
Abstract
Background
Glioblastoma (GB), an aggressive brain malignancy with a poor prognosis of 1.5–2 years, rarely exhibits extracranial metastasis (ECM). However, metabolic reprogramming has emerged as a key driver of GB progression and invasiveness. This study presents a rare case of recurrent GB with scalp metastasis, exploring how metabolic shifts enable GB cells to evade treatment and adapt to hostile environments, offering insights for developing innovative therapies.
Methods
Tandem mass spectrometry (MS/MS) was employed to analyze amino acid profiles in both the recurrent and metastatic stages of GB. Systems biology approaches were used to uncover genetic alterations and metabolic reprogramming associated with the progression from recurrence to metastasis.
Results
Our analysis revealed distinct amino acid utilization patterns in a patient with a molecular phenotype of wild-type IDH-1&2, TERT mutation, non-mutated BRAF and EGFR, and non-methylated MGMT. During recurrence and metastasis, significant differences in amino acid profiles were observed between blood and cerebrospinal fluid (CSF) samples. Additionally, protein-protein interaction (PPI) analysis identified key genomic drivers potentially responsible for the transition from recurrent to metastatic GB.
Conclusions
Beyond established risk factors such as craniotomy, biopsies, ventricular shunting, and radiation therapy, our findings suggest that metabolic reprogramming plays a crucial role in the transition from recurrent to metastatic GB. Targeting these metabolic shifts could provide new avenues for managing and preventing extracranial metastasis in GB, making this an important focus for future research.
Similar content being viewed by others

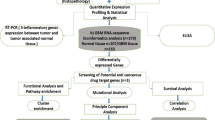

Introduction
Glioblastoma (GB) is adults’ most common malignant tumour of the central nervous system (CNS), with a median survival rate of approximately 14.6 months [1, 2]. The majority of patients with glioblastoma face progression of the disease locally, while the occurrence of extracranial metastasis remains exceptionally uncommon, affecting only about 0.4–0.5% of cases [3]. Among this rare subset, the prognosis is notably grim, with a median survival of just 1.5 months following the onset of metastasis [4]. The most commonly affected sites include the lungs, pleura, lymph nodes, bone marrow, bones, and liver [5]. Although there has been an increase in reported cases of extracranial metastasis in recent years, the underlying causes and mechanisms remain unclear and are not yet fully understood.
Nearly a century ago, Otto Warburg’s groundbreaking work identified aerobic glycolysis as a hallmark of cancer, laying the foundation for studying cancer metabolism [6]. While early research focused heavily on central carbon metabolism pathways such as the citric acid cycle and pentose phosphate pathway, recent advances have highlighted the critical role of non-carbon metabolism in supporting cancer cell growth and survival. Among these, amino acid metabolism has emerged as a key area of interest, given its profound influence on cellular processes [7, 8].
In cancer cells, amino acid metabolism is reprogrammed to support critical functions: protein synthesis, nucleic acid biosynthesis, energy production, redox balance, ammonia detoxification, epigenetic regulation, and activation of pathways like mTORC and autophagy, highlighting its role in sustaining tumor growth and survival [9, 10]. These multifaceted roles underscore the significance of disrupted amino acid metabolism in cancer and highlight its potential as a target for therapeutic interventions, particularly in the evolving context of the tumor microenvironment [11,12,13].
Cell transformation, or tumorigenesis, involves metabolic reprogramming, enabling cancer cells, including GB, to adapt energy production for enhanced survival, invasiveness, and therapy resistance [10, 14, 15]. Extensive evidence shows that multiple energy pathways are altered during cellular transformation, enabling cancer cells to meet their high demands for growth and repair [14]. Among the critical molecules involved in this process are amino acids (AAs), which play diverse and essential roles in cellular function. Beyond serving as the “building blocks” of proteins, amino acids contribute to tumor progression in several ways: (a) certain AAs are converted into α-keto acids, which are further transformed into glucose, fats, or ketone bodies to fuel energy production [15] ;(b) some AAs act as neurotransmitters or hormone precursors, influencing cellular communication and behavior [16] ; (c) glycine, serine, methionine, and histidine provide one-carbon units essential for DNA synthesis and antioxidant defenses, protecting cancer cells from oxidative damage [16]; and (d) AAs serve as epigenetic modifiers, influencing which genes are turned on or off and, consequently, affecting cell phenotypes [14]. These metabolic shifts in amino acid utilization are particularly prominent in GB, where they promote tumor survival and invasion and contribute to resistance against standard treatments such as chemotherapy and radiation [17]. This highlights the critical role of amino acid metabolism in the progression of GB, particularly in the transition from recurrence to metastasis.
In this study, we report a case of extracranial GB metastasis and identify a shift in amino acid consumption preferences during the progression from recurrence to metastasis. Additionally, we apply systems biology to analyze the metabolite-related pathways involved in this transition.
Method
Genetic profile
The genetic profiling of the selected genes (IDH-1&2, TERT, BRAF, EGFR, and MGMT) was conducted following the methodology detailed in our previous work [1], which thoroughly describes the techniques utilized.
Cerebrospinal fluid (CSF) sample
Informed consent was obtained, and the patient was positioned in the left lateral decubitus position. The L4-L5 interspinous space was identified, anesthetized with 10 mL of lidocaine, and prepared with antiseptic. A sterile drape was applied, and a 21-gauge spinal needle was inserted, bevel aligned parallel to the Dural fibers, and angled cephalad. After feeling the characteristic “pop” indicating Dural penetration, CSF was confirmed via intermittent withdrawal. CSF was collected, the stylet reinserted, the needle removed, and the site dressed with a sterile bandage. The patient was positioned supine for recovery.
Amino acid analysis in CSF was conducted using tandem mass spectrometry (Shimadzu CLAM2040, MS/MS) with CHROMSYSTEM MS/MS kits. A 10 µL CSF aliquot was mixed with 200 µL of internal standard solution, vortexed, and centrifuged at 6000 rpm for 10 minutes at 4 °C. The 150 µL supernatant was evaporated at 45 °C under nitrogen gas for 20 minutes, treated with 50 µL each of acetyl chloride and 1-butanol, shaken, and incubated at 65 °C for 15 minutes. After drying under nitrogen at 45 °C, 100 µL of 75% acetonitrile was added, shaken, and 10 µL was injected into the mass spectrometer. The analysis ran at a 150 µL/min flow rate for 2.074 minutes using multiple reaction monitoring (MRM) [18].
