
Abstract
Nicotinamide adenine dinucleotide (NAD+) is a coenzyme involved in a plethora of physiological reactions, with a key relevance in supporting mitochondrial function. Due to its critical role in these cellular processes, declining levels of NAD+ are associated with general aging and chronic disorders, including cognitive decline, sarcopenia, and metabolic diseases. These conditions are also typified by loss of mitochondrial health through dysfunction of homeostatic components such as mitophagy, unfolded protein response, and the antioxidant system. Therefore, raising cellular NAD+ through vitamin B3 family precursors or via drug-based interventions has become a broadly used strategy to restore mitochondrial and organismal homeostasis, with NAD+ precursors becoming a popular supplementation approach. As increasing components of the NAD+ biology are unraveled, this comprehensive review summarizes the advances in mechanisms of NAD+ metabolism and its modulation via compound-based strategies. Furthermore, it highlights the role of NAD+ in mitochondrial homeostasis in aging and disease conditions, the latest results of NAD+-boosting therapeutics in clinical trials, and areas of further translational development.
Similar content being viewed by others

Introduction
Nicotinamide adenine dinucleotide (NAD+) is an essential redox coenzyme that exists between the oxidized form NAD+ and its reduced form (NADH)1. NAD+ and NADH ratios are particularly important for processes including energy generation, metabolic homeostasis, mitochondrial function, and DNA repair; hence, NAD+ levels are tightly controlled in terms of production and consumption to ensure maintenance of cellular health. For instance, anabolism consumes energy in the form of ATP or NADH to synthesize biomolecules such as DNA and proteins, while catabolic reactions break down macromolecules, releasing energy in the form of ATP or NADH, which can be reutilized to generate NAD+2. NAD+ is involved in the catabolism of glucose in glycolysis, resulting in the production of ATP. Additionally, NAD+ is required for fatty acid metabolism during β-oxidation, glutaminolysis generating α-ketoglutarate, mitochondrial shuttling of metabolites, and lactic acid fermentation3,4,5. Apart from redox reactions, NAD+ is also a donor of ADP-ribose through enzymes such as poly (ADP-ribose) polymerases (PARPs) involved in DNA repair6. NAD+ is also a precursor to cyclic ADP-ribose (cADPR), a secondary signaling molecule generated by enzymes such as cluster of differentiation 38 (CD38)7, and it can be hydrolyzed by sirtuins, which deacetylate histones and regulate gene expression7. The phosphorylated form of NAD+, NADP/NADPH, parallels NAD+/NADH by functioning as a redox coenzyme in the pentose phosphate pathway, generation of acetyl coenzyme A (acetyl-CoA) for lipid and cholesterol biosynthesis, and in the glutathione and thioredoxin antioxidant systems4,5,8,9.
Given the importance of the cofactor in energy metabolism, NAD+ is especially important in mitochondrial function, and in fact both NAD+ and mitochondria are concentrated in energetically demanding tissues10,11,12. Mitochondria are essential organelles, whose dysfunction is central to major diseases13,14, as they are the center for aerobic ATP generation from oxidative phosphorylation (Oxphos) in the electron transport chain (ETC). NAD+/NADH is involved in this process within the mitochondria, where NAD+ is reduced to NADH in the tricarboxylic acid (TCA) cycle and is subsequently oxidized to NAD+ in the ETC for ATP generation. NAD+ levels are limiting in this reaction and determine the efficiency of mitochondrial energy production15. NAD+ also plays a critical role in other aspects of mitochondrial function, which must be maintained to ensure cellular health through several dynamic and coordinated homeostatic pathways. These include regulation of mechanisms controlling reactive oxygen species (ROS), which cause macromolecular damage16,17, mitochondrial turnover or mitophagy18, and the mitochondrial unfolded protein response (UPRmt), in which mitochondrial damage can be prevented by the removal of aggregated or unfolded proteins19.
Given the important central role of this cofactor and its relevance to mitochondrial function, this review will provide an extensive summary of the NAD+ homeostasis and modulation approaches, of the various mechanisms of NAD+-mediated mitochondrial regulation, and how the functional link between NAD+ and mitochondria has been investigated in the context of preclinical and clinical settings of aging and disease.
NAD+ homeostasis
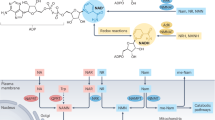
The NAD+ molecule is composed of a pyridine, derived from NAD+ precursors such as tryptophan (Trp) or nicotinamide (NAM)20, a purine, and a 5-phospho-d-ribosyl-1,α-disphosphate (PRPP) component21. For the pathways using precursors that require a phosphoribosyl moiety, such as NAM and nicotinic acid (NA), PRPP is required22. PRPP is an intermediate in purine nucleotide synthesis, generated from ribose 5-phosphate from the pentose phosphate pathway21. The purine component in NAD+ is adenine, which is incorporated from ATP23; its availability for NAD+ synthesis can be partially controlled by modulation of purine nucleotide phosphorylase (PNP), which plays a role in purine salvage pathways where purine bases are recycled into nucleotides24 and in metabolism of the NAD+ precursor nicotinamide riboside (NR)25. Multiple pathways of NAD+ synthesis have been identified, which are differentially active in tissues and utilize various molecular precursors to ensure maintenance of NAD+ homeostasis and counteract NAD+ turnover. Synthesis of the complete NAD+ molecule is achieved via different biochemical pathways, namely the de novo synthesis, Preiss–Handler, salvage pathway, and the nicotinamide riboside kinase (NRK)-mediated salvage26,27,28,29,30 (Fig. 1). NAD+ can be generated de novo in the kynurenine pathway, where Trp is oxygenated to N′-formylkynurenine by tryptophan 2,3-dioxygenase (TDO) or indoleamine 2,3-dioxygenase (IDO)31; N′-formylkynurenine is then hydrolyzed into kynurenine by arylformamidase (AFMID)32. Kynurenine is subsequently converted to NAD+ in a chain of reactions with the rate-limiting step by quinolinic phosphoribosyltransferase (QPRT), which converts quinolinic acid into nicotinic acid mononucleotide (NAMN)26, finally entering the Preiss–Handler pathway. The Preiss–Handler pathway also converts NA into NAMN by the rate-limiting nicotinic acid phosphoribosyltransferase (NAPRT). NAMN is then converted into nicotinic acid adenine dinucleotide (NAAD) by nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1), which leads to NAD+ via NAD+ synthase (NADS)28,29. Enzymes that consume NAD+ generate NAM as a product, and this can be recycled in the salvage pathway, providing the primary NAD+ synthesis pathway in mammals30. NAM generates nicotinamide mononucleotide (NMN) through nicotinamide phosphoribosyltransferase (NAMPT) in a rate-limiting reaction that determines the efficiency of NAD+ synthesis33. NMN is then converted into NAD+ by NMNAT1-3. NMNAT1 is ubiquitously expressed, NMNAT2 is primarily expressed in the brain, and NMNT3 is expressed in the lung and spleen34. The NAD+ precursor NR is also metabolized through salvage, but is first phosphorylated by nicotinamide riboside kinase 1 or 2 (NRK1/2) into NMN27. A recently discovered route of NAD+ synthesis utilizes reduced forms of NR and NMN, namely dihydronicotinamide riboside (NRH) and dihydronicotinamide mononucleotide (NMNH). NMNH is dephosphorylated into NRH to be transported into the cell through equilibrative nucleoside transporters (ENTs)35, where it is phosphorylated back into NMNH by adenosine kinase (AK)36. Unlike the other pathways, NRH and NMNH are condensed into NADH, possibly by NMNAT35. Another NAD+ precursor, nicotinic acid riboside (NAR), a deamidated form of NR, is phosphorylated by NRK1 into NAMN, where it enters Preiss–Handler37. Alternatively, NAR can be hydrolyzed by uridine hydrolase (URH1) into NA38. However, it is a poorer precursor as it requires methyl ester modification for transport into the cell37,38.

The figure summarizes the different biochemical routes for NAD+ formation. NAD+ can be synthesized de novo from tryptophan (Trp), which merges with the Preiss–Handler pathway, which also uses nicotinic acid (NA) for the processing of deamidated precursors. The salvage pathway utilizes amidated precursors and allows recycling of the nicotinamide (NAM) byproduct of NAD+-consuming enzymes. Reduced NMN (NMNH) and NR (NRH) are proposed to proceed through NADH to generate NAD+.
Only the liver and kidneys can produce NAD+ de novo starting from Trp39. Most tissues may rely on salvage utilizing NAM in circulation, as seen by expression of NAMPT and NMNAT1-340. For metabolism of NR, NRK1 is expressed in most tissues but is absent in the spleen and white adipose tissue, while NRK2 is only present in skeletal muscle, heart, and white adipose tissue (WAT)40. The Preiss–Handler pathway can take place in several tissues, such as the brain, heart, kidney, small intestine, and liver, as shown by the expression of NAPRT and NADS11; however, these organs also utilize the machinery for salvage. Lung, WAT, testis, and skeletal muscle preferentially rely on salvage and have low expression of proteins involved in the Preiss–Handler pathway41, with the muscle in particular relying on both NRK1 and NRK2 for supporting its mitochondrial metabolism and fiber type composition42. Salvage occurs at a lower rate in the blood compared to other tissues, where Preiss–Handler may even be the preferred pathway11. When looking at the endogenous NAD+ fluxes at the systemic level, most organs do not synthesize NAD+ de novo, but can utilize NAM made in the liver39. Tissues doing this may rely on NAMPT obtained exogenously from other cells in the body. Extracellular NAMPT (eNAMPT) is secreted from adipocytes and immune cells43,44 via exosomes, and enters the circulation, allowing transport of this enzyme to other tissues. Although primarily known as a pro-inflammatory cytokine that binds to cell membrane receptors to potentiate intracellular signaling45, eNAMPT can also be taken up by cells such as neurons and increase NAD+ synthesis46. eNAMPT may also allow extracellular conversion of NAM to NMN, as eNAMPT is catalytically active43.
Counteracting its biosynthesis, NAD+ is also utilized in numerous reactions leading to its degradation or conversion into other metabolites. NAD+-consuming enzymes include NAD+ glycohydrolases (NADases) such as CD38 and sterile alpha and TIR motif containing 1 (SARM1), deacylases (sirtuins), and ADP-ribosyl transferases (PARPs). The enzymes metabolize NAD+ and release NAM as a byproduct47. NAD+ is a substrate for sirtuins, which regulate enzymes through post-translational deacylation by transferring the acetyl group of a target protein to NAD+7,48. Sirtuins also perform other post-translational reactions such as NAD+-dependent desuccinylation, diacylation, and demalonylation, and are important in regulating metabolism, allowing metabolic plasticity7. PARPs catalyze reversible ADP-ribosylation of macromolecules6,49. PARPs are most commonly known for DNA repair, although they also function in transcription, cell cycle regulation, and proteasome regulation7. For example, PARP1 binds a DNA single strand break, then utilizes NAD+ to ADP-ribosylate macromolecules in the vicinity, a signal for recruitment of DNA repair machinery6. The NADase CD38 is involved in the immune response by producing second messengers ADPR, 20-deoxy-ADPR (2dADPR), NAADP, and cADPR. The enzyme mediates calcium channel opening, which leads to T-cell activation7; it is also a major NAD+ consumer with a lower Km than most NAD+-consuming enzymes40 and is implicated in the decline of NAD+ levels observed during aging and disease50,51,52. Thus, through high consumption of NAD+, CD38 also indirectly impacts the activity of other NAD+-consuming enzymes such as sirtuins52. SARM1, another NADase, rapidly consumes NAD+ following axon damage, resulting in neuronal degeneration7,53,54.
The NAD+-consuming enzymes produce NAM as a byproduct, of which high concentrations inhibit the activity of NAD+-consuming enzymes, providing feedback regulation. For example, NAM can inhibit the activity of human sirtuin 1 (SIRT1) and PARPs in vitro55,56. NAM levels are regulated by nicotinamide N-methyltransferase (NNMT), which converts NAM to methylnicotinamide (meNAM)57 (Fig. 1). NNMT permanently removes NAM by transferring the methyl group from S-adenosyl methionine (SAM) to NAM to produce meNAM and S-adenosyl-l-homocysteine (SAH)57. MeNAM can be excreted in the urine or is further oxidized into N-methyl-6-pyridone 3-carboxamide (me-6PY or 2PY) or N-methyl-4-pyridone 3-carboxamide (me-4PY or 4PY)58.
In addition to the endogenous cellular pathways, recent studies have also investigated the role of the gut microbiome in affecting NAD+ levels. Nearly half of the bacteria comprising the human gut microbiome are unable to produce NAD+ de novo and rely on NAD+ precursors available in the environment for their metabolic functions59,60. Consequently, studies have focused on how NAD+ precursors impact the biodiversity and composition of the gut microbiome and the resulting effects of this. Lozada-Fernández et al.61 demonstrated that NR supplementation altered the gut microbiome in mice and demonstrated that the gut microbiome change was able to impact weight gain in mice fed a high-fat diet. In another study, Yu et al.62 illustrated the change in microbiome composition with an increase in Actinobacteria and Deferribacteres following NR supplementation on alcohol-exposed mice. This study also suggested the protective effect NR supplementation had on alcohol-induced liver injury, whose mechanism could involve the gut microflora-bile acid axis. Interestingly, a recent study showed that oral supplementation of NR altered the intestinal microbial composition in rats and mice but not humans63, highlighting the need for further studies on this matter to assess the impact of NAD+ metabolism modulation by the microbiome on health outcomes.
