
Abstract
Background
We address the therapeutic relevance of glioblastoma (GBM)’s molecular characterization, focusing on whether a proneural-vs-mesenchymal classification may describe GBM heterogeneity, correlate to stemness and CD44 (a marker often, but conflictingly, used in GBM), and have a prognostic value in terms of temozolomide (TMZ) resistance.
Methods
We molecularly profiled 4 patient-derived GBM cells (PD-GBM) and a reference cell line (U87), validating the results in public datasets (PD-GBM, TCGA-GBM), then studying the subtype-specific response to TMZ in vitro, both under normoxia and simulated hypoxia.
Results
Firstly, the proneural subtype transcriptionally and phenotypically correlates with higher stemness. Secondly, CD44 inversely correlates to stemness, acting as a mesenchymal marker both intra-tumorally (FACS-sorted CD44LOW cells had higher stemness than CD44HIGH) and inter-tumorally (also in larger datasets). Further, mesenchymal PD-GBM cells are also enriched in other hyaluronic acid (HA) surface receptors, proneural in matrix HA receptors. Finally, although TMZ stimulates a general mesenchymal shift in all cells, there are proneural-specific responses: this subtype resists TMZ better under hypoxia (vs. normoxia) in a first treatment, and in a second treatment (vs. single) under normoxia.
Conclusion
PD-GBM show clear proneural/stemness and mesenchymal/CD44 correlations, which may bear prognostic significance, as more proneural/stem GBMs appeared more capable to develop TMZ resistance.
Similar content being viewed by others

Background
Glioblastoma (GBM) is highly heterogeneous, both inter- [1] and intra-tumorally [2], and various stratification approaches have been attempted to link molecular descriptions to clinical outcomes, but with mixed success to date.
At a genomic level, recognized GBM driver mutations occur in EGFR, PTEN, P53 [3, 4], and most importantly in IDH, the latter being a WHO keystone for diagnosis of gliomas: IDH is wild-type in GBM, while it is mutated in lower-grade gliomas and secondary GBM [5, 6]. This induces a hypermethylated (G-CIMP) phenotype, a completely different epigenetic status, and is linked to a more favorable prognosis [4]. However, besides IDH, no single genomic alteration to date has been directly associated to GBM survival.
Transcriptomic stratifications [1, 2, 7, 8] offer a better description of a tumor phenotype, thus, in principle, providing better diagnostic/prognostic results. Verhaak’s classification into classical/proneural/mesenchymal subtypes [1, 2] is the most widely adopted, although recently it has been often reduced to two subtypes only (proneural and mesenchymal [9, 10]), which are also increasingly considered as extremes of a ‘single axis of variation’ [11,12,13,14]. The proneural subtype features alterations in PDGFRA, and a gene expression reminiscent of neural/oligodendrocyte progenitors [1, 8], while the mesenchymal subtype expresses markers typical of differentiated astrocytes and of inflammatory signaling [10, 15,16,17]. Indeed, epithelial-to-mesenchymal transition (EMT) (“proneural-to-mesenchymal” in GBM) is a well-characterized mechanism of GBM recurrence [18, 19].
Conflicting evidence makes it currently impossible to associate a clear prognostic value to transcriptomic subtypes [20, 21], and it does not come as a surprise that they are not included in GBM’s clinical stratification [6]. For example, the historical association of proneural GBM to younger patients and better prognosis [7, 22] was due to a high proportion of IDH mutant/G-CIMP cases; this is no longer valid, as they are not classified as GBM. The mesenchymal subtype has a more aggressive phenotype (therapy-resistance, angiogenesis, and invasiveness [10, 17, 23] and is enriched in recurrent tumors [18, 23]. Yet, its correlation with poorer survival is not absolute, and has been shown only in intra-tumoral homogeneous cohorts [2].
There are, however, descriptors that may concur to GBM stratification and have prognostic potential. Firstly, tumoral stemness: “cancer stem cells” (CSC) are often connected to therapy resistance and recurrence [24]. Unfortunately, identification and quantification of such cells is difficult, and surface markers are not sufficiently specific [25]. For example, CD44 is often reported as a stemness marker in GBM [24] (and other tumors [26]), but this correlation is still debated [16, 27]: in GBM CD44 is also involved in mesenchymal features – e.g. invasiveness [28], EMT [10], interactions with immune cells [16] – and thus is also widely used as a mesenchymal marker [8, 10, 11, 16, 18]. A transcriptomic signature of GBM stemness has been proposed [29], based on the expression of a core set of 4 “master regulator” transcription factors. This signature may add to GBM transcriptional stratification, but to date, no attempt has been made to connect it to GBM’s molecular subtype stratification.
Another GBM descriptor with potential prognostic value is hypoxia, as several hypoxia-dependent pathways have been shown to directly induce therapy resistance [30]. Further, hypoxia’s spatial (and temporal) heterogeneity may connect to that of GBM: spatial transcriptomics has recently shown hypoxia as a central driver of histological organization in GBM, with its main niches (hypoxic/necrotic vs perivascular/neural) closely corresponding to a structured zonation of transcriptomic cellular states [31]. However, a clear connection between hypoxia and subtype/stemness has not been drafted yet; for example, hypoxia is conflictingly reported as either a stemness-promoting [32] or mesenchymal-inducing [33] factor.
Temozolomide (TMZ) is GBM’s standard-of-care drug [34], but has a limited efficacy due to the development of heterogeneous forms of resistance [35]. If a link could be established between the heterogeneity of GBM resistance and that in its molecular descriptors, this would be of high clinical relevance. It must be acknowledged that this relation – if possible - may not encompass the whole GBM’s resistance development, which also roots in metabolic [9, 36] and epigenetic adaptations, chiefly the de-alkylating activity of MGMT [37], or other methylases and histone deacetylases [30], all extremely plastic mechanisms [2, 25, 36].
In this study, we aimed to address the possible correlation between GBM’s molecular descriptors among themselves and with the in vitro TMZ response, using patient-derived cells.
Firstly, we have integrated the proneural vs mesenchymal axis with additional malignancy-related indicators, i.e., stemness score and expression of CD44; the latter is also a key mediator of the interactions with hyaluronic acid (HA), the main component of the brain parenchymal extracellular matrix (ECM) known to enhance GBM progression [38]. Secondly, we have looked for correlations between this stratification and TMZ resistance under either normoxic or hypoxic conditions. Methodologically, we employed 4 patient-derived cultures (PD-GBM), which retain key molecular features of the original tumors as well as their drug responsiveness [21, 39, 40]. A widely employed cell line (U87) was used as a control to ensure reproducibility of our data, and large transcriptomic datasets were employed for further orthogonal validation.
Materials and methods
General methods
Conditions for cell culture, flow cytometry, qPCR, and stock solutions preparation are in Supplementary Information.
Genomics/transcriptomics
Cells were seeded at 160,000 cells/well in 6-well plates and maintained in culture medium for 24 h, replacing it with TMZ-containing medium (20 µM for GBM1, 400 µM for GBM2, GBM5, GBM6, and U87) or complete medium as a control and incubated for 72 h.
RNA-Sequencing. Total RNA extraction was performed with PureLink RNA Mini kit (12183018 A, Invitrogen); RNA quantity/quality was assessed with a NanoDrop 1000 Spectrophotometer (Thermo Scientific) and on a Bioanalyzer 2100 (Agilent). RNA-Seq libraries were prepared using the Illumina Stranded mRNA Prep Library kit, and paired-end sequencing was run on a NovaSeq 6000 platform (Illumina).