Dried blood spot (DBS) sample
DBS samples were collected via heel prick during recurrent and metastatic phases, dried on Whatman 903 cards, and stored at −20 °C. For analysis, blood was extracted from the cards and processed. The MS/MS system was verified using electrospray ionization (ESI) and multiple reaction monitoring (MRM) with commercially available standards. A 3.2 mm blood disc was punched out, treated with 100 µl of internal standard, shaken for 30 minutes at 7000 rpm, and evaporated. After adding Reagent B and incubating at 60 °C for 20 minutes, the sample was dried, treated with Reagent C, shaken, and 20 µl of the supernatant was injected into the MS/MS system (Supplementary Fig. S1).
Metabolite set enrichment analysis (MSEA)
To explore the biological significance of key metabolites, we performed a metabolite set enrichment analysis (MSEA) using MetaboAnalyst 5.0, considering SMPDB and KEGG databases for Recurrent DBS, Metastasis DBS, and Metastasis CSF samples. We then compared metabolite patterns across these sample types to identify those involved in the progression from recurrence to metastasis. We conducted an integrative pathway analysis of significant metabolites and proteins associated with glioblastoma development and metastasis to gain further insight, focusing on essential proteins involved in GB.
Glioblastoma-related key protein identification
Pivotal genes involved in GB were identified through two strategies. First, a literature review highlighted genes influencing key pathways like homeostasis, cellular metabolism, telomere maintenance, chemoresistance, and epigenetic regulation. Six genes were selected for further analysis (Supplementary Fig. S2 A). The second strategy involved searching the KEGG database for pathways related to glioblastoma, including cholesterol signaling, cytoskeleton remodeling, metabolism, fatty acid metabolism, and autophagy. A list of 1943 genes was compiled (Supplementary Fig. S2 B) and used to reconstruct a protein-protein interaction network using GeneMANIA and Cytoscape. An additional six genes were identified through topological analysis and databases like ONcoDB and the Human Protein Atlas (Supplementary Figure S2 C&D). Combining both strategies, 11 final genes were selected, with EGFR common to both.
Joint pathway analysis
After identifying key glioblastoma-related proteins, we conducted an integrative pathway analysis of significant metabolites and proteins using MetaboAnalyst 5.0 for Recurrent DBS, Metastasis DBS, and Metastasis CSF samples. The analysis was based on three criteria: degree, betweenness, and closeness, and followed a three-stage process. First, we selected the top 20 pathways for each criterion. Second, we integrated the results for each sample, and third, we removed non-significant pathways using the false discovery rate (FDR). Ultimately, we identified 31 pathways for Recurrent DBS, 33 for Metastasis DBS, and 31 for Metastasis CSF, with several common and exclusive pathways in each sample.
Statement of translational relevance
This study uncovers novel metabolic shifts and genetic alterations during the transition from recurrent to metastatic glioblastoma, particularly in a rare case with scalp metastasis. By identifying key drivers of this aggressive behavior, our findings offer potential new targets for therapeutic intervention, providing a critical advancement in managing extracranial metastasis in glioblastoma.
Results
Case description
A 33-year-old woman, previously healthy with no significant family or psychosocial issues, presented at Sina Hospital’s emergency department four years ago with severe headaches and sleepiness. A non-contrast CT scan revealed a left temporoparietal hemorrhagic lesion (Wernicke’s area; speech perception and processing area), causing a pressure effect on cerebral tissues and significant brain swelling. Her initial Karnofsky Performance Scale (KPS) score was 70. Emergency surgery was performed to maximize safe tumor removal, achieving complete resection. Following surgery, she underwent combined treatment for GB with concurrent temozolomide (TMZ) and radiation therapy (RT), followed by maintenance TMZ cycles, as per the Stupp protocol [19, 20]. Her KPS improved to 90 shortly after surgery and reached 100 during the follow-up.
After eighteen months, she developed new neurological symptoms, including right-sided weakness, memory problems, and difficulty speaking. MRI revealed multifocal recurrence in the left frontal and temporal lobes, including near the surgical site. Due to the involvement of critical brain areas, she underwent awake craniotomy and complete resection of all lesions, including an extra-neural GB metastasis in the scalp. The pathological analysis confirmed wild-type IDH-1&2, mutated TERT, non-mutated BRAF, EGFR, and non-methylated MGMT (Supplementary S3-S7). The pathological diagnosis was a malignant tumor (Fig. 1), and immunohistochemical (IHC) examination (Fig. 2) confirmed that the diagnosis was consistent with the recurrence and metastasis of GB; the examination also demonstrated the following: β-catenin (negative), Desmin (negative), glial fibrillary acidic protein (GFAP) (positive), Myogein (negative), p53 (negative), SMA (Alpha Smooth Muscle Actin) (negative), and Vimentin (positive). In our IHC analysis, GB samples exhibited positive staining for GFAP and vimentin, which are associated with the mesenchymal transition. These markers’ expression indicates cellular plasticity and has been linked to increased invasive potential and metastatic behavior in glioblastoma. The positivity for GFAP and vimentin in our samples supports the hypothesis that these cells are undergoing a metastatic transition, consistent with the aggressive nature of GB progression [21,22,23,24].

Axial (a), sagittal (b), and coronal (c) views of brain MRI before the second surgery, along with the postoperative counterparts (d–f). In the preoperative imaging, multicentric GB involving left posterior frontal and left temporoparietal regions concomitant with metastasis to the scalp of the left frontal area is seen. Compared to postoperative imaging, gross total removal (GTR) of the lesions has been obtained. g Hypercellular neoplastic glial tissue (blue arrow) with palisading necrosis (black arrow) and vascular proliferation (yellow arrow) (hematoxylin-eosin, x200). h Hypercellular neoplastic glial tissue (blue arrow) with vascular proliferation (yellow arrow) and palisading necrosis (black arrow) (hematoxylin-eosin, x200). i Neoplastic glial tissue (blue arrow) with invasion to muscle bundles (black arrow) (hematoxylin-eosin, x200). j Neoplastic glial tissue (blue arrow) invades muscle bundles (black arrow) (hematoxylin-eosin, x200).