NAD+ homeostasis is not only regulated at the systemic level, but the pathways involved in synthesis and consumption also present dynamic and complex cellular locations and regulation. Extracellular NAD+ can enter cells via pore-forming channel connexin 43 (Cx43), a bi-directional NAD+ transporter allowing exogenous NAD+ transportation into the cell, and transportation out for utilization of NAD+ by CD3864. Alternatively, extracellular NAD+ can be utilized by NAD+ glycohydrolases, generating NAM, NR, or NMN outside of the cells, which can then be transported intracellularly for NAD+ synthesis65,66. Several membrane-bound proteins suggested to play a role in this process include CD73, able to convert NMN into NR67, and NAD+ glycohydrolases such as CD38 that convert NAD+ into NAM and ADP-ribose. Precursors are then transported into cells through unspecific ENTs, such as members of the SLC29A family for NR, while NMN requires dephosphorylation into NR before import into the cell by ENTs68. However, tracer experiments have also shown that NMN at low levels can enter cells intact in the kidney and white adipose tissue69. SLC12A8 has been proposed as an NMN transporter into the cell70, but this is still debated71,72. NAM transporters have also been investigated and recently identified as SLC29A1 and SLC29A2, which are also able to transport NR73. NA can instead be transported by SLC5A8 and SLC22A1374,75, while SLC7A5 and SLC36A4 are required for uptake of Trp71,72.
NAD+ is concentrated in the mitochondria, with lower concentrations in the nucleus and cytoplasm76,77. Mitochondrial NAD+ is utilized in essential reactions such as TCA, fatty acid oxidation, and Oxphos4. Due to the interconnection of glycolysis and Oxphos, there is dynamic interchange between cytoplasmic and mitochondrial NAD+, as depletion of cytoplasmic NMNAT2 also depletes mitochondrial NAD+77. Recently, a key finding uncovered SLC25A51 as a mitochondrial transporter for intact NAD+ in human cell lines78,79; additionally NAD+ can be generated in the mitochondria from NMNAT380, whose activity has been shown to be required in cells77, but dispensable in vivo as NAD+ can be replenished through other pathways such as import from the cytosol81. Cytosolic NAD+ is synthesized by NMNAT2 localized in the cytoplasm and Golgi, where it is essential for glycolysis82. Preiss–Handler and de novo do not occur within the mitochondria, based on the nuclear or cytosolic localization of QPRT, NAPRT, and NADS48. NAMPT and NRK1/2 are also localized to the nucleus and cytosol48,83,84, suggesting that only the final step of salvage is performed in the mitochondria, with a mitochondrial NMN transporter, Slc25a45, recently proposed85. NADH levels can be increased in mitochondria by malate-aspartate and the glycerol-3-phosphate redox shuttles14. Nuclear NAD+ is generated by nuclear NMNAT1 and is largely utilized by PARPs for DNA damage response and nuclear sirtuins such as SIRT1, SIRT6 or SIRT7 for epigenetic deacetylation82,86. Cytosolic and nuclear NAD+ are often considered a shared pool as NAD+ diffuses through nuclear pores58, and importantly, the concentrations of NAD+ in the cytosol, nucleus, and mitochondria mirror the Km of NAD+-consuming enzymes localized to the corresponding compartment, hence these enzymes are sensitive to compartmental fluctuations in NAD+ levels77.
Overall, it is clear that NAD+ fluxes within the cell are complex and dynamic, but the concentrated levels of NAD+ in the mitochondria indicate major impacts of the cofactor in this organelle in particular, which will be summarized below.
Mitochondrial homeostasis regulation by NAD+
Mitochondria are the source of high-yield energy production through Oxphos and the ETC to generate ATP, of which levels must be maintained for cellular homeostasis. Following the catabolism of glucose in the cytoplasm as part of glycolysis, the generated pyruvate is transported to the mitochondrial matrix, where it feeds into the TCA, undergoing oxidation to reduce NAD+ to NADH, which is the main electron donor of the ETC87. Given the importance of this organelle, maintenance of proper mitochondrial function and structure is essential, and dysfunction is a pathological hallmark of metabolic, muscular, and neurodegenerative conditions, which can be associated with aging in multiple tissues88,89. There are several processes that contribute to mitochondrial quality control, including UPRmt, mitophagy, mitochondrial membrane dynamics, and mitochondrial biogenesis13,89; NAD+ is a crucial cofactor for both mitochondrial bioenergetics and for the above-mentioned homeostasis pathways (Fig. 2).

The figure summarizes the different contributions of NAD+ to mitochondrial homeostasis. ATP generation from the ETC involves NAD+ as a critical electron acceptor, and homeostasis pathways of the organelle are regulated by NAD+ primarily through sirtuins. Mitophagy is mediated by membrane proteins and receptors that direct the organelle for autophagy, and SIRT3 can increase PINK1 expression through FOXO3α. UPRmt regulates protein folding and degradation to prevent mitochondrial proteostatic stress, and SIRT3 increases the activities of the chaperone HSP10 and LON protease. Antioxidant systems such as SOD and catalase (CAT), whose expression is also regulated by the SIRT3-FOXO3a axis, control ROS levels. The mitochondrial membrane undergoes fusion for the transfer of macromolecules, and fission for replication and mitophagy. SIRT3 increases expression of FIS1 and DRP1 through FOXO3α, while SIRT2 increases levels of MFN2. Finally, mitochondrial biogenesis is regulated by PGC-1α, which can be activated by SIRT1, triggering DNA replication and increased protein expression. In contrast, SIRT7 inhibits NRF1 to reduce mitochondrial biogenesis.
NAD+/NADH ratios reflect redox status and play an integral part in the ETC, with NADH being the primary electron donor. A depletion in NAD+ results in bioenergetic failure of mitochondria and eventually cell death90,91. Titov et al.92 showed that enzymatically increasing the NADH-to-NAD+ conversion exclusively in the mitochondria of HeLa cells improves ETC function, highlighting the importance of maintaining high NAD+ levels. In fact, NADH is an inhibitor of the TCA enzyme isocitrate dehydrogenase93, a protective mechanism to prevent overloading of the ETC and excessive ROS generation. This further indicates that maintenance of NAD+/NADH redox balance is required for optimal energy production. Indeed, high NAD+ levels and NAD+/NADH ratios are associated with increased energy production in humans, improved mitochondrial membrane potential, and decreased mitochondrial mass through mitophagy in vitro, suggesting improved efficiency of mitochondria94,95,96,97.
On top of the role of NAD+ as a direct substrate for the TCA and ETC, much of the influence of NAD+ on mitochondrial homeostasis is through the NAD+-consuming enzymes sirtuins. Mitochondrial sirtuin 3 (SIRT3) is a major modulator of mitochondrial metabolism and homeostasis, regulating UPRmt, mitochondrial biogenesis, mitophagy, fusion/fission, and antioxidant expression98. The effects of SIRT3 are through deacetylation and activation of the stress responder FOXO3α. The SIRT3/FOXO3α axis upregulates the PINK1/PARKIN axis to increase mitophagy when SIRT3 expression is increased99. FOXO3α also upregulates the expression of dynamin-related protein 1 (DRP1), mitochondrial fission 1 protein (FIS1), and mitofusin 2 (MFN2) to regulate mitochondrial fission100. SIRT3 also deacetylates targets such as the chaperone protein Hsp10 and the LON protease, involved in the UPRmt process98. During protein folding stress, the sirtuin SIRT7 also plays an important role by contributing to repressing nuclear respiratory factor 1 (NRF1) activity, a major transcriptional regulator of mitochondrial genes; this results in reduced transcription of mitochondrial ribosomal proteins and mitochondrial transcription factors, and subsequently halting protein translation101,102. SIRT1, localized in the nucleus, deacetylases and activates proliferator-activated receptor gamma coactivator-1 (PGC-1α), which leads to increased mitochondrial biogenesis103. SIRT2, present both in nuclear and cytoplasmic compartments, can increase MFN2 and decrease DRP1, thus promoting mitochondrial fusion104,105. Mitochondria also host antioxidant enzymes such as superoxide dismutase (SOD) and catalase as the first line of defense against ROS by converting superoxide to H2O2 and subsequently to water. Expression of SOD and catalase is indirectly regulated by SIRT3 through deacetylation of FOXO3α106,107. FOXO3α also prevents pro-apoptotic BAX translocation to the mitochondria, hence protecting cells from apoptosis100.
Given the role of NAD+ in modulating the processes described above, NAD+-boosting interventions are being investigated for supporting mitochondrial homeostasis. For instance, treatment with the NRK-salvage precursor NR in mice, C. elegans, and mammalian cells resulted in increased mitochondrial function and biogenesis, UPRmt, and antioxidant response gene expression108,109,110. In C. elegans, NR and NMN improved mitophagy through pink1 and Parkin homologs pdr-1111,112. NMN supplementation in mice also improved mitochondrial morphology through DRP1, and reduced ROS through antioxidant response expression113. Treatment with NR or the PARP inhibitor Olaparib also improved mitochondrial morphology in aged nematodes and mammalian muscle cells during proteotoxic stress114. NAM treatment increased mitochondrial NAD+/NADH ratios and improved mitochondrial membrane potential in human primary cells115.
As exemplified above, these and other studies have shown the mechanistic role of NAD+ in mitochondrial function in cellular and animal models. The relevance of NAD+-boosting strategies for mitochondrial homeostasis and implications for health benefits in the context of aging and disease in preclinical and clinical studies will be discussed in the “Preclinical and clinical applications of NAD+-modulating interventions for mitochondrial homeostasis and health benefits” section.
Interventions to modulate NAD+ levels
Several strategies have been developed to modulate NAD+ metabolism, both for mechanistic investigation purposes and to assess their potential therapeutic effects to improve health in aging or disease. The methods range from dietary or intravenous administration of NAD+ and precursors to inhibition of NAD+-consuming enzymes with small molecules, to activation of the NAD+ synthesis enzymes.
NAD+ and precursors supplementation
NAD+ can be directly supplemented intravenously to increase cellular NAD+ levels, which has been observed in healthy and elderly cohorts, suggesting improvements in cognitive function and increased expression of antioxidant genes, although these trials were conducted in small cohorts66,116,117. Cellular studies have shown that exogenous NAD+ can impact mitochondrial activity, such as boosting Krebs cycle and the ETC, but also impair genomic DNA replication and induce DNA replication stress due to impairment of pyrimidine biosynthesis118. Thus, direct NAD+ administration may exhibit unique effects compared to NAD+ precursors and modulators, warranting further investigation, especially given the rise of wellness and anti-aging clinics offering intravenous NAD+ therapy, while clinical evidence is still scarce119.
NAD+ precursors include both natural and synthetic compounds that can modulate the NAD+ pool via the above-mentioned biosynthesis pathways in the “NAD+ homeostasis” section. The majority of these compounds are Generally Recognised as Safe (GRAS) and are available on the market as supplements that can raise NAD+ levels in human subjects120,121,122; the most marketed supplements include NAM, NMN, and NR. Boosting NAD+ via the de novo pathway through Trp supplementation is not as popular, as quinolinic acid generated from Trp conversion is neurotoxic, and kynurenines have immunomodulatory roles58. Boosting NAD+ levels through the Preiss–Handler pathway is achievable through NA supplementation28,29; however, its utilization is limited due to side effects like skin flushing123. The salvage pathway utilizes the precursors NAM, NMN, and NR. Unlike NAM, NR, and NMN do not require ribose incorporation for NAD+ formation49. However, studies in mice show NR is more effectively taken up than NMN in some tissues, such as skeletal muscle39, highlighting the importance of the choice of precursor treatment depending on the intended target organ. Additionally, it is important to note that NR readily degrades in human and rodent plasma, and NMN degrades in human plasma, suggesting low systemic stability of the molecules when administered orally68,124,125.
The method of administration of precursors also influences NAD+ fluxes in the body, as uncovered by precursor tracer experiments in mice. A summary of possible NAD+ metabolite fluxes in the human body based on the mice studies summarized below is proposed in Fig. 3. Intravenous NR or NMN administration in mice results in rapid conversion into circulating NAM and NAD+ boosting in liver, kidney, skeletal muscle, while NAD+ in these organs after oral administration increases to a lower magnitude39,69. For oral administration in mice, NR and NMN are first metabolized by the liver or GI tract into NAM, which can then be taken up by other tissues. When provided intravenously, skeletal muscle is able to use circulating NR without hepatic metabolism, but not NMN; this may be because NMN first requires dephosphorylation into NR before uptake into cells39. Supporting this, the tracer experiments in mice show that NMN is first converted into either NAM or NR before NAD+ synthesis in most tissues69.

The figure provides an overview of NAD+ metabolism at the systemic level. Oral precursors (green arrow) are metabolized by the liver into NAM, which can be used by other tissues in the salvage pathway. Microbes in the GI tract can metabolize circulating NR into NAM, which in turn is subsequently metabolized into NA, bypassing the salvage pathway. Intravenous administration (blue arrow) of NR allows transport and utilization of the precursor by tissues through the circulatory system, while administration of NMN first requires metabolism by the liver. Once orally or intravenously administered, precursors are metabolized by the liver into NAM, which enters the circulation and can be utilized by tissues through salvage (black arrow). Further details in the main text39,69,126,127.
