
Abstract
Conversion of polyethylene (PE) into ethylene and propylene will enable closed-loop recycling of plastics. Conventional catalytic cracking of PE is restricted by kinetic entanglement between the formation of main products and by-products, limiting ethylene and propylene yields to less than 25%. Here we address this challenge with a kinetic decoupling–recoupling (KDRC) strategy, achieving yields of ethylene and propylene up to 79% from PE conversion using a tandem reactor with dual zeolite catalysts. Reaction kinetics analysis, synchrotron-based vacuum ultraviolet photoionization mass spectrometry and in situ neutron powder diffraction reveal that KDRC decouples kinetics of PE cracking to intermediates (butenes and pentenes) in the first stage and synchronizes this process with dimerization–β-scission reactions in the second stage. This synchronization minimizes by-products and enhances ethylene and propylene production substantially. Combined with high catalytic stability, this KDRC strategy represents a robust pathway to combating plastic pollution via a circular economy.

Similar content being viewed by others

Main
Large quantities of plastic products are used and discarded every year. Waste polyolefin plastics, primarily polyethylene (PE) and polypropylene (PP), pose increasing environmental and health risks due to their widespread distribution, durability and resistance to degradation1. Chemical recycling emerges as a promising solution to mitigate the increasing plastic wastes2,3,4,5,6,7. Polyolefins account for over 50% of plastic wastes, and extensive research has been conducted to convert polyolefins into fuels8,9,10,11,12,13,14,15,16 and valuable chemicals17,18,19,20,21,22,23,24,25.
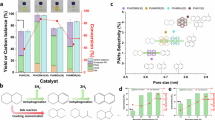
The conversion of polyolefins into bulk chemicals of high demands (for example, ethylene and propylene) is essential to the closed-loop recycling of plastics26,27,28,29,30,31,32,33. However, such conversion is extremely challenging. Several strategies have been explored. For example, it was discovered that the reaction of PE or PP with excess ethylene (15–20 bar) could efficiently enhance the yield of propylene or iso-butene using suitable catalysts26,27. Direct cracking of polyolefins yields a wide range of products, with yields of ethylene and propylene of approximately 23% (Fig. 1a and Supplementary Table 1)33,34,35,36,37. Recent studies using electrified spatiotemporal heating techniques have cracked polyolefins to ethylene and/or propylene with yields of 36–45% by optimizing the transient heating time28,29 (Fig. 1b and Supplementary Table 1). Direct cracking of polyolefins involves a series of reactions—including cracking, dimerization, oligomerization, isomerization, hydride transfer and aromatization—and the reaction pathway is difficult to control, making it challenging to achieve high selectivities34,35,36,37,38,39,40,41. Further research is needed to elucidate the impact of reaction kinetics on product selectivity in plastics conversion.

a, Conventional catalytic cracking of polyolefins to light olefins. b, Previous work using pulsed heating techniques to crack polyolefins to ethylene and/or propylene with or without catalysts28,29. c, Current work applying the KDRC strategy to yield ethylene and propylene from polyolefins. The KDRC strategy aligns butene and pentene formation with dimerization–β-scission, boosting ethylene and propylene yield.
Here, we report a kinetic decoupling–recoupling (KDRC) strategy to manipulate the kinetics of reaction pathways, and in the absence of noble metals, external H2 or olefins, this strategy has afforded yields of ethylene and propylene up to 79% (carbon molar yield) from PE conversion over zeolite catalysts (Fig. 1c and Supplementary Table 1). Direct observation of the important C8 intermediate and the location of acid sites of catalysts have been achieved using synchrotron-based vacuum ultraviolet photoionization mass spectrometry (SVUV-PIMS) and neutron powder diffraction (NPD), respectively, revealing molecular details of the KDRC strategy.
Results
KDRC strategy for selective production of ethylene and propylene
The layered self-pillared zeolite (LSP-Z100) features anMFI/MEL (framework type code of zeolite) intergrowth layered structure with a high external surface area and a well-developed mesoporous network9 (Supplementary Table 2 and Supplementary Fig. 1a,b). This structure generates numerous open-framework tri-coordinated Al sites, which serve as strong Lewis acid sites9. Owing to its combination of strong acidity and mesoporosity, LSP-Z100 exhibits higher activity in PE conversion than both conventional mesoporous and microporous zeolites (Supplementary Fig. 2 and Supplementary Table 3). We recently reported that LSP-Z100 can effectively crack PE to gasoline via hydride abstraction, β-scission, isomerization and hydride transfer in a batch reactor9. Here, to terminate the reaction before hydride transfer to yield ethylene and propylene, we used a fixed-bed reactor with a N2 flow to reduce the catalyst contact time. As a result, a notable amount of alkenes (carbon molar yield of 94%) was observed on the LSP-Z100 zeolite at 260 °C for 2 h, with a PE/catalyst ratio of 8:1 (Extended Data Fig. 1a and Supplementary Table 4). However, the alkenes produced are mainly butenes and pentenes (combined 46%) with only 5% of desired ethylene and propylene (Fig. 2a). Despite optimization of various reaction conditions, butenes and pentenes remained predominant products, with yields stabilizing at around 40–50% (Extended Data Fig. 1a,b and Supplementary Tables 4 and 5). To further convert these intermediates, we introduced a second catalyst, phosphorus-modified HZSM-5 (P-HZSM-5) (Fig. 2b and Supplementary Table 3), which possesses a microporous framework (Supplementary Table 2 and Supplementary Fig. 1c,d) and moderated acidity (Supplementary Fig. 3), making it suitable for converting butenes and pentenes into smaller molecules such as ethylene and propylene41,42. Indeed, this dual catalyst system achieved higher yields of ethylene and propylene (24%) and lower yields of butenes and pentenes (23%) at 540 °C (Fig. 2b, Extended Data Fig. 1c and Supplementary Table 6). However, increased amounts of by-products, such as alkanes and aromatics, were observed (Fig. 2a,b).

a, Conversion of HDPE over LSP-Z100 in a single-stage reactor. C2=C3= represents ethylene and propylene. C4=C5= represents butene and pentene. C>6 bar includes both alkanes and alkenes with carbon numbers greater than or equal to six. C20–C50 represents C2–C5 alkanes. ‘Arom.’ represents aromatics. Reaction conditions: 100 mg of HDPE and 12.5 mg of LSP-Z100 at stage I, reaction temperature, 260 °C; reaction time, 2 h; N2 flow rate, 10 sccm. b, Product distributions of HDPE conversion in a dual-catalyst, single-stage system (HDPE, 100 mg; LSP-Z100, 12.5 mg; P-HZSM-5, 100 mg). c, Rate constants of PE cracking on LSP-Z100. d, Concentrations of products from PE cracking on LSP-Z100 at different temperatures. e, Simplified reaction scheme proposed for PE conversion. Full reaction schemes are shown in Extended Data Fig. 2. f, Production rates of main products (ethylene and propylene) and by-products (alkanes and aromatics) against concentration of butene and pentene. g, Rate constants of dimerization–β-scission on P-HZSM-5. h, The yield of ethylene and propylene obtained from KDRC strategy. The detailed product yields are presented in Extended Data Table 1. Reaction conditions: 100 mg of HDPE and 12.5 mg of LSP-Z100 at stage I, 100 mg of P-HZSM-5 at stage II; reaction time, 2 h; N2 flow rate, 10 sccm. i, The optimized catalytic results via KDRC strategy. Reaction conditions: temperature-programmed reaction (TPR) from 260 °C to 300 °C in 2 h at stage I; 50 mg each of P-LSP-Z100 and P-HZSM-5 at stage II (540 °C); other conditions are the same as in h.
