
Abstract
Captured CO2 can either be stored or converted into a carbon-containing product. Both processes require energy to liberate CO2 from the CO2 capture solution. One way to bypass the energy-intensive recovery of CO2 is to integrate CO2 capture and conversion. For example, OH–-based capture solutions react with CO2 to form (bi)carbonate-rich liquids, which can then be electrochemically converted into upgraded carbon-containing products while regenerating OH– for further CO2 capture. However, bicarbonate electrolyzers designed to mediate this process are often characterized by high voltages (e.g., >3 V at 100 mA cm–2). We report here a bicarbonate electrolyzer that operates at 100 mA cm–2 with an applied voltage of 2.7 V at 50 °C. This lower voltage was made possible by designing a bicarbonate electrolyzer with a cationic exchange membrane that uses a neutral anolyte (3 M KHCO3(aq)) to minimize the thermodynamic potential for the oxygen evolution reaction. This bicarbonate electrolyzer enables a carbon capture and utilization energy efficiency for an electrochemical process that is comparable to thermochemical CO2 conversion for syngas production.
Similar content being viewed by others

Introduction
Carbon capture and utilization (CCU) is typically a sequential process that captures and upgrades CO2 from a point source or air into value-added fuels and chemicals1,2. One method of upgrading the captured CO2 is to electrochemically reduce it at a cathode in the presence of a proton source3,4,5,6. The challenge is that the captured CO2 needs to be released from the CO2 capture solution, then purified and pressurized prior to delivery into an electrolyzer (see “CO2 Electrolyzer Pathway”; Fig. 1). These steps are capital- and energy-intensive.

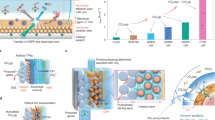
a Flow diagrams of four different carbon capture and utilization (CCU) pathways. The differentiating feature of each pathway is the utilization step (highlighted in yellow) where CO2 is converted into syngas with a 2:1 H2:CO molar ratio thermochemically [Reverse Water Gas Shift Pathway] or electrochemically [Bicarbonate Electrolyzer (this work), Solid Oxide Electrolysis Cell (SOEC), and CO2 Electrolyzer Pathways]. b CCU energy efficiencies and total energy consumption of four different pathways. The total energy consumption is divided into five different steps: CO2 capture and solvent regeneration; CO2 pressurization; CO2 conversion; product separation; and water electrolysis for H2 production. CCU energy efficiency is defined as the ratio of the minimum theoretical energy required to convert CO2 into 1 mol of syngas, to the total energy actually required to capture and convert CO2 into 1 mol of syngas. The input parameters for all pathways are summarized in Table S2.
This situation motivated us to develop a bicarbonate electrolyzer to couple the CO2 capture and upgrading processes (“Bicarbonate Electrolyzer Pathway”; Fig. 1)7,8,9. By using alkaline solutions (e.g., KOH10 or K2CO311) in the carbon capture unit to react with CO2 to form “reactive carbon solutions” (e.g, (bi)carbonates; Eq. 1), we recognized that an electrolyzer that leverages both pH and electrochemistry could convert these reactive carbon solutions into useful products (e.g., CO) while regenerating the carbon capture solution12. A key feature of the bicarbonate electrolyzer is that it delivers protons (H+) to react with (bi)carbonate ions to form high concentrations of CO2 in situ (Eq. 2). This in situ generated CO2 (“i-CO2”) is then electrochemically reduced into carbon-containing products such as CO, just as what would occur in an electrolyzer fed with gaseous CO213,14,15,16. Importantly, bicarbonate electrolyzers convert more of the CO2 feedstock into CO than gas-fed CO2 electrolyzers17,18. Bicarbonate electrolyzers are also less sensitive to impurities and oxygen than electrolyzers fed with gaseous CO219,20.
To deliver acid into the cathode compartment, we previously built bicarbonate electrolyzers containing bipolar membranes (BPMs) to separate the cathode and anode7,21,22,23. A BPM consists of a cation exchange membrane (CEM) pressed against an anion exchange membrane (AEM)24,25,26. Water is dissociated into OH– and H+ at a catalyst layer at the interface of these two membrane layers, and OH– and H+ are delivered into the anode and cathode compartments, respectively24,25,27. The challenge is that commercially available BPMs substantially increase voltage at meaningful current densities28.