The gut microbiome also plays a role in precursor metabolism. Precursors entering the gut are absorbed by the upper gastrointestinal tract during oral administration and are unavailable to the microbes in the large intestine. However, microbes in the colon can synthesize NAD+ de novo from fiber, and microbes in the ileum and jejunum are able to utilize circulating NAM. Interestingly, the microbes deamidate NAM into NA, therefore bypassing salvage. The synthesized NA can then re-enter circulation for Preiss–Handler or be utilized by gut microbes126. The gut microbiota also plays a significant role in late-phase oral NR metabolism (3 h post-administration), which is converted to NAM by the enzyme BST1, and then deamidated into NA by microbes, allowing Preiss–Handler synthesis by capable tissues127. In the early phase (1 h post-administration), NR is rapidly absorbed by the small intestine and enters salvage127. NA generated by gut microbes is also able to maintain a local NAD+/NAM pool that can be a source of circulating NA even in the absence of dietary precursor administration126,127.
Due to the poor bioavailability and interconversion of these precursors, more potential NAD+ precursors and intervention strategies are sought that have higher potency and stability. NRH is a new NAD+ precursor that is stable in mouse plasma and is not degraded into NAM, unlike NR125. Intravenous administration in mice showed that NRH is more potent at raising NAD+ levels in the liver and muscle than NR and NMN128. However, in the kidney tissue, which expresses high levels of NRK1, the effects of NR and NRH are comparable125,128. Similarly, NMNH, a reduced form of NMN, more potently increased NAD+ levels in the blood, liver, muscle, brown adipose tissue, brain, kidney, and heart compared to NMN, independently of NRKs and NAMPT35,129. Since the reduced precursors are hypothesized to synthesize NAD+ through formation of NADH, administration of NRH and NMNH may offer an NAD+ boosting avenue that is independent of both salvage and Preiss–Handler pathways, and is unaffected by the microbiome58. Another natural compound, trigonelline, a methylated form of nicotinic acid, is a newly discovered NAD+ precursor able to increase NAD+ in multiple species, with preclinical implications for use in sarcopenia, and for muscle and cognitive decline in aging124,130,131.
Modulation of enzymes involved in NAD+ consumption and synthesis
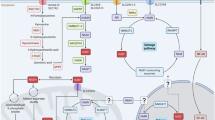
In addition to NAD+ precursor-based strategies, halting of processes linked to NAD+ consumption or biosynthesis represents a complementary and therapeutically appealing approach to alter or restore NAD+ homeostasis in context-specific applications. These include inhibition strategies of NAD+ consuming enzymes, such as CD38 and PARPs, or modulation of key NAD+ synthesis proteins such as NAPRT and NAMPT, as summarized below and in Fig. 4.

The figure summarizes several ways to control NAD+ and downstream effectors. These include: (1) Modulating NAD+ synthesis via activators or inhibitors of NAPRT, NAMPT, and amino-carboxy-muconate-semialdehyde decarboxylase (ACMSD); (2) Enhancing NAD+-dependent deacylation through sirtuins via compound interventions; (3) Compound-based inhibition of NAD+ consumption by targeting CD38, PARPs, NNMT, and SARM1. These approaches can yield benefits in diverse conditions such as metabolic disorders, inflammation, and neurological diseases. Frequently reported compounds and associated applications are indicated for each strategy. For more compounds studied across various health conditions, refer to the main text. CIPN chemotherapy-induced peripheral neuropathy, NAFLD non-alcoholic fatty liver disease. Created in BioRender. https://BioRender.com/0mrb07s.
CD38 inhibitors
CD38 is a multifunctional enzyme that catalyzes the conversion of NAD+ to cyclic ADP-ribose and ADP-ribose through a common covalent intermediate132,133. This intermediate is responsible for cyclization, hydrolysis, and base-exchange reactions133. CD38 has experimentally been proven to play a role in the regulation of cellular metabolism, inflammation, and oxidative stress134. Involved in cellular signaling and regulating calcium levels, CD38 degrades NAD+, using it as a co-substrate for its enzymatic activity135,136. CD38 inhibition, resulting in increased NAD+ levels, may slow aspects of the aging trajectory and enhance immune function, making it a promising area of research for various diseases137,138. To this aim, there has been a blooming effort in the development of potential CD38 inhibitors, which fall into different categories, such as small-molecule compounds, natural inhibitors, and antibody approaches.
Nicotinamide deoxyriboside derivatives, such as nicotinamide 2′-deoxyriboside and 5-methylnicotinamide 2′-deoxyriboside, act as mechanism-based small molecule inhibitors of CD38 by forming stable covalent intermediates132. Yet another small molecule that has been studied extensively as a highly potent small molecule inhibitor of both human and murine CD38 is 78c, which has been shown to be 10-fold less potent against the cyclase activity than the hydrolase activity of recombinant human CD38139. Interestingly, 78c showed no activity against ADP-ribosyl cyclase at concentrations up to 50 nM, while it exhibited no modulatory effects on CD157, suggesting its specificity toward CD38139.
Several natural CD38 Inhibitors are also currently under investigation. Cyanidins are recognized as natural CD38 inhibitors140. In a study involving rats with bovine type II collagen-induced arthritis, the administration of cyanidin-3-O-glucoside resulted in a reduction of the proportion of CD38+ natural killer cells in both peripheral blood and synovial fluid. Additionally, this treatment alleviated joint inflammation and contributed to a partial restoration of joint mobility in arthritic mice141 (Fig. 4).
Apigenin, a flavonoid widely distributed in plants such as parsley, celery, and chamomile, shows the ability to inhibit CD38142. Research indicates that apigenin effectively inhibits CD38 in mice, leading to significant increases in NAD+ levels143,144. Furthermore, apigenin treatment has been shown to reduce global protein acetylation while improving glucose and lipid homeostasis in obese mice143. It also ameliorates oxidative stress and inflammation in various tissues, including the kidneys of diabetic rats143,144,145 (Fig. 4).
Another interesting compound, quercetin, has been shown to inhibit the activity of CD38 by binding to its active site, which results in increased intracellular NAD+143. Quercetin inhibited CD38 NADase activity and ADP-ribosyl-cyclase activity with an IC50 of 13.8 ± 2.1 μmol/L and 15.6 ± 3.5 μmol/L, respectively. Additionally, a dose-dependent NAD+ increase was observed in A549 cells treated with quercetin. However, quercetin was not determined to be as potent as apigenin, which requires a lower concentration to achieve the same effect143.
Isatuximab (SAR650984) and Daratumumab are instead monoclonal antibodies that target CD38, which is also highly expressed on multiple myeloma (MM) cells. Isatuximab binds to the CD38 epitope via its gamma heavy chains, inducing cell death through mechanisms such as antibody-dependent cellular cytotoxicity (ADCC) and apoptosis146,147. Daratumumab targets a different epitope on CD38 and induces cell death through ADCC, complement-dependent cytotoxicity (CDC), and direct damage to myeloma cells. Both antibodies are effective in treating relapsed/refractory MM and can be used in combination with other therapies148,149. MOR202 and Mezagitamab bind with high specificity to CD38 on the surface of multiple myeloma cells150,151 inducing apoptosis through ADCC and antibody-dependent cellular phagocytosis (ADCP)152,153. TAK-079 is a human IgG1 monoclonal antibody that depletes CD38+ immunosuppressive regulatory T cells and myeloid-derived suppressor cells151,154,155. However, the effects of these compounds on NAD+ modulation have not been studied yet.
TNB-738 is a biparatopic antibody specifically designed to inhibit CD38 by binding to two distinct epitopes on the CD38 molecule simultaneously, thereby enhancing its inhibitory effect on CD38’s enzymatic activity156. Unlike other anti-CD38 antibodies, TNB-738 does not induce cell death and immunosuppression, leading to a more balanced immune response. TNB-738 has also been shown to boost NAD+ levels, making it a promising therapeutic option for metabolic and inflammatory diseases associated with NAD+ deficiencies, including aging156.
PARP inhibitors
PARP is a family of proteins involved in DNA repair, genomic stability, and programmed cell death. However, their activity consumes significant amounts of NAD+, leading to a depletion of cellular NAD+ levels and impacting energy metabolism and cell survival58. For instance, excessive activation of PARP1 in response to chronic DNA damage and inflammation accelerates features of aging. This is particularly evident in the context of Louis-Bar syndrome, where neurodegeneration and other aging symptoms appear earlier than expected157. Studies in the field of accelerated aging have established a link between PARP activation and impaired mitochondrial health, which is associated with reduced NAD+ levels resulting from PARP hyperactivation158. Research suggests that inhibiting PARP1 can enhance the functionality of senescent cells by increasing NAD+ levels and SIRT1 activity159. Consequently, targeting PARP enzymes may offer a promising therapeutic strategy for addressing age-related diseases and disorders linked to accelerated aging.
Olaparib is a compound that effectively inhibits PARP1 and PARP2, binding to the catalytic domain and preventing the transfer of ADP-ribose units from NAD+ to target proteins, thus lowering NAD+ consumption160,161. Olaparib has been studied for its neuroprotective effects in a mouse model of Huntington’s disease by modulating inflammasome activation162. In addition, it protects against chronic hypoxia/reoxygenation-induced retinal injury through various signaling mechanisms163. Olaparib treatment also replenished NAD+ levels, enhanced mitochondrial function, and promoted fatty acid oxidation, thereby reversing non-alcoholic fatty liver disease (NAFLD) in mice fed a high-fat, high-sucrose diet164 (Fig. 4).
Another potent oral PARP1/2 inhibitor, niraparib (MK–4827) binds to the active site of PARP enzymes, and studies indicate that Niraparib can restore NAD+ levels and improve mitochondrial function165,166. Niraparib showed promising activity in preclinical models and phase I clinical trials, achieving its pharmacodynamic target for PARP inhibition in epithelial ovarian cancer, particularly in patients who have responded to platinum-based chemotherapy167 (Fig. 4).
3,4-Dihydro-5 [4-(1-piperindinyl)butoxy]-1 (2H)-isoquinoline (DPQ) specifically inhibits PARP1 and has been reported to reduce tumor formation and size in skin carcinogenesis models168. It also attenuated lipopolysaccharide-induced acute lung injury in mice by inhibiting NF-κB-mediated inflammatory responses169. While it decreases ROS generation by enhancing antioxidant enzyme activity, DPQ has also been associated with detrimental effects, including increased DNA damage and apoptosis in cardiomyocytes170.
3-aminobenzamide (3-AB) is a competitive inhibitor of PARP enzymes, particularly PARP1171,172. PARP1 inhibition using 3-AB has shown protective effects in various models of ischemia-reperfusion injury. In a rat model of focal cerebral ischemia, 3-AB reduced lesion volumes and attenuated NMDA-induced neurotransmitter dysregulation173. Similarly, in myocardial ischemia-reperfusion, 3-AB suppressed PARP1 activation, reduced infarct size, and improved cardiac function174. Moreover, in an in vitro model of blast-induced auditory hair cell damage, 3-AB inhibition of PARP1 reduced oxidative stress markers, upregulated antioxidant defenses, and helped maintain cell viability by preserving ATP pools175. 3-AB is an early discovered PARP inhibitor, and its specificity is lower than newer PARP inhibitors developed for clinical use176. 3-AB can have off-target effects and less potency, making it mainly a research tool rather than a therapeutic agent177. Overall, these studies highlight the complex role of PARPs and the impact of their inhibition in different pathological conditions.
Sirtuin modulation
Modulating sirtuin activity by targeting NAD+-dependent deacylation may hold therapeutic potential for impacting mitochondrial homeostasis and other cellular pathways related to aging and disease. Research has identified several natural compounds, often linked to senolytic effects, that can enhance the activity of SIRT1 and SIRT2. For instance, while quercetin has been reported to activate both enzymes, curcumin has shown potential as an activator of SIRT1, promoting its deacetylase activity and contributing to anti-inflammatory effects178. Similarly, fisetin, a flavonoid found in strawberries and other fruits, activates SIRT1 by increasing NAD+ levels while also reducing oxidative stress and inflammation179 (Fig. 4). Additionally, berberine, an alkaloid found in several plants including goldenseal and barberry, further supports SIRT1 activation through its ability to induce NAD+ biosynthetic pathways and improve metabolic health180.