Short-read Whole Genome Sequencing. Genomic DNA (gDNA) was extracted using Purelink Genomic DNA mini kit (K1820, Invitrogen). Paired-end sequencing was performed with the Illumina NovaSeq 6000 platform, generating 151 bp-sized reads. Barcode adapters were used for library preparation and sequencing of the gDNA.
Transcriptomics and genomics analysis details are in Supplementary Information.
Cellular assays
FACS sorting of GMB1
GBM1 cells (adherent culture) were detached, counted, and incubated with PE-conjugated anti-CD44 antibody (12-0441-82, Invitrogen) at 1:200 dilution. Gating was performed using FSC-A and SSC-A/SSC-W scatter to acquire at least 10,000 viable single cells. CD44 signal on PE-A channel was gated using negative controls (not-stained cells). Cells were sorted into CD44HIGH and CD44LOW and harvested in complete medium with 1% Penicillin-Streptomycin (Gibco), and then kept in standard culture. One week later, GBM1 CD44HIGH and GBM1 CD44LOW cells were analyzed for their CD44 expression (at least 10,000 viable cells), cell eccentricity/size, and surface coverage (see next section).
TMZ toxicity
Cells were seeded at 5000 cells/well in 96-well plates and cultured for 24 h in 100 µL of complete medium, replacing it with 200 µL of medium containing 3000 to 23.4 µM TMZ (T2577, Sigma-Aldrich) (1:2 serial dilutions) ± 100 µM CoCl2; three viability indicators were assessed:
A) Confluence. In an Incucyte Live Cell Analysis System (S3, Sartorius), 2 images/well were acquired every 6 h over 72 h (phase contrast, 10x objective). The Cell confluence mask quantified the % of the field covered by cells, normalizing it to time 0 (“surface coverage”); the slope of surface coverage 0-24 h linear fit, normalized vs the no-TMZ control, provides a “normalized growth rate”. Cell eccentricity and cell size were also quantified with this mask.
B) MTS assay (G3582, Promega) was performed at 72 h, following manufacturer’s instructions and recording the 490 nm absorbance (Synergy H1 plate reader, Biotek) after 1 h incubation at 37 °C.
C) BCA assay (23225, Thermo Scientific). After MTS absorbance readings, wells were washed twice with 100 µL of cold PBS (Gibco), lysed with 75 µL cold RIPA buffer, and frozen at −80 °C for at least 24 h. BCA assay was performed according to the manufacturer’s instructions, reading 562 nm absorbance after 1 h incubation at 37 °C. The IC50 values were obtained by fitting the normalized protein content values with a DoseResponse function (OriginLab, version 2020) of the form \({Protein}\; {content}={\rm{A}}1+\frac{{\rm{A}}2-{\rm{A}}1}{1+{10}^{\left(\right.\log ({IC}50)-\log ([{TMZ}])p}}\), where \({\rm{A}}1\) and \({\rm{A}}2\) are bottom and top asymptotes, and \(p\) the slope of the sigmoidal function. The ratio between MTS absorbance and protein content, which is essentially the mitochondrial reductase activity normalized against the cell number, after a further normalization against controls, was used as an indication of cellular metabolic state.
Colony formation assay
Cells were seeded at 200 cells/well in 96-well plates in complete medium and without Matrigel coating (Corning). Images (acquired with Incucyte system, as described above) at day 14 and day 0 provide the counts respectively of colonies (aggregates of min. 1800 µm2) and initial single cells, and their ratio expresses the % of colony-forming cells.
Migration assay
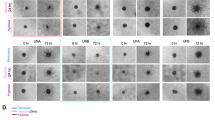
Cells were seeded at 30,000 cells/well in Imagelock 96-well plates (BA-04856, Sartorius) under adherent conditions. The next day, all wells were simultaneously scratched with an Incucyte®Woundmaker to create wounds; wells were washed with medium, and then cultured in fresh complete medium with or without 20 or 75 µM TMZ, 0.25 mg/mL 220 kDa HA (Fidia S.p.A, Italy), or 100 µM CoCl2 for 72 h. Images were acquired every 3 h (Scratch Wound Incucyte mode; 10x, phase contrast), and Relative Wound Density (RWD) values were obtained at every time point by comparing the confluence in the wound area relative to the confluence outside it (RWD = 0 at time zero, RWD = 100% when the cell density inside the wound is the same as the cell density outside the initial wound), as recommended by the Incucyte manual.
Results
Molecular profiling of a panel of 4 PD-GBM
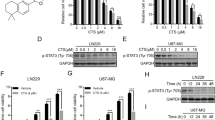
We comparatively stratified our panel of 4 PD-GBM (clinically relevant data in Fig. 1a) and the commercial U87 cell line. We used both Verhaak’s classification [1, 2] and an alternative 2-subtype “mesenchymal vs proneural” (MES vs PN) z-score [10], which was calculated as the difference in average expression of proneural (DLL3, OLIG2, ASCL1, and NCAM1) and mesenchymal (YKL40, SERPINE1, TIMP1, and TGFBI) genes. In brief, MES vs PN score stratifies our cellular models in a simpler fashion than Verhaak’s subtypes (in addition to a clearer correlation with both stemness and CD44 expression; see later), and was employed for the rest of the study.

a Clinically relevant information for the PD-GBM panel, GBM-specific genetic alterations from DNA-seq data, MGMT expression (from both DNA- and RNA-seq), and molecular subtype classification based on their transcriptomes (Verhaak’s subtype assigned using SVM algorithm from GlioVis, https://gliovis.bioinfo.cnio.es/; MES vs PN subtype based on the 8-genes score from Bhat [10]). PN=Proneural, CLA=Classical, MES=Mesenchymal; Mut: mutation; Del: homozygous deletion; Amp: amplification; LOH : loss of heterozygosity; chr: chromosome. In line with the last WHO classification of GBM, we have only included IDH wild-type tumors [6]. b PCA of RNA-seq data from cells treated for 72 h with near-IC50 TMZ (corresponding to 20 µM TMZ for GBM1 and 400 µM TMZ for the other cell types: see Fig. 4a for actual IC50 values) or untreated (Ctrl), n = 3 samples per condition. c Left. z-scores (from RNA-Seq) for stemness signature, MES vs PN, and CD44. Right: Pearson’s correlation analysis: r values (color scale) and p values (numbers; white asterisk: two-tailed p-value <0.05) are shown for each correlation. The top graph refers to our panel of 4 PD-GBM + U87 (RNA-Seq of n = 5 cell cultures, untreated, in technical triplicate), the mid graph to the PD-GBM cultures (n = 59) in Lazzarini et al. [43], and the bottom graph to TCGA-GBM tissue samples (n = 160).
DNA alterations
Our panel has genomic features typical of GBM: Whole Genome Sequencing (WGS) analysis detected alterations in all 3 canonical GBM oncogenic pathways (see Supplementary Table S1–S3 and Fig. S1), namely those of p53 (MDM2-4, TP53), Rb (CCND2, CDKN2A, RB1), and PI3K (PTEN, EGFR, NF1). Key tumor suppressors PTEN and CDKN2A [4] were mutated and/or deleted in all 5 samples, and TP53 in GBM2 and GBM6 (Fig. 1a). Further, we found EGFR gain/amplification (observed in >70% of GBM patients [4]) in GBM1, GBM5, and GBM6 (Fig. 1a).