To put this high voltage penalty into perspective, we calculated the energy required to capture and convert CO2 into syngas when using a bicarbonate electrolyzer (“Bicarbonate Electrolyzer Pathway”; Fig. 1). We targeted syngas with a H2:CO molar ratio of 2:1, a feedstock for the Fischer-Tropsch process that produces hydrocarbons and oxygenates29. We then benchmarked this CCU energy efficiency against three other CCU pathways widely considered to be commercially viable: the reverse water gas shift (“RWGS Pathway”); high-temperature CO2 electrolysis with a solid oxide electrolysis cell (“SOEC Pathway”); and low-temperature CO2 electrolysis (“CO2 Electrolyzer Pathway”; Fig. 1)30,31,32. All of these three pathways require a purified gaseous CO2 feed. We define CCU energy efficiency as the ratio of the minimum theoretical energy required to convert CO2 into 1 mol of syngas to the total energy actually required to capture and convert CO2 into 1 mol of syngas (see Fig. 1 and Supporting Information). The CCU energy efficiencies of each of these pathways are primarily differentiated by the utilization step (the conversion of CO2 into CO); the CO2 capture (using amines), solvent regeneration, and pressurization steps were held at parity for the analysis. All four pathways necessitate the use of a water electrolyzer to generate H2(g) to maintain the required 2:1 H2:CO molar ratio, as these specific utilization steps produce CO with high selectivity. The Bicarbonate Electrolyzer Pathway assumes CO2 capture by alkaline solutions that are fed directly into the bicarbonate electrolyzer. Note that the selectivity for CO formation along each pathway governs how much CO2 must be captured to produce 1 mol of CO, thus the energy required for the same step may differ for each pathway. We assumed unreacted CO2 leaving each CO2 conversion step would be recycled back as the inlet feedstock. The input parameters for all pathways used in the analysis are summarized in Tables S1 and S2.
This analysis shows CCU energy efficiencies of 51%, 47%, and 40% for the SOEC (cell voltage, Ecell = 1.5 V; Faradaic efficiency of CO formation, FECO = 100%; CO2 utilization efficiency = 50%), RWGS (CO selectivity = 100%; CO2 conversion = 40%), and CO2 Electrolyzer Pathways (Ecell = 3 V; FECO = 90%; CO2 utilization efficiency = 20%), respectively. In an optimistic future scenario where the energy requirement for solvent regeneration is much lower, the Bicarbonate Electrolyzer Pathway is still a relatively energy efficient pathway (Figure S1). The CCU energy efficiency for the pathway with a bicarbonate electrolyzer that uses a BPM (Ecell = 4 V; FECO = 90%; CO2 utilization efficiency = 50%) would be 44% (not shown in Fig. 1; see Supporting Information for the analysis)21,22,23. This analysis indicated that bicarbonate electrolyzers need to operate at Ecell < 3.1 V (in cases where FECO = 90%) to achieve comparable energy efficiencies to the RWGS or SOEC Pathways (Figure S2). The voltage of bicarbonate electrolyzers needs to be lowered substantially from the current benchmark of 4 V to <3.1 V.
We therefore set out to build a bicarbonate electrolyzer with a CEM instead of a BPM to increase the energy efficiency of the CO2 activation step. CEMs mediate the transport of cations (e.g., H+) to the cathode for the requisite reaction with bicarbonate to form i-CO2 (Eq. 2). We previously showed that a Nafion™ CEM does indeed lower the bicarbonate electrolyzer voltage, but we needed to feed H2(g) into the anode compartment17. This configuration resulted in a strikingly low full cell voltage of 1.7 V at 100 mA cm–2. However, the use of a gaseous H2 feedstock poses greater scale-up challenges and requires additional energy and capital costs. There is emerging work showing voltages below 4 V at 100 mA cm–2 through advances in cathode catalyst design33,34, or electrolyzer architecture35,36,37; however, either these voltages still remain higher than the 3.1 V target established by our energy analysis or require additional energy to source H2(g).
In this report, we show that a bicarbonate electrolyzer with a CEM separating a IrO2 nanoparticle anode spray-coated on a carbon paper substrate (IrO2/C) and a silver nanoparticle cathode spray-coated on a carbon paper substrate (Ag/C) can operate at 2.7 V at 100 mA cm–2. The higher energy efficiency of the bicarbonate electrolyzer is achieved with: (i) the use of a neutral anolyte (3 M KHCO3) that decreases the thermodynamic potential for the oxygen evolution reaction (OER) relative to that in strongly acidic media; and (ii) the use of a CEM to avoid the high voltage penalty associated with BPMs for dissociating water. Importantly, the Bicarbonate Electrolyzer Pathway with this new bicarbonate electrolyzer design exhibits a high CCU energy efficiency of 53% (Fig. 1) using only liquid feedstocks. This value is comparable to the SOEC Pathway and represents a 1.25-fold improvement over the CO2 Electrolyzer Pathway, where the maximum CCU energy efficiency is modeled to be merely 40%.