Focusing on SIRT3, recent studies indicate that its activation is generally protective; however, there are scenarios where inhibiting SIRT3 might be beneficial181. For example, in certain neurodegenerative diseases, modulating SIRT3 activity can help manage mitochondrial dysfunction and oxidative stress182. Specifically, SIRT3 deacetylates and inhibits aldehyde dehydrogenase 2, which prevents the conversion of acetaldehyde to acetate, potentially leading to acetaldehyde toxicity183. Additionally, SIRT3 deacetylates ceramide synthases, promoting ceramide accumulation and inhibiting complex III activity184. SIRT3 lysine deacetylase inhibitors specifically target SIRT3 without affecting its structurally similar counterparts, SIRT1 and SIRT2185,186. Yet another compound, Honokiol (HKL), has been shown to protect against doxorubicin-induced cardiotoxicity by activating SIRT3, which helps reduce ROS production, prevent mitochondrial fragmentation, and promote mitochondrial fusion187. Similarly, the small molecule activator 2-APQC effectively enhances SIRT3 activity, alleviating cardiac hypertrophy and myocardial fibrosis induced by isoproterenol (ISO). By maintaining mitochondrial homeostasis and reducing oxidative stress, 2-APQC emerges as a potential candidate for treating heart failure and related cardiac conditions188. Fucoidan, a group of sulfated polysaccharides from brown algae, acts as an SIRT3 activator, inhibiting cellular senescence and the harmful effects of senescence-associated secretory phenotypes (SASPs)189,190. Specifically, research indicates that fucoidan can protect against cellular senescence by regulating key proteins involved in the cell cycle and apoptosis, particularly in mesenchymal stem cells exposed to stressors like p-cresol191 (Fig. 4). Similar to SIRT3, SIRT6 is also activated by fucoidans; in addition to its crucial role in maintaining genomic stability and regulating inflammation, SIRT6 also directly deacetylates NAMPT, enhancing its activity and promoting NAD+ synthesis via the salvage pathway. By enhancing SIRT6 activity, fucoidans improve DNA repair mechanisms and reduce tissue senescence, particularly in the kidney and lung, thereby extending healthspan in aged mice192,193.
Another sirtuin, SIRT7, has been studied in relation to age-related mitochondrial stress. Its overexpression in aged mice alleviated mitochondrial protein-folding stress, resulting in enhanced neurogenesis and improved cognitive outcomes. This suggests that SIRT7 may be a potential target for therapeutic intervention aimed at preserving cognitive function in the elderly102. In parallel, other studies have explored the broader biological functions of SIRT7, particularly its role in immune regulation and mitochondrial homeostasis. For instance, SIRT7 has been identified as a key player in the pathogenesis of immune-mediated intestinal inflammation, specifically in inflammatory bowel disease (IBD). In this case, inhibition rather than activation of this protein reduced colonic mucosal immune activation. These findings position SIRT7 as a potential therapeutic target for treating IBD, with the inhibition of its activity offering a novel approach to attenuating intestinal inflammation194; however, this example, combined with the others above, also highlights that activation or inhibition of these enzymes is beneficial in a context-dependent manner.
NNMT inhibitors
NNMT has garnered significant attention due to its role in various metabolic processes and its potential as a therapeutic target195. Inhibition of NNMT has several implications for health, particularly in the context of metabolic disorders and aging191. Elevated NNMT activity is associated with metabolic syndrome, which includes conditions like obesity, type 2 diabetes, hypertension, and hyperlipidemia195,196. Nicotinamide analogs such as 6-methylaminonicotinamide form one class of NNMT inhibitors that work by mimicking the natural substrate, NAM, and competing for the enzyme’s active site197. Another NAM analog-based NNMT inhibitor, 6-methoxy NAM (also known as JBSNF000088), has shown promise in decreasing body weight and enhancing insulin sensitivity in mice models of obesity induced by a high-fat diet198. Another approach to modulate NNMT involves bisubstrate inhibitors, which are designed to resemble both the substrate (NAM) and the cofactor (SAM). These inhibitors bind to the enzyme’s active site, effectively blocking its activity. Bisubstrate inhibitors such as NS1, LL320, and II399 based on adenosyl scaffolds have shown promise in this regard199,200,201. Covalent inhibitors represent a different strategy, where the inhibitor forms a permanent bond with the enzyme, leading to irreversible inhibition. Compounds such as alpha-chloroacetamide-based inhibitors achieve this by covalently binding to a cysteine residue in the SAM-binding pocket of NNMT202. Additionally, methylated quinolines such as 5-amino-1-methylquinolinium have been identified as NNMT inhibitors through ligand-based in silico methods and biological evaluations203. 5-amino-1-methylquinolinium has been found to reduce adipocyte size and lower plasma total cholesterol levels in diet-induced obese mice and improve muscle strength in aged mice204,205 (Fig. 4). Other small molecule inhibitors have also been reported, including those derived from amino adenosine and other nicotinamide-related compounds206,207,208.
NAMPT modulators
As described above, NAMPT is a key enzyme in the NAD+ biosynthesis pathway via salvage, converting NAM into NMN209. By maintaining NAD+ levels, NAMPT supports sirtuin activity, highlighting its potential as a therapeutic target for age-related diseases210,211. NAMPT overexpression can extend cell proliferation capabilities and enhance stress resistance through SIRT1-mediated p53 degradation212. Moreover, NAMPT activity is crucial for the beneficial effects of calorie restriction, including improved oxidative stress management, mitochondrial biogenesis, and metabolic adaptation, as reported in a study involving Sprague-Dawley rats213. These findings suggest that optimal NAMPT levels and activity are critical for healthy aging and overall organismal homeostasis, and both upregulation and targeted inhibition may offer potential interventions depending on the specific context and tissue.
NAMPT inhibitors are primarily explored for their anti-cancer properties, as they reduce NAD+ levels and impair energy metabolism and survival of cancer cells214. Inhibitors like FK866 and CHS828, which are based on heterocyclic structures and act by binding to the active site of NAMPT215,216, are being investigated for their potential to selectively target cancer cells by exploiting their high NAD+ turnover214. Recent studies suggest that combining NAMPT inhibitors with other therapies could enhance their efficacy and reduce resistance in cancer treatment, as shown in various preclinical animal models217,218. Notably, FK866 has also been shown to have potential for brain health; low-dose NAMPT inhibition with FK866 can alleviate age-related cognitive impairment by enhancing autophagy and reducing neuroinflammation219. Another study has highlighted FK866’s potential in treating neurodegenerative diseases by modulating NAD+ metabolism and reducing oxidative stress to support neuronal health220. FK866 was again administered at a low dose (0.5 mg/Kg) to aged mouse models over four weeks, leading to significant improvements in cognitive function and locomotor activity. More research is needed to fully understand the implications of NAMPT inhibition and the use of FK866 for potential therapeutic benefits related to aging.
Conversely to the use of inhibitors, due to the interest in promoting maintenance of NAD+ levels in aging and age-associated conditions, the development of NAMPT activators has been the emphasis of recent investigation. Several studies have optimized urea-containing compounds such as SBI-797812 and 1-(2-phenyl-1,3-benzoxazol-6-yl)-3-(pyridin-4-methyl)urea (DS68702229) as potent NAMPT activators, which have been shown to increase NAD+ levels in vitro and in vivo, with potential applications in treating metabolic disorders, cardiovascular diseases, and neurodegeneration preclinically221,222.
Structure-activity relationship studies have led to the discovery of non-pyridyl class NAMPT activators, which suggest a through-water interaction mechanism within the NAMPT active site223. These activators have shown neuroprotective efficacy in animal models of chemotherapy-induced peripheral neuropathy (CIPN). Additionally, other types of NAMPT activators, such as NAT-5r and SBI-797812, have demonstrated effectiveness in a mouse model for CIPN, which may indicate their therapeutic promise in cognitive disorders224 (Fig. 4). While NAMPT activators show promise in preclinical studies, they are yet to enter clinical trials.
NAPRT modulators
Apart from the targets discussed above, one of the less explored yet crucial enzymes in NAD+ biosynthesis is NAPRT, the rate-limiting enzyme of the Preiss–Handler pathway. By maintaining NAD+ levels, NAPRT helps protect cells from oxidative stress, which is linked to aging and various diseases225,226. Selective inhibition of NAPRT can modulate NAD+ levels, providing therapeutic benefits in cancer treatment and neuroprotection227,228. 2-hydroxynicotinic acid (2-OHNA) inhibits NAPRT by binding to its active site, preventing the conversion of NA to NAD+; this depletion of NAD+ impairs cancer cell metabolism and may lead to cell death229 (Fig. 4). In particular, combining 2-OHNA with other NAD+ biosynthesis inhibitors like FK866 effectively kills NAPRT-proficient cancer cells resistant to single-agent treatments217.
Recent studies have reported the development of new chemical scaffolds as NAPRT inhibitors, demonstrating comparable anti-cancer activity to 2-OHNA with improved solubility and favorable drug-like profiles. These inhibitors target NAPRT-proficient cancer cells, particularly those resistant to FK866217. Another study identified potential inhibitors through in silico screening, discovering several chemotypes that inhibited NAPRT at micromolar concentrations; the combination of these new inhibitors with FK866 significantly reduced NAD+ levels and cell viability in ovarian cancer cells230.
As for NAMPT, in addition to inhibitors, there is ongoing development of potential NAPRT activators. Recently, a structure-based screening has led to the discovery of the first NAPRT activators based on 1,2-dimethylbenzimidazole scaffolds231, but evidence for these compounds in boosting NAD+ metabolism is still lacking. Further studies will be needed to investigate the physiological relevance of inhibiting or activating NAPRT in non-cancer chronic conditions, such as aging and age-associated diseases.
Sterile alpha and toll-interleukin receptor-containing motif (SARM1) inhibitors
Building on the therapeutic potential of modulating NAD+ pathways, attention has also turned to SARM1, a pivotal enzyme in axon degeneration, functioning through its NAD+ hydrolase activity. Upon nerve injury, SARM1 undergoes a conformational change that activates its NADase activity, leading to the rapid depletion of NAD+232. A recent screening-based study has identified several promising SARM1 inhibitors, including non-competitive inhibitors such as berberine chloride and zinc chloride233, suggesting the presence of an allosteric binding pocket that can be targeted for therapeutic development. A subsequent study identified the compound N-(4-(2-(4-(trifluoromethyl)phenyl)thiazol-4-yl)phenyl)acetamide as a notable SARM1 inhibitor, which demonstrated significant efficacy in a mouse model of CIPN (Fig. 4). This compound not only inhibited SARM1’s NADase activity effectively but also exhibited a favorable safety profile, making it a strong candidate for further development233. Hence, by targeting the enzymatic activity of SARM1, inhibitors may effectively prevent the depletion of NAD+, thereby maintaining cellular energy homeostasis and enhancing neuronal resilience, potentially expanding the treatment landscape for various neurological conditions.
α-Amino-β-carboxymuconate-ε-semialdehyde decarboxylase (ACMSD) inhibitors
ACMSD plays a crucial role in the kynurenine pathway, serving as a branching point between NAD+ and acetyl-CoA production234. ACMSD catalyzes the decarboxylation of aminocarboxymuconate-semialdehyde (ACMS) into aminomuconic semialdehyde, which can then enter the citric acid cycle as acetyl-CoA, contributing to cellular respiration. Alternatively, in the absence of ACMSD activity, ACMS can spontaneously decay into quinolinic acid, a biosynthetic precursor to NAD+ (see Fig. 1). ACMSD inhibition has been found to protect against acute kidney injury and NAFLD by increasing NAD+ levels in a tissue-specific manner. This modulation impacts a range of physiological processes, including neuroprotection, mitochondrial function, and oxidative stress defense. Inhibition of ACMSD with compounds TES-991 and TES-1025 has been shown to increase NAD+ levels in both cells and mice235 (Fig. 4). TES-1025, particularly, favors the flux from Trp through ACMS toward quinolinic acid, thereby enhancing NAD+ production236. The FDA-approved drug diflunisal can also inhibit ACMSD, providing a structural base from which derivatives can be designed237.
In addition to the approaches above, novel targets are being investigated to regulate the NAD+ pool. For instance, the effectiveness of precursor administration on NAD+ levels could be improved by PNP inhibition25. PNP, an enzyme that is part of the nucleotide metabolism via degradation of deoxyguanosine, is involved in NR metabolism in mammalian cells by metabolizing NR to NAM. When PNP was inhibited pharmacologically by the compound immucillin H (ImmH), intracellular cleavage of NR to NAM was prevented, and NAD+ synthesis was potentiated from NR both in vitro and in vivo25. By inhibiting PNP, ImmH also leads to an accumulation of deoxyguanosine triphosphate, which can induce apoptosis in certain cell types, such as T cells; hence, this compound has been initially studied as a drug therapy against certain types of cancers, such as T-cell leukemia and lymphoma238,239. The new findings for ImmH in the context of NAD+ synthesis potentially provide another strategy for indirect NAD+ upregulation that requires further investigation.
Preclinical and clinical applications of NAD+-modulating interventions for mitochondrial homeostasis and health benefits
NAD+ levels and mitochondrial homeostasis have been linked to symptoms of aging as well as other pathologies, including cancer, progressive muscle diseases, and neurodegenerative diseases. This recognition encouraged preclinical studies aiming to investigate both mechanistic aspects and intervention strategies to characterize and target this biological axis in conditions affecting muscle, brain, the immune system, and metabolic diseases. Findings from these investigations, particularly those involving natural NAD+ precursors, have reached the first clinical testing; this has contributed to acquiring a better physiological understanding of their metabolism in humans, while also highlighting the challenges and limitations of the current trials. This chapter will focus on NAD+-modulating interventions that have been more frequently indicated to exert health benefits through regulation of mitochondrial homeostasis; an overview of preclinical and clinical studies performed can be found in Table 1 and Table 2, respectively.
