a year is called reinfection9. Regardless of terminology, treatment-resistant infections are associated with increased morbidity and mortality and can have significant quality of life impacts10. Often necessitating repeated or long-term antibiotic usage, they may also contribute to the growing issue of classical AMR11, which is predicted to cause ~10 million deaths attributable to or associated with AMR globally in 20502.
Mechanisms of treatment resistance
Traditionally, antimicrobial resistance is classified as intrinsic or acquired and is measured using the minimum inhibitory concentration (MIC) assay. MIC is currently the gold-standard methodology to guide clinical treatment decisions (Table 1). Using dilutions into liquid or agar, or gradient strips, visible inhibition of bacterial growth can be observed. The resulting clinical breakpoints indicate whether isolates are susceptible or resistant against a panel of antibiotics, often with designations for more intermediate or dose-dependent phenotypes depending on the framework12,13.
Less discussed is the subset of refractory phenotypes that may be masked by the MIC assay14. This section will describe these phenotypes and address the mechanisms responsible where known (summarised in Table 2 with additional examples). It should be noted that while the literature describes a vast array of mechanisms, here we focus on a selection deem to be particularly noteworthy within the context of this review. We also stress that there are multiple mechanisms (not all described in this review) that could result in a particular phenotype.
In bacteria that test susceptible to an antibiotic in an MIC assay, strategic fluctuations in gene expression can allow short-term survival at the population level. Sometimes referred to as phenotypic ‘resilience’, this category includes tolerance, persistence and heteroresistance1. Tolerance is the ability of an entire bacterial population to survive transient exposure to a bactericidal antibiotic14, featuring slower bacterial killing compared with a fully susceptible population. Antibiotic persistence describes a subset of the population that can survive bactericidal antibiotic concentrations, typically represented by a biphasic kill curve. In contrast, heteroresistance occurs when a subset of cells within the population exhibits a higher MIC than the majority15, allowing it to grow despite antibiotic pressure.
These temporary behaviours (Fig. 2) are regulated by a complex programme of upstream signalling systems and stress responses (blue box). These are in turn influenced by environmental signals (green box). Ultimately, the net result of these triggers may manifest in situations (pink box) that result in resilience behaviours, sometimes in tandem with more physical obstructions to antibiotics.

Environmental cues, signalling pathways and phenotypes can influence one another and thereby be modulated in real time (grey arrows).
Two-component systems
Bacteria rely on two-component systems (TCSs) to sense and adapt to their environment, making TCSs critical for niche adaptation and downstream gene regulation. These signalling systems are key players in AMR across diverse environmental contexts, but are less understood in the context of resilience phenotypes. TCSs can respond directly to antibiotic exposure, but environmental stressors may also induce transient changes in key global regulators, resulting in cell surface modifications, increased drug efflux, antibiotic-degrading enzyme production, and biofilm formation16. Typically, TCSs consist of a homodimeric histidine kinase and a cognate response regulator.
For example, in Listeria monocytogenes, the TCS LisRK confers selective antibiotic tolerance, particularly to ampicillin, likely by regulating penicillin-binding protein expression and modulating cell envelope properties17. In Mycobacterium tuberculosis, the TCS TcrXY, an acid-sensing system, is essential for establishing persistent infection, and silencing TcrXY attenuates persistence during chronic mouse lung infection18. Similarly, populations of Escherichia coli under nitrogen starvation conditions form a higher percentage of metabolically dormant cells through the TCS NtrBC19.
TCSs also facilitate major pathoadaptive phenotypes, such as biofilms (see Biofilms), which play a role in treatment resistance. The TCS LisRK mentioned above also promotes adhesion during biofilm formation. Another example is the Gac-Rsm two-component system in Pseudomonas aeruginosa. The Gac-Rsm cascade is a central regulatory network that mediates the phenotypic switch between acute and chronic infections20, controlled by the sensor kinases RetS and LadS, which modulate the activation of the GacS/GacA TCS21. The net result is the activation of several virulence factors involved in the lifestyle transition of P. aeruginosa, such as utilization of the bacterial T6SS nano-weapon, biofilm formation and motility-related adaptations22.
Toxin/-Anti-toxin systems and stress response
Another proposed mechanism of bacterial persistence involves toxin/anti-toxin (TA) modules23. For example, E. coli mutants demonstrating persistence against penicillin had a gain-of-function allele, hipA7, later shown to be a toxin causing self-killing and growth arrest, relieved by the presence of its cognate anti-toxin15. Furthermore, overproduction in E. coli of the RelE toxin, an inhibitor of translation, caused a sharp increase in persister cells (defined as tolerant and often quiescent)24. MazF, a type II TA system in E. coli, was additionally shown to induce growth inhibition and persister generation25.
These TA modules are regulated by certain stress responses, highlighting environmental influence. These include the SOS response (facilitating DNA repair after damage), and the stringent response (which adjusts cellular behaviour during nutrient scarcity)26. Oxidative stress and antibiotic exposure can trigger the SOS response by upregulating genes involved in the SOS response, many of which are known to include toxin-antitoxin (TA) systems27. These upstream cues result in inhibition of cellular functions including transcription, translation, DNA replication and energy production, all of which result in cell dormancy. Like the SOS response, the stringent response is also modulated by certain environmental stresses including pH variation, nutrient limitation and heat shock. These triggers activate production of (p)ppGpp, mainly via SpoT and RelA, which have also been shown to activate TA modules28. Quorum sensing has further been linked to the regulation of TA systems via Rpos29.
Downstream phenotypes
How do these regulatory networks manifest in practice? Below, we review five cases of treatment resistance where resilience mechanisms are in play, sometimes in tandem with more physical obstructions to antibiotics. Similar phenotypes to the ones described in these cases might also be achieved by other mechanisms, but here we focus on a few discrete examples.
Biofilms
Chronic infections are often linked to microbial biofilms, which pose significant clinical challenges, whether occurring in native anatomical sites or on prosthetic material30. Biofilms consist of bacterial communities embedded in an extracellular polymeric substance (EPS) comprised of polysaccharides, proteins, and extracellular nucleic acids (eNAs), a structure enhancing bacterial survival and resilience to antibiotics across various infection contexts31. Specifically, positively charged antibiotics such as aminoglycosides may bind to, and become blocked by, negatively charged eNAs. In addition, polysaccharides can act as a permeability barrier against antibiotic penetration32. Thus, the biofilm structure itself can resist antibiotics even when antimicrobial tests indicate susceptibility16.
Bacterial populations within biofilms can also temporarily endure high doses of antibiotics by slowing metabolic processes, with some cells growing very slowly or becoming inactive (persister cells). The presence of highly metabolically active cells in the top layers and slower/inactive ones residing deeper33 mirrors how nutrients and oxygen are distributed and affect how different parts of the biofilm respond to antibiotic treatment16. Hypoxic pockets deep within the biofilm promote persister formation16,34. Dormant subpopulations, which are not actively dividing, complicate treatments, as reduced activity can present fewer active targets for antibiotics, which often oppose active metabolic processes, e.g. beta-lactams and cell division35. Persister cells can resume normal growth when treatment stops, leading to relapse or recrudescence. Important in the clinical setting, biofilms are often independent compartments with different pharmacodynamics compared with that of the surrounding tissue36.
Increasing evidence suggests that polymicrobial interactions and bacterial mutualism play a role in response to antibiotic tolerance and resistance. These interspecies interactions can be outlined as collective resistance, collective tolerance and exposure protection, which have been previously described37. In the context of this review, we argue that MICs are additionally limited because they test antimicrobial susceptibility in single species during planktonic (free-floating) growth, absent from the host microenvironment.
Efflux pumps
Whether in planktonic populations or biofilms, efflux pumps contribute significantly to antibiotic treatment failure and multidrug resistance by regulating the intracellular environment38. These transmembrane proteins expel toxic substances, such as antibiotics and harmful metabolites, and are predominantly found in Gram-negative bacteria. While efflux pumps are central to classical AMR, they may also play a role in persistence and tolerance phenotypes. For example, E. coli persisters exposed to beta-lactam antibiotics exhibited reduced drug accumulation due to increased efflux activity, with upregulation of the tolC, acrA, and acrB multidrug efflux genes. The high expression of tolC specifically enhanced bacterial persistence39. TolC activity also contributed to delafloxacin persistence in E. coli, with induced expression of acrAB efflux genes following fluoroquinolone treatment40. Notably, deletion of acrB led to reduced survival rates of persister cells. Additionally, efflux pump genes in Streptococcus pyogenes persisters were upregulated when exposed to penicillin, along with a concurrent downregulation of genes related to protein synthesis and cell growth41.