Results
Electrochemical generation of i-CO2 using a CEM
An acidic anolyte is typically used with cation exchange membranes to create the acidic environment required for i-CO2 generation in the cathode compartment38,39. However, in this configuration, the anolyte becomes more acidic over time because more water molecules are transported per 1 mol of proton migrated from the anode to the cathode compartment (i.e., electro-osmotic drag)40,41. This increase in acidity leads to an increase in cell voltage over time38,39. This situation prompted us to test neutral anolytes with a CEM. The relatively lower pH of the neutral medium decreases the thermodynamic potential for OER42.
We first performed two-electrode galvanostatic electrolysis in a two-compartment H-cell using 3 M KHCO3 for both the anolyte and catholyte solutions. The IrO2/C anode was compressed against a Nafion™ N117 CEM using a hydraulic press prior to H-cell assembly to simulate the zero-gap environment in a bicarbonate electrolyzer (Fig. 2). Porous carbon paper was used as the cathode. A stream of N2 gas was passed through the headspace of the cathode compartment at 80 sccm to transport the CO2 product from the cathode chamber to the gas chromatograph (GC). We initially applied no current to measure the amount of CO2 degassed from the catholyte by purging the stream of N2 gas into the cathode compartment. We observed that 1.5 sccm of CO2 was released from the catholyte by purging the N2 gas to the cathode compartment. Next, we applied a current of 100 mA to investigate whether CO2 could be generated electrochemically. Under this current, we observed the formation of CO2 bubbles at the surface of the CEM facing the cathode compartment (Fig. 2). A GC analysis confirmed an increase in CO2 flow rate (Fig. 2). Further increasing the current to 200 mA resulted in an additional increase in CO2 flow rate. These results indicate that the CO2 was generated electrochemically in the H-cell.

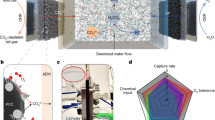
a A two-compartment H-cell setup to visualize CO2 bubbles formed at the membrane|catholyte interface. The IrO2/C anode (light gray) and the cation exchange membrane (green; Nafion™ N117) were compressed against each other to simulate the zero-gap environment in a bicarbonate electrolyzer. A carbon paper (dark gray; Freudenberg H23) was used as the cathode. b CO2 bubbles formed at the membrane|catholyte interface and accumulate on top. c The CO2 flow rate from the H-cell outlet increases with increasing current.
Zero-gap bicarbonate electrolyzer design and performance
We then performed a control experiment where the IrO2/C anode was separated from the CEM and placed in the anode compartment (Fig. S3). All other components and experimental conditions were kept the same as the previous H-cell experiment. CO2 bubbles did not form at the surface of the CEM when the anode and the membrane were separated from each other. This experiment signaled to us that it is only when the anode is in physical contact with the CEM that the protons are transferred from the anode to the cathode compartment.
With confirmation that CO2 could be sourced from an anode immersed in a neutral anolyte, we designed and built a zero-gap flow reactor for electrolyzing 3 M KHCO3 solutions. This solution was supplied to both the anolyte and catholyte from the same reservoir (Fig. 3). This configuration enables the anolyte and catholyte solutions to be mixed after being passed through the flow cell, thereby rebalancing the ionic concentrations of each electrolyte prior to entering the electrolyzer again. The electrolyzer consisted of grade 3 titanium anode and cathode flow plates with serpentine patterns sandwiching the membrane electrode assembly (MEA) (Fig. 3). The MEA was composed of iridium oxide nanoparticles spray-coated on a carbon paper with a microporous layer (anode) and silver nanoparticles spray-coated on a carbon paper (cathode) sandwiching a Nafion™ NR211 CEM. Nafion™ NR211 was selected because it is relatively thin (25.4 µm), which helps minimize the membrane Ohmic resistance and reduce the cell voltage. We added a microporous layer to the anode because this design has been shown to facilitate OER effectively in bicarbonate anolytes43. However, the microporous layer was not added to the cathode because this layer inhibits aqueous bicarbonate transport, decreasing FECO21,23. The geometric surface area of the anode and cathode were both 4 cm2. A 3 M KHCO3 solution was delivered to both cathode and anode flow plates at 100 mL min–1. The reservoir was sparged with 175 sccm of N2 gas to purge gaseous products from the electrolyzer into a GC. The GC and 1H NMR were each used to determine the Faradaic efficiencies of gaseous and liquid products, respectively, at various current densities. The gaseous product stream was analyzed once after 5 min of electrolysis. Liquid products were collected after 20 min of electrolysis for 1H NMR analysis. All experiments were performed in triplicate, unless otherwise noted. We conducted experiments using an electrolyzer with a BPM to benchmark the performance of an electrolyzer with a CEM. For electrolysis experiments with a BPM, a 1 M KOH solution was fed to the anode compartment and a 3 M KHCO3 solution was delivered to the cathode compartment. Nickel foam and silver nanoparticle spray-coated on carbon paper were used as the anode and cathode, respectively, in the bicarbonate electrolyzer configured with a BPM. Other conditions were kept the same as the electrolysis experiments with a CEM. CO and H2 were the only gaseous reduction products detected by the GC analysis. No significant quantities of liquid products were detected by 1H NMR analysis.