Gain of the chromosome 7 and hemizygous loss of chromosome 10, a common early event in GBM tumorigenesis [14], was detected in GBM5 and GBM6; the other PD-GBMs show a different CNV pattern in the same chromosomes: partial (GBM1) or absent (GBM2) gain in chromosome 7 and, in both cases, homozygous loss of the entire 10q region (GBM1) or most of it (GBM2) (Supplementary Fig. S1). Interestingly, MGMT is in this region: indeed, it is deleted in homozygosis (GBM1, GBM2) or heterozygosis (GBM5, GBM6) in the PD-GBMs (and diploid in U87). However, from RNA-Seq and qPCR data, MGMT is only expressed in GBM6, and thus is silenced (likely through promoter methylation [37]) in GBM5 and U87 (Supplementary Fig. S2).
Transcriptomic-based subtype stratification
Our panel is representative of GBM’s molecular subtypes: applying Verhaak’s classification, U87 and GBM6 are mesenchymal, GBM2 and GBM5 classical, GBM1 proneural (Fig. 1a). Instead, using the MES vs PN z-score, U87, GBM2, and GBM6 are mesenchymal, GBM1 and GBM5 proneural (Fig. 1a and Supplementary Table S4). Of note, since OLIG2 will be later used to stratify transcriptional stemness, we also calculated the score with PDGFRA – the most established proneural marker [1] – in its stead, with no significant difference (Supplementary Table S4, S5).
The GBM1-GBM5 (proneural) and GBM2-GBM6 (mesenchymal) clustering was confirmed by principal component analysis (PCA) of transcriptomic data (Fig. 1b). U87 clustered on the same side as GBM2-GBM6, in a way confirming their mesenchymal nature, but clearly separating from the patient-derived cells; this may be due to their nature as a cell line, as much as their culture in serum-containing medium. Treatment with TMZ (triangles in Fig. 1b) is visible in the PCA plot as a small shift for each GBM, but does not affect the general clustering.
DNA alterations/molecular subtype correlations
Some DNA alterations and clinical features often show a degree of association with transcriptomic-based subtypes [1, 4]. The concomitant EGFR gain/amplification and lack of TP53 mutations – as found in GBM5 - are more frequent in Verhaak’s classical subtype [1]. Interestingly, PDGFRA has high expression in GBM5 (Fig. S3) (contributing to its low MES vs PN score) despite being affected by hemizygous loss (as the entire chromosome 4, Fig. S1). It is noteworthy that PDGFRA amplification is a typical proneural feature [1, 14], its heterozygous deletion must be compensated for by an elevated PDGFRA-driven oncogenic signaling.
GBM2, classified as Verhaak’s classical, presents genomic alterations pretty uncommon for this subtype: it lacks gains in chromosome 7, including EGFR region, and has mutated TP53 (Figs. 1a, S1). Conversely, high expression of mesenchymal genes such as ANGPT1-2, NFKB family, and NFKB-induced transcription factors STAT3, WWTR1 (RNA-Seq data) [10] makes this PD-GBM more compatible with a mesenchymal subtype (as it results from MES vs PN score).
GBM6 has clear mesenchymal traits: at a genomic level, RB1 deletion [1]; at a transcriptomic level, high expression of ANGPT1, NF-kB pathway, STAT3, and WWTR1. Accordingly, both MES vs PN and Verhaak classified GBM6 as mesenchymal.
U87 was previously classified as mesenchymal using Verhaak’s system [41], and we confirmed that they bear mesenchymal-associated NF1 mutation [42] and the overall assignment with both Verhaak and MES vs PN scores.
In short, in our panel, DNA alterations support equally well Verhaak’s and MES vs PN classifications; however, they do not necessarily align to transcriptomic subtypes.
Transcriptomic-based stemness and CD44
Stemness is a key feature of GBM, but typically not directly addressed through transcriptomic-based classification system(s). For its quantitative assessment, we have employed a 4-gene signature based on master regulators of GBM stem state reprogramming: POU3F2, SOX2, SALL2, and OLIG2 [29]. The resulting stemness z-score (Fig. 1c, top, and Supplementary Table S4) not only set apart higher-stemness GBM1 and GBM5, lower-stemness GBM2 and GBM6, and the lowest-stemness U87 (as expected for serum-cultured cell lines), but also correlated precisely and inversely with the MES vs PN one (Pearson r = -0.98). Interestingly, CD44 expression (RNA-Seq values in Supplementary Table S6) inversely correlated with stemness and directly with the MES vs PN z-score, strongly hinting at the mesenchymal association of this receptor.
We have validated these correlations in larger, publicly available transcriptomic datasets: a panel of 59 PD-GBM [43] and the TCGA-GBM dataset (n = 160 tumor samples) [44] (Fig. 1c). Both proneural/stemness and CD44/mesenchymal correlations were statistically significant in the two datasets. Of note, the CD44/stemness inverse correlation was not confirmed in TCGA, possibly because of the variable presence of CD44+ tumor-associated cells in tissue samples (GBM-associated macrophages and T cells express CD44 comparably to malignant cells [16]), absent from in vitro cultures.
In short, both in our panel and in public transcriptomic datasets MES vs PN score correlates inversely to stemness and directly to CD44 expression.
CD44 as a marker: in a proneural population, CD44LOW have higher stemness and plasticity than CD44HIGH
A well-known approach to increase stemness features in patient-derived cells is to promote their growth as spheroids (low-adhesion culture) [45]. The proneural GBM1 and GBM5 grew free-floating, round spheroids, whereas mesenchymal GBM2 and GBM6 formed plastic-anchored, smaller spheroids, surrounded by adherent cells (see Supplementary Fig. S4).
This was mirrored by a different CD44 expression: GBM1 and GBM5 originally expressed less CD44 (see Supplementary Table S6) and further decreased in spheroids (Fig. 2a, left), whereas no significant change was seen in mesenchymal GBMs.

a Left. CD44 mRNA expression measured by qPCR after 72 h of culture under adherent or spheroid conditions (i.e., with or without Matrigel coating, respectively); fold change to adherent, mean ± SD; n = 3, *p < 0.005, two-tailed t-test. Right. GBM1’s CD44 expression measured through flow cytometry using a PE-conjugated anti-CD44 antibody (12-0441-82, Invitrogen); n = 3, two-tailed t-test *p < 0.005. b Left. GBM1 sorting into CD44LOW and CD44HIGH: dot-plot showing the intensity gates over the PE channel. Middle. CD44 mRNA expression of the two subpopulations measured by qPCR after 72 h in adherent or spheroid culture; fold change to CD44HIGH, mean ± SD; n = 2 in technical triplicate, one-way ANOVA and multiple-comparisons, each vs CD44HIGHadherent, *p < 0.05. Right. Correlation between the z-scores of stemness and CD44. The signature gene expression was measured by qPCR (n = 2 in technical triplicate), and z-scores were calculated using qPCR fold change of each gene, in the total population of 4 cell types (CD44LOW and CD44HIGH, each in adherent and spheroid conditions). c Top left. Representative images of CD44LOW and CD44HIGH cells (Incucyte live cell imaging, 12 h after plating; 10x objective, phase confluence mask in yellow; scale bar 200 µm. Top right. Surface coverage (proportional to cell count), average cell area, and average eccentricity were quantified in a 72 h time course through a live cell imaging system (Incucyte) (10x objective, phase confluence mask with cell-by-cell analysis, mean ± SEM, Standard Error of the Mean, of n = 24 wells. *p < 0.05 two-way ANOVA). Bottom. Normalized distribution of CD44 median fluorescent intensity (left) and corresponding dot-plots (right) for CD44LOW and CD44HIGH 1 week after sorting, in comparison to unsorted GBM1 (in black).