IHC staining of the patient’s recurrent and metastatic glioblastoma (GB) tumor samples highlights the expression of markers associated with mesenchymal transition (EMT), including GFAP and vimentin. These markers indicate cellular plasticity, correlated with enhanced invasive potential and metastatic behavior in GB. Panels (a, h) show beta-catenin with negative (0) staining, indicating no detectable expression in the analyzed tumor cells, while panels (b, i) demonstrate desmin with negative (0) staining, reflecting its absence in the tumor. Panels (c, j) display strong positive (+3) staining for GFAP, suggesting potential involvement in tumor invasiveness. Panels (d, k) show myogenin with negative (0) staining, indicating its absence in the tumor, and panels (e, l) depict p53 with negative (0) staining, indicating no overexpression in the analyzed samples. Panels (f, m) highlight SMA with negative (0) staining, reflecting no significant expression. In contrast, panels (g, n) show vimentin with strong positive (+3) staining, supporting its role as a marker of mesenchymal transition in glioblastoma. Based on standardized scoring, each marker’s staining intensity is categorized as Negative (0) or Positive (+3). The combined positive staining for GFAP and vimentin provides evidence supporting mesenchymal transition and enhanced invasive and metastatic potential in GB. GFAP Glial Fibrillary Acidic Protein, SMA Spinal Muscular Atrophy, IDH1&2 Isocitrate dehydrogenase 1, WT wild-type, TERT Telomerase reverse transcriptase, MUT Mutated, EGFR Epidermal growth factor receptor.
Following surgery, she received further treatment, including radiation therapy and concurrent TMZ with bevacizumab, which initially improved her speech and mobility. However, four months later, the tumor recurred, necessitating another craniotomy for complete mass removal. Despite initial improvement, her KPS declined to 40 within a month, and she passed away approximately two years after her initial diagnosis Table 1.
Analysis of amino acid profiles in blood and CSF of a patient with recurrent and metastatic glioblastoma
In this study, the DBS sample from the recurrent stage showed decreased concentrations of Arginine, Methionine, Phenylalanine, Tyrosine, and Valine compared to the reference range, while Glutamic acid levels were elevated. The concentrations of the remaining amino acids were within normal limits [25] (Supplementary S8). In the metastasis stage, DBS samples showed decreased concentrations of Alanine and Glycine. In contrast, Glutamic acid remained elevated, with other amino acids within the normal range. In the metastasis CSF sample, Alanine, Glutamic acid, Methionine, Phenylalanine, Valine, Glycine, Leucine, Isoleucine, Histidine, Lysine, Tryptophan, and Threonine were all at the upper limits of the reference range (Table 2).
Metabolic profile and exclusive metabolite-related pathways for each sample
A simultaneous review of the metabolite set enrichment analysis results (Table 3) and the joint pathway analysis of metabolites and proteins (Table 4) reveals that aminoacyl-tRNA biosynthesis plays a pivotal role across all three samples. Notably, phenylalanine, tyrosine, tryptophan, arginine, and their respective biosynthetic pathways were exclusively observed in the recurrent DBS samples. In contrast, aspartate and glutamate were found only in the metastasis DBS samples. The metastasis CSF samples exclusively contained valine, leucine, isoleucine, and their biosynthesis. Furthermore, glycine, serine, and alanine and their biosynthesis, along with glutathione and its metabolism, were common to both the metastasis DBS and metastasis CSF samples. These results are crucial for improving diagnostic methods and deepening our understanding of the mechanical processes contributing to disease progression.
Discussion
Various studies have demonstrated the significance of metabolic reprogramming at different stages of glioblastoma (GB) creation, progression, and development. Research in this field has led to the classification of metabolic pathways into five primary groups: Glycolysis (Warburg Effect) and the Pentose Phosphate Pathway (PPP) [26,27,28,29,30], Amino Acid Metabolism [31,32,33], Lipid Metabolism [34,35,36], Nucleotide Metabolism [37, 38], and the TCA Cycle and Oxidative Phosphorylation [39,40,41,42,43]. Additionally, several biological processes contribute to the driving forces behind metabolic reprogramming. These include hypoxia [44,45,46,47], the tumor microenvironment and extracellular matrix [48,49,50], interactions with the immune system [51,52,53,54,55], the presence of stem cells and tunneling nanotubes (TNTs) [56,57,58,59], effects of therapeutic drugs [60, 61], and various epigenetic factors [62, 63].
Glioblastoma (GB) metastasis beyond the central nervous system (CNS) is exceptionally rare, mainly due to the protective function of the blood-brain barrier (BBB), the lack of lymphatic drainage, and the typically short overall survival (OS) of this aggressive malignancy [64]. Previous studies have reported that the average interval from initial diagnosis to extracranial tumor dissemination is around 11 months [65, 66]. Metastasis to vertebral sites, however, tends to occur later, with a median time of around 26 months [65, 66].
The mechanisms behind GB metastasis outside the CNS remain unclear but are believed to involve a mix of iatrogenic, genetic, and molecular factors, requiring further research [64]. Identified risk factors for extracranial metastasis include craniotomy, stereotactic biopsy, ventricular shunting, younger age, radiation therapy, prolonged survival, tumor recurrence, and sarcomatous components [64]. Over 90% of patients with extracranial metastases have had craniotomy, suggesting that glial cell dissemination through the bloodstream during surgery is a likely pathway for spread [67, 68].
GB cells may also metastasize via cerebrospinal fluid through peritoneal shunts or by seeding soft tissues through craniotomy defects. Chronic wound infections and tumor resection may increase the risk of extracranial metastasis due to direct surgical seeding [69, 70]. The mechanisms driving the osteolytic metastasis of GB are hypothesized to involve complex, bidirectional interactions between brain tumor cells and bone tissue [71].
Despite extensive research on molecular variants associated with GB and its subtypes, a critical gap persists in identifying genomic drivers enabling GB metastasis. Through protein-protein interaction network reconstruction, we identified six key genes—Ubiquitin C (UBC), Fibronectin 1 (FN1), Epidermal Growth Factor Receptor (EGFR), Catenin Beta 1 (CTNNB1), Jun Proto-Oncogene (JUN), and Mitogen-Activated Protein Kinase 1 (MAPK1)—that may play pivotal roles in GB invasion (Supplementary S9-S11).