a Exploded view of the bicarbonate electrolyzer reported in this work. The anolyte and catholyte are mixed after each circulation to reset ion concentrations. b Full cell voltages (Ecell) of bicarbonate electrolyzers that use a cation exchange membrane (CEM) or bipolar membrane (BPM) at various current densities and, c Faradaic efficiency of CO (FECO) of a bicarbonate electrolyzer with a CEM at various current densities. Bicarbonate electrolysis was conducted at two different temperatures: room temperature and 50 °C for both BPM-based and CEM-based electrolyzers. The FECO values for BPM are in the Supporting Information (Figure S4). Both electrolyzers yielded equivalent FECO values (within experimental error).
The bicarbonate electrolyzer with the CEM yielded an Ecell of 2.9 ± 0.1 V at an applied current density of 100 mA cm–2 (Fig. 3). This value is much lower than the 4.0 ± 0.3 V measured for the same electrolyzer containing the BPM. Both electrolyzers yielded similar FECO values at 100 mA cm–2: 65% for the electrolyzer with the BPM (Fig. S4), and 61% for the electrolyzer with the CEM (Fig. 3). The CO2 utilization efficiencies of both electrolyzers were also similar at 100 mA cm–2: 49% ± 5% for the CEM; and 47% ± 1% for the BPM. As in the H-cell experiment, i-CO2 increased with increasing current density, further indicating that i-CO2 is electrochemically generated within the electrolyzer (Figure S5).
Our energy consumption analysis assumed a 90% FECO in the bicarbonate electrolyzer. On an industrial scale, when electrolyzers with large active areas operate at high current densities, significant heat is generated due to resistance (Joule heating). We explored the possibility of utilizing this waste heat to increase the FECO of the bicarbonate electrolyzer reported in this work. We also performed electrolysis experiments at various current densities with the electrolyte temperature set at 50 °C, to better understand how temperatures of an electrolyzer operating at commercial conditions impact voltage (Fig. 3). With an inlet electrolyte temperature of 50 °C, we measured an Ecell of 2.7 V ± 0.1 V at 100 mA cm–2. This value is 0.2 V lower than the Ecell measured at room temperature. The FECO value increased from 61% at room temperature to 84% at 50 °C.
We then evaluated the stability of the bicarbonate electrolyzer containing the CEM over 16 h at room temperature. This experiment was performed at room temperature and used an electrolyte sparged with 100 sccm of CO2 to simulate the CO2 capture process. Over the course of the 16-hour experiment, both the anolyte and catholyte were fed to the electrolyzer from the same reservoir to facilitate the mixing of the anolyte and catholyte solutions. All other experimental conditions were otherwise kept the same as the 5-min electrolysis experiments. The Ecell were measured to be 3.0 ± 0.1 V over the course of the 16-hour experiment (Fig. 4). Moreover, the bulk pH of the electrolyte solution and the rate of i-CO2 formation remained constant at 8 and 5.3 sccm, respectively, for the duration of the experiment (Fig. 4). The only parameter that changed substantially was FECO and CO2 utilization efficiency, which began to decline after 30 min of electrolysis (Fig. 4).

Over the course of the 16-hour experiment, both the anolyte and catholyte were fed to the electrolyzer from the same reservoir to facilitate the mixing of the anolyte and catholyte solutions. The stability experiment was performed at room temperature. a Ecell remains relatively stable throughout the 16-hour electrolysis, but FECO begins to decline after 30 min. b The pH of the electrolyte and the rate of in situ CO2 (i-CO2) generation remain steady during the sustained 16-hour electrolysis. c CO2 utilization begins to decline after 30 min of electrolysis indicating that the cathode is deactivating.
Discussion
The positive electrolysis results notwithstanding, we observed a marked decrease in FECO after 30 min of electrolysis, despite sustained i-CO2 formation rates (Fig. 4). We attribute the decrease in FECO to the instability of the anode material or the anode flow plate, which would diminish CO2 utilization efficiency. To test this hypothesis, we performed X-ray fluorescence (XRF) analysis on both the pre- and post-electrolysis silver cathode, which had been spray-coated onto a carbon paper substrate (Fig. S6). The XRF analysis indicates that iridium anode material and titanium from the flow plate deposited onto the cathode. An increase in iridium and titanium concentrations in the electrolyte, measured by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES; Table S3), also corroborates this observation.