Due to this possibility to easily modulate CD44 expression, we focused on the proneural subtype, and specifically on GBM1 (confirming qPCR data at the protein level with flow cytometry, Fig. 2a, right): CD44 levels in GBM5 are very low, barely detectable. Due to their broad CD44 distribution (in black in Fig. 2a), it was possible to sort GBM1 into CD44LOW and CD44HIGH fractions (Fig. 2b, left). The two subpopulations
A) differed in CD44 expression (decreasing for both in spheroid-forming conditions; Fig. 2b, middle) and in stemness, with the two parameters again clearly showing an inverse correlation (Fig. 2b, right).
B) were phenotypically different: after one week, CD44HIGH were visually more elongated, spindle-shaped, and with longer protrusions (Fig. 2c, top left), and thus more eccentric, larger, and also more proliferative (Fig. 2c, top right) than CD44LOW, which all tally with a more differentiated/mesenchymal-like state.
C) differed in ‘plasticity’: one week after sorting, CD44HIGH cells substantially maintained their CD44 distribution (71% still above the “CD44-HIGH” expression gate after 1 week, Fig. 2c, bottom). On the contrary, CD44LOW shifted theirs: half (52%) moved above the expression gate, making the whole population indistinguishable from unsorted cells (Fig. 2c, bottom). This shift may be a typical stemness feature, i.e., contemporaneous self-renewal (retaining CD44LOW cell compartment) and generation of a more differentiated population (CD44HIGH).
In brief, the inverse correlation between CD44 expression and stemness appears to be valid also at an intra-tumoral level.
Subtype-dependent interactions with hyaluronic acid (HA)
CD44 is the main receptor for HA, chiefly responsible to its endocytosis [46, 47], and is overexpressed in solid tumors, including GBM [28]. Using the Gene Ontology term HA binding (GO:0005540), we have extended our study to other cell-membrane (Surface HA binding) and matrix HA binders (Matrix HA binding) (Fig. 3a and Supplementary Table S7); the former are typically involved in cell adhesion, migration, and matrix degradation, and the latter (mostly proteoglycans) in matrix build-up.

a Genes belonging to the “HA binding” GO term (GO:0005540, from The Gene Ontology Resource) grouped according to where they act: cell surface vs. matrix (extracellular). Left. Heatmap of LOG10 expression (normalized read counts) for each gene. Right. Pearson correlation analysis of the molecular stratification signatures (as in Fig. 1) and the HA binding gene sets (surface HA binding and matrix HA binding). R value is shown in each square, * p-value of the correlation<0.05. Bottom. Graphical summary: mesenchymal GBM are enriched in genes coding for cell surface HA-binding proteins, and proneural GBM in genes coding for matrix HA-binding proteins. b Left. Scratch wound assay: cell migration (Relative Wound Density, RWD, %) data as a function of time, +/− 20 µM TMZ treatment and +/− 0.25/mL HA (mean ± SEM of n = 6 wells per condition); Right. RWD % data were fitted as \({RWD}=a* (1-{e}^{-{bt}})\), where \(a\) and \(b\) are respectively the max wound closure and the wound closure rate; in this graph, the overall migration speed was calculated as \(V=a* {b}\) (RWD/time) and normalized against controls without TMZ (non-normalized \(V\) values are reported in Supplementary Fig. S6) to obtain a relative migration speed (in the plot: V ± Standard Error as obtained from the fitting; two-tailed unpaired t-test, ***p < 0.005).
RNA-Seq showed a general enrichment of Surface HA binding genes (not only CD44 but also e.g. LAYS, CEMIP, SUSD5 or STAB1), in mesenchymal GBM2 and GBM6, and of several Matrix HA binding genes (BCAN, NCAN and ACAN) in proneural GBM1 and GBM5 (Fig. 3a). These correlations were statistically significant also in larger datasets (Supplementary Fig. S5).
We further looked at the effects of HA on migration speed (Fig. 3b and Supplementary Fig. S6), specifically employing sub-IC50 TMZ (20 µM) to ‘stress’ the PD-GBM without affecting their number. Although individually neither HA nor TMZ appears to have any effect, their combination reduced migration speed in mesenchymal GBM2 and GBM6 (Fig. 3b, right), which tallies with their higher expression of (mobility-related) HA surface receptors.
In short, interactions with HA appear to preferentially occur in the matrix for proneural and on the cell surface for mesenchymal GBM.
Subtype-dependent responses to TMZ
In our panel, mesenchymal and proneural subtypes differ in stemness, CD44 expression, and their interactions with HA. We have then assessed any difference in TMZ response: early (0–24 h) growth rates (proliferation) were unaffected in proneural GBM1 and GBM5 by TMZ doses from 23 up to 1500 µM TMZ, while mesenchymal GBM2 and GBM6 progressively decreased their proliferation with increasing TMZ concentration (Fig. 4a). In this context, U87 behaved more similarly to proneural GBMs.

a Left: Cell growth (time-dependent “surface coverage” i.e. confluence normalized to time zero) in the presence of different TMZ concentrations (i.e. six 1:2 serial dilutions from 1500 to 23 µM TMZ); the plot shows mean ± SEM of one experiment per cell culture. (Of note, GBM1 appears to have a very proliferative population fraction, so sensitive to TMZ to disappear already at 23 µM). Right: growth rates, calculated as the 0–24 h slope in surface coverage graphs; each TMZ concentration is normalized to untreated ctrl. In the inset, “absolute growth rate without TMZ” is the non-normalized growth rate of untreated cells. b Left: Representative 72 h phase contrast images of GBM1 and GBM2 showing the effect of 93 µM TMZ administered either as a 1st dose (top images, controls are untreated) or as a 2nd dose (bottom images, controls are treated with only the 1st dose of 75 µM and no 2nd dose). Scale bar 100 µm. Top right: scheme of 1st and 2nd dose TMZ experiments. BCA provides the total protein content, i.e., surviving adherent cells; this is unaffected by morphological or metabolic contributions (see Supplementary Fig. S7), unlike MTS or MTS/BCA. 75 µM = 13.99 μg/mL, is the max TMZ concentration observed in patients’ plasma, about 10x that in cerebrospinal fluid [51]. Bottom: summary of IC50 (for 1st and 2nd dose), calculated from sigmoidal curve fitting of BCA dose-response plots (see Supplementary Fig. S7). The GBM5 2nd dose IC50 is estimated as > 3 mM, since at the highest concentration used, these cells still retained >50% viability. Two notes: (1) the low 1st dose IC50 of GBM1 (42 µM) is likely due to a drug-sensitive subpopulation, whereas 30-40% of the cells survived to up to more than 200 µM TMZ; the removal of this by the 1st TMZ treatment (see first graph in Fig. 5a, left) is a contributor to the higher IC50, but likely a minor one. (2) In the 2nd treatment, GBM6 maintained the same IC50, most likely due to their expression of MGMT (See Fig. 1a).
Assessment of viability at later time points (72 h) showed a further difference: although - in agreement with literature [21] - IC50s did not directly correlate to subtype (Supplementary Table S10), proneural PD-GBMs were more prone to develop resistance: after a first 75 µM TMZ treatment, their IC50 (measured at the end of a 2nd TMZ exposure, see “Experimental protocol” inset in Fig. 4b) increased drastically, while no significant effect was recorded for the mesenchymal subtype, which maintain similar IC50 (or slightly decrease it, as in GBM2) (Fig. 4b).