FN1, in particular, has been suggested as a diagnostic marker to differentiate GB from low-grade astrocytoma, with its expression playing a functional role in the progression of malignant gliomas through the TGF-β-induced epithelial-mesenchymal transition (EMT) pathway [72]. Hendrych et al. identified genetic alterations in NF1, NOTCH3, AIRDA1, and MTOR in cases of glioblastoma metastasizing to the spine. Interestingly, while the BRAF mutation is commonly found in primary tumors, it is absent in metastatic lesions, where NF1 mutations are detected, indicating that tumor cells lacking the BRAF mutation may acquire metastatic potential [73]. Moreover, the Epidermal Growth Factor (EGF) has been shown to promote glioblastoma metastasis by inducing matrix metalloproteinase-9 (MMP-9) through an EGFR-dependent mechanism [74]. In the context of metastasis, the aberrant activation of MAPK signaling in glioblastoma cells is believed to enhance their invasive capacity, potentially leading to the formation of secondary tumors at distant sites, although extracranial metastasis remains a rare occurrence [75, 76].
In recurrent GB, metabolism shifts toward increased glycolysis, altered lipid and amino acid metabolism, and enhanced hypoxia-driven pathways. Metastatic GB adapts further, utilizing diverse energy sources, boosting antioxidant defenses, and remodeling the extracellular matrix, demonstrating distinct metabolic strategies at each stage [77]. In other words, transitioning from recurrence to metastasis is a journey from mere survival to profound adaptation.
Our results revealed that phenylalanine, tyrosine, and arginine pathways were unique to the recurrent DBS samples, while aspartate and glutamate were exclusive to metastasis DBS. Notably, many tumours rely on arginine, as they lack the ability to synthesize it [78, 79]. Arginine deprivation causes elongated cell morphology and loss of intracellular lamellipodia, inhibiting cell motility, adhesion, and invasion. In GB, arginylation is crucial for actin assembly, and arginine deprivation reduces N-terminal arginylation of β-actin, impairing these processes [80] (Supplementary S12).
Glutamine is the most depleted amino acid in tumor cells. It serves as a crucial metabolic fuel, meeting the extensive energy demands of metastatic cells for ATP, biosynthetic precursors, and reductants [77]. Enhanced glutamine availability bolsters cancer cell invasiveness, facilitating distant metastasis. Brain metastatic tumor cells, exhibiting metabolic plasticity, can derive energy from non-glucose sources, notably utilizing glutamine and branched-chain amino acids (BCAAs) as alternative fuels [81, 82]. Brain metastases show a significant reliance on glutamine metabolism, with overexpression of xCT promoting cystine uptake and glutamine oxidation, thus aiding metabolic adaptation and redox balance [83]. Given the brain’s high demand for glutamate, cells require an amino group donor, primarily sourced from BCAAs, to sustain glutamate anabolism. Consequently, cerebral vascular endothelial cells express numerous neutral amino acid transporters to support substantial BCAA uptake [84, 85]. Additionally, aspartate metabolism has been linked to tumor metastasis, where metastatic cancer cells consume substantial glutamine to compensate for TCA cycle deficiencies, increasing their dependence on asparagine. Inhibiting asparagine synthetase (ASNS) can induce rapid apoptosis in these metastatic cells [86].
Serine and glycine are critical amino acids required substantially during metastasis to support biosynthetic processes such as glycolysis, glutathione synthesis, and nucleotide production [87]. In the microenvironment of brain metastases, the levels of serine and glycine are notably lower than in plasma [88], likely due to the high demand of metastatic tissues for these amino acids [89]. Tryptophan metabolism is also crucial in glioblastoma progression, exhibiting distinct alterations between recurrent and metastatic stages. In recurrent GB, Indoleamine 2,3-dioxygenase (IDO) upregulation shifts tryptophan metabolism toward kynurenine production, activating the hydrocarbon receptor (AhR) pathway to enhance immune suppression and treatment resistance. During metastasis, the kynurenine pathway demonstrates metabolic flexibility, adapting tryptophan degradation to different microenvironments, thereby further promoting immune evasion, tumor invasion, and colonization [90, 91].
In summary, this case-based report explores and compares the shifting amino acid profiles observed in recurrent and metastatic phases of GB. Our findings suggest a preference for specific amino acids associated with recurrence and metastasis. Notably, phenylalanine, tyrosine, tryptophan, arginine, and their respective biosynthetic pathways were uniquely detected in recurrent samples. Conversely, aspartate and glutamate were exclusive to metastasis samples. Moreover, valine, leucine, isoleucine, and their biosynthetic pathways were uniquely identified in metastasis CSF samples. Glycine, serine, and alanine and their biosynthesis, glutathione, and metabolism were shared between metastasis DBS and CSF samples. These metabolic preferences might extend to other cancer types, warranting further investigation. While large-scale evaluations of these metabolic shifts are resource-intensive, small-scale case studies like this can offer valuable insights to inform and guide future research directions.
Emerging research highlights that modulating specific amino acid availability can significantly enhance cancer therapies’ efficacy. This positions amino acid metabolism as a promising avenue for therapeutic intervention in oncology. However, the metabolic intricacies of GB surpass those of many other cancers, driven by its pronounced intratumoral heterogeneity. Often described as a “tumor within a tumor,” GB harbors diverse cellular subpopulations, each contributing distinctively to disease progression, therapeutic resistance, and recurrence [92]. The GB tumor microenvironment is a complex and dynamic ecosystem comprising a wide array of elements such as bulk tumor cells, proliferative cell populations, brain tumor-initiating cells (BTICs), rare clones, resident microglia, and infiltrating immune cells derived from the bone marrow. Despite significant advances, the metabolic distinctions between these GB subtypes remain largely unexplored. Unraveling the metabolic dependencies unique to these subgroups can revolutionize therapeutic strategies, paving the way for more targeted and individualized treatment approaches for this highly aggressive and multifaceted disease [92].