From thermodynamic estimations, the conversion of PE to ethylene and propylene should occur at about 400 °C and 290 °C, respectively (Supplementary Note 1 and Supplementary Table 7); thus, the observed low yields of ethylene and propylene originate from the formation of various kinetically favored by-products. Therefore, a thorough analysis of the reaction kinetics of PE conversion was performed using lumped kinetic modeling43 (Extended Data Fig. 2a–d, Supplementary Note 2, Supplementary Figs. 4–6 and Supplementary Tables 8–13). The rate constants (ki) and reaction rates (ri) were determined from kinetic data using the fourth–fifth-order Runge–Kutta algorithm (ode45) and a genetic algorithm (ga) implemented in MATLAB44,45. At stage I with LSP-Z100, the rate constant k2 is higher than k1 (Fig. 2c and Supplementary Table 9), suggesting that butenes and pentenes are preferentially produced from PE cracking (Fig. 2d). At stage II with P-HZSM-5, butenes and pentenes can either undergo route 1, dimerizing to C8 (or C>8) followed by β-scission to yield ethylene and propylene, as widely reported in the literature41,42,43, or route 2, to form by-products (alkanes and aromatics) via hydrogen transfer and aromatization (Fig. 2e). Kinetic experiments further demonstrate that dimerization–β-scission (route 1) is a first-order reaction where the reaction rate is linearly dependent on the concentration of butenes and pentenes, whereas hydrogen transfer and aromatization (route 2) are second-order reactions with the reaction rate proportional to the square of the concentration of butenes and pentenes (Fig. 2f, Extended Data Fig. 2d, Supplementary Note 2 and Supplementary Tables 12 and 13). Thus, to achieve a faster rate of formation of ethylene and propylene compared with that of by-products, the concentration of butenes and pentenes in the reaction system must be controlled within a range of 0.004–0.008 molCH2 g−1, identified as a ‘kinetic sweet spot’ I (KSS I) (Fig. 2f and Extended Data Fig. 2e). This was achieved by tuning the reaction temperature of PE cracking at stage I, and a butene and pentene concentration of 0.005 molCH2 g−1 achieved at 260 °C with a N2 flow rate of 10 standard cubic centimeters per minute (sccm) (Fig. 2d,f). Conversely, at stage II, the production of ethylene and propylene from butene and pentene via route 1 requires that the C8 (or C>8) species preferentially undergo β-scission to form ethylene and propylene rather than the reversed reaction of dimerization to form butene and pentene. In other words, a higher k4 than k3 is essential (Fig. 2e,g). This only be achieved only at temperatures above 500 °C (KSS II), as shown by the temperature dependence of the reaction rate of these two competing reactions (Fig. 2g). The analysis of reaction kinetics clearly indicates that the reaction rate k2 at stage I should be decoupled for independent optimization, in order to synchronize with the reaction rates k3 and k4 at stage II (KDRC strategy), ultimately enabling a high yield of ethylene and propylene (Figs. 1c and 2e). The reactor has, therefore, been redesigned by controlling the temperature of the two catalysts separately: 260 °C for LSP-Z100 (stage I) and 540 °C for P-HZSM-5 (stage II) (Extended Data Fig. 3a) to hit the KSS I and II (Fig. 2f,g), respectively, and the yield of ethylene and propylene reached as high as 71% (Fig. 2h, Extended Data Fig. 3b, Extended Data Table 1, Supplementary Figs. 7 and 8 and Supplementary Table 14). The yield of ethylene and propylene can be further increased to 79% by using a programmed temperature control at stage I (ranging from 260 °C to 300 °C) and a dual-catalyst bed (P-LSP-Z100 + P-HZSM-5) at stage II at 540 °C (Fig. 2i, Extended Data Fig. 3c, Supplementary Tables 15 and 16 and Supplementary Fig. 9). Given that the system operates without noble metals, external H2 or added olefins, this finding is important for polyolefin conversion (Fig. 1 and Supplementary Table 1). The atom and mass balances for all reactions exceed 95% (Supplementary Table 17).
Studies of dimerization–β-scission and reaction intermediates
To elucidate the mechanism of the cracking of PE to butene followed by its dimerization to C8 and the subsequent β-scission to ethylene and propylene, deuterated PE (PE-d4) and 1-butene (1-C4H8) were co-fed to the catalyst bed as feedstocks and products detected by SVUV-PIMS (Fig. 3a and Supplementary Fig. 10). The detected alkene products showed hydrogen/deuterium (H/D) isotopologues (Fig. 3b), indicating dynamic intermolecular interactions. C4D8 was identified as a key intermediate derived from PE-d4 cracking, and both C4D8 and C4H8 are actively involved in the formation of desired alkenes (Fig. 3b and Supplementary Figs. 11 and 12).

a, Schematic setup for the transformation of 1-butene during PE-d4 cracking. b, Transformation of 1-butene during PE and PE-d4 conversion in a two-stage reactor. The mass spectrum in the m/z range of 26–50 from the PE-d4 + C4H8 system is zoomed in. Reaction conditions: 80 mg PE or PE-d4 and 10 mg LSP-Z100 at stage I, 30 mg of P-HZSM-5 at stage II; reaction temperature, 260 °C (stage I), 540 °C (stage II); 1.5% C4H8/N2 flow rate, 10 sccm; ionization energy 11 eV. c, Schematic setup for the temperature-programmed reactions (TPR) of 1-butene on P-HZSM-5. d–f, TPR of 1-butene over P-HZSM-5 in a single-stage reactor. Reaction conditions: P-HZSM-5, 30 mg; reaction temperature, ramp from 50 °C to 400 °C at a rate of 100 °C min−1; 1.5% C4H8/N2 flow rate, 20 sccm; ionization energy 11 eV.