Actually, proneural PD-GBMs cells that recovered from the first TMZ dose (72 h treatment + 72 h washout) showed strongly reduced proliferation, possibly causing their higher resistance. Conversely, mesenchymal PD-GBMs maintained some proliferative potential (especially MGMT-expressing GBM6) and a similar TMZ sensitivity (Supplementary Fig. S7).
In short, the less proliferative the proneural PD-GBMs are, the more likely they also are to develop resistance.
Effect of TMZ on subtype: a near-IC50 treatment leads to a “mesenchymal shift”
We aimed to assess any subtype-specific impact of TMZ at the transcriptomic level, keeping the drug concentration close to the IC50 to simulate a condition of significant cytotoxicity but not total eradication. For what concerns the subtype and stemness scores, we observed that:
A) in proneural PD-GBMs (and in U87), the MES vs PN z-score increased up to 2-fold, while remaining constant in mesenchymal PD-GBMs (Fig. 5a; Supplementary Fig. S3); further, CD44 increased in GBM1, which tallies with their increased mesenchymal character. Of note, GBM5 has an undetectable CD44 expression by RNA-Seq (and qPCR shows very low levels, Fig. 2a), thus – although likely – an analogous conclusion cannot be reached for them.

a Effect of TMZ on MES vs PN, stemness z-scores and on CD44 expression; RNA-Seq data, 72 h of near-IC50 TMZ treatment and untreated controls (GBM1: 20 µM; GBM2, GBM5, GBM6: 400 µM; n = 3 samples per condition; fold change: ratio of signatures z-scores in TMZ-treated versus untreated samples; SD is provided for CD44 transcript expression). b Y-axis: pathways enriched respectively in proneural and mesenchymal subtype, as obtained from GSEA analysis of the PD-GBM cell panel (“hallmarks” gene sets with FDR < 0.25 in the “mesenchymal vs proneural” comparison analysis). X-axis: Normalized Enrichment Score (NES) of pathways upon near-IC50 TMZ treatment, as assessed through GSEA analysis of each PD-GBM (“TMZ vs untreated” analysis). c Left: representative picture of spheroids culture with/without 75 µM TMZ treatment (72 h, followed by 72 h washout) (scale bar 200 µm). Middle: Effect of 75 µM TMZ treatment on stemness indicators: % of colony-forming cells (ratio no. spheroids at day 14/single cells at day 0, left y-axis); spheroids growth rate (fold change of spheroids area day 14/day 0, right y-axis). Data are the mean ± SD of n = 8 technical replicates per condition. Right: Correlation between stemness score in untreated GBM (x-axis: z-scores, as from Fig. 1) and stemness phenotype variation upon TMZ (y-axis: spheroid formation fold change with/without TMZ) (Pearson correlation: r = −0.98, *p < 0.05).
B) the stemness z-score decreased for all PD-GBM, but not for U87, possibly because the latter already had minimal expression of stemness genes (Supplementary Fig. S3).
In short, TMZ toxicity induces a “mesenchymal shift”. This was already described in proneural GBMs and correlated to the development of TMZ resistance [19]. We studied this issue at a whole-transcriptome level through comparative gene set enrichment analysis (GSEA) with the following aims:
(1) to identify the top-enriched pathways in mesenchymal and in proneural PD-GBMs (Fig. 5b, Supplementary Table S8).
MES-enriched pathways are known inducers of mesenchymal phenotype (EMT, TNFα signaling via NFKβ and TGFβ signaling [48]), and immune/cytokine-related pathways (Inflammatory, Interferon response, IL2/STAT5 and IL6/JAK/STAT3 signaling). This tallies with reports of a connection between immune signaling and mesenchymal signature [10, 16, 19]. Of note, this enrichment in the absence of immune cells supports the cell-intrinsic nature of this signaling in mesenchymal GBMs.
PN-enriched pathways, conversely, are stemness-promoting (Notch and Wnt/Beta catenin signaling) and oncogenic gene sets (E2F targets and Myc targets). To our knowledge, the latter have not been previously associated with this subtype. Importantly, the Myc gene is a master regulator of GBM stem cell properties [49], supporting the PN-stemness correlation.
The robustness of these subtype-specific pathways was also validated in the Lazzarini dataset (Supplementary Table S9).
2) to highlight the subtype-specific response of these pathways to TMZ treatment. The majority of MES-specific pathways increased, while PN-specific ones decreased (Fig. 5b). The “mesenchymal shift”, which was previously shown only for the MES vs PN 8-gene score (Fig. 5a), seems therefore to affect a larger proportion of the transcriptome, occurring not only in proneural GBM but also in the mesenchymal subtype.
Further, and specifically in proneural PD-GBMs, the decrease of PN-enriched pathways corresponds to a reduction in stemness, as witnessed by both a lower z-score (Fig. 5a), a lower tendency of forming spheroids, and a lower spheroid growth rate (Fig. 5c, left and middle). Actually, spheroid formation upon TMZ appeared to scale remarkably well with the initial stemness z-score: the more stem (and proneural) PD-GBM were, the higher was the stemness decrease (Fig. 5c, right).
Of note, no near-IC50 TMZ treatment produced any significant genomic alteration (CNV, see Supplementary Fig. S1), i.e., selection of specific gains/losses. However, in the treated samples, the chromosomal scatter plots suggest a lower heterogeneity, possibly due to the elimination of minor sub-populations.
(Simulated) hypoxia affects subtype-dependent TMZ response
Besides a generic hypoxia/therapy-resistance relation, there is yet no general consensus on how GBM subtypes may be differently affected by hypoxia, and how this may relate to drug response.
Here, we have employed simulated hypoxic conditions (CoCl2 rapidly induces a reproducible hypoxic response [50], by stabilizing hypoxia inducible factors 1α and 2α - HIF-1/2 α - also under normoxic conditions), and molecularly stratified the response to TMZ under hypoxia.
Effect of simulated hypoxia on viability indicators (no TMZ)
Simulated hypoxia (100 µM CoCl2) reduced growth and increased metabolic activity in proneural, but not mesenchymal GBMs (Fig. 6a). Further, CD44LOW GBM1 behaved similarly to their high-stemness parent GBM1, while GBM1 CD44HIGH behaved more like mesenchymal PD-GBMs (Fig. 6a, left); this is in agreement with their respective stemness scores (see Fig. 2b), and shows that the subtype/stemness stratification links to a different phenotypic response also to hypoxia.