Intracellular bacterial communities
Another way bacteria may evade antibiotics is via the formation of intracellular bacterial communities (IBC), which are reservoirs that result from bacterial invasion. For example, in urinary tract infections with uropathogenic E. coli (UPEC)42, IBCs are protected from antibiotics because the tight barrier function of the urothelium limits or prevents their penetration43; moreover, deeper reservoirs achieve a dormant state. Metabolic flux in UPEC has been shown to regulate the quiescent state via succinyl-coA resulting in antibiotic persistence44. There are many other examples of difficult-to-treat bacteria with obligate or facultative intracellular lifestyles, including Salmonella enterica, M. tuberculosis, L. monocytogenes and Chlamydia trachomatis (reviewed by Kamaruzzaman et al.13).
Membrane Vesicles
Bacteria can secrete spherical structures (20–400 nm) called membrane vesicles (MVs)45, which contain various cargoes and have a variety of functions46,47. Depending on treatment or environmental factors, MVs can be upregulated, allowing a nuanced survival response.
While MVs are known to transfer AMR genes48,49,50, they can also contribute to atypical treatment failure45, e.g. harbouring beta-lactamase enzymes in the case of E. coli51 and Neisseria gonorrhoeae52, or esterases associated with macrolide resistance in the case of Gram-negative bacteria (reviewed in53). MVs have also been implicated as decoys, decreasing the environmental antibiotic concentration and uptake of antibiotic by target bacteria54,55,56,57. The SOS response caused by antibiotic treatment can lead to increased MV secretion, as seen with P. aeruginosa responding to ciprofloxacin58, while different antibiotics deployed in sub-MIC concentrations can induce different quantities of MVs with different cargo59. Finally, MV cargo has been implicated in biofilm formation in Gram-negative bacteria such as Acinetobacter baumannii60 and H. pylori61 directly, and indirectly with P. aeruginosa62.
L-forms
L-forms are a transient, cell wall-deficient morphotypes of normally wall-possessing bacteria which can resist wall-targeting antibiotics, increasing their tolerance and persistence. L-form formation has been shown in Gram-negative bacteria, e.g. E. coli isolated from recurrent UTI (rUTI) patients. L-form switching was present in response to antibiotics that target the cell wall, and may also be a mechanism of tolerance or persistence in rUTI63.
Environmental factors are important for L-form maintenance, as these morphotypes can only survive in osmotically stabilised conditions64. Reactive oxygen species (ROS) also play a role in L-form growth, with increased ROS limiting L-form formation; however, addition of ROS scavengers or anaerobic conditions contributed to L-form proliferation65.
Role of the host microenvironment
The complex regulatory systems described in Mechanisms of treatment resistance section, and their practical outputs in Downstream phenotypes section, are broadly influenced by the larger environment (Fig. 2, green box). While the broader milieu can affect bacterial phenotype and contribute to classical AMR by providing selective pressures, less well understood is how the environment can affect treatment response in other ways. Fluctuations in host anatomy, physiology, metabolic function and cellular components can lead to homoeostatic imbalance, which in turn can affect drug pharmacokinetics/pharmacodynamics (PK/PD)4,66,67. Such fluctuations include blood composition, pH, microbiota and mucous composition, which are elements not usually reflected in in vitro diagnostic testing4.
Blood composition
Systemically administered antibiotics enter the bloodstream either directly (e.g. through the intravenous route) or indirectly following absorption (e.g. through the gastrointestinal tract), where they circulate until they are bound to blood cells or plasma proteins, which can prevent effective drug function68. As protein binding to drug increases, the overall pharmacological efficacy decreases as the amount of active drug left in circulation is lowered4,67. Another factor is that erythrocytes contain iron, which promotes bacterial growth. Indeed, the addition of lysed erythrocytes into cultures of E. coli, Staphylococcusaureus and P. aeruginosa raised the MIC of meropenem, ciprofloxacin and tigecycline68; moreover, significant differences in MIC for oxacillin, ampicillin and moxifloxacin occurred in S. aureus when cultured with albumin68. In addition, the inclusion of albumin to bacterial cultures led to a significant decrease in bacterial killing by moxifloxacin68. Similarly, E. coli and Streptococcus pneumoniae treated with serum albumin showed 8-fold higher MIC values for ceftriaxone69. Human serum has also been shown to trigger antibiotic tolerance in S. aureus70.
pH
Maintenance of an acid-base balance is a major component of homoeostasis, and variations of H+ concentration can alter protein structure and function, causing a difference in drug bioavailability4. Acidic environments occur naturally (e.g. in the vagina) as a defence mechanism to prevent bacterial colonisation. In addition, acid stress responses are essential for survival71,72. During infection, local pH can change due to the presence of bacteria, with concomitant alterations in cell metabolism affecting antibiotic function, which could affect treatment outcomes73.
It has been noted that fluctuating pH is not reflected in MIC tests, which could affect their results74. Indeed, urinary pH is known to affect antibiotic efficacy75. For example, in one study, patient-relevant acidification in MIC tests increased the susceptibility of 71% of the bacterial isolates76. In the case of ciprofloxacin, the impact of acidic pH on E. coli susceptibility was due to the positive and negative charge on the drug and its zwitterionic nature76. Another study focusing on UPEC resistance to trimethoprim sulfamethoxazole also reported that fluctuating urine pH affected the MIC77.
Other environmental factors
Aside from its pH, the composition of urine, including salt and urea, may affect the transcriptome of urinary pathogens16 as well as their response to antibiotics78. The gut microbiota is known to affect drugs generally, with most research focusing chemotherapeutics78; while not well-described, antibiotics may also be processed and altered by commensal microorganisms. Media mimicking mucus has been used to show how this milieu affects P. aeruginosa during cystic fibrosis infection79, while others have studied the effect of glucose on growth and co-culture of S. aureus and P. aeruginosa in artificial sputum medium80.
Beyond the MIC: Challenges and advances in more physiological antimicrobial susceptibility testing platforms
In addition to the gold-standard MIC, other methods are also employed in clinical settings, relying either on phenotypic outputs (e.g. growth, death), or on omics-based identification of resistance (Table 1). More recently, faster methods have been developed that detect more advanced parameters, although they are expensive. For example, the Accelerate Pheno System identifies bacteria by gel electrofiltration and fluorescence in situ hybridisation, measuring bacterial growth using microscopy to infer MIC values81. Resistell technology uses atomic force microscopy (AFM) to measure bacterial nanomotion, a proxy for viability1. Finally, the Sysmex PA-100 System deploys nanofluidics and imaging to determine cell growth. Yet none of these recapitulates a more physiological microenvironment82.
Media mimicking
The most basic improvement that could be made in planktonic antimicrobial sensitivity testing would involve supplementing, or fully replacing, the standard bacterial growth medium used for MIC (e.g. Mueller-Hinton broth) with the relevant bodily fluids or their synthetic equivalent83,84. Heithoff et al. showed that ~15% of MIC tested in more physiological media predicted a shift across the clinical breakpoint, altering susceptibility classification84. Similarly, by using sputum-mimicking media, a reduction in antimicrobial susceptibility of Mycobacterium abscessus was observed in comparison with standard medium85. As previously mentioned, the host microenvironment might trigger phenotypic adaptations that lead to bacterial tolerance or increased antimicrobial susceptibility. For example, the presence of serum in growth medium triggered tolerance of S. aureus against daptomycin by two distinct mechanisms, leading to peptidoglycan accumulation and increased cardiolipin concentration in the bacterial membrane70. Transcriptomics analysis of P. aeruginosa isolated directly from a human infection was compared with laboratory growth conditions to explore this issue. Key genes related to antimicrobial resistance were found to be upregulated in the human host infection in comparison with the in vitro settings, which potentially could explain why AST assays often underestimate resistance in the clinical settings86.