While dimerization–β-scission (Fig. 2e, route 1) has been widely reported as the pathway for ethylene and propylene production from butene over acidic zeolites, the experimental evidence of the presence of the C8 intermediate is missing. The SVUV-PIMS technique has been used owing to its unique advantages in the detection of transient reaction intermediates due to its tuneable photon energy, soft ionization and an exceptional signal-to-noise ratio (Supplementary Fig. 13). As shown in Fig. 3c, upon feeding C4H8 to a program-heated bed of P-HZSM-5 (Extended Data Fig. 4a), negligible C4H8 conversion was observed at 250 °C (Fig. 3d). At 300 °C, a clear signal of C8H16 appeared with concurrent detection of propylene and pentene (Fig. 3e, Extended Data Fig. 4b and Supplementary Figs. 14 and 15). This provides direct experimental evidence to confirm that the conversion of butene proceeds via the dimerization–β-scission pathway. Butene cracking experiments were conducted at 540 °C using P-HZSM-5 as the catalyst, yielding a propylene-to-ethylene molar ratio of approximately 2.4–2.8 in the products, further supporting the dimerization–β-scission pathway (Supplementary Figs. 16–18 and Supplementary Note 3). As the reaction temperature was further increased to 350 °C, a broader product distribution was observed due to the subsequent β-scission and oligomerization reactions (Fig. 3f). Based upon these results, we further investigated the reaction pathway starting from C4H8 and C4D8. The dimerization of butenes could yield 5-methylheptene and 4,5-dimethylhexene carbenium ions on acidic zeolites38, and four C8 carbenium (C8+) isomers were formed by the dimerization of C4H8 and C4D8 (Supplementary Fig. 19). The subsequent β-scission is categorized according to the type of carbenium ion involved (A–E types)39. B-type and C-type scissions, which are energetically favorable, predominantly cleave C8+ species into smaller fragments, such as C3 carbenium ion and pentene or C5 carbenium ion (C5+) and propylene. Further cleavage of C5+ yields C2HxDy (x,y = 0–4, m/z = 28–32) and C3HaDb (a,b = 0–6, m/z = 42–48) species as the final products, in excellent agreement with the observed mass spectrometry signals (Fig. 3b, Supplementary Fig. 20, Supplementary Table 18 and Supplementary Notes 3 and 4).
Determination of adsorption domains for reaction intermediate
The P-HZSM-5 catalyst, with reduced acidity (Supplementary Fig. 3), produced fewer by-products such as alkanes and aromatics compared to HZSM-5 (Supplementary Fig. 21). The formation of these by-products is known to occur via bimolecular reactions that require closely spaced acid sites46. To further investigate the effect of phosphorus modification on the spatial distribution of acid sites in HZSM-5, we conducted in situ NPD using deuterated pyridine (Py) as a probe molecule (Supplementary Note 5). Upon adsorption of Py, P-HZSM-5 and HZSM-5 showed negligible structural changes and three distinct binding sites for Py (I, II and III) were observed within both zeolites (Fig. 4a–h, Supplementary Figs. 22 and 23 and Supplementary Tables 19–21). Site I located within the sinusoidal channel and showed similar interactions between PyI and O5(T2) (N∙∙∙O5(T2) = 2.747(135) Å) for HZSM-5 and (N∙∙∙O5(T2) = 2.899(42) Å) for P-HZSM-5 (Fig. 4b,f), whereas PyII and PyIII resided within the straight channel and interacted differently to HZSM-5 and P-HZSM-5 (Supplementary Table 22). Within HZSM-5, PyII interacted with the bridging O(H) center via hydrogen bonding (N∙∙∙O21(T11) = 3.255(24) Å) (Fig. 4c), but this interaction was absent within P-HZSM-5, resulting in parallel orientation of adsorbed PyII molecules within the pore (Fig. 4g). For PyIII, the hydrogen bond between the N center of PyIII and the (O)T8 site was weaker in P-HZSM-5 (N∙∙∙O16(T8) = 3.210(27) Å) than that in H-ZSM-5 (N∙∙∙O15(T8) = 2.918(57) Å (Fig. 4d,h). These results are in excellent agreement with the NH3 temperature-programmed desorption analysis (Supplementary Fig. 3), confirming that P-HZSM-5 had a reduced density and strength of acid sites compared with pristine HZSM-5.

All models were obtained from Rietveld refinements based on in situ NPD data. Three distinct binding sites for pyridine-d5 and two distinct binding sites for octene have been observed in the sinusoidal channel (PyI and OctI) and straight channel (PyII, PyIII and OctI). a, Py@HZSM-5 (T12O24(C5D5N)0.53). b–d, Detailed views of PyI (b), PyII (c) and PyIII (d) in HZSM-5. e, Py@P-HZSM-5 (T12O24(C5D5N)0.56). f–h, Detailed views of PyI (f), PyII (g) and PyIII (h) in P-HZSM-5. i, Oct@HZSM-5 (T12O24(C8H16)0.423). j, Detailed views of OctII in HZSM-5. k, Oct@P-HZSM-5 (T12O24(C8H16)0.398). l, Detailed views of OctII in P-HZSM-5. Py and Oct molecules and the functional sites involved in the binding are highlighted by the use of an amplified ball-and-stick model (Al/Si, violet; C, gray; O, red; H, white; N, blue). The O(H)···N interactions and O(H)···C=C interactions are highlighted in cyan and orange, respectively. Owing to the uncertainty on locations of protons, all hydrogen bonds in this Article are described as the distance between the N/C and the Ozeolite centers.
The adsorption of the important C8 intermediate in these two zeolites was also investigated by in situ NPD and two adsorption sites identified for both systems (Fig. 4i–l, Supplementary Figs. 24–26 and Supplementary Tables 23 and 24). As shown in Extended Data Fig. 5, OctI located in the sinusoidal channel showed similar interactions to these two frameworks, which is consistent with pyridine-d5 adsorption. OctII was located near the intersection of straight channels and a clear difference was observed between these two zeolites. In HZSM-5, OctII interacted primarily with the framework O21(T11) through a dipole interaction between the C=C bond and bridging O(H) centers (C=C∙∙∙O21(T11) = 3.280(37) Å) (Fig. 4j and Supplementary Table 25), leaving the O15(T8) site (Fig. 4d) free to bind other molecules to promote hydrogen transfer, which eventually led to the formation of by-products. Meanwhile, in P-HZSM-5, OctII interacted with O19(T8) through a weaker dipole interaction (C=C∙∙∙O19(T8) = 3.484(46) Å) (Fig. 4l and Supplementary Table 25), and the binding of nearby molecule was hindered due to the absence of acidity at O21(T11) site (Fig. 4g), thus overcoming any bimolecular reactions and leading to β-scission as the main reaction pathway. The NPD study directly illustrates the observed high selectivity of ethylene and propylene over the P-HZSM-5 catalyst compared with pristine HZSM-5 (Supplementary Fig. 21).
Practical application viability
To further explore the practical prospects of this KDRC strategy, the stability of LSP-Z100 and P-HZSM-5 was tested through cycling experiments, and the yields of ethylene and propylene remained stable after five cycles (Fig. 5a). Characterizations of the used catalysts by X-ray powder diffraction, high resolution transmission electron microscope (HRTEM), N2 sorption, NH3 temperature-programmed desorption, nuclear magnetic resonance spectroscopy and thermal gravimetric analysis showed negligible changes in structure and acidic sites, confirming the excellent stability of these zeolites (Supplementary Note 6, Supplementary Figs. 27–39 and Supplementary Table 26). The effects of plastic additives on PE conversion were systematically investigated. Oxygen-containing additives had minimal impact, and phosphorus-containing additives led to moderate catalyst deactivation, while strongly basic nitrogen-containing additives markedly suppressed catalytic activity (Supplementary Figs. 40–45, Supplementary Table 27 and Supplementary Note 7), consistent with observations reported by Vlachos et al47. These results suggest that pretreatment to remove or neutralize basic nitrogen-containing compounds may be required when processing such plastics over acid catalysts. The KDRC strategy applied to real-world plastic waste containing oxygenated additives (Supplementary Table 28) resulted in only a slight reduction in conversion efficiency compared with reagent-grade polyolefins (Supplementary Table 29, entries 1, 2, 4, 6, 8 and 10). Upon optimization of reaction temperature and duration, conversion of all tested plastics approached 100%, with ethylene and propylene yield reaching approximately 70% (Fig. 5b, Supplementary Table 29, entries 3, 7 and 11, Supplementary Table 30 and Supplementary Fig. 46). In addition, the potential economic viability (Supplementary Note 8) and carbon emission (Supplementary Note 9) of this process were evaluated and compared with PE pyrolysis. The data show that promising economical profits and carbon emission reduction can be achieved simultaneously (Fig. 5c, Supplementary Fig. 47 and Supplementary Tables 31–33). We also ran the continuous flow mode where PE was continuously fed to the catalyst bed at a rate of 0.1 g h−1 and the selectivity of ethylene and propylene remained around 75% over an 8-h period (Fig. 5d,e and Supplementary Fig. 48). A gram-scale reaction was also performed to evaluate the practical applicability of the system. One gram of PE was efficiently converted within 4 h, yielding 57% ethylene and propylene (Supplementary Fig. 49). These results support the practical potential of the KDRC strategy for the conversion of PE to ethylene and propylene as bulk chemicals.