Several critical considerations and unanswered questions surround targeting amino acid metabolism in GB and other cancers. It is well-established that amino acids play central roles in tumor biology, regulating numerous processes such as signaling pathways, interactions within the tumor microenvironment, and epigenetic modifications. Moreover, clinical evidence supports those restricting specific amino acids may enhance cancer treatment outcomes [93, 94]. However, significant gaps in understanding remain. For instance, among the diverse effects mediated by individual amino acids, which specific role is most influential in driving tumor progression or suppression? Furthermore, is the altered amino acid metabolism observed across different cancers causally linked to their initiation and progression, or is it merely a secondary consequence of tumor development? Additionally, how do genetic variations influence amino acid metabolism across primary, recurrent, and metastatic stages of cancer? Addressing these questions is essential to fully harness the therapeutic potential of targeting amino acid metabolism in oncology.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Barzegar Behrooz A, Darzi Ramandi H, Latifi-Navid H, Peymani P, Tarharoudi R, Momeni N, et al. Genetic Prognostic Factors in Adult Diffuse Gliomas: A 10-Year Experience at a Single Institution. Cancers. 2024;16:2121.
Basso J, Matos AM, Ghavami S, Fortuna A, Vitorino R, Vitorino C. Are we better together? Addressing a combined treatment of pitavastatin and temozolomide for brain cancer. Eur J Pharmacol. 2024;985:177087 https://doi.org/10.1016/j.ejphar.2024.177087.
Clark C, Barzegar Behrooz A, da Silva Rosa SC, Jacobs J, Weng X, Srivastava, A, et al. BCL2L13 Influences Autophagy and Ceramide Metabolism without Affecting Temozolomide Resistance in Glioblastoma. bioRxiv. 2024. https://doi.org/10.1101/2024.08.23.609447.
Lun M, Lok E, Gautam S, Wu E, Wong ET. The natural history of extracranial metastasis from glioblastoma multiforme. J Neurooncol. 2011;105:261–73. https://doi.org/10.1007/s11060-011-0575-8.
Noch EK, Sait SF, Farooq S, Trippett TM, Miller AM. A case series of extraneural metastatic glioblastoma at Memorial Sloan Kettering Cancer Center. Neuro-oncology practice. 2021;8:325–36.
Alizadeh J, Kavoosi M, Singh N, Lorzadeh S, Ravandi A, Kidane B, et al. Regulation of Autophagy via Carbohydrate and Lipid Metabolism in Cancer. Cancers (Basel) 2023;15. https://doi.org/10.3390/cancers15082195.
Lieu EL, Nguyen T, Rhyne S, Kim J. Amino acids in cancer. Experimental & Molecular Medicine. 2020;52:15–30. https://doi.org/10.1038/s12276-020-0375-3
Karno B, Edwards DN, Chen J. Metabolic control of cancer metastasis: role of amino acids at secondary organ sites. Oncogene. 2023;42:3447–56. https://doi.org/10.1038/s41388-023-02868-3.
Wei Z, Liu X, Cheng C, Yu W, Yi P. Metabolism of Amino Acids in Cancer. Front Cell Dev Biol. 2021;8, Review. https://doi.org/10.3389/fcell.2020.603837.
Jacobs J, Iranpour R, Behrooz AB, da Silva Rosa SC, Ghavami S. The role of BCL2L13 in glioblastoma: turning a need into a target. Biochem Cell Biol. 2024;102:127–34. https://doi.org/10.1139/bcb-2023-0221.
Sohrabi A, Lefebvre AEYT, Harrison MJ, Condro MC, Sanazzaro TM, Safarians G, et al. Microenvironmental stiffness induces metabolic reprogramming in glioblastoma. Cell Rep. 2023;42. https://doi.org/10.1016/j.celrep.2023.113175.
Qiu H, Shao N, Liu J, Zhao J, Chen C, Li Q, et al. Amino acid metabolism in tumor: New shine in the fog? Clinical Nutrition. 2023;42:1521–30. https://doi.org/10.1016/j.clnu.2023.06.011.
Sivanand S, Vander Heiden MG. Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell. 2020;37:147–56. https://doi.org/10.1016/j.ccell.2019.12.011.
Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12:31–46. https://doi.org/10.1158/2159-8290.Cd-21-1059.
Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. https://doi.org/10.1016/j.cmet.2015.12.006.
Chen S, Jiang J, Shen A, Miao Y, Cao Y, Zhang Y, et al. Rewired Metabolism of Amino Acids and Its Roles in Glioma Pathology. Metabolites 2022;12. https://doi.org/10.3390/metabo12100918.
Zhou W, Wahl DR Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers (Basel) 2019;11. https://doi.org/10.3390/cancers11091231.
Rahmani P, Abolhasani R, Heidari G, Mohebbi A, Sayarifard F, Rabbani A, et al. Determination of carnitine ester profile in the children with type 1 diabetes: a valuable step towards a better management. Arch Physiol Biochem. 2022;128:1209–14. https://doi.org/10.1080/13813455.2020.1762662.
Alimohamadi M, Shirani M, Moharari RS, Pour-Rashidi A, Ketabchi M, Khajavi M, et al. Application of awake craniotomy and intraoperative brain mapping for surgical resection of insular gliomas of the dominant hemisphere. World Neurosurgery. 2016;92:151–8.
Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New England Journal of Medicine. 2005;352:987–96.
Radu R, Petrescu GED, Gorgan RM, Brehar FM. GFAPδ: A Promising Biomarker and Therapeutic Target in Glioblastoma. Front Oncol. 2022;12:859247 https://doi.org/10.3389/fonc.2022.859247.
Yung WK, Luna M, Borit A. Vimentin and glial fibrillary acidic protein in human brain tumors. J Neurooncol. 1985;3:35–38. https://doi.org/10.1007/bf00165169.
van Bodegraven EJ, van Asperen JV, Sluijs JA, van Deursen CBJ, van Strien ME, Stassen O, et al. GFAP alternative splicing regulates glioma cell-ECM interaction in a DUSP4-dependent manner. Faseb j. 2019;33:12941–59. https://doi.org/10.1096/fj.201900916R.
Zottel A, Novak M, Šamec N, Majc B, Colja S, Katrašnik M, et al. Anti-Vimentin Nanobody Decreases Glioblastoma Cell Invasion In Vitro and In Vivo. Cancers (Basel). 2023;15. https://doi.org/10.3390/cancers15030573.