As a potential case in point, in a side-by-side comparison, the Mast Uri® System, where patient urine is inoculated directly onto antibiotic-containing agar, was found to call resistance more frequently than did standard MIC87. Although the authors interpreted this finding as the Mast Uri over-estimating resistance, it is equally possible that the MIC was under-estimating it. The different results may stem from the fact that, unlike a standard MIC, where a colony is selected after overnight growth and adaptation to agar-containing standard culture medium, the inoculated bacteria come straight from the patient, where their metabolism is primed for the nutrient-poor urine environment.
While enriched medium can recapitulate some physiological aspects of planktonic conditions, the reality is that many infections in a clinical setting involve biofilms88, and of course, host cells, alongside other external factors such as fluid flow89. There is therefore great interest in developing more physiological platforms to test antibiotics in these more complicated microenvironments.
Biofilm susceptibility testing methods
Typically, acute infections are caused by planktonically growing bacteria, while biofilm-associated bacteria are involved in chronic infections. Indeed, in humans, 80% of chronic infections are thought to be caused by biofilm-associated bacteria90,91. Due to their unique features, biofilm-associated infections manifest between 100-1000-fold increased antimicrobial resistance compared with planktonically growing bacteria88,92.
Different types of biofilm models are summarized in Table 3 with their pros and cons, alongside examples, and illustrated in Fig. 3. Generally, these methods involve growing biofilms on different abiotic substrates, such as agar, plastic, glass or beads, which can be either static or embedded in a dynamic flow system, such as a bioreactor or microfluidic chip93,94. A significant challenge in biofilm susceptibility testing methods is standardization, as well as determining whether a given methodology yields clinically relevant results95. Neither the European Committee on Antimicrobial Susceptibility Testing (EUCAST) nor the Clinical and Laboratory Standards Institute (CLSI) have established standardized definitions of biofilm endpoint parameters/MIC96. In the biofilm field, different susceptibility endpoint parameters have been reported (Table 4). However, some of the terms are often used interchangeably in the literature, causing confusion16,96,97.

These model have been subcategorised depending on the complexity of the platform used to grow the bacterial biofilm (in vitro, microcosm, ex vivo, organoid and in vivo model).
The main limitation of biofilm testing methods is the uncertain validity of the model used94. Although biofilms have been studied extensively in vitro, they may not accurately mimic biofilms found in vivo in chronic infections93. The lack of chemical/physical host-microenvironment, alongside the relatively short infection set-up of in vitro experiments, are additional key differences98.
More complex infection models
In addition to studying biofilms in isolation, the next logical step is to include more physiological components. Table 5 and Fig. 3 summarise these types of models93,94,99,100, all of which can also foster biofilms. One such model is the microcosm model, which includes the nutrients and structures present in a normal infection. As an example, a saliva-derived polymicrobial biofilm model on a titanium surface was developed to test antimicrobial therapies; even when the treatments were efficacious, the polymicrobial communities re-established after two days with an altered microbial diversity101. Secondly, ex vivo models deploy tissue excised from an animal or human. For example, porcine skin was used to study S. aureus wound infections, and porcine lung, to study P. aeruginosa in cystic fibrosis; in both cases, the bacteria exhibited a higher antimicrobial susceptibility compared with their respective MICs, even when performed in mimicking host media102. Thirdly, organoid/microtissue models involve biomimetic microtissue or organoids which are grown and then infected experimentally. For instance, we developed the 3D human urine-tolerant urothelial model (3D-UHU), which allows infections in the presence of 100% urine103,104; this supports the formation of intracellular reservoirs as well as biofilms on the urothelial surface, and can be used to test antibiotic response. Sharma et al. reported a dynamic urothelial on-chip model incorporating flow and stretch parameters in which intracellular bacteria were more resistant to antibiotics105. Finally, in vivo models, where infected animals are studied, are arguably the most complex, and have been heavily relied upon, but for some parameters, such as direct host cell/pathogen interactions, their relevance may not be greater than human ex vivo or organoid/microtissue models94,106.
However, recapitulating more physiological infections, including model development, validation and real-world execution, are challenging94. To date, most biofilm therapy testing is still performed using simple in vitro biofilm models, or animal models that do not fully recapitulate the human host-microenvironment100. The discrepancies between antibiofilm drug efficacy and clinical trial performance strongly suggest that microenvironment matters in therapeutic development as well as in susceptibility testing95.
In choosing which system might be most appropriate, a further complication is that some bacterial infections, particularly those resulting in sepsis, involve multiple niches, with bacteria present in the bloodstream as well as in the originally infected site107. It is therefore a challenge to know which physiological testing platform might be the most appropriate. It must also be acknowledged that increased complexity will likely be more time- and resource-intensive, which may not be possible in some diagnostic settings.
Clinical relevance and conclusions
If a testing platform is not sufficiently nuanced to recapitulate the correct infection microenvironment, it seems logical that it might struggle to accurately predict clinical outcome. But what is the evidence for this? Taking a big-picture perspective, treatment failure rates can serve as a proxy that diagnostic techniques are not always optimal. For example, recurrence rates for urinary tract infection are worryingly high108. While uncomplicated UTI tends to be treated empirically and not all samples undergo AST, not all treatment failure is resistance-mediated.
In addition, it has been shown that the predictive value of AST methods on treatment success is relatively low on immunocompromised patients, patients treated with multiple antimicrobials, and patients with infections caused by polymicrobial communities109 (which demonstrate synergy in resistance110,111). A systematic review revealed lack of evidence of AST methods as a predictor of clinical outcomes in cystic fibrosis antimicrobial treatment112.
Given the documented discordance between AST and outcome, it would therefore be of great interest to examine studies that compared the predictive value of traditional testing to more advanced methods; unfortunately, such studies are vanishingly rare and more research is needed. As one example, a randomised controlled clinical trial to compare the utility of biofilm antimicrobial susceptibility testing versus MIC in treating pulmonary exacerbations in cystic fibrosis (CF) patients revealed no significant differences in pulmonary bacterial loads nor clinical outcomes4. The antimicrobial biofilm test was a Calgary device where biofilms of P. aeruginosa were grown in a rich media. Although this methodology offers high-throughput screening in clinical microbiology settings, it grossly oversimplifies the CF environment. Lung biopsies of people with CF reveal biofilms suspended in bronchial mucus, a sponge-like mass filled with mucus, alginate and/or lung fluid. These are substantially different to biofilms observed in vitro113. Hence, it is likely that in vitro biofilms do not accurately mimic CF biofilms and the antibiofilm susceptibility test performed offered no substantial advantages in comparison with traditional antimicrobial testing114,115. However, this example is valuable firstly in that it shows again that traditional testing cannot always predict treatment outcomes, and secondly that one cannot assume that only incremental complexity in modelling will make a difference.
In summary, recapitulating the host microenvironment to allow for more meaningful host-pathogen-drug interactions are key to advancing fundamental knowledge about the biology of treatment failure, and to increasing the pipeline for badly needed new alternatives to antibiotics. In parallel, the future of antimicrobial susceptibility testing likely lies in increasing the complexity and physiological relevance of platforms, while balancing these advances with the need to achieve high-throughput, point-of-care (or near point-of-care) timelines and manageable healthcare expense. We acknowledge that this balance will not be trivial to achieve. Finally, we need clinical studies and robust data to back up the ultimate clinical usefulness of newly developed tests.
Data availability
No datasets were generated or analysed during the current study.
References
Sturm, A. et al. Accurate and rapid antibiotic susceptibility testing using a machine learning-assisted nanomotion technology platform. Nat. Commun. 15, 2037 (2024).
Murray, C. J. L. et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
Darby, E. M. et al. Molecular mechanisms of antibiotic resistance revisited. Nat. Rev. Microbiol. 21, 280–295 (2023).
Yau, Y. C. W. et al. Randomized controlled trial of biofilm antimicrobial susceptibility testing in cystic fibrosis patients. J. Cyst. Fibros. 14, 262–266 (2015).
Cattamanchi, A., Kyabayinze, D., Hubbard, A., Rosenthal, P. J. & Dorsey, G. Distinguishing recrudescence from reinfection in a longitudinal antimalarial drug efficacy study: comparison of results based on genotyping of msp-1, msp-2, and glurp. https://doi.org/10.4269/ajtmh.2003.68.133 (2003).