a, Comparison of ethylene and propylene yield over five cycles of reaction. Reaction conditions: 100 mg of HDPE and 12.5 mg of LSP-Z100 at stage I, 100 mg of P-HZSM-5 at stage II; reaction temperature, 260 °C (stage I), 540 °C (stage II); reaction time, 2 h; N2 flow rate, 10 sccm. The catalyst recycling process is detailed in the Supplementary Information. b, Catalytic conversion of waste polyolefins and resulting yields of ethylene and propylene in a two-stage reactor. Reaction conditions for HDPE and low-density polyethylene (LDPE): 100 mg waste HDPE or LDPE and 12.5 mg LSP-Z100 at stage I, 50 mg each of P-LSP-Z100 and P-HZSM-5 at stage II; reaction temperature, 260 °C (stage I), 540 °C (stage II); reaction time, 2.5 h; N2 flow rate, 10 sccm. *Reaction condition for waste PP: 100 mg PP and 20 mg LSP-Z100 at stage I, 50 mg each of P-LSP-Z100 and P-HZSM-5 at stage II; reaction temperature, 280 °C (stage I), 540 °C (stage II); reaction time, 2 h; N2 flow rate, 10 sccm. c, Comparison of the CO2 emission from the production of 1 metric ton (MT) of ethylene and propylene using PE pyrolysis and KDRC strategy proposed in this work. d, A continuous process that converts PE to ethylene and propylene. Reaction conditions: 12.5 mg LSP-Z100 mixing with quartz sand at stage I, HDPE dosed at a rate of 100 mg h−1; 50 mg each of P-LSP-Z100 and P-HZSM-5 at stage II; reaction temperature, 260 °C (stage I), 540 °C (stage II); reaction time, 8 h; N2 flow rate, 10 sccm. e, The continuous flow reaction system and the quartz tube reactor.
Discussion
Overall, this study demonstrates an advance in polyolefin recycling, producing ethylene and propylene from PE direct cracking with high yields (79%) via the KDRC strategy in the absence of noble metals, external H2 or additional olefins. The success of KDRC strategy is attributed to two vital factors: (1) the use of LSP-Z100, which effectively cracks PE at stage I (Supplementary Fig. 2), and P-HZSM-5, which efficiently converts butene to ethylene and propylene at stage II; and (2) the decoupling of the complex reaction pathway into two separated stages, enabling independent control and synchronization of reactions at both stages. This approach minimizes side reactions, ensuring that PE conversion follows the desired primary reaction pathway. Initially, PE is mainly cracked into butene and pentene on LSP-Z100 zeolite at stage I. They subsequently dimerize to C8 (or C>8) intermediates on P-modified zeolite at stage II, which undergo β-scission to produce ethylene and propylene. Experimental observations of C8 intermediates and their interactions with acid sites validated this reaction mechanism. Owing to the high stability of the catalysts, the KDRC strategy offers a promising pathway for the closed-loop recycling of waste plastics.
Methods
Catalytic conversion of polyolefins
Polyolefin conversion experiments were conducted in a two-stage reactor equipped with independently temperature-controlled furnaces for each stage. The schematic design of the reactor is illustrated in Extended Data Fig. 3a. Stage I consisted of a quartz tube reactor containing an open-ended internal quartz tube with a diameter of 6 mm, which was used for loading polyolefins and catalysts. A quartz tube reactor in stage II has a diameter of 6 mm. The two stages were separated by a distance of 180 mm. In a typical experiment, 100 mg of PE and 12.5 mg of LSP-Z100 catalyst were mixed and placed in the inner quartz tube of stage I, while 100 mg of P-HZSM-5 catalyst was loaded into stage II (Fig. 2h). Under a nitrogen flow, both stages were heated to their target temperatures at a ramp rate of 10 °C min−1, and the reaction was maintained for 2 h. After the reaction, the liquid products, including those in cold trap and adsorbed on the quartz reactor wall, were thoroughly extracted with dichloromethane. The liquid products were identified by gas chromatography (GC)–mass spectrometry (Agilent 7890B-5977A MSD) with a DB-5ms column. For quantitative analysis of liquid products, n-pentadecane was used as the internal standard, and GC was performed using an Agilent 8890 system equipped with an HP-5ms column. Throughout the entire reaction period, gaseous products were collected in gas bags and subsequently analyzed using an Agilent 8890 GC system equipped with the following columns: an HP-PLOT Al2O3 column, an HP PONA column, a 5-Å molecular sieve column and two Hayesep Q columns. The use of multiple chromatographic columns enables effective separation of individual components and precise detection through dedicated detectors. Two flame ionization detectors were used for hydrocarbon analysis, while a thermal conductivity detector was used to detect hydrogen and nitrogen. Gas products were identified using standard gases. For quantitative analysis of gas products, nitrogen was used as the internal standard because its molar amount remains constant during the reaction and can be determined from the flow rate and reaction duration.
The mole and mass of each gas product were determined using the following equation:
Here, \({n}_{{\rm{N}}2}\) is the molar amount of N2. \({n}_{{i}}\), \({m}_{{i}}\) and \({M}_{{i}}\) denote the mole, mass and molecular weight of gas product i, respectively. \({A}_{{i}}\) and \({A}_{{\rm{N}}2}\) are the GC peak areas of product i and N2, respectively. \({f}_{i,{\rm{N}}2}\) is the relative response factor of product i to N2. \({V}_{{\rm{N}}2}\) is volume of N2 at standard temperature and pressure (0 °C and 1 atm). \({V}_{{\rm{m}}}\) is the molar volume at standard temperature and pressure.
The mole and mass of each liquid product were calculated as follows:
Here, \({n}_{{j}}\), \({m}_{{j}}\) and \({M}_{{j}}\) denote the mole, mass and molecular weight of liquid product j, respectively. \({n}_{{\rm{C}}15{\rm{H}}32}\) is the molar amount of added n-pentadecane. \({A}_{{j}}\) and \({A}_{{\rm{C}}15{\rm{H}}32}\) are the GC peak areas of product j and n-pentadecane, respectively. \({f}_{j,{\rm{C}}15{\rm{H}}32}\) is the relative response factor of product j to n-pentadecane.