To further support the hypothesis, we conducted a 2-hour bicarbonate electrolysis experiment in a zero-gap electrolyzer with a CEM at 100 mA cm–2 using a CO2-sparged cathode feed, but without mixing the 3 M KHCO3 anolyte and 3 M KHCO3 catholyte, and observed FECO values that remained steady for the duration of the experiment (Fig. S7). Future work will focus on addressing the durability of the anode for this electrolyzer design44,45. This work will also have to consider the use of capture promoters to increase the reaction rate between CO2 and OH– in the air capture unit12,46,47,48.
Here we demonstrate an energy-efficient bicarbonate electrolyzer that mediates the conversion of (bi)carbonate-enriched reactive carbon solutions into syngas without an obvious source of protons. The reported bicarbonate electrolyzer uses a neutral anolyte (i.e., 3 M KHCO3) to minimize the thermodynamic potential associated with the oxygen evolution reaction and a CEM to minimize the overpotentials associated with the Ohmic resistance. This configuration enables the use of liquid feedstocks, a feature that is consistent with commercial H2-producing electrolyzers. The applied voltage of this system is 2.7 V at 100 mA cm–2 with a Faradaic efficiency for CO of 84% at 50 °C with the balance product being H2. These remarkable outcomes culminate in a carbon capture and utilization (CCU) energy efficiency of 53% for the conversion of CO2 into syngas. This value is comparable to the highest efficiency CCU pathways that are currently being considered.
Methods
Materials
KHCO3 (Ph. Eur., BP, USP, E501, 99.5-101.0%, Sigma Aldrich, USA), KOH (ACS reagent, ≥85%, pellets, Sigma Aldrich, USA), ethylenediaminetetraacetic acid (EDTA; 99%, Sigma Aldrich, USA), and silver nanoparticles (<100 nm particle size, contains PVP as dispersant, 99.5% trace metals basis, Sigma Aldrich, USA) were purchased and used as received. NafionTM membranes, Fumasep-FBM® bipolar membranes (BPMs), Freudenberg H23 carbon paper, and NafionTM D2020 (20 wt% in a mixture of lower aliphatic alcohols and water) were purchased from Fuel Cell Store, USA. Nickel foam anodes were purchased from MTI Corporation (USA). IrOx anodes were purchased from Dioxide Materials.
Materials Preparation
Cathodes were prepared by spray-coating silver nanoparticles onto carbon papers. The cathode catalyst inks were prepared by mixing 27 mg of silver nanoparticles (<100 nm) with 10 μL of 20 wt% NafionTM (Nafion Dispersion D2020) in 9 mL of ethanol. The catalyst inks were sonicated for 20 min for even dispersion. Carbon papers were cut into 4 cm2 (2 × 2 cm). The cathode catalyst inks were then deposited onto these carbon papers using an air-brush until the catalyst loading was 2 mg cm–2 (gravimetric measurement). The air pressure for the air-brush was set to 20 psi. A hotplate below the cathode samples was heated to 150 °C to accelerate solvent evaporation during the spray-coating process. IrOx anode purchased from Dioxide Materials was cut into 2 × 2 cm and used as-received. Bipolar membranes were cut into 3.5 × 3.5 cm with a blade and were stored in 1 M NaCl prior to use. NafionTM membranes were also cut into 3.5 × 3.5 cm and were stored in deionized water prior to use.
Electrolysis in a flow cell
A detailed design of the flow cell used in this work is reported in our previous works with a slight modification7,22. The modification that was made involved the materials for the anode flow plate. In this work, we utilized a grade 3 titanium anode flow plate with serpentine flow pattern, whereas in our previous works, we had used a stainless steel anode flow plate with serpentine flow pattern. The membrane electrode assembly for the bicarbonate electrolyzer containing a cation exchange membrane consisted of a IrOx nanoparticle anode spray-coated on a carbon paper substrate, a silver cathode spray-coated on Freudenberg H23 carbon paper, and a cation exchange membrane. The membrane electrode assembly for the bicarbonate electrolyzer containing a bipolar membrane consisted of a nickel foam anode, a silver cathode spray-coated on Freudenberg H23, and a bipolar membrane. The entire assembly was tightened to a torque of 4.5 N⋅m for membrane electrode assemblies (MEAs) with bipolar membranes and 3 N⋅m for MEAs with cation exchange membranes.