Popovici, J. et al. Recrudescence, Reinfection, or Relapse? A More Rigorous Framework to Assess Chloroquine Efficacy for Plasmodium vivax Malaria. J. Infect. Dis. 219, 315–322 (2019).
Okoro, C. K. et al. High-resolution single nucleotide polymorphism analysis distinguishes recrudescence and reinfection in recurrent invasive nontyphoidal Salmonella Typhimurium disease. Clin. Infect. Dis. 54, 955–963 (2012).
McIvor, A., Koornhof, H. & Kana, B. D. Relapse, re-infection and mixed infections in tuberculosis disease. Pathog. Dis. 75 (2017).
Xu, L. et al. Global H. pylori recurrence, recrudescence, and re-infection status after successful eradication in pediatric patients: a systematic review and meta-analysis. J. Gastroenterol. 59, 668–681 (2024).
Grant, S. S. & Hung, D. T. Persistent bacterial infections, antibiotic tolerance, and the oxidative stress response. Virulence 4, 273–283 (2013).
Ku, J. H. et al. Antibiotic resistance of urinary tract infection recurrences in a large integrated US healthcare system. J. Infect. Dis. 230, e1344–e1354 (2024).
Kadeřábková, N., Mahmood, A. J. S. & Mavridou, D. A. I. Antibiotic susceptibility testing using minimum inhibitory concentration (MIC) assays. Npj Antimicrob. Resist. 2, 1–9 (2024).
Kamaruzzaman, N. F., Kendall, S. & Good, L. Targeting the hard to reach: challenges and novel strategies in the treatment of intracellular bacterial infections. Br. J. Pharmacol. 174, 2225–2236 (2017).
Brauner, A., Fridman, O., Gefen, O. & Balaban, N. Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 14, 320–330 (2016).
Balaban, N. Q., Merrin, J., Chait, R., Kowalik, L. & Leibler, S. Bacterial Persistence as a Phenotypic Switch. Science 305, 1622–1625 (2004).
Macia, M. D., Rojo-Molinero, E. & Oliver, A. Antimicrobial susceptibility testing in biofilm-growing bacteria. Clin. Microbiol. Infect. 20, 981–990 (2014).
Aslan, H. et al. Activation of the Two-Component System LisRK Promotes Cell Adhesion and High Ampicillin Tolerance in Listeria monocytogenes. Front. Microbiol. 12, 618174 (2021).
Stupar, M. et al. TcrXY is an acid-sensing two-component transcriptional regulator of Mycobacterium tuberculosis required for persistent infection. Nat. Commun. 15, 1615 (2024).
Brown, D. R. Nitrogen Starvation Induces Persister Cell Formation in Escherichia coli. J. Bacteriol. 201, e00622–18 (2019).
Moscoso, J. A. et al. The Diguanylate Cyclase SadC Is a Central Player in Gac/Rsm-Mediated Biofilm Formation in Pseudomonas aeruginosa. J. Bacteriol. 196, 4081–4088 (2014).
Song, H., Li, Y. & Wang, Y. Two-component system GacS/GacA, a global response regulator of bacterial physiological behaviors. Eng. Microbiol. 3, 100051 (2023).
Sultan, M., Arya, R. & Kim, K. K. Roles of Two-Component Systems in Pseudomonas aeruginosa Virulence. Int. J. Mol. Sci. 22, 12152 (2021).
Ronneau, S. & Helaine, S. Clarifying the Link between Toxin–Antitoxin Modules and Bacterial Persistence. J. Mol. Biol. 431, 3462–3471 (2019).
Keren, I., Shah, D., Spoering, A., Kaldalu, N. & Lewis, K. Specialized Persister Cells and the Mechanism of Multidrug Tolerance in Escherichia coli. J. Bacteriol. 186, 8172–8180 (2004).
Tripathi, A., Dewan, P. C., Siddique, S. A. & Varadarajan, R. MazF-induced Growth Inhibition and Persister Generation in Escherichia coli. J. Biol. Chem. 289, 4191–4205 (2014).
LeRoux, M., Culviner, P. H., Liu, Y. J., Littlehale, M. L. & Laub, M. T. Stress Can Induce Transcription of Toxin-Antitoxin Systems without Activating Toxin. Mol. Cell 79, 280–292.e8 (2020).
Kawano, M., Aravind, L. & Storz, G. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol. Microbiol. 64, 738–754 (2007).
Radzikowski, J. L. et al. Bacterial persistence is an active σS stress response to metabolic flux limitation. Mol. Syst. Biol. 12, 882 (2016).
Murakami, K. et al. Role for rpoS gene of Pseudomonas aeruginosa in antibiotic tolerance. FEMS Microbiol. Lett. 242, 161–167 (2005).
Mendhe, S., Badge, A., Ugemuge, S. & Chandi, D. Impact of Biofilms on Chronic Infections and Medical Challenges. Cureus https://doi.org/10.7759/cureus.48204 (2023).
Flemming, H.-C. & Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 8, 623–633 (2010).
Liu, H. Y., Prentice, E. L. & Webber, M. A. Mechanisms of antimicrobial resistance in biofilms. Npj Antimicrob. Resist. 2, 27 (2024).
Shree, P., Singh, C. K., Sodhi, K. K., Surya, J. N. & Singh, D. K. Biofilms: Understanding the structure and contribution towards bacterial resistance in antibiotics. Med. Microecol. 16, 100084 (2023).
Yan, J. & Bassler, B. L. Surviving as a Community: Antibiotic Tolerance and Persistence in Bacterial Biofilms. Cell Host Microbe 26, 15–21 (2019).
Wivagg, C. N., Bhattacharyya, R. P. & Hung, D. T. Mechanisms of β-lactam killing and resistance in the context of Mycobacterium tuberculosis. J. Antibiot. ((Tokyo)) 67, 645–654 (2014).
Hengzhuang, W., Wu, H., Ciofu, O., Song, Z. & Høiby, N. Pharmacokinetics/Pharmacodynamics of Colistin and Imipenem on Mucoid and Nonmucoid Pseudomonas aeruginosa Biofilms. Antimicrob. Agents Chemother. 55, 4469–4474 (2011).
Bottery, M. J., Pitchford, J. W. & Friman, V.-P. Ecology and evolution of antimicrobial resistance in bacterial communities. ISME J. 15, 939–948 (2021).
Nishino, K., Yamasaki, S., Nakashima, R., Zwama, M. & Hayashi-Nishino, M. Function and Inhibitory Mechanisms of Multidrug Efflux Pumps. Front. Microbiol. 12, 737288 (2021).
Pu, Y. et al. Enhanced Efflux Activity Facilitates Drug Tolerance in Dormant Bacterial Cells. Mol. Cell 62, 284–294 (2016).
Byrd, B. A. et al. The AcrAB-TolC Efflux Pump Impacts Persistence and Resistance Development in Stationary-Phase Escherichia coli following Delafloxacin Treatment. Antimicrob. Agents Chemother. 65, e00281–21 (2021).
Martini, C. L. et al. Cellular Growth Arrest and Efflux Pumps Are Associated With Antibiotic Persisters in Streptococcus pyogenes Induced in Biofilm-Like Environments. Front. Microbiol. 12, 716628 (2021).
Anderson, G., Dodson, K., Hooton, T. & Hultgren, S. Intracellular bacterial communities of uropathogenic in urinary tract pathogenesis. Trends Microbiol. 12, 424–430 (2004).
Robino, L. et al. Presence of intracellular bacterial communities in uroepithelial cells, a potential reservoir in symptomatic and non-symptomatic people. BMC Infect. Dis. 24, 590 (2024).
Morrison, J. J. et al. Metabolic flux regulates growth transitions and antibiotic tolerance in uropathogenic Escherichia coli. J. Bacteriol. 206, e00162–24 (2024).
MacNair, C. R. & Tan, M. The role of bacterial membrane vesicles in antibiotic resistance. Ann. N. Y. Acad. Sci. 1519, 63–73 (2023).
Schwechheimer, C. & Kuehn, M. J. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat. Rev. Microbiol. 13, 605–619 (2015).
Cao, Y. & Lin, H. Characterization and function of membrane vesicles in Gram-positive bacteria. Appl. Microbiol. Biotechnol. 105, 1795–1801 (2021).