The conversion of PE, yield of products, selectivity, carbon atom and mass balance were calculated as follows:
Here, \({m}_{\mathrm{PE},0}\) and \({m}_{\mathrm{PE},t}\) stand for the masses of the PE before and after the reaction, respectively. \({N}_{\mathrm{iorj}}\) denotes the carbon number of molecule i or j. As the mass balance exceeded 95% in each experiment, the yields calculated using the internal standard method were normalized accordingly.
Catalyst cycling experiments were conducted in the two-stage reactor discussed above. After the reaction, the catalysts together the internal quartz reactor were transferred to a tube furnace and calcined at 600 °C under an airflow to remove organic residues. The catalyst in stage I was then removed, remixed with fresh PE powder and reloaded into the internal quartz reactor for the subsequent cycle. The catalyst in stage II was directly used for the next cycle after calcination. After five cycling tests, the used catalysts were characterized to evaluate their stability.
The schematic of the continuous feeding experiment setup is shown in Supplementary Fig. 48. The diameter of the reactors in stages I and II is 10 mm and 6 mm, respectively. PE was fed continuously using a motor-controlled funnel. The rotation of motor was regulated by a programmed system, delivering approximately 50 mg of PE every 30 min, and the catalyst in stage I was mixed with PE under stirring. The reaction proceeded for 8 h, during which 600 mg of PE, dosed over 6 h, was fully converted.
Kinetic experiments
Before the kinetic experiments, we conducted experiments to assess the effects of external and internal diffusion limitations on PE conversion using LSP-Z100 and 1-butene conversion using P-HZSM-5. For LSP-Z100 particles smaller than 0.075 mm, the mass transfer limitations were minimal (Supplementary Fig. 4a). However, with larger particle sizes, PE conversion was hindered due to diffusion limitations of PE (Supplementary Fig. 4a). Therefore, LSP-Z100 particles smaller than 0.05 mm were used for kinetic studies. To enhance external diffusion, PE was premixed with LSP-Z100 before the reaction (Supplementary Fig. 4b). In the case of 1-butene conversion on P-HZSM-5, neither internal nor external diffusion had obvious effects on the reaction under the studied conditions (Supplementary Fig. 6).
For the kinetic experiments of PE conversion in stage I, the fixed amount of PE (100 mg) with LSP-Z100 (12.5 mg) was added in the internal tube reactor and loaded in stage I. The experiments were then conducted at four different temperatures (~260–400 °C) with different reaction times to obtain the conversion of PE and concentration of product for kinetic calculation. For the kinetic experiments of hydrocarbon conversion in stage II, the fixed amount of PE (100 mg) with LSP-Z100 (12.5 mg) was loaded in stage I. The temperature of stage I was kept at 260 °C to continuously supply hydrocarbons for stage II. P-HZSM-5 was loaded in stage II. Each kinetic experiment was conducted for 40 min to ensure that PE conversion in stage I reached approximately 60%. Hydrocarbon products from stage I were then converted in stage II at different temperatures (~400–540 °C) with different amounts of P-HZSM-5, so that the reaction contact time could be changed. The concentrations of products were obtained for kinetic calculation.
To simplify the kinetic calculation, lumped kinetic modeling43, which groups chemical compounds into distinct ‘lumps’, was used. Based on the catalytic performance of the zeolites, the proposed kinetic model includes six lumped components (Extended Data Fig. 2a,b). The feedstock, PE (lump 1), undergoes cracking, producing compounds categorized as lump 2 (ethylene and propylene), lump 3 (butene and pentene), lump 4 (C>6 alkenes and alkanes) and others (hydrogen, C2–C5 alkanes and aromatics) with interconversion occurring among these lumps through cracking and dimerization. These intermediates also participate in hydrogen transfer and aromatization reactions, generating by-products classified as lump 5 (butane, pentane and aromatics) and lump 6 (ethane and propane). In addition, cracking of lump 5 into lumps 2 and 6 was observed (Supplementary Fig. 5). Butenes primarily proceed via the dimerization–β-scission pathway with minimal monomolecular cracking38,39,41. Cracking of 1-butene and isobutene produced a propylene-to-ethylene molar of approximately 2.4–2.8, supporting the involvement of the dimerization–β-scission pathway (Supplementary Figs. 16–18 and Supplementary Note 3). Pentene can follow either monomolecular cracking or the dimerization–β-scission pathway48,49, as shown in Extended Data Fig. 2a,b and Supplementary Note 3. For simplicity, the monomolecular cracking of butene and pentene to ethylene and propylene is not shown in Fig. 2e. The reaction pathways leading to minor products such as H2, C2–C5 alkanes and aromatics were simplified, as these species account for less than 15% of the total yield (Fig. 2a, Extended Data Fig. 1a and Supplementary Table 4) and have negligible impact on the rate constants k1 and k2. In the case of hydrocarbon conversion on P-HZSM-5, the following assumptions were made: (1) the reactor is modeled as having ideal plug flow, where reaction products move axially with no radial mixing, and all reactants share the same contact time; and (2) the deactivation of catalysts was minimal and not considered.
For the PE conversion on LSP-Z100 in stage I, reaction rate \({r}_{{i}}\) was defined as follows:
where \({m}_{\mathrm{cat}}\) is the mass of the catalyst (g); \({m}_{{\rm{N}}2}\) is the mass of the nitrogen carrier gas in the reactor (g); \({m}_{0}\) is the initial mass of high-density polyethylene (HDPE) in the reactor (g); \({C}_{{i}}\) is the concentration of the carbon molar number of lump i (\({\mathrm{mol}}_{\mathrm{CH}2}\,{{\rm{g}}}^{-1}\)); t is the reaction time (s); n is the reaction order; \({n}_{0}\) is the initial carbon mole of HDPE (\({{\rm{mol}}}_{{\rm{CH}}2}\)); \({X}_{{i}}\) is the carbon mole fraction of lump i, based on the organic compounds, in CH2 equivalent units; and \({k}_{{i}}\) is the rate constant of each reaction, expressed in s−1 for first-order reactions or g mol−1 s−1 for second-order reactions.
Based on the proposed kinetic scheme and defined reaction rates, kinetic equations for PE conversion on LSP-Z100 in stage I are as follows:
For the hydrocarbon conversion on P-HZSM-5 in stage II, reaction rate \({r}_{{i}}\) was defined as follows:
\({{\rm{M}}}_{{\rm{gas}}}\) where Mgas is the mass flow rate of total hydrocarbons and nitrogen (g s−1); \({C}_{{i}}\) is the concentration of the carbon molar number of lump i (\({\mathrm{mol}}_{\mathrm{CH}2}\,{{\rm{g}}}^{-1}\)); \({m}_{\mathrm{cat}}\) is the mass of catalyst (g); n is the reaction order; \({F}_{0}\) is the carbon molar flow rate of total hydrocarbons (\({\mathrm{mol}}_{\mathrm{CH}2}\,{{\rm{s}}}^{-1}\)); \({X}_{{i}}\)is the carbon mole fraction of lump i, based on the organic compounds, in CH2 equivalent units; \({k}_{{i}}\) is the rate constant of each reaction (s−1 (first-order reaction) or g mol−1 s−1 (second-order reaction)).