The flow rate of N2 supply (Linde, 99.9%) was set to 175 sccm using a mass flow controller. A mass flow indicator (AALBORG) was placed at the outlet of the gas chromatograph (GC) analyzer to confirm the gas flow rate. A 50 mL 3 M KHCO3 catholyte solution was circulated at 100 mL min−1 for the duration of all experiments using a peristaltic pump (McMaster-Carr; 43205K11). 20 mM EDTA was added to the 3 M KHCO3 for all experiments to help prevent electrolyte impurities from depositing on the electrode surface. For the bicarbonate electrolyzer with a cation exchange membrane, a 50 mL of 3 M KHCO3 anolyte was circulated at 100 mL min–1. For the bicarbonate electrolyzer containing a bipolar membrane, a 50 mL of 1 M KOH anolyte was circulated at 100 mL min–1.
N2 was continuously purging the headspace of the catholyte reservoir (100-mL round bottom flask) to carry electrolysis products from the catholyte to the GC (SRI Instruments, SRI-8610C). The headspace of the electrolyzer outlet was connected to a condenser before being vented directly into the gas-sampling loop of the GC. The anode compartment was open to air, except when both the 3 M KHCO3 anolyte and catholyte were supplied from a shared reservoir. In this configuration, gaseous products from both the anode and cathode were directed to the GC.
The GC runs were initiated after 5 min of electrolysis unless otherwise stated. The GC was equipped with a packed MolSieve 5 A column and a packed HaySep D column. Argon (Linde, 99.999%) was used as the carrier gas. A flame ionization detector (FID) with a methanizer was used to quantify CO, CO2, CH4, and C2H4, while a thermal conductivity detector (TCD) was used to quantify H2. The GC was calibrated by injecting different calibration gas mixes from NorLAB containing CO, CH4, H2, C2H4, C2H6, CO2, and C3H8 at concentrations ranging from 100–50,000 ppm. A solution of 3 M KHCO3 was recirculated through the anode compartment at a flow rate of 100 mL min−1 using a peristaltic pump (McMaster-Carr; 43205K11). Electrochemical measurements were conducted at ambient conditions using a programmable power supply (CH instruments 660D with a 680 C Amp Booster). Current density is expressed as the total current divided by the geometric surface area of the electrodes (i.e., 4 cm2).
Product quantification
The Faradaic efficiencies (FE) of gas products were calculated by the equation below (Eq. 3):
where zi is the number of electrons per mole of gaseous product i involved in the reduction reaction, F is Faraday’s constant (F = 96,485 C mol−1), xi is the mole fraction of gas i in the gaseous mixture analyzed using GC, \(\dot{n}\) is the total molar flow rate in mol s−1, and I is the total current in A. The molar flow rate is derived from the volumetric flow rate Q by the ideal gas equation of state (\(\dot{n}={P}_{{atm}}Q/{RT}\)) with Patm being the atmospheric pressure in Pa, R the ideal gas constant of 8.314 J mol−1 K−1, and T the temperature in K. The volumetric flow rate was measured using a flow indicator located immediately upstream of the GC.
The in situ CO2 (i-CO2) generated was calculated assuming CO was the only reduced carbon product in this study. This is a fair assumption using an Ag electrocatalyst in our reactor conditions (Eq. 4).
where [CO] and [CO2]unreacted represent the concentrations of CO and CO2 measured by GC.
The carbon utilization was calculated by dividing the concentration of CO at the outlet by the total amount of CO2 generated in situ (Eq. 5):
Data availability
All data are provided in the manuscript and Supporting Information.
References
Welch, A. J., Dunn, E., DuChene, J. S. & Atwater, H. A. Bicarbonate or Carbonate Processes for Coupling Carbon Dioxide Capture and Electrochemical Conversion. ACS Energy Lett. 5, 940–945 (2020).
Alerte, T. et al. Downstream of the CO2 Electrolyzer: Assessing the Energy Intensity of Product Separation. ACS Energy Lett. 4405–4412 (2021).
Weekes, D. M., Salvatore, D. A., Reyes, A., Huang, A. & Berlinguette, C. P. Electrolytic CO2 Reduction in a Flow Cell. Acc. Chem. Res. 51, 910–918 (2018).
Ren, S. et al. Molecular electrocatalysts can mediate fast, selective CO2 reduction in a flow cell. Science 365, 367–369 (2019).
Higgins, D., Hahn, C., Xiang, C., Jaramillo, T. F. & Weber, A. Z. Gas-Diffusion Electrodes for Carbon Dioxide Reduction: A New Paradigm. ACS Energy Lett. 4, 317–324 (2019).
Lees, E. W., Mowbray, B. A. W., Parlane, F. G. L. & Berlinguette, C. P. Gas diffusion electrodes and membranes for CO2 reduction electrolysers. Nat. Rev. Mater. 1–10 (2021).
Li, T. et al. Electrolytic Conversion of Bicarbonate into CO in a Flow Cell. Joule 3, 1487–1497 (2019).