Hua, Y. et al. Outer membrane vesicles-transmitted virulence genes mediate the emergence of new antimicrobial-resistant hypervirulent Klebsiella pneumoniae. Emerg. Microbes Infect. 11, 1281–1292 (2022).
Lehmkuhl, J. et al. Role of membrane vesicles in the transmission of vancomycin resistance in Enterococcus faecium. Sci. Rep. 14, 1895 (2024).
Rumbo, C. et al. Horizontal Transfer of the OXA-24 Carbapenemase Gene via Outer Membrane Vesicles: a New Mechanism of Dissemination of Carbapenem Resistance Genes in Acinetobacter baumannii. Antimicrob. Agents Chemother. 55, 3084–3090 (2011).
Kim, S. W. et al. The Importance of Porins and β-Lactamase in Outer Membrane Vesicles on the Hydrolysis of β-Lactam Antibiotics. Int. J. Mol. Sci. 21, 2822 (2020).
Dhital, S. et al. Neisseria gonorrhoeae -derived outer membrane vesicles package β-lactamases to promote antibiotic resistance. microLife 3, uqac013 (2022).
Olovo, C. V., Wiredu Ocansey, D. K., Ji, Y., Huang, X. & Xu, M. Bacterial membrane vesicles in the pathogenesis and treatment of inflammatory bowel disease. Gut Microbes 16, 2341670 (2024).
Murray, B. O., Dawson, R. A., Alsharaf, L. M. & Anne Winter, J. Protective effects of Helicobacter pylori membrane vesicles against stress and antimicrobial agents. Microbiology 166, 751–758 (2020).
Kulkarni, H. M., Nagaraj, R. & Jagannadham, M. V. Protective role of E. coli outer membrane vesicles against antibiotics. Microbiol. Res. 181, 1–7 (2015).
Manning, A. J. & Kuehn, M. J. Contribution of bacterial outer membrane vesicles to innate bacterial defense. BMC Microbiol. 11, 258 (2011).
Andreoni, F. et al. Antibiotics Stimulate Formation of Vesicles in Staphylococcus aureus in both Phage-Dependent and -Independent Fashions and via Different Routes. Antimicrob. Agents Chemother. 63, e01439–18 (2019).
Liu, X. et al. Research Progress on Bacterial Membrane Vesicles and Antibiotic Resistance. Int. J. Mol. Sci. 23, 11553 (2022).
Li, Q. et al. Sub-MIC Antibiotics Modulate Productions of Outer Membrane Vesicles in Tigecycline-Resistant Escherichia coli. Antibiotics 13, 276 (2024).
Mendez, J. A. et al. Extracellular Proteome of a Highly Invasive Multidrug-resistant Clinical Strain of Acinetobacter baumannii. J. Proteome Res. 11, 5678–5694 (2012).
Yonezawa, H. et al. Analysis of outer membrane vesicle protein involved in biofilm formation of Helicobacter pylori. Anaerobe 17, 388–390 (2011).
Zhao, Z. et al. Regulation of the formation and structure of biofilms by quorum sensing signal molecules packaged in outer membrane vesicles. Sci. Total Environ. 806, 151403 (2022).
Mickiewicz, K. M. et al. Possible role of L-form switching in recurrent urinary tract infection. Nat. Commun. 10, 4379 (2019).
Kilcher, S. & Loessner, M. J. Engineering Bacteriophages as Versatile Biologics. Trends Microbiol. 27, 355–367 (2019).
Kawai, Y. et al. Cell Growth of Wall-Free L-Form Bacteria Is Limited by Oxidative Damage. Curr. Biol. 25, 1613–1618 (2015).
Day, N. J., Santucci, P. & Gutierrez, M. G. Host cell environments and antibiotic efficacy in tuberculosis. Trends Microbiol. 32, 270–279 (2024).
Walsh, J. et al. Impact of host and environmental factors on β-glucuronidase enzymatic activity: implications for gastrointestinal serotonin. Am. J. Physiol. -Gastrointest. Liver Physiol. 318, G816–G826 (2020).
Nussbaumer-Pröll, A. K. et al. Impact of erythrocytes on bacterial growth and antimicrobial activity of selected antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 38, 485–495 (2019).
Ahmed, H., Bergmann, F. & Zeitlinger, M. Protein Binding in Translational Antimicrobial Development-Focus on Interspecies Differences. Antibiotics 11, 923 (2022).
Ledger, E. V. K., Mesnage, S. & Edwards, A. M. Human serum triggers antibiotic tolerance in Staphylococcus aureus. Nat. Commun. 13, 2041 (2022).
Lin, Q., Pilewski, J. M. & Di, Y. P. Acidic Microenvironment Determines Antibiotic Susceptibility and Biofilm Formation of Pseudomonas aeruginosa. Front. Microbiol. 12, 747834 (2021).
Xu, Y. et al. An acid-tolerance response system protecting exponentially growing Escherichia coli. Nat. Commun. 11, 1496 (2020).
Loffredo, M. R. et al. The pH-Insensitive Antimicrobial and Antibiofilm Activities of the Frog Skin Derived Peptide Esc(1-21): Promising Features for Novel Anti-Infective Drugs. Antibiotics 13, 701 (2024).
Martín-Gutiérrez, G. et al. Urinary Tract Physiological Conditions Promote Ciprofloxacin Resistance in Low-Level-Quinolone-Resistant Escherichia coli. Antimicrob. Agents Chemother. 60, 4252–4258 (2016).
Cunha, B. A. An infectious disease and pharmacokinetic perspective on oral antibiotic treatment of uncomplicated urinary tract infections due to multidrug-resistant Gram-negative uropathogens: the importance of urinary antibiotic concentrations and urinary pH. Eur. J. Clin. Microbiol. Infect. Dis. 35, 521–526 (2016).
Herrera-Espejo, S. et al. Acidic Urine pH and Clinical Outcome of Lower Urinary Tract Infection in Kidney Transplant Recipients Treated with Ciprofloxacin and Fosfomycin. Antibiotics 13, 116 (2024).
Kincses, A. et al. The Relationship between Antibiotic Susceptibility and pH in the Case of Uropathogenic Bacteria. Antibiotics 10, 1431 (2021).
Abbott, I. J. et al. Evaluation of pooled human urine and synthetic alternatives in a dynamic bladder infection in vitro model simulating oral fosfomycin therapy. J. Microbiol. Methods 171, 105861 (2020).
Neve, R. L., Carrillo, B. D. & Phelan, V. V. Impact of Artificial Sputum Medium Formulation on Pseudomonas aeruginosa Secondary Metabolite Production. J. Bacteriol. 203, https://doi.org/10.1128/jb.00250-21 (2021).
Tognon, M., Köhler, T., Luscher, A. & van Delden, C. Transcriptional profiling of Pseudomonas aeruginosa and Staphylococcus aureus during in vitro co-culture. BMC Genomics 20, 30 (2019).
Chapot, V., Effenberg, L., Dohmen-Ruetten, J., Buer, J. & Kehrmann, J. Evaluation of the Accelerate Pheno System for Rapid Identification and Antimicrobial Susceptibility Testing of Positive Blood Culture Bottles Inoculated with Primary Sterile Specimens from Patients with Suspected Severe Infections. J. Clin. Microbiol. 59, https://doi.org/10.1128/jcm.02637-20 (2021).
Alonso-Tarrés, C. et al. Bacteriuria and phenotypic antimicrobial susceptibility testing in 45 min by point-of-care Sysmex PA-100 System: first clinical evaluation. Eur. J. Clin. Microbiol. Infect. Dis. 43, 1533–1543 (2024).
Ersoy, S. C. et al. Correcting a fundamental flaw in the paradigm for antimicrobial susceptibility testing. EBioMedicine 20, 173–181 (2017).
Heithoff, D. M. et al. Re-evaluation of FDA-approved antibiotics with increased diagnostic accuracy for assessment of antimicrobial resistance. Cell Rep. Med. 4, 101023 (2023).
Baker, E. J., Allcott, G., Molloy, A. & Cox, J. A. G. Cystic fibrosis sputum media induces an overall loss of antibiotic susceptibility in Mycobacterium abscessus. Npj Antimicrob. Resist. 2, 1–8 (2024).