Physician’s Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases; Springer Berlin, Heidelberg, https://doi.org/10.1007/978-3-642-40337-8.
Gao M, Huang J, Jiang X, Yuan Y, Pang H, Luo S, et al. Regulation of aerobic glycolysis to decelerate tumor proliferation by small molecule inhibitors targeting glucose transporters. Protein & Cell. 2020;11:446–51.
Papavassiliou KA, Papavassiliou AG. Transcription factors in glioblastoma–Molecular pathogenesis and clinical implications. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2022;1877:188667.
Delle Donne R, Iannucci R, Rinaldi L, Roberto L, Oliva MA, Senatore E, et al. Targeted inhibition of ubiquitin signaling reverses metabolic reprogramming and suppresses glioblastoma growth. Communications Biology. 2022;5:780.
Garcia JH, Jain S, Aghi MK. Metabolic drivers of invasion in glioblastoma. Frontiers in Cell and Developmental Biology. 2021;9:683276.
Sun M, Sheng H, Wu T, Song J, Sun H, Wang Y, et al. PIKE-A promotes glioblastoma growth by driving PPP flux through increasing G6PD expression mediated by phosphorylation of STAT3. Biochemical Pharmacology. 2021;192:114736.
Wang Q, Wu M, Li H, Rao X, Ao L, Wang H, et al. Therapeutic targeting of glutamate dehydrogenase 1 that links metabolic reprogramming and Snail-mediated epithelial–mesenchymal transition in drug-resistant lung cancer. Pharmacological Research. 2022;185:106490.
Jin J, Byun J-K, Choi Y-K, Park K-G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Experimental & Molecular Medicine. 2023;55:706–15.
Hou X, Chen S, Zhang P, Guo D, Wang B. Targeted arginine metabolism therapy: A dilemma in glioma treatment. Frontiers in Oncology. 2022;12:938847.
Weng X, Gonzalez M, Angelia J, Piroozmand S, Jamehdor S, Behrooz AB, et al. Lipidomics-driven drug discovery and delivery strategies in glioblastoma. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2024;167637.
Jin H-R, Wang J, Wang Z-J, Xi M-J, Xia B-H, Deng K, et al. Lipid metabolic reprogramming in tumor microenvironment: from mechanisms to therapeutics. Journal of Hematology & Oncology. 2023;16:103.
Darwish A, Pammer M, Gallyas F Jr, Vígh L, Balogi Z, Juhász K. Emerging lipid targets in glioblastoma. Cancers. 2024;16:397.
Spina R, Mills I, Ahmad F, Chen C, Ames HM, Winkles JA, et al. DHODH inhibition impedes glioma stem cell proliferation, induces DNA damage, and prolongs survival in orthotopic glioblastoma xenografts. Oncogene. 2022;41:5361–72.
Shi DD, Savani MR, Levitt MM, Wang AC, Endress JE, Bird CE, et al. De novo pyrimidine synthesis is a targetable vulnerability in IDH mutant glioma. Cancer Cell. 2022;40:939–956.e916.
Yang S, Zhao J, Cui X, Zhan Q, Yi K, Wang Q, et al. TCA-phospholipid-glycolysis targeted triple therapy effectively suppresses ATP production and tumor growth in glioblastoma. Theranostics. 2022;12:7032.
Bisht P, Kumar VU, Pandey R, Velayutham R, Kumar N. Role of PARP inhibitors in glioblastoma and perceiving challenges as well as strategies for successful clinical development. Frontiers in Pharmacology. 2022;13:939570.
Wang W, Cui J, Ma H, Lu W, Huang J. Targeting pyrimidine metabolism in the era of precision cancer medicine. Frontiers in Oncology. 2021;11:684961.
Fhu CW, Ali A. Fatty acid synthase: an emerging target in cancer. Molecules. 2020;25:3935.
Shen Q, Yang H, Kong Q-P, Li G-H, Li L. Metabolic Modeling Identifies a Novel Molecular Type of Glioblastoma Associated with Good Prognosis. Metabolites. 2023;13:172.
Zhang B, Chen Y, Shi X, Zhou M, Bao L, Hatanpaa KJ, et al. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cellular and Molecular Life Sciences. 2021;78:195–206.
Trejo-Solís C, Castillo-Rodríguez RA, Serrano-García N, Silva-Adaya D, Vargas-Cruz S, Chávez-Cortéz EG, et al. Metabolic Roles of HIF1, c-Myc, and p53 in Glioma Cells. Metabolites. 2024;14:249.
Domènech M, Hernández A, Plaja A, Martínez-Balibrea E, Balañà C. Hypoxia: the cornerstone of glioblastoma. International Journal of Molecular Sciences. 2021;22:12608.
Mangraviti A, Raghavan T, Volpin F, Skuli N, Gullotti D, Zhou J, et al. HIF-1α-targeting acriflavine provides long term survival and radiological tumor response in brain cancer therapy. Scientific Reports. 2017;7:14978.
Nowosad A, Marine J-C, Karras P. Perivascular niches: critical hubs in cancer evolution. Trends Cancer 2023.
Sohrabi A, Lefebvre AE, Harrison MJ, Condro MC, Sanazzaro TM, Safarians G, et al. Microenvironmental stiffness induces metabolic reprogramming in glioblastoma. Cell Rep. 2023;42.
Wang C, Zhao Q, Zheng X, Li S, Chen J, Zhao H, et al. Decellularized brain extracellular matrix slice glioblastoma culture model recapitulates the interaction between cells and the extracellular matrix without a nutrient-oxygen gradient interference. Acta Biomaterialia. 2023;158:132–50.
Ye Z, Ai X, Yang K, Yang Z, Fei F, Liao X, et al. Targeting microglial metabolic rewiring synergizes with immune-checkpoint blockade therapy for glioblastoma. Cancer Discovery. 2023;13:974–1001.
Safaee MM, Wang EJ, Jain S, Chen J-S, Gill S, Zheng AC, et al. CD97 is associated with mitogenic pathway activation, metabolic reprogramming, and immune microenvironment changes in glioblastoma. Scientific Reports. 2022;12:1464.