Kinetics equations for hydrocarbons conversion on P-HZSM-5 in stage II are as follows:
The complete raw kinetic datasets have been compiled in Supplementary Tables 8 and 11. For numerical solutions, we used the Ode45 function in MATLAB to solve the ordinary differential equations, coupled with a genetic algorithm for global optimization44,45. Local fine-tuning was performed using the lsqnonlin function to precisely estimate the reaction rate constants (k) in the kinetic model.
SVUV-PIMS
SVUV-PIMS experiments were conducted on the mass spectroscopy beamline (BL04B) at the National Synchrotron Radiation Laboratory in Hefei, China. The experimental setup (Supplementary Fig. 10a) included a catalytic reactor operating under near-vacuum conditions, a gas inlet system and an orthogonal time-of-flight mass spectrometer with tuneable synchronized vacuum ultraviolet light as the ionization source50,51.
To study the proposed reaction pathway, the PE-d4 (or PE) and LSP-Z100 catalyst were mixed with quartz sand and placed in stage I (Supplementary Fig. 10b). The P-HZSM-5 catalyst was loaded in stage II. After installing the reactor into the two-stage furnace, the residual gas was evacuated to maintain a pressure below 2 torr. A mass flow controller regulated the 1.5% 1-butene/N2 feed at a rate of 10 sccm. The reaction temperatures were set at 260 °C (stage I) and 540 °C (stage II), respectively. Upon initiating the catalytic reaction, the two-stage furnace was activated, and the reaction products were directed through a quartz nozzle into the ionization chamber. There, molecular beams were ionized by synchrotron vacuum ultraviolet light and analyzed by the orthogonal time-of-flight mass spectrometer.
To detect the C8 intermediates, only the P-HZSM-5 catalyst was loaded in stage II, with a mass flow controller regulating the 1.5% 1-butene/N2 feed at a rate of 10 or 20 sccm. A programmed temperature control was set at stage II (ramp from 50 °C to 400 °C at a rate of 100 °C min−1). The reaction intermediates and products were detected by in situ SVUV-PIMS.
NPD and Rietveld refinement
Structural determination of the positions of acidic sites and the binding location of 1-octene within HZSM-5 and P-HZSM-5 was performed using WISH, a long-wavelength powder and single-crystal neutron diffractometer at the ISIS Neutron and Muon Facility, Rutherford Appleton Laboratory (UK). This instrument features a solid methane moderator, delivering a high flux of cold neutrons with a wide bandwidth, which is directed to the sample via an elliptical guide. The WISH detectors consist of 1-m-long, 8-mm-diameter pixelated ³He tubes, positioned 2.2 m from the sample and arranged on a cylindrical locus covering a 2θ scattering angle range of 10–170°. Before NPD measurements, the pyridine-d5@zeolite and 1-octene@zeolite were prepared by charging activated zeolites with pyridine-d5 or 1-octene. Data collection was performed at a stable temperature of 100 K for pyridine-d5-loaded sample and of 7 K for 1-octene-loaded sample, maintained using a helium cryostat. Time-of-flight neutron diffraction patterns were recorded at these temperatures, spanning five banks with varied d-spacing ranges.
The crystallographic structures were obtained using Bruker TOPAS V6. Initially, Pawley refinements were conducted to extract nonstructural parameters, including background, peak profile and unit cell dimensions. These parameters served as the starting values for subsequent Rietveld refinements. For the Rietveld refinement, the initial structural model of the zeolite was constructed based on the crystal structure of orthorhombic ZSM-5, which comprises 12 T sites and 26 O sites. Bond distance restraints were applied to all T–O bonds during the refinement process. The Si/Al ratio is refined freely during the refinement and has good consistency with the experimental results (Supplementary Table 3).
For the pyridine-d5-loaded sample, the initial geometry of the pyridine-d5 molecule was modeled as a rigid body, with bond distances and angles derived from density functional theory calculations using ORCA 5.0 under the RI-B3LYP-D3(BJ)/def2-TZVP(-f) method. The initial positions of the pyridine-d5 molecules within the pores were determined using the nuclear residue density map, which identified three distinct types of pyridine-d5 molecule. Subsequently, 10,000 cycles of simulated annealing were conducted to refine the locations of the pyridine-d5 molecules, with the optimal configuration being selected. In the final stage, the rigid body model of the pyridine-d5 molecule was replaced with constraints on bond lengths and angles. All parameters, including the unit cell parameters, location, occupancy, atomic displacement parameters of the guest molecules, and the positional and atomic displacement parameters of the framework atoms, were refined against the NPD patterns from all five banks, with errors estimated.
For 1-octene, different conformations are expected to exist. To minimize thermal motion and stabilize the dominant conformation of adsorbed 1-octene, NPD data were collected at 7 K. Analysis of the neutron density map revealed the presence of one molecule in each of the straight and sinusoidal channels. The conformation of 1-octene can be described as a combination of the conformations of neighboring C–C pairs. For instance, in long-chain alkanes, the trans conformation typically has lower energy than the gauche conformation due to steric hindrance, resulting in a more linear molecular structure. Given that our catalyst can cleave PE effectively, the proportion of gauche conformations during the catalytic process is expected to be low. Therefore, we considered only the all-trans conformation and those with a single gauche conformation, specifically focusing on six possibilities (Supplementary Fig. 24). The location of the gauche conformation in these structures is defined by the corresponding C–C bond (with the double bond at the 1-position), and the relative energy of each conformation was calculated using ORCA 5.0 under the RI-B3LYP-D3(BJ)/def2-TZVP(-f) method.
Considering the 6 × 6 possible conformations of adsorbed 1-octene, 36 refined models were generated. The initial positions were carefully selected to avoid interference with the framework atoms, and the models were treated under rigid body conditions, focusing solely on the position and orientation of the entire molecule. Following the initial refinement, a 2,000-cycle simulated annealing process was conducted to optimize the locations of the 1-octene molecules. The conformation combination featuring 2–3 gauche conformation in the straight channels and 6–7 gauche conformation in the sinusoidal channels exhibited the best agreement factors for both HZSM-5 and P-HZSM-5. To confirm that the refinement process had achieved a global minimum, an additional 2,000-cycle simulated annealing was performed. In the final stage, C–C and C–H bond lengths were refined with soft restraints, while the dihedral angles were maintained as fixed. All parameters, including unit cell parameters, the location, occupancy, atomic displacement parameters of the guest molecules, and the positional and atomic displacement parameters of the framework atoms, were refined against the NPD patterns from all five banks, with associated errors estimated.
Data availability
All data are available in the Article or its Supplementary Information. Source data are provided with this paper, including the atomic coordinates of the optimized computational models. The crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2380347–2380350. These data can be obtained free of charge via The Cambridge Crystallographic Data Centre at www.ccdc.cam.ac.uk/data_request/cif.
References
Geyer, R., Jambeck, J. R. & Law, K. L. Production, use, and fate of all plastics ever made. Sci. Adv. 3, e1700782 (2017).