a Effect of CoCl2 treatment (72 h, 100 µM) on viability indicators: number of adherent cells via BCA assay (left y axis, black symbols) and normalized metabolic activity (MTS/BCA; right y axis, red symbols) plotted as a function of stemness z-score (x axis). MTS and BCA data are the mean ± SD of n = 2 biological replicates, each in technical triplicate; each parameter is normalized on ctrl, i.e., untreated cells. Please note that a lower BCA count means that cells proliferate less than controls, and does not imply direct toxicity. Stemness z-score was calculated from qPCR data (fold change of each gene, see Supplementary Information) in the population of 6 cell culture types showed. b Effect of 75 µM TMZ (with or without 100 µM CoCl2, left) and of 100 µM CoCl2 alone (right) on migration (scratch wound assay performed during the 72 h treatments). Relative Wound Density (RWD %) data (Supplementary Fig. S8) are fitted as described in the caption to Fig. 3 and relative migration speeds were obtained as the treated vs. untreated ratios. The fitting used n = 3 technical replicates from a single experiment; the controls were untreated normoxic samples for TMZ in normoxia and CoCl2 in hypoxia, and CoCl2-only samples for TMZ+CoCl2 in hypoxia (unpaired t-test, two-tailed, *p < 0.5, **p < 0.005). c Dose-response curves of cell viability indicators after 72 h of TMZ treatment in the presence/absence of simulated hypoxia (+/− CoCl2). BCA (in red, no. of attached, i.e. surviving, cells), MTS/BCA ratio (in black, metabolic activity of surviving cells) and surface coverage (in blue). Representative curves of GBM1 and GBM2 are shown (mean ± SD of one experiment in triplicate), complete data are in Supplementary Fig. S9). d Correlation between stemness z-score (x-axis) and sensitivity to TMZ (y-axis) in presence/absence of simulated hypoxia (CoCl2), expressed as ratio of IC50TMZ+CoCl2 /IC50TMZ.
Effect of simulated hypoxia on TMZ efficacy: cell migration
75 µM TMZ seems to increase cell migration under hypoxia (compared to normoxia), but only in proneural cells.
It must be noted that simulated hypoxia (CoCl2, no TMZ) slowed down cell migration, slightly but significantly, only in mesenchymal GBM2 and GBM6 (empty circles in Fig. 6b, right). We have previously seen that TMZ alone at 20 µM (sub IC50 for all cell types) had a negligible effect on migration (Fig. 3c). Here, we employed a higher concentration (75 µM) (Fig. 6b and Supplementary Fig. S8), closer to TMZ IC50 values and near the maximum drug concentration detected in patient’s plasma [51].
Looking separately at the effect of TMZ under normoxia (black circles in Fig. 6b, left) and under simulated hypoxia (red circles in Fig. 6b, left), it is difficult to highlight a trend; however, their comparison clearly shows that the hypoxic conditions significantly increased the migration rate only in proneural PD-GBM.
TMZ sensitivity in simulated hypoxia
Simulated hypoxia promoted TMZ resistance in proneural GBM.
We have compared the effect of TMZ in the presence or absence of CoCl2 (empty and full symbols in Fig. 6c) using three indicators: cell survival (BCA), metabolic activity (MTS/BCA), and surface coverage (confluence). In proneural GBM (GBM1 in Fig. 6c; GBM5 in Supplementary Fig. S9), the TMZ treatment in hypoxia left unaltered metabolism, mildly increased survival, and more strongly increased cell spreading (coverage) in comparison to TMZ+normoxia. In mesenchymal GBM (GBM2 in Fig. 6c; GBM6 in Supplementary Fig. S9), TMZ+hypoxia increased metabolism, and decreased both survival and spreading.
Lastly, we correlated the variations in IC50 (IC50TMZ vs IC50TMZ+CoCl2), i.e., the hypoxia effect on TMZ potency, with the stemness score (Fig. 6d); the graph clearly highlights a stratification: mesenchymal cells show lower stemness, and TMZ is more potent under hypoxia; proneural cells have higher stemness and are more resistant to TMZ under hypoxia.
In short, the activation of hypoxic signaling produces different effects on proneural and on mesenchymal PD-GBM, with the former apparently becoming more resistant.
Discussion
GBM cells are frequently described through a few, recurrent expression signatures; the most popularly used subtypes are proneural and mesenchymal, also used to illustrate the “proneural to mesenchymal” transition upon treatment [7, 13]. We support the concept that the transcriptomic variance can be mainly addressed through a proneural vs mesenchymal spectrum (MES vs PN score), here assigned through a reduced 8-gene signature (as from Bhat [10]). We demonstrated this point experimentally using a panel of 4 PD-GBM cultures, and further validated it in larger transcriptomic datasets of cells [43] and tissue samples (TCGA). On the basis of both CNV and SNV, and of transcriptomic subtypes (both Verhaak’s and MES vs PN), we align with the notion that genomic alterations show some statistical association with subtypes [1, 4], but are not univocal indicators.
Stemness is potentially another important descriptor, be it expressing the average expression of stemness features, or the proportion of stem-like cells in the tumoral population. However, its integration with molecular subtypes has not found much consensus. Yet, some studies have noticed that the GBM proneural subtype often presents higher stemness traits [14, 52,53,54], and that it may be hierarchically upstream of the mesenchymal [7, 14].
Here, we align with this perspective: firstly, we found that proneural GBM express higher levels of genes from a stemness signature of 4 transcriptional factors [29] - POU3F2, SOX2, SALL2 and OLIG2 - that are not simple “stemness markers” in GBM, but hierarchically high, master regulators of stemness. Secondly, in our PD-GBM panel, we have also demonstrated the higher spheroid-forming capacity of proneural GBM (vs. mesenchymal), thereby aligning a transcriptomic score with a phenotypic feature.
Our study has also focused on the role of CD44, which is often - and conflictingly – used as a marker both for stem-like and mesenchymal GBM phenotypes. In our PD-GBM panel and in the validation datasets, CD44 expression appeared to closely correlate with the mesenchymal score and inversely with stemness (the latter is not valid in TCGA, possibly because of the presence of non-tumoral cells in these surgical specimens). Although CD44 signaling notoriously also promotes stemness-associated features (e.g., radiation-resistance and hypoxic response [10, 55]), our results cast significant doubts on its frequent use as a putative GBM stemness marker; if a CD44/stemness correlation exists, indeed it would appear to be inverse. This was further validated by sorting one of our PD-GBM models into CD44LOW and CD44HIGH subpopulations: the former is indeed more reminiscent of a stem-related phenotype, while the latter shows more distinct mesenchymal traits. Interestingly, there may be a wider association between expression of HA-binding proteins and the MES vs PN score: surface receptors (putatively degrading/internalizing HA) correlate to mesenchymal score, while HA-binding ECM components (mainly neural-specific chondroitin sulfate proteoglycans) correlate to proneural score, possibly making the former more sensitive than the latter to HA direct signalling. Indeed, we have seen that the TMZ + HA combination affected the migration speed of mesenchymal but not proneural PD-GBMs.
We have also highlighted some proneural vs mesenchymal differences in their response to TMZ. Firstly, in the first 24 hours the proliferation of proneural PD-GBMs was unaffected, while that of mesenchymal ones decreased in a TMZ dose-responsive fashion. Of note, and in line with other literature reports [21, 56], this effect does not correlate to the original proliferation rate, or the IC50TMZ of each PD-GBM.
Secondly, despite the initial lack of effects mentioned before, a sub-IC50 (75 µM) 72 h TMZ dose made proneural (but not mesenchymal) PD-GBM less proliferative; this was accompanied by a higher resistance to a second TMZ dose, which may be due to TMZ predominantly acting on proliferating cells.
Thirdly, near-IC50 72 h TMZ treatment produced a mesenchymal shift mirrored by a reduction in stemness phenotype, which is most apparent in proneural GBM.
This is quite peculiar: the mesenchymal score does not correlate to differential TMZ resistance among the initial subtypes (=no prognostic value for a first treatment), but for a given subtype it may contain insightful information on the outcome of successive ones (=indicative of acquired resistance).
Our data do not allow to further draft a mechanistic correlation between MES shift and increased TMZ resistance, although it has been reported that several mesenchymal-induced transcripts directly promote it. For example, CEBPB, a known inducer of mesenchymal reprogramming [57], is upregulated by TMZ treatment in our panel(Supplementary Fig. S10) and contributes to TMZ resistance [58] and radio-resistance [10].