Preclinical studies
Age-related declining NAD+ levels in aging have been observed in rodents and different human tissues, such as muscle, liver, and brain58,240,241. Similarly, NAD+ levels appear reduced in preclinical models of age-associated diseases, including ataxia-telangiectasia (A-T), mitochondrial myopathies, and Alzheimer’s disease (AD), and disease-matched clinical samples114,242,243,244,245,246. Interestingly, interventions that boost NAD+ levels resulted in extended lifespan in worms and mice108,109,111,247, suggesting a therapeutic potential for raising NAD+ in various pathological conditions or age-associated diseases. A recurring theme across these studies is that mitochondrial dysfunction parallels NAD+ metabolism alterations, contributing to the detrimental phenotypes in different tissues or at the organism level.
Muscle disorders
For instance, multiple studies have observed alterations at the molecular level in NAD+ metabolism and mitochondrial homeostasis in skeletal muscle, associated with muscle loss and functional decline. These conditions include aging and age-associated chronic muscle conditions such as cachexia248, sarcopenia124,249,250, inclusion body myositis (IBM)114,251,252, and neuromuscular diseases like Duchenne’s muscular dystrophy (DMD)253,254.
In the case of aging, it is known that over time, mitochondrial dysfunction occurs in parallel with NAD+ depletion in muscle tissues, and often this depletion has been linked to muscle wasting or loss of muscle function240,241. Interestingly, treating mice with the salvage precursor NR resulted in NAD+ repletion in muscle stem cells (MuSCs) of young and aged mice108. This was mirrored by reduced MuSC senescence and increased mitochondrial respiration and membrane potential in MuSCs, paralleled by improved fitness and exercise performance. Additionally, Nampt knockout mice models demonstrated that NAD+ depletion in muscle leads to progressive loss of muscle function, while NR treatment restored mitochondrial function and induced muscle remodeling and restoration255. Another effect of aging is capillary loss, accompanied by a decline in SIRT1 activity and NAD+ levels; administering the precursor NMN to aged mice in combination with exercise training restored the ability to generate new blood vessels256. While NMN increased oxygen consumption and endurance, it is important to note that NMN did not affect the activity of mitochondrial Oxphos complexes or key enzymes of the TCA cycle256.
Next to NAD+ precursors, modulation of NAD+-consuming enzymes showed promising results. Chronic treatment with PARP inhibitors increased NAD+ levels and mitochondrial respiration in 10-week-old mice and triggered UPRmt and increased mitochondrial respiration and lifespan in worms109,257. Similarly, inhibition of CD38 in aged mice boosted NAD+ levels, improving exercise capacity, but in this case, no significant changes in mitochondrial biogenesis-related genes were observed138.
Cancer cachexia (CC) is a muscle condition affecting patients undergoing anti-cancer treatment. CC is characterized by weight loss, primarily from muscle and body fat, resulting in muscle wasting258. Specifically, 80% of gastric or pancreatic cancer patients and 50% of lung cancer patients develop CC258. Recent studies have observed reduced NAD+ levels and mitochondrial Oxphos protein levels in skeletal muscle cells of CC mice models248,258. Preventative NR supplementation prior to induced cancer cachexia, in mice, increased NAMPT and NAD+ levels; they only tested mitofusin-2 levels, an indicator for mitochondrial activity, which was unaltered259. Moreover, treatment of CC mice with NA rescued NAD+ levels in skeletal muscle, improved muscle function, and increased Oxphos complexes expression, yet NA treatment had no effect on mitochondrial respiration248. Another strategy involves the use of PARP inhibitor BGP-15, which might protect against CC, to enhance mitochondrial homeostasis260,261. Indeed, preventive injections with BGP-15 during chemotherapy-induced CC in healthy mice showed improved skeletal muscle mitochondrial viability and protection against ROS generation260,261.
Sarcopenia is a progressive skeletal muscle disorder characterized by a decline in muscle mass, strength, and gait speed124,249,250. While a study found declined levels of NAD+ and NAMPT levels and mitochondrial function in human sarcopenic muscle249, another study demonstrated no notable difference in NAD+ or NAMPT levels in sarcopenic muscle of human patients262. Moreover, this study reported that NR supplementation did not impact NAD+ levels in the muscle of Nampt knockout mice; however, at 4 weeks of age, these mice demonstrated increased ATP levels in their skeletal muscle262. While no further (pre)clinical interventions with NAD+ modulators have been reported for this condition263,264, a Singaporean cohort study showed lower concentrations of circulating trigonelline, the N-methylated form of NA, in sarcopenic subjects124. Importantly, systemic trigonelline correlated positively with muscle health clinical readouts in the same cohort and with muscle strength in an Iranian cohort124. Treating primary muscle cells, C. elegans, and aged mice with trigonelline resulted in increased NAD+ levels and enhanced mitochondrial function through the Preiss–Handler pathway, and improved muscle aging phenotypes124. Thus, trigonelline might be an interesting therapeutic strategy as an NAD+ modulator to explore in sarcopenia and possibly other chronic conditions.
Another class of muscle diseases characterized by mitochondrial dysfunction is IBM, which primarily affects older individuals. IBM is characterized by primary pathological hallmarks, including protein amyloid aggregates in muscle fibers, inflammation, and loss of skeletal muscle114,251. While specific therapies for IBM are still lacking, NAD+ boosters have shown promising results in various models, including nematodes with amyloid aggregation, aged rodents, and human muscle cells from IBM patients. Treatment with either NR or the PARP inhibitor Olaparib resulted in improvements at the mitochondrial and proteostasis level; in fact, these treatments promoted UPRmt and mitophagy, restoring mitochondrial and organismal function, and even stimulated the reduction of amyloid deposits in these models114.
Duchenne muscle dystrophy (DMD) is a genetic muscle disease manifesting clinically during childhood. It is characterized by frameshifting or nonsense mutations in the dystrophin protein gene, which is essential for maintaining the membrane integrity of muscle fibers265. These mutations result in progressive muscle wasting and weakness253,254,265. Research on skeletal muscle biopsies from DMD patients has revealed decreased NAD+ levels, increased PARP activity253 and dysfunction of mitochondrial metabolism254,266. To improve mitochondrial metabolism, studies explored boosting NAD+ levels by administering NR to mdx mice, a DMD mouse model254. Treated mice showed decreased susceptibility to muscle damage, improved muscle strength, reduced PARP activity, increased NAD+ levels, and elevated mitochondrial function254, thus suggesting a potential therapeutic benefit of NR supplementation in DMD. However, in another study reporting administration of NR or the CD38 inhibitor GSK987A to mdx mice for 20 weeks267, treated mice with the CD38 inhibitor showed increased NAD+ levels in muscle tissue, but no improvement was found in muscle function after treatment with either the inhibitor or with NR. More recently, another study explored NAM supplementation in a golden retriever muscular dystrophy model, demonstrating increased NAD+ levels in skeletal muscles and improved skeletal muscle function266. These partially conflicting results highlight the challenge of identifying effective DMD amelioration strategies and underline the importance of further research in NAD+ boosting interventions for this condition.
Neurodegenerative diseases
Similar to muscle diseases, neurodegenerative conditions present altered mitochondrial homeostasis and NAD+ metabolism, which have been thoroughly investigated as targets for NAD+ modulation-based approaches13,268,269. Alzheimer’s disease (AD), the most common form of dementia worldwide270,271, is characterized at the molecular level by the accumulation of Aβ plaques, tau neurofibrillary tangles (NFTs), declined NAD+ levels, mitochondrial stress, and impaired mitophagy, occurring first in the medial temporal lobe and eventually spreading to temporal and parietal cortices, and cortex112,242,243,270,271,272. Preclinical studies found that treating neuronal, nematode, or rodent AD models with NR resulted in induction of a mitochondrial stress response, including UPRmt and mitophagy, reduced proteotoxic stress, and decreased Aβ accumulation243. Additionally, NR treatment led to an improvement in the cognitive functions of mice242,243. Alternatively, studies using tau-driven AD models of worms and mice explored the effects of NMN treatment. NMN induced mitophagy, which in turn contributed to reducing Aβ and tau aggregates and deposits112,273; additionally, NMN treatment led to reduced mitochondrial ROS production and enhanced oxygen consumption rate112.
Another AD study, focused on targeting CD38, showed that CD38 knockdown in APPSwe mutation-expressing transgenic mice enhanced NAD+ levels and reduced Aβ plaques in brain extracts, mirrored by some improvement of cognitive performance270,274. Additionally, inhibition of CD38 in the AD mouse model restored NAD+ levels, decreased mitochondrial ROS production and plaque depositions in the hippocampus and cortex, and improved cognitive abilities158. Another strategy is utilizing PARP inhibitors, which resulted in rescuing NAD+ levels and decreasing Aβ aggregation in brain samples of AD flies, however, this study did not include any measurements of mitochondrial function275. Given the beneficial effects of several precursors and inhibitors summarized above, these strategies may be promising for finding a therapeutic strategy for AD, either as lead molecules or in combination approaches with other classes of molecules or drugs, with several studies having been launched at the clinical level.
Like AD, altered NAD+ and mitochondrial metabolism are common features of Parkinson’s disease (PD)270,276. In humans, lower levels of the de novo NAD+ synthesis precursor Trp have been observed in blood samples of PD patients, indicating disruption of the kynurenine pathway276,277. Preclinical studies in fly models of PD suggest beneficial effects from NAD+ modulators. Treatment of Drosophila flies with kynurenine pathway inhibitors and the NAD+ precursors NR or NAM increased the lifespan of these animals, suggesting an advantage in combining different therapeutic strategies; however, this study did not assess any mitochondrial function277. Treatment with only NR in fly models of PD increased NAD+ levels, markers of UPRmt, mitophagy, and improved climbing function278. Moreover, recent evidence indicates that mitochondrial complex I deficiency, linked to increased oxidative stress, promotes PD279,280. Therefore, to enhance mitochondrial complex I activity, NAD+ levels were boosted in fly models of PD with NAM, leading to reduced oxidative stress, including lower ROS levels and DNA damage, as well as improved climbing ability281. Another characteristic of PD is oxidative DNA damage, which triggers PARP activation, leading to the depletion of NAD+ levels40,279,282,283,284. Studies in fly models of PD treated with NAM, NAD+, or through PARP mutations demonstrated increased NAD+ levels, which also rescued mitochondrial function283,284,285. Moreover, mice models of PD were treated with 20 µg of NAD+ injection once, before injection with 6-hydroxydopamine to induce PD. Four weeks after the NAD+ injection, the treated mice showed reduced loss of neurons286. On the other hand, another study found that 3-month treatment with NR in a PD mouse model resulted in increased NMN and NAD+ metabolism marker NAAD but did not increase NAD+ levels287. Behavioral analysis revealed initial improvement; however, a progressive decline was observed after 10 weeks. Additionally, NR-treated mice exhibited a greater loss of dopamine neurons, which are associated with the clinical motor symptoms of PD287. These results highlight the need for further studies focusing on the long-term effects of NAD+ modulators in ameliorating PD phenotypes.
Another neurodegeneration condition is ataxia-telangiectasia (A-T), a disorder caused by mutations in the ataxia-telangiectasia-mutated (ATM) kinase gene. This mutation leads to detrimental phenotypes such as premature aging, senescence, immune deficiency, and neurodegeneration111,288. ATM-deficient mice, worms, and cell models demonstrated accumulation of cytoplasmic DNA, hyperactivation of PARP1, and low NAD+ levels111,288. To restore NAD+ levels, ATM-deficient models were treated with NR, which stimulated PINK1-dependent mitophagy, reduced cytoplasmic DNA levels, and activated the NAD+-dependent SIRT1 signaling pathway, which in turn further promoted mitochondrial mitophagy and healthy morphology111,288. Additionally, NR treatment led to improved health and lifespan in the same animal models111,288. Another strategy involved the use of SIRT activator SRT1720 and the PARP inhibitor Olaparib, which enhanced SIRT1 activity and increased NAD+111. Overall, increasing NAD+ levels stimulated mitochondrial function, promoting mitophagy and preventing cellular senescence.
Axon degeneration is a common feature observed in multiple neurodegenerative diseases, including traumatic brain injury and AD. The NAD+-consuming enzyme SARM1 promotes programmed axon degeneration, also known as Wallerian degeneration53,54,289. In healthy axons, SARM1 is inactivated; however, in the case of injury or disease, loss of NMNAT activity leads to NAD+ depletion and NMN accumulation, which in turn activates SARM153,54,289. Interestingly, while NMN is known to be beneficial for aging-associated declines, this data suggests that NMN might have a neurotoxic effect in cases of axonal degeneration54. Another study demonstrated that NAMPT inhibition combined with NAR supplementation resulted in reduced NMN levels and SARM activity, and delayed axon degeneration290. Moreover, Cd38 knockout mice treated with NR demonstrated delayed axon degeneration in the brain stem291. These studies suggest that strategies combining NAD+ boosting with inhibitors of NAD+-consuming enzymes could be a promising approach to mitigate axon degeneration.
Metabolic diseases
In addition to muscle and neurodegenerative conditions, alterations in NAD+ levels and mitochondrial homeostasis are also observed in aging-associated metabolic diseases, particularly those conditions impacting highly mitochondrially-active tissues such as the kidney and liver292,293,294. Aging and chronic ailments like diabetes decrease the ability of the kidneys to recover from injuries, resulting in chronic kidney disease (CKD). Several studies suggest that the renal proximal tubule (PT) plays a critical role in CKD. The PT is rich in mitochondria since it needs ATP production for reabsorption and transportation against gradients292,293,294. This high energy demand makes the kidneys vulnerable to stress, and accumulating data suggests that changes in NAD+ metabolism are linked to CKD progression292,295,296. In preclinical studies, healthy mice, CKD rats, and diabetic mice treated with NA, NAM, or NMN showed increased NAD+ levels in the kidney297,298,299. In another study, rats with diabetic kidney disease (DKD) were treated with a CD38 inhibitor, resulting in increased renal NAD+ levels, which in turn decreased mitochondrial oxidative stress144.