Kesarwani P, Prabhu A, Kant S, Chinnaiyan P. Metabolic remodeling contributes towards an immune-suppressive phenotype in glioblastoma. Cancer Immunology, Immunotherapy. 2019;68:1107–20.
Yang R, Wang M, Zhang G, Li Y, Wang L, Cui H. POU2F2 regulates glycolytic reprogramming and glioblastoma progression via PDPK1-dependent activation of PI3K/AKT/mTOR pathway. Cell Death & Disease. 2021;12:433.
Pires‐Afonso Y, Muller A, Grzyb K, Oudin A, Yabo YA, Sousa C, et al. Elucidating tumour‐associated microglia/macrophage diversity along glioblastoma progression and under ACOD1 deficiency. Molecular Oncology. 2022;16:3167–91.
Biserova K, Jakovlevs A, Uljanovs R, Strumfa I. Cancer stem cells: significance in origin, pathogenesis and treatment of glioblastoma. Cells. 2021;10:621.
Wang Z, Zhang H, Xu S, Liu Z, Cheng Q. The adaptive transition of glioblastoma stem cells and its implications on treatments. Signal Transduction and Targeted Therapy. 2021;6:124.
Nakhle J, Khattar K, Özkan T, Boughlita A, Abba Moussa D, Darlix A, et al. Mitochondria transfer from mesenchymal stem cells confers chemoresistance to glioblastoma stem cells through metabolic rewiring. Cancer Research Communications. 2023;3:1041–56.
Li H-Y, Feng Y-H, Lin C-L, Hsu T-I. Mitochondrial mechanisms in temozolomide resistance: Unraveling the complex interplay and therapeutic strategies in glioblastoma. Mitochondrion. 2024;75:101836.
Hao Z, Wang J, Lv Y, Wu W, Zhang S, Hao S, et al. Identification of MGMT promoter methylation as a specific lipid metabolism biomarker, reveals the feasibility of atorvastatin application in glioblastoma. Metabolism. 2024;153:155794.
Nguyen TT, Shang E, Shu C, Kim S, Mela A, Humala N, et al. Aurora kinase A inhibition reverses the Warburg effect and elicits unique metabolic vulnerabilities in glioblastoma. Nature Communications. 2021;12:5203.
Gunn K, Myllykoski M, Cao JZ, Ahmed M, Huang B, Rouaisnel B, et al. (R)-2-hydroxyglutarate inhibits KDM5 histone lysine demethylases to drive transformation in IDH-mutant cancers. Cancer Discovery. 2023;13:1478–97.
Sun X, Johnson J, St. John JC. Global DNA methylation synergistically regulates the nuclear and mitochondrial genomes in glioblastoma cells. Nucleic Acids Research. 2018;46:5977–95.
Kurdi M, Baeesa S, Okal F, Bamaga AK, Faizo E, Fathaddin AA, et al. Extracranial metastasis of brain glioblastoma outside CNS: Pathogenesis revisited. Cancer Rep (Hoboken). 2023;6:e1905 https://doi.org/10.1002/cnr2.1905.
Goodwin CR, Liang L, Abu-Bonsrah N, Hdeib A, Elder BD, Kosztowski T, et al. Extraneural Glioblastoma Multiforme Vertebral Metastasis. World Neurosurg. 2016;89:578–582.e573. https://doi.org/10.1016/j.wneu.2015.11.061.
Pietschmann S, von Bueren AO, Kerber MJ, Baumert BG, Kortmann RD, Müller K. An individual patient data meta-analysis on characteristics, treatments and outcomes of glioblastoma/ gliosarcoma patients with metastases outside of the central nervous system. PLoS One. 2015;10:e0121592 https://doi.org/10.1371/journal.pone.0121592.
Huang P, Allam A, Taghian A, Freeman J, Duffy M, Suit HD. Growth and metastatic behavior of five human glioblastomas compared with nine other histological types of human tumor xenografts in SCID mice. J Neurosurg. 1995;83:308–15. https://doi.org/10.3171/jns.1995.83.2.0308.
Oktay K, Yildirim DC, Acikalin A, Ozsoy KM, Cetinalp NE, Erman T. Extensive Extraneural Metastases of Cerebral Glioblastoma in a Pediatric Patient: An Extreme Case Report and Comprehensive Review of the Literature. Pediatr Neurosurg. 2021;56:300–5. https://doi.org/10.1159/000515348.
Newton HB, Rosenblum MK, Walker RW. Extraneural metastases of infratentorial glioblastoma multiforme to the peritoneal cavity. Cancer. 1992;69:2149–53. 10.1002/1097-0142(19920415)69:8<2149::aid-cncr2820690822>3.0.co;2-g
Forsyth TM, Bi WL, Abedalthagafi M, Dunn IF, Chiocca EA. Extracranial growth of glioblastoma multiforme. J Clin Neurosci. 2015;22:1521–3. https://doi.org/10.1016/j.jocn.2015.03.018.
Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101.
Chen CW, Yang CH, Lin YH, Hou YC, Cheng TJ, Chang ST, et al. The Fibronectin Expression Determines the Distinct Progressions of Malignant Gliomas via Transforming Growth Factor-Beta Pathway. Int J Mol Sci. 2021;22. https://doi.org/10.3390/ijms22073782.
Hendrych M, Solar P, Hermanova M, Slaby O, Valekova H, Vecera M, et al. Spinal Metastasis in a Patient with Supratentorial Glioblastoma with Primitive Neuronal Component: A Case Report with Clinical and Molecular Evaluation. Diagnostics (Basel) 2023;13. https://doi.org/10.3390/diagnostics13020181.
Chen XC, Wei XT, Guan JH, Shu H, Chen D. EGF stimulates glioblastoma metastasis by induction of matrix metalloproteinase-9 in an EGFR-dependent mechanism. Oncotarget. 2017;8:65969–82. https://doi.org/10.18632/oncotarget.19622.
Kciuk M, Gielecińska A, Budzinska A, Mojzych M, Kontek R. Metastasis and MAPK Pathways. Int J Mol Sci 2022;23. https://doi.org/10.3390/ijms23073847.
Chen X, Hao A, Li X, Ye K, Zhao C, Yang H, et al. Activation of JNK and p38 MAPK Mediated by ZDHHC17 Drives Glioblastoma Multiforme Development and Malignant Progression. Theranostics. 2020;10:998–1015. https://doi.org/10.7150/thno.40076.