It is noteworthy that the mesenchymal shift should be taken in a comparative manner: for example, transcriptional stemness of proneural GBM decreased upon treatment but yet remained relatively high in absolute terms, which suggests that their potential to keep a stem-like population may only be transiently affected, and fits well with the general concept that GBM stem-like populations survive therapies and participate in tumor recurrence.
Finally, we identified different subtype-specific responses also under simulated hypoxia: in this condition, proneural but not mesenchymal PD-GBM increased metabolic activity and decreased proliferation. This different adaptation possibly affects their response to treatment: indeed, under hypoxia, TMZ was less effective (higher IC50) in proneural and more effective in mesenchymal PD-GBM, compared to treatment in normoxia.
In summary, our data support:
1) the proneural/mesenchymal signature both as a sufficient descriptor for GBM’s transcriptional variability, degree of stemness and response to TMZ (both in normoxia and hypoxia), and as a tool to establish further correlations, e.g., with HA-binding genes, or prospectively for the patient-stratified application of new GBM drugs.
(2) CD44 expression level (and potentially other cell-surface HA receptors) is considered as mesenchymal rather than as a stem/proneural marker.
(3) a seemingly incongruent description of the development of resistance to TMZ. Treatments apparently promote two phenomena in proneural GBM, which at the same time become more resistant and acquire mesenchymal features; however, the latter per se does not imply resistance development (in the same in vitro settings). This may be explained by the higher stemness potential of proneural cells, which show higher plasticity in response to stimuli such as chemotherapy and hypoxia.
Limitations of the study
This study is mainly correlative (subtype - stemness - HA-binding genes), using larger datasets to validate data from an admittedly limited number of cellular models. Therefore, it is impossible (a) to fully generalize a subtype-dependent behavior in TMZ resistance, due to the rather small number of patient-derived GBM cultures, and (b) to reach firm mechanistic conclusions, due to the lack of experimental perturbation of key components of transcriptomic signatures or the HA pathway.
Data availability
Transcriptomic data are available at the Sequence Read Archive (SRA) NCBI database under the project accession PRJNA1268952.
References
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110.
Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor evolution of Glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32:42–56.e6.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77.
Reuss DE. Updates on the WHO diagnosis of IDH-mutant glioma. J Neurooncol. 2023;162:461–9.
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23:1231–51.
Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–73.
Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for Glioblastoma. Cell. 2019;178:835–49.e21.
Mao P, Joshi K, Li J, Kim S-H, Li P, Santana-Santos L, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci. 2013;110:8644–9.
Bhat KPL, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in Glioblastoma. Cancer Cell. 2013;24:331–46.
Wang L, Babikir H, Müller S, Yagnik G, Shamardani K, Catalan F, et al. The phenotypes of proliferating Glioblastoma cells reside on a single axis of variation. Cancer Discov. 2019;9:1708–19.
Richards LM, Whitley OKN, MacLeod G, Cavalli FMG, Coutinho FJ, Jaramillo JE, et al. Gradient of Developmental and Injury Response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat Cancer. 2021;2:157–73.
Bv H, Jolly MK Proneural-mesenchymal antagonism dominates the patterns of phenotypic heterogeneity in glioblastoma. iScience. 2024;27.
Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, et al. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26:288–300.
Piao Y, Liang J, Holmes L, Henry V, Sulman E, de Groot JF. Acquired resistance to Anti-VEGF therapy in Glioblastoma Is associated with a Mesenchymal transition. Clin Cancer Res. 2013;19:4392–403.
Xiao Y, Yang K, Wang Z, Zhao M, Deng Y, Ji W, et al. CD44-Mediated poor prognosis in glioma is associated with M2-polarization of tumor-associated macrophages and immunosuppression. Front Surg. 2021;8:775194.
Joseph JV, Conroy S, Tomar T, Eggens-Meijer E, Bhat K, Copray S, et al. TGF-β is an inducer of ZEB1-dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis. 2014;5:e1443–e.
Wang L, Jung J, Babikir H, Shamardani K, Jain S, Feng X, et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat Cancer. 2022;3:1534–52.
Kim Y, Varn FS, Park S-H, Yoon BW, Park HR, Lee C, et al. Perspective of mesenchymal transformation in glioblastoma. Acta Neuropathol Commun. 2021;9:50.
Teo WY, Sekar K, Seshachalam P, Shen J, Chow WY, Lau CC, et al. Relevance of a TCGA-derived Glioblastoma Subtype Gene-Classifier among Patient Populations. Sci Rep. 2019;9:7442.
Ntafoulis I, Kleijn A, Ju J, Jimenez-Cowell K, Fabro F, Klein M, et al. Ex vivo drug sensitivity screening predicts response to temozolomide in glioblastoma patients and identifies candidate biomarkers. Br J Cancer. 2023;129:1327–38.
Chin L, Meyerson M, Aldape K, Bigner D, Mikkelsen T, VandenBerg S, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8.
Tso CL, Freije WA, Day A, Chen Z, Merriman B, Perlina A, et al. Distinct transcription profiles of primary and secondary glioblastoma subgroups. Cancer Res. 2006;66:159–67.
Bradshaw A, Wickremsekera A, Tan ST, Peng L, Davis PF, Itinteang T. Cancer stem cell hierarchy in Glioblastoma Multiforme. Front Surg. 2016;3:21.
Dirkse A, Golebiewska A, Buder T, Nazarov PV, Muller A, Poovathingal S, et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat Commun. 2019;10:1787.
Yan Y, Zuo X, Wei D. Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem Cells Transl Med. 2015;4:1033–43.
Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11:254–67.
Safarians G, Sohrabi A, Solomon I, Xiao W, Bastola S, Rajput BW, et al. Glioblastoma spheroid invasion through soft, brain-like matrices depends on Hyaluronic Acid-CD44 interactions. Adv Health Mater. 2023;12:e2203143.
Suvà ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, et al. Reconstructing and reprogramming the tumor-propagating potential of Glioblastoma stem-like Cells. Cell. 2014;157:580–94.
Uribe D, Niechi I, Rackov G, Erices JI, San Martín R, Quezada C. Adapt to Persist: Glioblastoma microenvironment and epigenetic regulation on cell plasticity. Biology. 2022;11.
Greenwald AC, Darnell NG, Hoefflin R, Simkin D, Mount CW, Gonzalez Castro LN, et al. Integrative spatial analysis reveals a multi-layered organization of glioblastoma. Cell. 2024;187:2485–501.e26.
Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1α. Oncogene. 2009;28:3949–59.
Marallano VJ, Ughetta ME, Tejero R, Nanda S, Ramalingam R, Stalbow L, et al. Hypoxia drives shared and distinct transcriptomic changes in two invasive glioma stem cell lines. Sci Rep. 2024;14:7246.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96.
Singh N, Miner A, Hennis L, Mittal S. Mechanisms of temozolomide resistance in glioblastoma - a comprehensive review. Cancer Drug Resist. 2021;4:17–43.
Rabé M, Dumont S, Álvarez-Arenas A, Janati H, Belmonte-Beitia J, Calvo GF, et al. Identification of a transient state during the acquisition of temozolomide resistance in glioblastoma. Cell Death Dis. 2020;11:19.
Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350–4.
Pibuel MA, Poodts D, Díaz M, Hajos SE, Lompardía SL. The scrambled story between hyaluronan and glioblastoma. J Biol Chem. 2021;296:100549.