Similarly, non-alcoholic fatty liver disease (NAFLD) is characterized by increased liver fat and is commonly associated with obesity or diabetes patients300. Studies have indicated that, in human NAFLD patients, increased PARP activity could potentially lead to NAD+ depletion and mitochondrial dysfunction164. Therefore, to test this theory, NAFLD mice were treated with PARP inhibitors or NR. This treatment restored NAD+ levels, improving mitochondrial function164.
Another therapeutic strategy for kidney or liver diseases is increasing NAD+ production by inhibiting ACMSD in the de novo NAD+ synthesis pathway235,301. In studies with mouse models of NAFLD and acute kidney injury (AKI), treatment with ACMSD inhibitors increased the transcription of mitochondrial and β-oxidation genes, as well as restoring NAD+ levels in the kidney and liver235. Recently, ACMSD inhibitors were investigated in the human liver organoid model of metabolic dysfunction-associated steatohepatitis (MASH), an advanced form of liver fibrosis characterized by mitochondrial dysfunction. ACMSD inhibition increased NAD+ levels and improved mitochondrial homeostasis302. These studies indicate the potential of utilizing the de novo pathway, a less extensively studied pathway, as a therapeutic strategy for metabolic diseases
Barth syndrome is a rare disease caused by mutations in the tafazzin gene encoding for cardiolipin (CL), a phospholipid specific for the mitochondria303. Defective CL leads to mitochondrial dysfunction, which is associated with disrupted NAD+ metabolism304,305. Preclinical studies in flies demonstrated that supplementation with NR or NMN improved mitochondrial respiration and membrane potential, and exercise endurance was improved304,305.
Next to pharmacological or NAD+ precursor modulators, it is possible to raise NAD+ levels through exercise302,306,307, however, not all preclinical studies investigating exercise have linked increased NAD+ levels to mitochondrial function. In young and aged rats, treadmill exercise for a duration of 6 weeks led to increased SIRT1 activity, NAMPT and NAD+ levels, and decreased mitochondrial generation of ROS302. In another study, a mouse model of Huntington’s disease (HD) underwent 12 weeks of treadmill exercise, 3 times a day. It was found that exercise increased NAD+ levels, enhancing mitochondrial complex activities in the brain and resulting in improved motor performance307. However, another study using rats showed that protein levels of NAMPT, COX1, and PGC-1a were unchanged in skeletal muscles after 4 weeks of treadmill exercise, and NAD+ levels or other mitochondrial readouts were not included308. Similarly, a study based on swimming exercise for a total of four hours showed an increase in NAD+ levels, yet also in this case, no measurements of mitochondrial function were performed309, reinforcing the need for more exercise-based investigations to further define how NAD+ and mitochondrial homeostasis are perturbed and connected during exercise.
Overall, preclinical studies have shown that NAD+ and mitochondrial metabolism play a crucial and intertwined role in aging and age-associated diseases, as well as being possible therapeutic targets for ameliorating these conditions. Nevertheless, not all NAD+ modulation strategies (as described in the “Interventions to modulate NAD+ levels” section) have been studied for their possible effects on mitochondrial health yet; additionally, more research is required to investigate differences between NAD+ modulators within different tissues, different models or pathologies, administration or exercise regimens, and how to successfully translate these findings to humans, while several clinical trials have already been conducted.
Clinical studies
As summarized above, a multitude of preclinical studies have investigated the therapeutic effects of stimulating NAD+ metabolism to ameliorate symptoms of aging and various pathologies, yielding promising results. These findings have stimulated clinical development attempts, particularly for NAD+ modulators deemed suitable for human trials in terms of safety and upscaling, such as natural precursors. However, the extent of clinical testing differs between precursors. For instance, a review we conducted at this time of ClinicalTrials.gov.org using NAD+ as “other term” input and the following precursors as intervention reveals that there are 4 clinical trials investigating NA, 7 trials focused on NAM, 8 trials administering NAD+, 16 trials studying NMN, and over 60 trials exploring NR with NAD+ measurements included as readout. When reviewing the database with NAD+ precursors as interventions yet with the “other term” input using “aging”, it indicates that there are currently no ongoing or published clinical trials for NA, 2 trials exploring NAM, 4 trials examining NAD+, 5 trials investigating NMN, and 18 trials focused on NR in elderly cohorts.
One key factor in evaluating clinical translation is determining the optimal dose and safety of that specific therapy. Given the current clinical landscape, the precursor NR is still the most utilized NAD+ precursor and regulator overall. For instance, it has been demonstrated that NR supplementation is safe up to 3000 mg daily, with readouts reported for NAD+ levels, mitochondrial function, and clinical phenotypes measured in several trials246,310,311,312,313.
One clinical study investigated the effects of NR supplementation on skeletal muscle in 12 healthy elderlies314. They demonstrated that a three-week supplementation of 1000 mg NR per day increased muscle NAD+ metabolome, specifically NAAD and products of NAM clearance pathways, yet NAD+ levels were not altered. Additionally, mitochondrial energy metabolism and inflammatory cytokines pathways were unaffected or downregulated, and no changes in functional readouts like hand grip strength were observed.
In another study, six weeks of 1000 mg NR supplementation per day for CKD patients improved mitochondrial metabolism, yet no effects were observed in the physical endurance of skeletal muscle315.
On the other hand, another study found no effect of 2000 mg NR per day supplementation in obese or overweight men on NAD+ metabolite or mitochondria in skeletal muscle, although muscle NAMPT levels were decreased316.
Another clinical trial utilized NR to help ameliorate heart failure (HF), which is also characterized by a decline in NAD+ levels in whole blood317,318. Therefore, 30 patients diagnosed with HF were given 1000 mg NR twice a day for 12 weeks, which increased whole blood NAD+ levels and corresponded with higher peripheral blood mononuclear cells (PMBCs) mitochondrial respiration and reduced ROS production318. Although NR has shown promising results in this context, further studies are necessary to establish the efficacy and safety of NR in HF patients.
NR supplementation has also been investigated in relation to neurodegenerative diseases. In the NADPARK trial319, 15 PD patients were treated with 1000 mg NR per day for 30 days. NR treatment increased cerebral NAD+ levels and NAD+ metabolome in CSF, skeletal muscle, and PBMCs. Additionally, in muscle tissue, mitochondrial translation and respiratory genes were also induced, and moreover, patients demonstrated a mild clinical improvement. Another study determined that 3000 mg NR supplementation per day in 20 PD patients without moderate or severe side effects for four weeks is well tolerated313. NR supplementation increased NAD+ levels and improved clinical symptoms, suggesting NR’s potential as a safe therapeutic strategy for PD patients. However, more research should focus on the long-term effects of NR or other NAD+ precursors in neurodegenerative patients. Another study using NR supplementation showed that doses up to 1000 mg per day for 10 weeks were well tolerated in patients affected by mild cognitive impairments (MCI)246. Additionally, blood NAD+ levels were significantly increased; however, NR supplementation did not alter cognitive functions.
The longest clinical trial for NR, involving 10 A-T patients, assessed the effects of up to 500 mg per day of NR supplementation over two years. To investigate whether NR supplementation affected neurological symptoms for A-T patients, whole blood NAD+ concentrations on seven different time points over two years were measured320. The study found increased NAD+ levels in blood and, more importantly, no notable side effects were observed, suggesting that a chronic long-term NR treatment is safe. Additionally, NR treatment enhanced neuromotor coordination and improved eye coordination. Further research should assess a larger cohort size to confirm that A-T disease progression is attenuated upon NR supplementation.
Recent trials have explored other precursors beyond NR. To date, the most promising clinical trial focuses on the progressive muscle disease mitochondrial myopathy244. In this trial, patients received NA starting with 250 mg–750 or 1000 mg per day over a course of four months, with the last follow-up 10 months later. NA supplementation increased NAD+ levels and metabolites in muscle, inducing mitochondrial biogenesis and Oxphos activity, and improved shoulder and knee extension strength. However, the administered NA dose was based on efficiency in hypercholesterolemia studies, therefore, an optimal NA dose for mitochondrial myopathy has to be determined, especially considering that another study found an association between metabolites of excess NA, 2PY, and 4PY, and cardiovascular disease (CVD)245. Investigating two different cohorts (combined n = 3163), it was validated that individuals with high 2PY or 4PY levels have an increased risk of major adverse cardiovascular events. Therefore, it is recommended to focus on a titrated approach to find the optimum NA supplementation without promoting excessive NAD+ catabolism metabolite generation. Furthermore, in a study of NA supplementation with a dose of 207.5 mg NA per day for 32 days to older adults with impaired physical function, no improvements in NAD+ or mitochondrial functions in skeletal muscle were observed321.
NMN is well tolerated in healthy participants who received 300, 600, or 900 mg per day for a total of 60 days322,323, with all NMN doses increasing blood NAD+ levels, and no adverse events were observed. Additionally, a 10-week study in overweight or obese post-menopausal women with prediabetes found that 250 mg NMN per day partially increased muscle insulin signaling and sensitivity, but no change in mitochondrial oxidative capacity was found324. Another study with NMN indicated that NMN supplementation of 250 mg per day in aged men was well tolerated, leading to increased NAD+ levels in whole blood and improved muscle performance; however, this study did not provide mitochondrial function measurements325.
Alternative to supplementation of precursors, as described in the “NAD+ and precursors supplementation” section, it is possible to administer NAD+ itself through intravenous infusions66,116,117. These initial studies reported increased NAD+ and NAM levels in plasma, and in urine, the excreted NAD+ levels increased after 6 h66, but it must be noted that these trials utilized small cohorts and short testing periods, and only a minor trend for cognitive improvement was observed117. Furthermore, evidence for changes in the level of blood or tissue mitochondrial function or metabolic readouts is currently lacking for these studies.
As for preclinical studies, clinical trials are investigating the effects exercise has on NAD+ metabolism. Two studies showed that NAMPT protein expression is enhanced in skeletal muscle following exercise326,327; additionally, acute exercise lasting 1 h increased NAD+, NAMPT, and ATP levels in PBMCs while decreasing serum NAM levels, suggesting an increase in NAD+ salvage pathway326. Similarly, 12 weeks of aerobic and resistance training increased NAMPT levels in skeletal muscle; however, this study did not assess NAD+ levels or mitochondrial function328. In another trial, full-body resistance training for 10 weeks also led to increased NAD+, NADH levels, and SIRT activity in muscle329. This study also reported elevated citrate synthase activity, which was speculated to reflect enhanced mitochondrial biogenesis. Another trial linked NAD+ metabolism to mitochondrial function by demonstrating that athletes exhibited higher NAMPT levels in skeletal muscle and increased expression of the mitochondrial biogenesis marker PCG-1a. Notably, NAD+ levels were not determined in this study330. NAD+ levels were measured in another clinical trial investigating the difference between different groups of the elderly, based on their amount of exercise activity, and a younger cohort331; older athletic participants who had at least 3 h of exercise per week lasting one year, exhibited similar NAD+ levels as healthy young participants. Moreover, muscle biopsies from older athletic participants showed improved mitochondrial respiration compared to age-matched and physically impaired participants331.
Altogether, initial reports of NAD+ precursors and exercise in clinical trials, while encouraging in certain instances, are still too inconclusive to directly link an increase in NAD+ levels and mitochondrial metabolism to established health benefits in humans. Moreover, some NAD+ precursors have shown side effects, such as the use of NA at certain doses that might be limited due to skin flushing or induction of insulin resistance332,333. Therefore, further investigation is required to determine optimal safe and efficacious doses of the currently clinically available NAD+ precursors. Nevertheless, all these clinical trials underline the potential of utilizing NAD+ modulators or identifying possible exercise regimens to optimize NAD+ metabolism in a wide range of age-associated diseases, including cardiovascular conditions.
Discussion
This review covers the importance of maintaining NAD+ levels for cellular homeostasis and mitochondrial maintenance, as well as evaluating established and novel NAD+ boosting strategies from simple supplementation of precursors to modulation of enzymes by drugs. However, when it comes to clinically applying these strategies, the task of simply boosting NAD+ comes with challenges. One of which is the stability and bioavailability of orally administered NAD+ precursors, which induce slow and suboptimal NAD+ increases39,69. Intravenous administration would overcome these problems, as it allows instant circulation of the precursors rather than initial metabolism by the gut microbiota and liver. However, exposure of compounds like NR and NMN to serum results in their low stability and degradation in the blood68,124. The reduced precursors NRH and NMNH show promise in overcoming this, although preclinical evidence for health benefits is still limited, and whether their efficacy on functional readouts would be superior to the classic counterparts NR and NMN is also unclear. Of note, NMNH has been recently declared self-affirmed GRAS, which should accelerate its clinical development. Intravenous administration of direct NAD+ has been carried out in humans66,116,117, but the human trials were conducted in extremely small cohorts; furthermore, exogenous NAD+ impacts processes such as DNA replication118. Sublingual NAD+ for absorption into circulation is another new commercially proposed method; however, the evidence for efficacy on NAD+ or health benefits is still largely lacking, raising the need to properly assess different delivery methodologies. Another issue with clinical translation is patient genetic variation in the expression of NAD+ synthesis genes, or lifestyle conditions changes, including exercise regimens, that would influence the effectiveness of interventions. For example, NAMPT expression changes during diseases such as cancer, diabetes, and fatty liver disease211, and eNAMPT levels decrease with age328. In liver cancer, NAPRT expression reflects the ability of the organ to synthesize NAD+334, thus highlighting the importance of tissue gene expression to be considered when targeting a particular organ.