Wang Z, Wu X, Chen HN, Wang K. Amino acid metabolic reprogramming in tumor metastatic colonization. Front Oncol. 2023;13:1123192 https://doi.org/10.3389/fonc.2023.1123192.
Allen MD, Luong P, Hudson C, Leyton J, Delage B, Ghazaly E, et al. Prognostic and therapeutic impact of argininosuccinate synthetase 1 control in bladder cancer as monitored longitudinally by PET imaging. Cancer Res. 2014;74:896–907. https://doi.org/10.1158/0008-5472.Can-13-1702.
Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, et al. Autophagy maintains tumour growth through circulating arginine. Nature. 2018;563:569–73. https://doi.org/10.1038/s41586-018-0697-7.
Pavlyk I, Rzhepetskyy Y, Jagielski AK, Drozak J, Wasik A, Pereverzieva G, et al. Arginine deprivation affects glioblastoma cell adhesion, invasiveness and actin cytoskeleton organization by impairment of β-actin arginylation. Amino Acids. 2015;47:199–212. https://doi.org/10.1007/s00726-014-1857-1.
Chen J, Lee HJ, Wu X, Huo L, Kim SJ, Xu L, et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. 2015;75:554–65. https://doi.org/10.1158/0008-5472.Can-14-2268.
Albrecht J, Sidoryk-Węgrzynowicz M, Zielińska M, Aschner M. Roles of glutamine in neurotransmission. Neuron Glia Biol. 2010;6:263–76. https://doi.org/10.1017/s1740925x11000093.
Parida PK, Marquez-Palencia M, Nair V, Kaushik AK, Kim K, Sudderth J, et al. Metabolic diversity within breast cancer brain-tropic cells determines metastatic fitness. Cell Metab. 2022;34:90–105.e107. https://doi.org/10.1016/j.cmet.2021.12.001.
Yudkoff M, Nissim I, Daikhin Y, Lin ZP, Nelson D, Pleasure D, et al. Brain glutamate metabolism: neuronal-astroglial relationships. Dev Neurosci. 1993;15:343–50. https://doi.org/10.1159/000111354.
del Amo EM, Urtti A, Yliperttula M. Pharmacokinetic role of L-type amino acid transporters LAT1 and LAT2. Eur J Pharm Sci. 2008;35:161–74. https://doi.org/10.1016/j.ejps.2008.06.015.
Luo M, Brooks M, Wicha MS. Asparagine and Glutamine: Co-conspirators Fueling Metastasis. Cell Metab. 2018;27:947–9. https://doi.org/10.1016/j.cmet.2018.04.012.
Levine EM, Simmonds S. Effect of cultural conditions on growth and metabolite uptake by serine-glycine auxotrophs of Escherichia coli. J Bacteriol. 1962;84:683–93. https://doi.org/10.1128/jb.84.4.683-693.1962.
Dolgodilina E, Imobersteg S, Laczko E, Welt T, Verrey F, Makrides V. Brain interstitial fluid glutamine homeostasis is controlled by blood-brain barrier SLC7A5/LAT1 amino acid transporter. J Cereb Blood Flow Metab. 2016;36:1929–41. https://doi.org/10.1177/0271678x15609331.
Venneti S, Thompson CB. Metabolic Reprogramming in Brain Tumors. Annu Rev Pathol. 2017;12:515–45. https://doi.org/10.1146/annurev-pathol-012615-044329.
Panitz V, Končarević S, Sadik A, Friedel D, Bausbacher T, Trump S, et al. Tryptophan metabolism is inversely regulated in the tumor and blood of patients with glioblastoma. Theranostics. 2021;11:9217–33. https://doi.org/10.7150/thno.60679.
Xu Y, Zhang H, Sun Q, Geng R, Yuan F, Liu B, et al. Immunomodulatory Effects of Tryptophan Metabolism in the Glioma Tumor Microenvironment. Front Immunol. 2021;12:730289 https://doi.org/10.3389/fimmu.2021.730289.
Agnihotri S, Zadeh G. Metabolic reprogramming in glioblastoma: the influence of cancer metabolism on epigenetics and unanswered questions. Neuro-Oncology. 2015;18:160–72. https://doi.org/10.1093/neuonc/nov125.
Safrhansova L, Hlozkova K, Starkova J. Chapter Two—Targeting amino acid metabolism in cancer. In: Buqué A, Galluzzi L. Editors. International Review of Cell and Molecular Biology. Vol. 373; Cambridge, Massachusetts: Academic Press, 2022; pp 37–79.
Chen J, Cui L, Lu S, Xu S. Amino acid metabolism in tumor biology and therapy. Cell Death & Disease. 2024;15:42 https://doi.org/10.1038/s41419-024-06435-w
Acknowledgements
We appreciate Sina Hospital for providing the required testing facilities and equipment for the study. The authors acknowledge CHATGPT 4o for professional English editing.
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
ABB drafted and edited the manuscript with input from all authors. HL-N contributed to the systems biology analysis, data interpretation, manuscript drafting, and editing. NZ, SP, MB, and FJ assisted with data curation. MA and NA contributed to the pathological evaluation and immunohistochemistry (IHC) analysis. FM prepared the pathology slides and performed the IHC staining. SA also prepared pathology slides, conducted the IHC tests, and verified the results. AN and SS participated in manuscript review and editing. JL contributed to the MS/MS analysis, interpretation of results, preparation of pathology slides, and drafting of the MS/MS methods section. EN was involved in pathological analysis. AP-R supervised the project and contributed to writing, reviewing, and editing the manuscript. SG provided overall project supervision and finalized the manuscript draft. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate:
The patient involved in the study gave informed consent. The Ethics and Research Committee of Tehran University of Medical Science, Neurosurgical Department of Sina Hospital (IR.TUMS.SINAHOSPITAL.REC.1399.111) approved this study, and all methods were in compliance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Behrooz, A.B., Latifi-Navid, H., Zolfaghari, N. et al. Metabolic reprogramming in glioblastoma: a rare case of recurrence to scalp metastasis. BJC Rep 3, 27 (2025). https://doi.org/10.1038/s44276-025-00134-5
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44276-025-00134-5

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}