Sun, J. et al. Catalytic upcycling of polyolefins. Chem. Rev. 124, 9457–9579 (2024).
Van Geem, K. M. Plastic waste recycling is gaining momentum. Science 381, 607–608 (2023).
Yan, N. Recycling plastic using a hybrid process. Science 378, 132–133 (2022).
Lee, K., Jing, Y., Wang, Y. & Yan, N. A unified view on catalytic conversion of biomass and waste plastics. Nat. Rev. Chem. 6, 635–652 (2022).
Jing, Y. et al. Towards the circular economy: converting aromatic plastic waste back to arenes over a Ru/Nb2O5 catalyst. Angew. Chem., Int. Ed. 60, 5527–5535 (2021).
Hancock, J. N. & Rorrer, J. E. Hydrogen-free catalytic depolymerization of waste polyolefins at mild temperatures. Appl. Catal. B 338, 123071 (2023).
Zhang, W. et al. Low-temperature upcycling of polyolefins into liquid alkanes via tandem cracking-alkylation. Science 379, 807–811 (2023).
Cen, Z. et al. Upcycling of polyethylene to gasoline through a self-supplied hydrogen strategy in a layered self-pillared zeolite. Nat. Chem. 16, 871–880 (2024).
Torres, C. & Rorrer, J. E. Layered self-pillared zeolites convert polyethylene to gasoline. Nat. Chem. 16, 841–842 (2024).
Singh, S., Martínez-Ortigosa, J., Ortuño, N., Polshettiwar, V. & García-Martínez, J. Enhanced efficiency in plastic waste upcycling: the role of mesoporosity and acidity in zeolites. Chem. Sci. 15, 20240–20250 (2024).
Rorrer, J. E. et al. Role of bifunctional Ru/acid catalysts in the selective hydrocracking of polyethylene and polypropylene waste to liquid hydrocarbons. ACS Catal. 12, 13969–13979 (2022).
Rorrer, J. E., Beckham, G. T. & Román-Leshkov, Y. Conversion of polyolefin waste to liquid alkanes with Ru-based catalysts under mild conditions. JACS Au 1, 8–12 (2021).
Jia, X., Qin, C., Friedberger, T., Guan, Z. & Huang, Z. Efficient and selective degradation of polyethylenes into liquid fuels and waxes under mild conditions. Sci. Adv. 2, e1501591 (2016).
Chu, M. et al. Layered double hydroxide derivatives for polyolefin upcycling. J. Am. Chem. Soc. 146, 10655–10665 (2024).
Hu, P. et al. Stable interfacial ruthenium species for highly efficient polyolefin upcycling. J. Am. Chem. Soc. 146, 7076–7087 (2024).
Zhang, F. et al. Polyethylene upcycling to long-chain alkylaromatics by tandem hydrogenolysis/aromatization. Science 370, 437–441 (2020).
Du, J. et al. Efficient solvent- and hydrogen-free upcycling of high-density polyethylene into separable cyclic hydrocarbons. Nat. Nanotechnol. 18, 772–779 (2023).
Xu, Z. et al. Chemical upcycling of polyethylene, polypropylene, and mixtures to high-value surfactants. Science 381, 666–671 (2023).
Sullivan, K. P. et al. Mixed plastics waste valorization through tandem chemical oxidation and biological funneling. Science 378, 207–211 (2022).
Li, H. et al. Hydroformylation of pyrolysis oils to aldehydes and alcohols from polyolefin waste. Science 381, 660–666 (2023).
Ciccia, N. R. et al. Diverse functional polyethylenes by catalytic amination. Science 381, 1433–1440 (2023).
Shi, J. X. et al. Chemical modification of oxidized polyethylene enables access to functional polyethylenes with greater reuse. J. Am. Chem. Soc. 145, 21527–21537 (2023).
Chen, L. et al. Selective, catalytic oxidations of C–H bonds in polyethylenes produce functional materials with enhanced adhesion. Chem 7, 137–145 (2021).
Soltani, M. & Rorrer, J. E. Converting waste plastic to liquid organic hydrogen carriers. Angew. Chem., Int. Ed. 62, e202314530 (2023).
Conk, R. J. et al. Catalytic deconstruction of waste polyethylene with ethylene to form propylene. Science 377, 1561–1566 (2022).
Conk, R. J. et al. Polyolefin waste to light olefins with ethylene and base-metal heterogeneous catalysts. Science 385, 1322–1327 (2024).
Dong, Q. et al. Depolymerization of plastics by means of electrified spatiotemporal heating. Nature 616, 488–494 (2023).
Selvam, E., Yu, K., Ngu, J., Najmi, S. & Vlachos, D. G. Recycling polyolefin plastic waste at short contact times via rapid joule heating. Nat. Commun. 15, 5662 (2024).
Li, L. et al. Converting plastic wastes to naphtha for closing the plastic loop. J. Am. Chem. Soc. 145, 1847–1854 (2023).
Epps, T. H. I., Korley, L. T. J., Yan, T., Beers, K. L. & Burt, T. M. Sustainability of synthetic plastics: considerations in materials life-cycle management. JACS Au 2, 3–11 (2022).
Korley, L. T. J., Epps, T. H., Helms, B. A. & Ryan, A. J. Toward polymer upcycling—adding value and tackling circularity. Science 373, 66–69 (2021).
Zou, L. et al. Chemical recycling of polyolefins: a closed-loop cycle of waste to olefins. Natl. Sci. Rev. 10, nwad207 (2023).
Lin, Y.-H., Sharratt, P. N., Garforth, A. A. & Dwyer, J. Catalytic conversion of polyolefins to chemicals and fuels over various cracking catalysts. Energy Fuels 12, 767–774 (1998).
Matsuura, S., Hashimoto, T. & Ishihara, A. Catalytic cracking of low-density polyethylene over zeolite-containing hierarchical two-layered catalyst with different mesopore size using Curie point pyrolyzer. Fuel Process. Technol. 227, 107106 (2022).
Rodríguez, E. et al. Towards waste refinery: Co-feeding HDPE pyrolysis waxes with VGO into the catalytic cracking unit. Energy Convers. Manage. 207, 112554 (2020).
Xiao, H. et al. Hydrogen and aromatics recovery through plasma-catalytic pyrolysis of waste polypropylene. J. Cleaner Prod. 350, 131467 (2022).
Iwase, Y. et al. Shape-selective catalysis determined by the volume of a zeolite cavity and the reaction mechanism for propylene production by the conversion of butene using a proton-exchanged zeolite. J. Phys. Chem. C 116, 5182–5196 (2012).
Buchanan, J. S., Santiesteban, J. G. & Haag, W. O. Mechanistic considerations in acid-catalyzed cracking of olefins. J. Catal. 158, 279–287 (1996).
Yang, J. et al. Selective electrified polyethylene upcycling by pore-modulated pyrolysis. Nat. Chem. Eng. 2, 424–435 (2025).
Lin, L. et al. Acid strength controlled reaction pathways for the catalytic cracking of 1-butene to propene over ZSM-5. J. Catal. 309, 136–145 (2014).
Blasco, T., Corma, A. & Martineztriguero, J. Hydrothermal stabilization of ZSM-5 catalytic-cracking additives by phosphorus addition. J. Catal. 237, 267–277 (2006).