The vast number of preclinical studies on NAD+ modulation via precursors or modulators of NADase enzymes has established a tight functional relationship between NAD+ metabolism and mitochondrial homeostasis. Furthermore, it has emerged that supporting or inducing mitochondrial bioenergetics, stress responses, mitophagy, and other adaptation mechanisms is critical to mediate some of the health or longevity benefits observed in studies using NAD+ modulation strategies. Although this is consistent and mechanistically validated across different preclinical models, as reviewed in the “Preclinical and clinical applications of NAD+-modulating interventions for mitochondrial homeostasis and health benefits” section, the correspondence between NAD+ changes and mitochondrial or metabolic alterations is more complex in human studies. For instance, NAD+ levels and mitochondrial function have been shown to decline in sarcopenia muscle in one cohort249, while another study did not observe a difference in NAD+ levels in sarcopenic muscle, and even NR supplementation had no effect on NAD+ levels262. Instead, in mitochondrial myopathy patients, utilizing compounds like NA increased both muscle and blood NAD+ levels, muscle strength, and induced mitochondrial biogenesis and respiration244. Studies with NR supplementation in PD or HF patients have provided indications of impacting mitochondrial function in peripheral tissues, although evidence for impacting mitochondria in the primary target tissues was not reported318,319. Additionally, in the context of relatively healthy cohorts like aged or overweight individuals, or even in obese cohorts, NAD+ modulation by NR did not correspond to significant changes in mitochondrial homeostasis314,316,321,335,336. Only a trial by Lapatto et al.312 showed increased muscle mitochondrial biogenesis with NR supplementation, although this was not placebo-controlled. Finally, a large proportion of the other studies reviewed herein, including the ones focused on exercise, did not include or report on mitochondrial readouts following NAD+ modulation. Therefore, this highlights the need for further establishing and validating the crosstalk between NAD+ and the different features of mitochondrial function as a panel of possible molecular efficacy readouts in clinical settings.
In terms of new research avenues in the NAD+ field, it is also emerging that apart from its effects on the mitochondria, NAD+ plays a multitude of roles in regulating both metabolic and non-metabolic processes. One of which was discussed earlier, on NAD+/NMN levels regulating axon degeneration through SARM1. While the implications for SARM1 in regulating axonal fate are well-established, the sensing mechanisms of this protein complex toward NAD+, NMN, or even possibly toward other NAD+ metabolites are not fully understood, and may suggest that also the other NAD+ metabolites could regulate more biological processes than initially thought54. Another relatively novel role of NAD+ is involvement in forming non-canonical caps on the 5′ end of RNA that mark RNA for degradation in eukaryotes337,338,339. These structures are formed by RNA polymerase transcription initiation using the adenine moiety of NAD+ rather than ATP, resulting in NAD+-capping of the RNA molecule. NAD+ is also involved in metabolic switching, or the Warburg effect, in which the main source of energy production is anaerobic glycolysis through fermentation of pyruvate to lactic acid, as opposed to mitochondrial oxidative phosphorylation340. Studies indicate that cells will make a glycolytic switch when NAD+ demand is high and exceeds ATP demand, therefore skewing the NAD+ use for this pathway341. This metabolic switching is regulated by sirtuins, namely SIRT3 and SIRT2 repress the Warburg effect342,343,344,345, while SIRT5 maintains glycolysis346. NAD+ is also involved in nucleotide and amino acid synthesis, such as the folate-dependent serine catabolism347, as well as genomic DNA replication, as reviewed above118.
Collectively, these studies indicate that modulation of NAD+ levels affects additional critical cellular pathways other than mitochondrial homeostasis. These new discoveries, together with the in-depth preclinical characterization of NAD+ modulators and ongoing developments in their translation in humans, will open further avenues of study of this essential cofactor and its clinical therapeutic potential.
Data availability
No datasets were generated or analyzed during the current study.
References
Chini, C. C. S., Zeidler, J. D., Kashyap, S., Warner, G. & Chini, E. N. Evolving concepts in NAD+ metabolism. Cell Metab. 33, 1076–1087 (2021).
Judge, A. & Dodd, M. S. Metabolism. Essays Biochem. 64, 607–647 (2020).
Kalyanaraman, B. et al. A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biol. 14, 316–327 (2018).
Xie, N. et al. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 5, 227 (2020).
Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid. Redox Signal. 10, 179–206 (2008).
Dasovich, M. & Leung, A. K. L. PARPs and ADP-ribosylation: Deciphering the complexity with molecular tools. Mol. Cell 83, 1552–1572 (2023).
Zeidler, J. D. et al. The CD38 glycohydrolase and the NAD sink: implications for pathological conditions. Am. J. Physiol. Cell Physiol. 322, C521–C545 (2022).
Encyclopedia of Food and Health (Elsevier, 2016).
Brody, T. Nutritional Biochemistry (Academic Press, San Diego, 1999).
Fernández-Vizarra, E., Enríquez, J. A., Pérez-Martos, A., Montoya, J. & Fernández-Silva, P. Tissue-specific differences in mitochondrial activity and biogenesis. Mitochondrion 11, 207–213 (2011).
Mori, V. et al. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS ONE 9, e113939 (2014).
McLaughlin, K. L. et al. Novel approach to quantify mitochondrial content and intrinsic bioenergetic efficiency across organs. Sci. Rep. 10, 17599 (2020).
Sorrentino, V., Menzies, K. J. & Auwerx, J. Repairing mitochondrial dysfunction in disease. Annu. Rev. Pharmacol. Toxicol. 58, 353–389 (2018).
Stein, L. R. & Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 23, 420–428 (2012).
Pittelli, M. et al. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol. Pharm. 80, 1136–1146 (2011).
Fiers, W., Beyaert, R., Declercq, W. & Vandenabeele, P. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene 18, 7719–7730 (1999).
Juan, C. A., Pérez de la Lastra, J. M., Plou, F. J. & Pérez-Lebeña, E. The chemistry of reactive oxygen species (ROS) revisited: outlining their role in biological macromolecules (DNA, lipids and proteins) and induced pathologies. Int. J. Mol. Sci. 22, 4642 (2021).
Ma, K. et al. Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 8, 467 (2020).
Shpilka, T. & Haynes, C. M. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 19, 109–120 (2018).
Combs, G. F. The Vitamins: Fundamental Aspects in Nutrition and Health (Elsevier/AP, Amsterdam, 2012).
Hove-Jensen, B. et al. Phosphoribosyl diphosphate (PRPP): biosynthesis, enzymology, utilization, and metabolic significance. Microbiol Mol. Biol. Rev. 81, e00040–16 (2017).
Hara, N., Yamada, K., Shibata, T., Osago, H. & Tsuchiya, M. Nicotinamide phosphoribosyltransferase/visfatin does not catalyze nicotinamide mononucleotide formation in blood plasma. PLoS ONE 6, e22781 (2011).
Pinson, B., Ceschin, J., Saint-Marc, C. & Daignan-Fornier, B. Dual control of NAD+ synthesis by purine metabolites in yeast. eLife 8, e43808 (2019).
Tsui, M. et al. Purine nucleoside phosphorylase deficiency induces p53-mediated intrinsic apoptosis in human induced pluripotent stem cell-derived neurons. Sci. Rep. 12, 9084 (2022).
Kropotov, A. et al. Purine nucleoside phosphorylase controls nicotinamide riboside metabolism in mammalian cells. J. Biol. Chem. 298, 102615 (2022).
Bender, D. A. & Olufunwa, R. Utilization of tryptophan, nicotinamide and nicotinic acid as precursors for nicotinamide nucleotide synthesis in isolated rat liver cells. Br. J. Nutr. 59, 279–287 (1988).
Bieganowski, P. & Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117, 495–502 (2004).
Preiss, J. & Handler, P. Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates. J. Biol. Chem. 233, 488–492 (1958).
Preiss, J. & Handler, P. Biosynthesis of diphosphopyridine nucleotide. II. Enzymatic aspects. J. Biol. Chem. 233, 493–500 (1958).
Zhang, X. et al. Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J. Biol. Chem. 278, 13503–13511 (2003).
Efimov, I. et al. Heme-containing dioxygenases. In Advances in Inorganic Chemistry Vol. 64 33–51 (Elsevier, 2012).
Han, Q., Robinson, H. & Li, J. Biochemical identification and crystal structure of kynurenine formamidase from Drosophila melanogaster. Biochem. J. 446, 253–260 (2012).
Revollo, J. R., Grimm, A. A. & Imai, S. I. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 279, 50754–50763 (2004).
Lau, C., Niere, M. & Ziegler, M. The NMN/NaMN adenylyltransferase (NMNAT) protein family. Front. Biosci. Landmark Ed. 14, 410–431 (2009).
Zapata-Pérez, R. et al. Reduced nicotinamide mononucleotide is a new and potent NAD+ precursor in mammalian cells and mice. FASEB J. 35, e21456 (2021).
Yang, Y., Zhang, N., Zhang, G. & Sauve, A. A. NRH salvage and conversion to NAD+ requires NRH kinase activity by adenosine kinase. Nat. Metab. 2, 364–379 (2020).
Orlandi, I., Alberghina, L. & Vai, M. Nicotinamide, nicotinamide riboside and nicotinic acid-emerging roles in replicative and chronological aging in yeast. Biomolecules 10, 604 (2020).
Belenky, P., Christensen, K. C., Gazzaniga, F., Pletnev, A. A. & Brenner, C. Nicotinamide riboside and nicotinic acid riboside salvage in fungi and mammals. Quantitative basis for Urh1 and purine nucleoside phosphorylase function in NAD+ metabolism. J. Biol. Chem. 284, 158–164 (2009).
Liu, L. et al. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab. 27, 1067–1080.e5 (2018).
Katsyuba, E., Romani, M., Hofer, D. & Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2, 9–31 (2020).
Wagner, S., Manickam, R., Brotto, M. & Tipparaju, S. M. NAD+ centric mechanisms and molecular determinants of skeletal muscle disease and aging. Mol. Cell Biochem. 477, 1829–1848 (2022).
Sonntag, T. et al. Nicotinamide riboside kinases regulate skeletal muscle fiber-type specification and are rate-limiting for metabolic adaptations during regeneration. Front. Cell Dev. Biol. 10, 1049653 (2022).
Revollo, J. R. et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 6, 363–375 (2007).
Lundt, S. & Ding, S. NAD+ metabolism and diseases with motor dysfunction. Genes12, 1776 (2021).
Wen, F. et al. Drug discovery targeting nicotinamide phosphoribosyltransferase (NAMPT): updated progress and perspectives. Bioorg. Med. Chem. 99, 117595 (2024).
Yoshida, M. et al. Extracellular vesicle-contained eNAMPT delays aging and extends lifespan in mice. Cell Metab. 30, 329–342.e5 (2019).
Covarrubias, A. J., Perrone, R., Grozio, A. & Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 22, 119–141 (2021).
Nikiforov, A., Dölle, C., Niere, M. & Ziegler, M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem. 286, 21767–21778 (2011).
Canto, C. NAD+ precursors: a questionable redundancy. Metabolites 12, 630 (2022).
Camacho-Pereira, J. et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 23, 1127–1139 (2016).
Chini, E. N., Chini, C. C. S., Espindola Netto, J. M., de Oliveira, G. C. & van Schooten, W. The pharmacology of CD38/NADase: an emerging target in cancer and diseases of aging. Trends Pharmacol. Sci. 39, 424–436 (2018).
Shi, B. et al. Targeting CD38-dependent NAD+ metabolism to mitigate multiple organ fibrosis. iScience 24, 101902 (2021).
Hopkins, E. L., Gu, W., Kobe, B. & Coleman, M. P. A novel NAD signaling mechanism in axon degeneration and its relationship to innate immunity. Front. Mol. Biosci. 8, 703532 (2021).
Loreto, A., Antoniou, C., Merlini, E., Gilley, J. & Coleman, M. P. NMN: The NAD precursor at the intersection between axon degeneration and anti-ageing therapies. Neurosci. Res. 197, 18–24 (2023).
Bitterman, K. J., Anderson, R. M., Cohen, H. Y., Latorre-Esteves, M. & Sinclair, D. A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 277, 45099–45107 (2002).
Domínguez-Gómez, G. et al. Nicotinamide sensitizes human breast cancer cells to the cytotoxic effects of radiation and cisplatin. Oncol. Rep. 33, 721–728 (2015).
Roberti, A., Fernández, A. F. & Fraga, M. F. Nicotinamide N-methyltransferase: At the crossroads between cellular metabolism and epigenetic regulation. Mol. Metab. 45, 101165 (2021).
Migaud, M. E., Ziegler, M. & Baur, J. A. Regulation of and challenges in targeting NAD+ metabolism. Nat. Rev. Mol. Cell Biol. 25, 822–840 (2024).