Cornforth, D. M. et al. Pseudomonas aeruginosa transcriptome during human infection. Proc. Natl. Acad. Sci. USA 115, E5125–E5134 (2018).
Cottell, J. L. & Webber, M. A. Experiences in fosfomycin susceptibility testing and resistance mechanism determination in Escherichia coli from urinary tract infections in the UK. J. Med. Microbiol. 68, 161–168 (2019).
Ciofu, O., Moser, C., Jensen, P. Ø & Høiby, N. Tolerance and resistance of microbial biofilms. Nat. Rev. Microbiol. 20, 621–635 (2022).
Padron, G. C., Shuppara, A. M., Palalay, J.-J. S., Sharma, A. & Sanfilippo, J. E. Bacteria in fluid flow. J. Bacteriol. 205, e00400–e00422 (2023).
Perry, E. K. & Tan, M.-W. Bacterial biofilms in the human body: prevalence and impacts on health and disease. Front. Cell. Infect. Microbiol. 13, 1237164 (2023).
Römling, U. & Balsalobre, C. Biofilm infections, their resilience to therapy and innovative treatment strategies. J. Intern. Med. 272, 541–561 (2012).
Sharma, D., Misba, L. & Khan, A. U. Antibiotics versus biofilm: an emerging battleground in microbial communities. Antimicrob. Resist. Infect. Control 8, 76 (2019).
Guzmán-Soto, I. et al. Mimicking biofilm formation and development: Recent progress in in vitro and in vivo biofilm models. iScience 24, 102443 (2021).
Rumbaugh, K. P. & Whiteley, M. Towards improved biofilm models. Nat. Rev. Microbiol. 23, 57–66 (2025).
Coenye, T. Biofilm antimicrobial susceptibility testing: where are we and where could we be going?. Clin. Microbiol. Rev. 36, e00024–23 (2023).
Thieme, L. et al. MBEC Versus MBIC: the Lack of Differentiation between Biofilm Reducing and Inhibitory Effects as a Current Problem in Biofilm Methodology. Biol. Proced. Online 21, 18 (2019).
Cruz, C. D., Shah, S. & Tammela, P. Defining conditions for biofilm inhibition and eradication assays for Gram-positive clinical reference strains. BMC Microbiol. 18, 173 (2018).
Bjarnsholt, T. et al. The in vivo biofilm. Trends Microbiol. 21, 466–474 (2013).
Cometta, S., Hutmacher, D. W. & Chai, L. In vitro models for studying implant-associated biofilms - A review from the perspective of bioengineering 3D microenvironments. Biomaterials 309, 122578 (2024).
Vyas, H. K. N., Xia, B. & Mai-Prochnow, A. Clinically relevant in vitro biofilm models: a need to mimic and recapitulate the host environment. Biofilm 4, 100069 (2022).
Han, Q. et al. Regrowth of microcosm biofilms on titanium surfaces after various antimicrobial treatments. Front. Microbiol. 10, 2693 (2019).
Maset, R. G. et al. Combining SNAPs with antibiotics shows enhanced synergistic efficacy against S. aureus and P. aeruginosa biofilms. Npj Biofilms Microbiomes 9, 1–17 (2023).
Jafari, N. V. & Rohn, J. L. An immunoresponsive three-dimensional urine-tolerant human urothelial model to study urinary tract infection. Front. Cell. Infect. Microbiol. 13, 1128132 (2023).
Flores, C. et al. A human urothelial microtissue model reveals shared colonization and survival strategies between uropathogens and commensals. Sci. Adv. 9, eadi9834 (2023).
Sharma, K. et al. Dynamic persistence of UPEC intracellular bacterial communities in a human bladder-chip model of urinary tract infection. eLife 10, e66481 (2021).
Murray, B. O. et al. Recurrent Urinary Tract Infection: A Mystery in Search of Better Model Systems. Front. Cell. Infect. Microbiol. 11, 691210 (2021).
Mayr, F. B., Yende, S. & Angus, D. C. Epidemiology of severe sepsis. Virulence 5, 4–11 (2014).
Foxman, B. Urinary Tract Infection Syndromes: Occurrence, Recurrence, Bacteriology, Risk Factors, and Disease Burden. Infect. Dis. Clin. North Am. 28, 1–13 (2014).
Doern, G. V. & Brecher, S. M. The Clinical Predictive Value (or Lack Thereof) of the Results of In Vitro Antimicrobial Susceptibility Tests. J. Clin. Microbiol. 49, S11–S14 (2011).
Cui, S. & Kim, E. Quorum sensing and antibiotic resistance in polymicrobial infections. Commun. Integr. Biol. 17, 2415598 (2024).
Yu, V. L. et al. An International Prospective Study of Pneumococcal Bacteremia: Correlation with In Vitro Resistance, Antibiotics Administered, and Clinical Outcome. Clin. Infect. Dis. 37, 230–237 (2003).
Somayaji, R. et al. Antimicrobial susceptibility testing (AST) and associated clinical outcomes in individuals with cystic fibrosis: A systematic review. J. Cyst. Fibros. 18, 236–243 (2019).
Bjarnsholt, T. et al. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr. Pulmonol. 44, 547–558 (2009).
Harrington, N. E., Sweeney, E. & Harrison, F. Building a better biofilm - Formation of in vivo-like biofilm structures by Pseudomonas aeruginosa in a porcine model of cystic fibrosis lung infection. Biofilm 2, 100024 (2020).
Baltimore, R. S., Christie, C. D. & Smith, G. J. Immunohistopathologic localization of Pseudomonas aeruginosa in lungs from patients with cystic fibrosis. Implications for the pathogenesis of progressive lung deterioration. Am. Rev. Respir. Dis. 140, 1650–1661 (1989).
M02 Ed14 | Performance Standards for Antimicrobial Disk Susceptibility Tests, 14th Edition. Clinical & Laboratory Standards Institute https://clsi.org/standards/products/microbiology/documents/m02/
Kowalska-Krochmal, B. & Dudek-Wicher, R. The Minimum Inhibitory Concentration of Antibiotics: Methods, Interpretation, Clinical Relevance. Pathogens 10, 165 (2021).
Balouiri, M., Sadiki, M. & Ibnsouda, S. K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm. Anal. 6, 71 (2016).
Gajic, I. et al. Antimicrobial Susceptibility Testing: A Comprehensive Review of Currently Used Methods. Antibiotics 11, 427 (2022).
Ishak, A., Mazonakis, N., Spernovasilis, N., Akinosoglou, K. & Tsioutis, C. Bactericidal versus bacteriostatic antibacterials: clinical significance, differences and synergistic potential in clinical practice. J. Antimicrob. Chemother. 80, 1–17 (2025).
Zhang, Y., Kepiro, I., Ryadnov, M. G. & Pagliara, S. Single Cell Killing Kinetics Differentiate Phenotypic Bacterial Responses to Different Antibacterial Classes. Microbiol. Spectr. 11, e03667–22 (2023).
Levison, M. E. & Levison, J. H. Pharmacokinetics and Pharmacodynamics of Antibacterial Agents. Infect. Dis. Clin. North Am. 23, 791–vii (2009).
Bjarnsholt, T. et al. The importance of understanding the infectious microenvironment. Lancet Infect. Dis. 22, e88–e92 (2022).
Hassall, J., Coxon, C., Patel, V. C., Goldenberg, S. D. & Sergaki, C. Limitations of current techniques in clinical antimicrobial resistance diagnosis: examples and future prospects. Npj Antimicrob. Resist. 2, 16 (2024).
M26-A: Methods for Determining Bactericidal Activity of Antimicrobial Agents; Approved Guideline.
Foerster, S., Unemo, M., Hathaway, L. J., Low, N. & Althaus, C. L. Time-kill curve analysis and pharmacodynamic modelling for in vitro evaluation of antimicrobials against Neisseria gonorrhoeae. BMC Microbiol. 16, 216 (2016).
Antibiotic tolerance among clinical isolates: mechanisms, detection, prevalence, and significance | Clinical Microbiology Reviews. https://journals.asm.org/doi/10.1128/cmr.00106-24.
Mueller, M., De La Peña, A. & Derendorf, H. Issues in Pharmacokinetics and Pharmacodynamics of Anti-Infective Agents: Kill Curves versus MIC. Antimicrob. Agents Chemother. 48, 369–377 (2004).