Xiang-hai, Chun-ming, M., Li, X. & Jin-sen, G. Studies on the kinetics of heavy oil catalytic pyrolysis. Ind. Eng. Chem. Res. 42, 6012–6019 (2003).
Shampine, L. F. & Reichelt, M. W. The MATLAB ODE suite. SIAM J. Sci. Comput. 18, 1–22 (1997).
Goldberg, D. E. Genetic Algorithms (Pearson Education India, 2013).
Pine, L. A., Maher, P. J. & Wachter, W. A. Prediction of cracking catalyst behavior by a zeolite unit cell size model. J. Catal. 85, 466–476 (1984).
Ngu, J. et al. Catalytic deconstruction of organic additive-containing plastics. Nat. Chem. Eng. 2, 220–228 (2025).
Lin, L. F. et al. Acid strength controlled reaction pathways for the catalytic cracking of 1-pentene to propene over ZSM-5. ACS Catal. 5, 4048–4059 (2015).
Miyaji, A. et al. Selective production of ethylene and propylene via monomolecular cracking of pentene over proton-exchanged zeolites: Pentene cracking mechanism determined by spatial volume of zeolite cavity. J. Catal. 302, 101–114 (2013).
Li, Y. & Qi, F. Recent applications of synchrotron VUV photoionization mass spectrometry: insight into combustion chemistry. Acc. Chem. Res. 43, 68–78 (2010).
Wen, W. et al. Formation and fate of formaldehyde in methanol-to-hydrocarbon reaction: in situ synchrotron radiation photoionization mass spectrometry study. Angew. Chem. Int. Ed. 59, 4873–4878 (2020).
Acknowledgements
The work was supported by the National Natural Science Foundation of China (grant no. 22293012 to L.L., grant no. 22293015 to B.H. and grant no. 22121002 to B.H.), BNLMS and Peking University. We are grateful to the National Synchrotron Radiation Laboratory, University of Science and Technology of China and STFC/ISIS Neutron and Muon Facility for access to beamline BL04B and WISH, respectively. We thank D. Crawshaw for their assistance in NPD collection.
Author information
Authors and Affiliations
Contributions
Conceptualization: L.L. and T.B. Methodology: T.B., L.L., X.H., Y.C., S.Y., Y.P., C. Liu, P.M., Q.L., N.W. and J.X. Investigation: T.B., L.L., Y.C., X.Z., Z.C., C. Luo, W.W. and H.Z. Visualization: T.B., L.L., X.H. and S.Y. Funding acquisition: L.L. and B.H. Project administration: L.L., X.H., S.Y. and B.H. Supervision: L.L., X.H., S.Y. and B.H. Writing—original draft: L.L., T.B. and X.H. Writing—review and editing: L.L., X.H., S.Y. and B.H.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Engineering thanks Javier Garcia-Martinez and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Catalytic performance of HDPE conversion in a one-stage reactor.
a, Product distribution of HDPE conversion at different temperatures in a one-stage reactor. Reaction conditions: HDPE, 100 mg; LSP-Z100, 12.5 mg; reaction time, 2 h; N2 flow rate, 10 sccm. b, Product distribution of HDPE conversion at different mass ratio of HDPE to catalyst in a one-stage reactor. Reaction condition: HDPE, 100 mg; LSP-Z100, 50 mg / 25 mg / 12.5 mg / 6.25 mg; reaction temperature, 260 °C; reaction time, 2 h; N2 flow rate, 10 sccm. Note that at the 16:1 ratio, the HDPE conversion was 76.4% in two hours, while the rest of the ratios had PE conversion of 100%. c, Product distribution of HDPE conversion at different temperatures in a one-stage reactor with two-catalyst bed. Reaction condition: 100 mg of HDPE and 12.5 mg of LSP-Z100 in the first bed, 100 mg of P-HZSM-5 in the second bed; reaction time, 2 h; N2 flow rate, 10 sccm.
Extended Data Fig. 2 Kinetic studies for decoupling-coupling strategy.
a, Kinetic scheme proposed for PE conversions over LSP-Z100 in Stage I. b, Kinetic scheme proposed for hydrocarbons conversion over P-HZSM-5 in Stage II. c, Simulated vs experimental concentration of each lump in PE conversions on LSP-Z100. d, Simulated vs experimental concentration of each lump in hydrocarbons conversion on P-HZSM-5. e, Production rate difference between main products (ethylene and propylene) and by-products (alkanes and aromatics) against concentration of butene and pentene. Extended Data Fig. 2e is derived from Fig. 2f. The y-axis represents the difference between the formation rates of the main products and the by-products.
Extended Data Fig. 3 Catalytic performance via KDRC strategy in a two-stage reactor.
a, Schematic structure of a two-stage reactor. b, Product distribution of PE conversion in a two-stage reactor. Reaction conditions: 100 mg of HDPE and 12.5 mg of LSP-Z100 at Stage I, 100 mg of P-HZSM-5 at Stage II; reaction temperature of Stage II, 540 °C; reaction time, 2 h; N2 flow rate, 10 sccm. c, Product distribution of PE conversion at optimised reaction conditions in a two-stage reactor. Reaction conditions: 100 mg of HDPE and 12.5 mg of LSP-Z100 at Stage I; temperature programmed reaction (TPR) from 260 °C to 300 °C in 2 h at Stage I; 50 mg each of P-LSP-Z100 and P-HZSM-5 at Stage II; reaction temperature of Stage II, 540 °C; reaction time, 2 h; N2 flow rate, 10 sccm.
Extended Data Fig. 4 Studies of temperature-programmed reaction of 1-butene by SVUV-PIMS.
a, The temperature ramp for temperature-programmed reaction of 1-butene on P-HZSM-5. b, Product distribution of temperature-programmed reaction of 1-butene on P-HZSM-5. Reaction conditions: P-HZSM-5, 30 mg; temperature ramp, 100 °C/min, from 50 to 400 °C; 1.5% 1-C4H8/N2 flow rate, 10 sccm; ionization energy 11 eV.
Extended Data Fig. 5 Views of crystal structures of octene-loaded zeolites.
a, Detailed views of OctI in HZSM-5. b, Detailed views of OctI in P-HZSM-5.
Supplementary information
Supplementary Information (download PDF )
Supplementary Methods, Notes 1–9, Figs. 1–49, Tables 1–33 and References.
Supplementary Data 1 (download ZIP )
Computational data file of the considered conformations of 1-octene.
Supplementary Data 2
Crystallographic information file for HZSM-5 and P-HZSM-5 zeolites with adsorbed 1-octene or pyridine, together with the corresponding checkCIF reports.
Source data
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Extended Data Table 1 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bi, T., Chen, Y., Lin, L. et al. Closed-loop recycling of polyethylene to ethylene and propylene via a kinetic decoupling–recoupling strategy. Nat Chem Eng 2, 650–661 (2025). https://doi.org/10.1038/s44286-025-00290-y
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44286-025-00290-y
This article is cited by
-
Current perspective on heterogeneous thermal catalytic approaches for closed-loop polyolefin plastic recycling
Discover Chemistry (2026)
-
Deep cracking of polyethylene to light olefins
Nature Chemical Engineering (2025)