Teng J, Hejazi S, Hiddingh L, Carvalho L, de Gooijer MC, Wakimoto H, et al. Recycling drug screen repurposes hydroxyurea as a sensitizer of glioblastomas to temozolomide targeting de novo DNA synthesis, irrespective of molecular subtype. Neuro-Oncology. 2017;20:642–54.
Skaga E, Kulesskiy E, Fayzullin A, Sandberg CJ, Potdar S, Kyttälä A, et al. Intertumoral heterogeneity in patient-specific drug sensitivities in treatment-naïve glioblastoma. BMC Cancer. 2019;19:628.
Breznik B, Motaln H, Vittori M, Rotter A, Turnsek TL. Mesenchymal stem cells differentially affect the invasion of distinct glioblastoma cell lines. Oncotarget. 2017;8:25482–99.
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7.
Lazzarini E, Silvestris DA, Benvenuto G, Osti D, Fattore L, Paterra R, et al. Genome-wide profiling of patient-derived glioblastoma stem-like cells reveals recurrent genetic and transcriptomic signatures associated with brain tumors. J Neurooncol. 2023;163:47–59.
Network CGAR. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8.
Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of Tumorigenic, stem-like neural precursors from human Glioblastoma. Cancer Res. 2004;64:7011–21.
Knudson W, Chow G, Knudson CB. CD44-mediated uptake and degradation of hyaluronan. Matrix Biol. 2002;21:15–23.
Rios de la Rosa JM, Tirella A, Tirelli N. Receptor-targeted drug delivery and the (many) problems we know of: The case of CD44 and hyaluronic acid. Adv Biosyst. 2018;2:1800049.
Yan T, Tan Y, Deng G, Sun Z, Liu B, Wang Y, et al. TGF-β induces GBM mesenchymal transition through upregulation of CLDN4 and nuclear translocation to activate TNF-α/NF-κB signal pathway. Cell Death Dis. 2022;13:339.
Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, et al. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One. 2008;3:e3769.
Muñoz-Sánchez J, Chánez-Cárdenas ME. The use of cobalt chloride as a chemical hypoxia model. J Appl Toxicol. 2019;39:556–70.
Ostermann S, Csajka C, Buclin T, Leyvraz S, Lejeune F, Decosterd LA, et al. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin Cancer Res. 2004;10:3728–36.
Cusulin C, Chesnelong C, Bose P, Bilenky M, Kopciuk K, Chan JA, et al. Precursor states of brain tumor initiating cell lines are predictive of survival in Xenografts and associated with Glioblastoma Subtypes. Stem Cell Rep. 2015;5:1–9.
Schulte A, Günther HS, Phillips HS, Kemming D, Martens T, Kharbanda S, et al. A distinct subset of glioma cell lines with stem cell-like properties reflects the transcriptional phenotype of glioblastomas and overexpresses CXCR4 as therapeutic target. Glia. 2011;59:590–602.
Wang Z, Wang Y, Yang T, Xing H, Wang Y, Gao L, et al. Machine learning revealed stemness features and a novel stemness-based classification with appealing implications in discriminating the prognosis, immunotherapy and temozolomide responses of 906 glioblastoma patients. Brief Bioinform. 2021;22.
Pietras A, Katz AM, Ekström EJ, Wee B, Halliday JJ, Pitter KL, et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014;14:357–69.
Nam Y, Koo H, Yang Y, Shin S, Zhu Z, Kim D, et al. Pharmacogenomic profiling reveals molecular features of chemotherapy resistance in IDH wild-type primary glioblastoma. Genome Med. 2023;15:16.
Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–25.
Gao Y, Liu B, Feng L, Sun B, He S, Yang Y, et al. Targeting JUN, CEBPB, and HDAC3: a novel strategy to overcome drug resistance in hypoxic Glioblastoma. Front Oncol. 2019;9:33.
Quagliariello V, Gennari A, Jain SA, Rosso F, Iaffaioli RV, Barbarisi A, et al. Double-responsive hyaluronic acid-based prodrugs for efficient tumour targeting. Mater Sci Eng C Mater Biol Appl. 2021;131:112475.
Gangemi RMR, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, et al. SOX2 silencing in Glioblastoma tumor-initiating cells causes stop of proliferation and loss of Tumorigenicity. Stem Cells. 2009;27:40–8.
Rancati S, Pereira RC, Schlich M, Sgroi S, Beatini S, La Rosa L, et al. Synergic microRNAs suppress human glioblastoma progression by modulating clinically relevant targets. Mol Ther Nucleic Acids. 2025;36:102763.
Acknowledgements
The authors thank Fidia S.p.A. (Abano Terme, Italy) for the kind donation of hyaluronic acid with \(\bar{{M}_{w}}\) = 220 kDa (characterization in a previous paper of Tirelli’s group [59]. The authors are also indebted to Dr. Ennio Albanesi (IIT Neurofacility, Genova, Italy), Dr.s Paolo Inglese and Diego Vozzi (IIT Genomic Facility, Genova, Italy) for preparing RNA libraries and running the RNA-Sequencing, Sara Trova and the technicians of the IIT Genomic facility in Aosta for DNA library preparation and sequencing, and Debora Charrance for bioinformatics support.
Funding
Funding is gratefully acknowledged from (1) Associazione Italiana Ricerca Cancro (AIRC) through the IG-2020 grant “Hyaluronic acid-based carriers. Targeting mechanisms and application in a combination therapy approach”, project code 24628; (2) the National Center for Gene Therapy and Drugs based on RNA Technology”, codice CN00000041 finanziato dall’Unione Europea – NextGenerationEU PNRR MUR - M4C2 – Investimento 1.4 - Avviso “Potenziamento strutture di ricerca e creazione di “campioni nazionali di R&S” CUP J33C22001130001; (3) the project 5000genomi@VdA, co-funded by “Fondo Europeo di Sviluppo Regionale” (FESR CUP B68H19005520007 and FESR CUP B68H19005520007); (4) “Programma Investimenti per la crescita e l’occupazione 2014/20” (European Social Fund, ESF CUP B65F19001200009); the project Genomics 2.0@vda founded by “Programma Regionale Fondo sociale europeo Plus 2021/2027” (European Social Fund, ESF CUP J51B24000170002); (5) the project “National Centre for HPC, Big Data and Quantum Computing” (CN00000013 - Spoke8).
Author information
Authors and Affiliations
Contributions
SR and NT conceived the study. SR designed all experiments, with contributions by MV and SG. SR, MC, and GD performed all cell experiments. SR (cell culture), AC, FF, and FL (genomic) and PL (transcriptomic) analyzed the experimental results. SR and NT interpreted the results and wrote the first draft of the manuscript, with FF, AC, and MV contributing to the genomic section. SR, NT, AC, SG, and MV contributed to the final version of the manuscript, which was read and approved by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations: PD-GBM cultures originate from surgical samples obtained after informed consent at the Neurosurgery Department of the San Martino Hospital (Genova, Italy), and then processed by the group of Antonio Daga in the Department of Translational Oncology of the same hospital, as described in previous literature [60], and used by the group of Davide De Pietri Tonelli at IIT [61].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rancati, S., Campolungo, M., Dalmolin, D.G. et al. Molecular subtypes and the (in vitro) response of glioblastoma to temozolomide. BJC Rep 4, 24 (2026). https://doi.org/10.1038/s44276-026-00228-8
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44276-026-00228-8