Pimlott, D. J. D., Kim, Y. & Berlinguette, C. P. Reactive Carbon Capture Enables CO2 Electrolysis with Liquid Feedstocks. Acc. Chem. Res. 57, 1007–1018 (2024).
Namdari, M., Kim, Y., Pimlott, D. J. D., Jewlal, A. M. L. & Berlinguette, C. P. Reactive carbon capture using electrochemical reactors. Chem. Soc. Rev. 54, 590–600 (2025).
Keith, D. W., Holmes, G., St Angelo, D. & Heidel, K. A Process for Capturing CO2 from the Atmosphere. Joule 2, 1573–1594 (2018).
Benson, H. E., Field, J. H. & Jimeson, R. M. Carbon dioxide absorption employing hot potassium carbonate solutions. Chem. Eng. Prog. 50, 356–364 (1954).
Kim, Y. et al. Integrated CO2 capture and conversion to form syngas. Joule 8, 3106–3125 (2024).
Rosen, B. A. et al. Ionic Liquid–Mediated Selective Conversion of CO2 to CO at Low Overpotentials. Science 334, 643–644 (2011).
Li, Y. C. et al. Electrolysis of CO2 to Syngas in Bipolar Membrane-Based Electrochemical Cells. ACS Energy Lett. 1, 1149–1153 (2016).
Vermaas, D. A. & Smith, W. A. Synergistic Electrochemical CO2 Reduction and Water Oxidation with a Bipolar Membrane. ACS Energy Lett. 1, 1143–1148 (2016).
Salvatore, D. A. et al. Electrolysis of Gaseous CO2 to CO in a Flow Cell with a Bipolar Membrane. ACS Energy Lett. 3, 149–154 (2018).
Zhang, Z. et al. Conversion of Reactive Carbon Solutions into CO at Low Voltage and High Carbon Efficiency. ACS Cent. Sci. 8, 749–755 (2022).
Ozden, A. et al. Carbon-efficient carbon dioxide electrolysers. Nat. Sustain. 5, 563–573 (2022).
Pimlott, D. J. D., Jewlal, A., Mowbray, B. A. W. & Berlinguette, C. P. Impurity-Resistant CO2 Reduction Using Reactive Carbon Solutions. ACS Energy Lett. 8, 1779–1784 (2023).
Pimlott, D. J. D., Jewlal, A., Kim, Y. & Berlinguette, C. P. Oxygen-Resistant CO2 Reduction Enabled by Electrolysis of Liquid Feedstocks. J. Am. Chem. Soc. 145, 25933–25937 (2023).
Lees, E. W. et al. Electrodes Designed for Converting Bicarbonate into CO. ACS Energy Lett. 5, 2165–2173 (2020).
Zhang, Z. et al. Porous metal electrodes enable efficient electrolysis of carbon capture solutions. Energy Environ. Sci. 15, 705–713 (2022).
Kim, Y., Lees, E. W. & Berlinguette, C. P. Permeability Matters When Reducing CO2 in an Electrochemical Flow Cell. ACS Energy Lett. 7, 2382–2387 (2022).
Oener, S. Z., Foster, M. J. & Boettcher, S. W. Accelerating water dissociation in bipolar membranes and for electrocatalysis. Science 369, 1099–1103 (2020).
Blommaert, M. A. et al. Insights and Challenges for Applying Bipolar Membranes in Advanced Electrochemical Energy Systems. ACS Energy Lett. 6, 2539–2548 (2021).
Chen, L., Xu, Q., Oener, S. Z., Fabrizio, K. & Boettcher, S. W. Design principles for water dissociation catalysts in high-performance bipolar membranes. Nat. Commun. 13, 3846 (2022).
Oener, S. Z., Ardo, S. & Boettcher, S. W. Ionic Processes in Water Electrolysis: The Role of Ion-Selective Membranes. ACS Energy Lett. 2, 2625–2634 (2017).
Salvatore, D. & Berlinguette, C. P. Voltage Matters When Reducing CO2 in an Electrochemical Flow Cell. ACS Energy Lett. 5, 215–220 (2020).
Rostrup-Nielsen, J. R. Syngas in perspective. Catal. Today 71, 243–247 (2002).
Jarvis, S. M. & Samsatli, S. Technologies and infrastructures underpinning future CO2 value chains: A comprehensive review and comparative analysis. Renew. Sustain. Energy Rev. 85, 46–68 (2018).
Hauch, A. et al. Recent advances in solid oxide cell technology for electrolysis. Science 370, (2020).
Li, C. S., Frankhouser, A. D. & Kanan, M. W. Carbonate-catalyzed reverse water-gas shift to produce gas fermentation feedstocks for renewable liquid fuel synthesis. Cell Rep. Phys. Sci. 3, 101021 (2022).