Gumbo, T. et al. Hollow-fibre system model of tuberculosis reproducibility and performance specifications for best practice in drug and combination therapy development. J. Antimicrob. Chemother. 78, 953–964 (2023).
Hammond, R. J. H. Using Hollow Fiber to Model Treatment of Antimicrobial-Resistant Organisms. Methods Mol. Biol. Clifton NJ 2833, 57–64 (2024).
Kloprogge, F., Hammond, R., Kipper, K., Gillespie, S. H. & Della Pasqua, O. Mimicking in-vivo exposures to drug combinations in-vitro: anti-tuberculosis drugs in lung lesions and the hollow fiber model of infection. Sci. Rep. 9, 13228 (2019).
Narasimhan, V. et al. Nucleic Acid Amplification-Based Technologies (NAAT)—Toward Accessible, Autonomous, and Mobile Diagnostics. Adv. Mater. Technol. 8, 2300230 (2023).
Luo, J., Yu, J., Yang, H. & Wei, H. Parallel susceptibility testing of bacteria through culture-quantitative PCR in 96-well plates. Int. J. Infect. Dis. 70, 86–92 (2018).
Framing Bacterial Genomics for Public Health (Care) | Journal of Clinical Microbiology. https://journals.asm.org/doi/10.1128/jcm.00135-21.
Weinmaier, T. et al. Validation and Application of Long-Read Whole-Genome Sequencing for Antimicrobial Resistance Gene Detection and Antimicrobial Susceptibility Testing. Antimicrob. Agents Chemother. 67, e01072–22 (2022).
Tamae, C. et al. Determination of antibiotic hypersensitivity among 4,000 single-gene-knockout mutants of Escherichia coli. J. Bacteriol. 190, 5981–5988 (2008).
Suzuki, S., Horinouchi, T. & Furusawa, C. Prediction of antibiotic resistance by gene expression profiles. Nat. Commun. 5, 5792 (2014).
Ellington, M. J. et al. The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: report from the EUCAST Subcommittee. Clin. Microbiol. Infect. 23, 2–22 (2017).
Yoon, E.-J. & Jeong, S. H. MALDI-TOF mass spectrometry technology as a tool for the rapid diagnosis of antimicrobial resistance in bacteria. Antibiot. Basel Switz. 10, 982 (2021).
Wieser, A., Schneider, L., Jung, J. & Schubert, S. MALDI-TOF MS in microbiological diagnostics-identification of microorganisms and beyond (mini review). Appl. Microbiol. Biotechnol. 93, 965–974 (2012).
Oviaño, M. & Rodríguez-Sánchez, B. MALDI-TOF mass spectrometry in the 21st century clinical microbiology laboratory. Enfermedades Infecc. Microbiol. Clin. Engl. Ed 39, 192–200 (2021).
Idelevich, E. A. & Becker, K. Matrix-assisted laser desorption ionization-time of flight mass spectrometry for antimicrobial susceptibility testing. J. Clin. Microbiol. 59, e0181419 (2021).
Fedrigo, N. H. et al. Pharmacodynamic evaluation of fosfomycin against Escherichia coli and Klebsiella spp. from urinary tract infections and the influence of pH on fosfomycin activities. Antimicrob. Agents Chemother. 61, e02498–16 (2017).
Debets-Ossenkopp, Y. J. & MacLaren, D. M. Effect of an acidic environment on the susceptibility of helicobacterpyiori to trospectomycin and other antimicrobial agents. Eur. J. Clin. Microbiol. Infect. Dis. 14, 353–355 (1995).
Aust, A.-C. et al. Influence of kidney environment parameters on antibiotic efficacy against uropathogenic Escherichia coli. Eur. Urol. Focus 10, 742–750 (2024).
Withman, B., Gunasekera, T. S., Beesetty, P., Agans, R. & Paliy, O. Transcriptional responses of uropathogenic Escherichia coli to increased environmental osmolality caused by salt or urea. Infect. Immun. 81, 80–89 (2013).
Landry, R. M., An, D., Hupp, J. T., Singh, P. K. & Parsek, M. R. Mucin–Pseudomonas aeruginosa interactions promote biofilm formation and antibiotic resistance. Mol. Microbiol. 59, 142–151 (2006).
Vasiljevs, S., Gupta, A. & Baines, D. Effect of glucose on growth and co-culture of Staphylococcus aureus and Pseudomonas aeruginosa in artificial sputum medium. Heliyon 9, e21469 (2023).
Dörr, T., Lewis, K. & Vulić, M. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 5, e1000760 (2009).
Peyrusson, F. et al. Intracellular Staphylococcus aureus persisters upon antibiotic exposure. Nat. Commun. 11, 2200 (2020).
Michiels, J. E., Van Den Bergh, B., Verstraeten, N., Fauvart, M. & Michiels, J. In vitro emergence of high persistence upon periodic aminoglycoside challenge in the ESKAPE pathogens. Antimicrob. Agents Chemother. 60, 4630–4637 (2016).
Brinkman, F. S. L., Macfarlane, E. L. A., Warrener, P. & Hancock, R. E. W. Evolutionary Relationships among Virulence-Associated Histidine Kinases. Infect. Immun. 69, 5207–5211 (2001).
Murtha, A. N. et al. High-level carbapenem tolerance requires antibiotic-induced outer membrane modifications. PLoS Pathog. 18, e1010307 (2022).
Nalca, Y. et al. Quorum-sensing antagonistic activities of azithromycin in Pseudomonas aeruginosa PAO1: a global approach. Antimicrob. Agents Chemother. 50, 1680–1688 (2006).
Hentzer, M. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 22, 3803–3815 (2003).
Leung, V. & Lévesque, C. M. A stress-inducible quorum-sensing peptide mediates the formation of persister cells with noninherited multidrug tolerance. J. Bacteriol. 194, 2265–2274 (2012).
Muthuramalingam, M., White, J. C., Murphy, T., Ames, J. R. & Bourne, C. R. The toxin from a ParDE toxin-antitoxin system found in Pseudomonas aeruginosa offers protection to cells challenged with anti-gyrase antibiotics. Mol. Microbiol. 111, 441–454 (2019).
Choudhary, E., Sharma, R., Kumar, Y. & Agarwal, N. Conditional silencing by CRISPRi reveals the role of DNA gyrase in formation of drug-tolerant persister population in Mycobacterium tuberculosis. Front. Cell. Infect. Microbiol. 9, 70 (2019).
Bryson, D., Hettle, A. G., Boraston, A. B. & Hobbs, J. K. Clinical mutations that partially activate the stringent response confer multidrug tolerance in Staphylococcus aureus. Antimicrob. Agents Chemother. 64, e02103–e02119 (2020).
Dutta, N. K. et al. Inhibiting the stringent response blocks Mycobacterium tuberculosis entry into quiescence and reduces persistence. Sci. Adv. 5, eaav2104 (2019).
Anderl, J. N., Zahller, J., Roe, F. & Stewart, P. S. Role of Nutrient limitation and stationary-phase existence in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob. Agents Chemother. 47, 1251–1256 (2003).
Adams, K. N. et al. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145, 39–53 (2011).
Srinivasan, V. B. & Rajamohan, G. KpnEF, a New Member of the Klebsiella pneumoniae cell envelope stress response regulon, Is an SMR-type efflux pump involved in broad-spectrum antimicrobial resistance. Antimicrob. Agents Chemother. 57, 4449–4462 (2013).
Kaiser, P. et al. Cecum lymph node dendritic cells harbor slow-growing bacteria phenotypically tolerant to antibiotic treatment. PLoS Biol. 12, e1001793 (2014).
Maredia, R. et al. Vesiculation from Pseudomonas aeruginosa under SOS. Sci. World J. 2012, 1–18 (2012).
Kim, S. W. et al. Significant increase in the secretion of extracellular vesicles and antibiotics resistance from methicillin-resistant Staphylococcus aureus induced by ampicillin stress. Sci. Rep. 10, 21066 (2020).
Zheng, Y., Cai, Y., Sun, T., Li, G. & An, T. Response mechanisms of resistance in L-form bacteria to different target antibiotics: Implications from oxidative stress to metabolism. Environ. Int. 187, 108729 (2024).