Song, H. et al. Integrated carbon capture and CO production from bicarbonates through bipolar membrane electrolysis. Energy Environ. Sci. 17, 3570–3579 (2024).
Li, Y. C. et al. CO2 Electroreduction from Carbonate Electrolyte. ACS Energy Lett. 4, 1427–1431 (2019).
Pang, X., Verma, S., Liu, C. & Esposito, D. V. Membrane-free electrochemical CO2 conversion using serially connected porous flow-through electrodes. Joule 6, 2745–2761 (2022).
Zhang, Z., Xi, D., Ren, Z. & Li, J. A carbon-efficient bicarbonate electrolyzer. Cell Rep. Phys. Sci. 4, 101662 (2023).
Ahmadi, R. et al. Reactive carbon electrolysis at 3 V, in an electrolyzer containing abipolar membrane operating under forward bias. ACS Energy Lett. https://doi.org/10.1021/acsenergylett.5c02914 (2025).
Lee, G. et al. CO2 electroreduction to multicarbon products from carbonate capture liquid. Joule 7, 1277–1288 (2023).
Xiao, Y. C. et al. Direct carbonate electrolysis into pure syngas. Ees. Catal. 1, 54–61 (2023).
Ren, X. & Gottesfeld, S. Electro-osmotic Drag of Water in Poly(perfluorosulfonic acid) Membranes. J. Electrochem. Soc. 148, A87 (2001).
Maier, M. et al. Mass transport in PEM water electrolysers: A review. Int. J. Hydrog. Energy 47, 30–56 (2022).
Rossmeisl, J., Qu, Z.-W., Zhu, H., Kroes, G.-J. & Nørskov, J. K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607, 83–89 (2007).
Kutz, R. B. et al. Sustainion Imidazolium-Functionalized Polymers for Carbon Dioxide Electrolysis. Energy Technol. 5, 929–936 (2017).
Xu, Y., Wang, C., Huang, Y. & Fu, J. Recent advances in electrocatalysts for neutral and large-current-density water electrolysis. Nano Energy 80, 105545 (2021).
Teuku, H., Alshami, I., Goh, J., Masdar, M. S. & Loh, K. S. Review on bipolar plates for low-temperature polymer electrolyte membrane water electrolyzer. Int. J. Energy Res. 45, 20583–20600 (2021).
Hu, G., Smith, K. H., Wu, Y., Kentish, S. E. & Stevens, G. W. Screening Amino Acid Salts as Rate Promoters in Potassium Carbonate Solvent for Carbon Dioxide Absorption. Energy Fuels 31, 4280–4286 (2017).
Cullinane, J. T. & Rochelle, G. T. Carbon dioxide absorption with aqueous potassium carbonate promoted by piperazine. Chem. Eng. Sci. 59, 3619–3630 (2004).
Gutiérrez-Sánchez, O. et al. Bifunctional Artificial Carbonic Anhydrase for the Integrated Capture and Electrochemical Conversion of CO2. ACS Sustain. Chem. Eng. 10, 13865–13876 (2022).
Acknowledgements
The authors would like to thank Dr. Monika Stolar for valuable discussions and assistance with manuscript writing. SR is grateful for financial support from UBC’s International Doctoral Fellowship Program. The authors are grateful to the Natural Sciences and Engineering Research Council of Canada (RGPIN-2018-06748; STPGP 521255), Canadian Foundation for Innovation (229288), Canadian Institute for Advanced Research (BSE-BERL-162173), and the Canada Research Chairs, for financial support. This research was undertaken thanks in part to funding from the Canada First Research Excellence Fund, Quantum Materials and Future Technologies Program. AS is funded by TotalEnergies.
Author information
Authors and Affiliations
Contributions
Conceptualization: C.P.B., Y.K.; Methodology: Y.K., A.M.L.J., E.W.L., Z.B.Z., S.R., B.A.W.M., A.S.; Investigation: Y.K., C.D., A.V.; Visualization: Y.K.; Funding acquisition: C.P.B.; Supervision: C.P.B.; Writing – original draft: Y.K., E.W.L.; Writing – review & editing: All authors.
Corresponding author
Ethics declarations
Competing interests
Y.K. and C.P.B. have filed a US provisional patent application and subsequent PCT application on the work presented in this manuscript. The bicarbonate electrolyzer has been patented (e.g., US Patent No. US12006580) and licensed, and C.P.B. is a founder of a company commercializing the technology. The remaining authors declare no other competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, Y., Jewlal, A.M.L., Lees, E.W. et al. Electrolytic pathway for upgrading waste CO2 into syngas with a carbon capture and utilization energy efficiency greater than 50%. npj Mater. Sustain. 4, 4 (2026). https://doi.org/10.1038/s44296-025-00090-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44296-025-00090-8