Merritt, J. H., Kadouri, D. E. & O’Toole, G. A. Growing and analyzing static biofilms. Curr. Protoc. Microbiol. 0 1, Unit-1B.1 (2005).
Crivello, G., Fracchia, L., Ciardelli, G., Boffito, M. & Mattu, C. In vitro models of bacterial biofilms: innovative tools to improve understanding and treatment of infections. Nanomaterials 13, 904 (2023).
Straub, H. et al. A microfluidic platform for in situ investigation of biofilm formation and its treatment under controlled conditions. J. Nanobiotechnol.18, 166 (2020).
Gomes, I. B. et al. Standardized reactors for the study of medical biofilms: a review of the principles and latest modifications. Crit. Rev. Biotechnol. 38, 657–670 (2018).
Lee, J.-H., Kaplan, J. B. & Lee, W. Y. Microfluidic devices for studying growth and detachment of Staphylococcus epidermidis biofilms. Biomed. Microdev.10, 489–498 (2008).
Blanco-Cabra, N. et al. A new BiofilmChip device for testing biofilm formation and antibiotic susceptibility. Npj Biofilms Microbiomes 7, 1–9 (2021).
Sriramulu, D. D., Lünsdorf, H., Lam, J. S. & Römling, U. Microcolony formation: a novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J. Med. Microbiol. 54, 667–676 (2005).
Palmer, K. L., Aye, L. M. & Whiteley, M. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J. Bacteriol. 189, 8079–8087 (2007).
Sun, Y., Dowd, S. E., Smith, E., Rhoads, D. D. & Wolcott, R. D. In vitro multispecies Lubbock chronic wound biofilm model. Wound Repair Regen. Off. Publ. Wound Heal. Soc. Eur. Tissue Repair Soc. 16, 805–813 (2008).
Werthén, M. et al. An in vitro model of bacterial infections in wounds and other soft tissues. APMIS Acta Pathol. Microbiol. Immunol. Scand. 118, 156–164 (2010).
Charles, C. A., Ricotti, C. A., Davis, S. C., Mertz, P. M. & Kirsner, R. S. Use of tissue-engineered skin to study in vitro biofilm development. Dermatol. Surg. Off. Publ. Am. Soc. Dermatol. Surg. Al 35, 1334–1341 (2009).
high-throughput microfluidic dental plaque biofilm system to visualize and quantify the effect of antimicrobials | Journal of Antimicrobial Chemotherapy | Oxford Academic. https://academic.oup.com/jac/article-abstract/68/11/2550/828884?redirectedFrom=fulltext.
Samarian, D. S., Jakubovics, N. S., Luo, T. L. & Rickard, A. H. Use of a high-throughput in vitro microfluidic system to develop oral multi-species biofilms. J. Vis. Exp. JoVE 52467 https://doi.org/10.3791/52467 (2014).
Lamret, F. et al. Human Osteoblast-Conditioned Media Can Influence Staphylococcus aureus Biofilm Formation. Int. J. Mol. Sci. 23, 14393 (2022).
Chutipongtanate, S. & Thongboonkerd, V. Systematic comparisons of artificial urine formulas for in vitro cellular study. Anal. Biochem. 402, 110–112 (2010).
Ipe, D. S. & Ulett, G. C. Evaluation of the in vitro growth of urinary tract infection-causing gram-negative and gram-positive bacteria in a proposed synthetic human urine (SHU) medium. J. Microbiol. Methods 127, 164–171 (2016).
Rimbi, P. T. et al. Enhancing a multi-purpose artificial urine for culture and gene expression studies of uropathogenic Escherichia coli strains. J. Appl. Microbiol. 135, lxae067 (2024).
Townsend, E. M., Moat, J. & Jameson, E. CAUTI’s next top model - Model dependent Klebsiella biofilm inhibition by bacteriophages and antimicrobials. Biofilm 2, 100038 (2020).
Zaborskyte, G., Wistrand-Yuen, E., Hjort, K., Andersson, D. I. & Sandegren, L. Modular 3D-printed peg biofilm device for flexible setup of surface-related biofilm studies. Front. Cell. Infect. Microbiol. 11, 802303 (2022).
Alves, D. R. et al. Development of a High-Throughput ex-Vivo Burn Wound Model Using Porcine Skin, and Its Application to Evaluate New Approaches to Control Wound Infection. Front. Cell. Infect. Microbiol. 8, 196 (2018).
Maset, R. G. et al. Combining SNAPs with antibiotics shows enhanced synergistic efficacy against S. aureus and P. aeruginosa biofilms. NPJ Biofilms Microbiomes 9, 36 (2023).
Wurbs, A. et al. A human ex vivo skin model breaking boundaries. Sci. Rep. 14, 24054 (2024).
Du, Q. et al. Candida albicans promotes tooth decay by inducing oral microbial dysbiosis. ISME J. 15, 894–908 (2021).
de Poel, E. et al. FDA-approved drug screening in patient-derived organoids demonstrates potential of drug repurposing for rare cystic fibrosis genotypes. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 22, 548–559 (2023).
Sollier, J. et al. Revitalizing antibiotic discovery and development through in vitro modelling of in-patient conditions. Nat. Microbiol. 9, 1–3 (2024).
Aguilar, C. et al. Organoids as host models for infection biology – a review of methods. Exp. Mol. Med. 53, 1471–1482 (2021).
Swart, A. L. et al. Pseudomonas aeruginosa breaches respiratory epithelia through goblet cell invasion in a microtissue model. Nat. Microbiol. 9, 1725–1737 (2024).
Meirelles, L. A. et al. Pseudomonas aeruginosa faces a fitness trade-off between mucosal colonization and antibiotic tolerance during airway infection. Nat. Microbiol. 9, 3284–3303 (2024).
Han, X. et al. Creating a more perfect union: modeling intestinal bacteria-epithelial interactions using organoids. Cell. Mol. Gastroenterol. Hepatol. 12, 769–782 (2021).
Anonye, B. O. et al. Probing Clostridium difficile Infection in Complex Human Gut Cellular Models. Front. Microbiol. 10, 879 (2019).
Dash, S. K., Marques, C. N. H. & Mahler, G. J. Small intestine on a chip demonstrates physiologic mucus secretion in the presence of Lacticaseibacillus rhamnosus biofilm. Biotechnol. Bioeng. 1–12 (2025).
Łaniewski, P. & Herbst-Kralovetz, M. M. Bacterial vaginosis and health-associated bacteria modulate the immunometabolic landscape in 3D model of human cervix. Npj Biofilms Microbiomes 7, 1–17 (2021).
Redman, W. K. et al. Efficacy and safety of biofilm dispersal by glycoside hydrolases in wounds. Biofilm 3, 100061 (2021).
Nissanka, M. C., Dilhari, A., Wijesinghe, G. K. & Weerasekera, M. M. Advances in experimental bladder models: bridging the gap between in vitro and in vivo approaches for investigating urinary tract infections. BMC Urol. 24, 206 (2024).
Conover, M. S., Flores-Mireles, A. L., Hibbing, M. E., Dodson, K. & Hultgren, S. J. Establishment and Characterization of UTI and CAUTI in a Mouse Model. J. Vis. Exp. JoVE e52892, https://doi.org/10.3791/52892 (2015).
Acknowledgements
We thank the EPSRC for their generous funding (EP/V026623/1). The funder played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript. We also thank Richard P. Grant for helpful discussion. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Author information
Authors and Affiliations
Contributions
J.R. and R.G.M. conceived the article, and J.R. oversaw the funding application. J.R., R.G.M., V.C., N.Y., D.B., J.Y., B.O.M., and A.A.J. researched, co-wrote, co-edited and approved the manuscript, figures and tables.
Corresponding authors
Ethics declarations
Competing interests
Author JR holds share options in AtoCap Ltd. and declares no non-financial competing interests. All other authors declare no financial or non-financial competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Garcia-Maset, R., Chu, V., Yuen, N. et al. Effect of host microenvironment and bacterial lifestyles on antimicrobial sensitivity and implications for susceptibility testing. npj Antimicrob Resist 3, 42 (2025). https://doi.org/10.1038/s44259-025-00113-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44259-025-00113-3