Gazzaniga, F., Stebbins, R., Chang, S. Z., McPeek, M. A. & Brenner, C. Microbial NAD metabolism: lessons from comparative genomics. Microbiol. Mol. Biol. Rev. 73, 529–541 (2009). Table of Contents.
Magnúsdóttir, S., Ravcheev, D., de Crécy-Lagard, V. & Thiele, I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front. Genet. 6, 148 (2015).
Lozada-Fernández, V. V. et al. Nicotinamide riboside-conditioned microbiota deflects high-fat diet-induced weight gain in mice. mSystems 7, e0023021 (2022).
Yu, X. et al. Effect of nicotinamide riboside on lipid metabolism and gut microflora-bile acid axis in alcohol-exposed mice. Food Sci. Nutr. 9, 429–440 (2021).
Peluso, A. A. et al. Oral supplementation of nicotinamide riboside alters intestinal microbial composition in rats and mice, but not humans. NPJ Aging 9, 7 (2023).
Bruzzone, S., Guida, L., Zocchi, E., Franco, L. & De Flora, A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 15, 10–12 (2001).
Wei, W., Graeff, R. & Yue, J. Roles and mechanisms of the CD38/cyclic adenosine diphosphate ribose/Ca(2+) signaling pathway. World J. Biol. Chem. 5, 58–67 (2014).
Grant, R. et al. A pilot study investigating changes in the human plasma and urine NAD+ metabolome during a 6 h intravenous infusion of NAD. Front. Aging Neurosci. 11, 257 (2019).
Grozio, A. et al. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J. Biol. Chem. 288, 25938–25949 (2013).
Ratajczak, J. et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 7, 13103 (2016).
Sauve, A. A. et al. Triple-Isotope Tracing for Pathway Discernment of NMN-Induced NAD+ Biosynthesis in Whole Mice. Int. J. Mol. Sci. 24, 11114 (2023).
Grozio, A. et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 1, 47–57 (2019).
Kanai, Y. et al. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem. 273, 23629–23632 (1998).
Pillai, S. M. & Meredith, D. SLC36A4 (hPAT4) is a high affinity amino acid transporter when expressed in Xenopus laevis oocytes. J. Biol. Chem. 286, 2455–2460 (2011).
Chen, M. et al. SLC29A1 and SLC29A2 are human nicotinamide cell membrane transporters. Nat. Commun. 16, 1181 (2025).
Ohkubo, M., Ohta, K., Inoue, K. & Yuasa, H. Nicotinate uptake by two kinetically distinct Na÷-dependent carrier-mediated transport systems in the rat small intestine. Drug Metab. Pharmacokinet. 27, 255–262 (2012).
Bahn, A. et al. Identification of a new urate and high affinity nicotinate transporter, hOAT10 (SLC22A13). J. Biol. Chem. 283, 16332–16341 (2008).
Alano, C. C. et al. Differences among cell types in NAD(+) compartmentalization: a comparison of neurons, astrocytes, and cardiac myocytes. J. Neurosci. Res. 85, 3378–3385 (2007).
Cambronne, X. A. et al. Biosensor reveals multiple sources for mitochondrial NAD+. Science 352, 1474–1477 (2016).
Kory, N. et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci. Adv. 6, eabe5310 (2020).
Luongo, T. S. et al. SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature 588, 174–179 (2020).
Berger, F., Lau, C., Dahlmann, M. & Ziegler, M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 280, 36334–36341 (2005).
Yamamoto, M. et al. Nmnat3 is dispensable in mitochondrial NAD level maintenance in vivo. PLoS ONE 11, e0147037 (2016).
Zhu, Y., Liu, J., Park, J., Rai, P. & Zhai, R. G. Subcellular compartmentalization of NAD+ and its role in cancer: A sereNADe of metabolic melodies. Pharmacol. Ther. 200, 27–41 (2019).
Fletcher, R. S. et al. Nicotinamide riboside kinases display redundancy in mediating nicotinamide mononucleotide and nicotinamide riboside metabolism in skeletal muscle cells. Mol. Metab. 6, 819–832 (2017).
Svoboda, P. et al. Nuclear transport of nicotinamide phosphoribosyltransferase is cell cycle-dependent in mammalian cells, and its inhibition slows cell growth. J. Biol. Chem. 294, 8676–8689 (2019).
Chen, L. et al. Quantitative dynamics of intracellular NMN by genetically encoded biosensor. Biosens. Bioelectron. 267, 116842 (2025).
Michishita, E., Park, J. Y., Burneskis, J. M., Barrett, J. C. & Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 16, 4623–4635 (2005).
Nolfi-Donegan, D., Braganza, A. & Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 37, 101674 (2020).
Gorman, G. S. et al. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2, 16080 (2016).
Yusri, K., Kumar, S., Fong, S., Gruber, J. & Sorrentino, V. Towards healthy longevity: comprehensive insights from molecular targets and biomarkers to biological clocks. Int. J. Mol. Sci. 25, 6793 (2024).
Alano, C. C. et al. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 30, 2967–2978 (2010).
Yang, H. et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130, 1095–1107 (2007).
Titov, D. V. et al. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352, 231–235 (2016).
Liu, Y., Hu, L., Ma, T., Yang, J. & Ding, J. Insights into the inhibitory mechanisms of NADH on the αγ heterodimer of human NAD-dependent isocitrate dehydrogenase. Sci. Rep. 8, 3146 (2018).
Cuenoud, B. et al. Brain NAD is associated with ATP energy production and membrane phospholipid turnover in humans. Front. Aging Neurosci. 12, 609517 (2020).
Jang, S., Kang, H. T. & Hwang, E. S. Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 287, 19304–19314 (2012).
Yuan, X. et al. NAD+/NADH redox alterations reconfigure metabolism and rejuvenate senescent human mesenchymal stem cells in vitro. Commun. Biol. 3, 774 (2020).
Wang, H. et al. Nicotinamide mononucleotide supplementation improves mitochondrial dysfunction and rescues cellular senescence by NAD+/Sirt3 pathway in mesenchymal stem cells. Int. J. Mol. Sci. 23, 14739 (2022).
Meng, H. et al. SIRT3 regulation of mitochondrial quality control in neurodegenerative diseases. Front. Aging Neurosci. 11, 313 (2019).
Huang, L. et al. Effect of Sirt3 on retinal pigment epithelial cells in high glucose through Foxo3a/ PINK1-Parkin pathway mediated mitophagy. Exp. Eye Res. 218, 109015 (2022).
Tseng, A. H. H., Shieh, S.-S. & Wang, D. L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 63, 222–234 (2013).
Mohrin, M. et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 1374–1377 (2015).
Wang, C.-L. et al. The mitochondrial unfolded protein response regulates hippocampal neural stem cell aging. Cell Metab. 35, 996–1008.e7 (2023).
Zhou, Y., Wang, S., Li, Y., Yu, S. & Zhao, Y. SIRT1/PGC-1α signaling promotes mitochondrial functional recovery and reduces apoptosis after intracerebral hemorrhage in rats. Front. Mol. Neurosci. 10, 443 (2017).
Lemos, V. et al. The NAD+-dependent deacetylase SIRT2 attenuates oxidative stress and mitochondrial dysfunction and improves insulin sensitivity in hepatocytes. Hum. Mol. Genet. 26, 4105–4117 (2017).
Cha, Y. et al. SIRT2 regulates mitochondrial dynamics and reprogramming via MEK1-ERK-DRP1 and AKT1-DRP1 axes. Cell Rep. 37, 110155 (2021).
Fasano, C., Disciglio, V., Bertora, S., Lepore Signorile, M. & Simone, C. FOXO3a from the nucleus to the mitochondria: a round trip in cellular stress response. Cells 8, 1110 (2019).
Sundaresan, N. R. et al. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 119, 2758–2771 (2009).
Zhang, H. et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443 (2016).
Mouchiroud, L. et al. The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441 (2013).
Cantó, C. et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 15, 838–847 (2012).
Fang, E. F. et al. NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 24, 566–581 (2016).
Fang, E. F. et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412 (2019).
Klimova, N., Long, A. & Kristian, T. Nicotinamide mononucleotide alters mitochondrial dynamics by SIRT3-dependent mechanism in male mice. J. Neurosci. Res. 97, 975–990 (2019).
Romani, M. et al. NAD+ boosting reduces age-associated amyloidosis and restores mitochondrial homeostasis in muscle. Cell Rep. 34, 108660 (2021).
Song, S. B. et al. Modulation of mitochondrial membrane potential and ROS generation by nicotinamide in a manner independent of SIRT1 and mitophagy. Mol. Cells 40, 503–514 (2017).
Braıdy, N. Intravenous NAD+ effectively increased the NAD metabolome, reduced oxidative stress and inflammation, and increased expression of longevity genes safely in elderly humans. J. Cell. Neurosci. Oxid. Stress 10, 779–779 (2018).
Gibson, S. B., Mestayer, R., Berg, J., Grant, R. & Dyess, G. Intravenous administration of nicotinamide adenine dinucleotide improves cognitive performance in human subjects: implications for clinical populations. Arch. Phys. Med. Rehabil. 102, e42 (2021).
Munk, S. H. N. et al. NAD+ regulates nucleotide metabolism and genomic DNA replication. Nat. Cell Biol. 25, 1774–1786 (2023).
Radenkovic, D., Reason & Verdin, E. Clinical evidence for targeting NAD therapeutically. Pharmaceuticals 13, 247 (2020).
Song, Q. et al. The safety and antiaging effects of nicotinamide mononucleotide in human clinical trials: an update. Adv. Nutr. 14, 1416–1435 (2023).
Damgaard, M. V. & Treebak, J. T. What is really known about the effects of nicotinamide riboside supplementation in humans. Sci. Adv. 9, eadi4862 (2023).
Henderson, J. D., Quigley, S. N. Z., Chachra, S. S., Conlon, N. & Ford, D. The use of a systems approach to increase NAD+ in human participants. NPJ Aging 10, 7 (2024).
Bodor, E. T. & Offermanns, S. Nicotinic acid: an old drug with a promising future. Br. J. Pharmacol. 153, S68–S75 (2008).
Membrez, M. et al. Trigonelline is an NAD+ precursor that improves muscle function during ageing and is reduced in human sarcopenia. Nat. Metab. 6, 433–447 (2024).
Giroud-Gerbetant, J. et al. A reduced form of nicotinamide riboside defines a new path for NAD+ biosynthesis and acts as an orally bioavailable NAD+ precursor. Mol. Metab. 30, 192–202 (2019).
Chellappa, K. et al. NAD precursors cycle between host tissues and the gut microbiome. Cell Metab. 34, 1947–1959.e5 (2022).
Yaku, K. et al. BST1 regulates nicotinamide riboside metabolism via its glycohydrolase and base-exchange activities. Nat. Commun. 12, 6767 (2021).
Yang, Y., Mohammed, F. S., Zhang, N. & Sauve, A. A. Dihydronicotinamide riboside is a potent NAD+ concentration enhancer in vitro and in vivo. J. Biol. Chem. 294, 9295–9307 (2019).
Liu, Y. et al. Reduced nicotinamide mononucleotide (NMNH) potently enhances NAD+ and suppresses glycolysis, the TCA cycle, and cell growth. J. Proteome Res. 20, 2596–2606 (2021).
Aktar, S., Ferdousi, F., Kondo, S., Kagawa, T. & Isoda, H. Transcriptomics and biochemical evidence of trigonelline ameliorating learning and memory decline in the senescence-accelerated mouse prone 8 (SAMP8) model by suppressing proinflammatory cytokines and elevating neurotransmitter release. GeroScience https://doi.org/10.1007/s11357-023-00919-x (2023).
Farid, M. M., Yang, X., Kuboyama, T. & Tohda, C. Trigonelline recovers memory function in Alzheimer’s disease model mice: evidence of brain penetration and target molecule. Sci. Rep. 10, 16424 (2020).
Sauve, A. A. & Schramm, V. L. Mechanism-based inhibitors of CD38: a mammalian cyclic ADP-ribose synthetase. Biochemistry 41, 8455–8463 (2002).
Sauve, A. A., Munshi, C., Lee, H. C. & Schramm, V. L. The reaction mechanism for CD38. A single intermediate is responsible for cyclization, hydrolysis, and base-exchange chemistries. Biochemistry 37, 13239–13249 (1998).
Hogan, K. A., Chini, C. C. S. & Chini, E. N. The multi-faceted ecto-enzyme CD38: Roles In Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 10, 1187 (2019).
Aksoy, P., White, T. A., Thompson, M. & Chini, E. N. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem. Biophys. Res. Commun. 345, 1386–1392 (2006).
Piedra-Quintero, Z. L., Wilson, Z., Nava, P. & Guerau-de-Arellano, M. CD38: An Immunomodulatory Molecule In Inflammation And Autoimmunity. Front. Immunol. 11, 597959 (2020).
Liu, Q. et al. Covalent and noncovalent intermediates of an NAD utilizing enzyme, human CD38. Chem. Biol. 15, 1068–1078 (2008).
Tarragó, M. G. et al. A Potent And Specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD+ decline. Cell Metab. 27, 1081–1095.e10 (2018).
Peclat, T. R. et al. CD38 inhibitor 78c increases mice lifespan and healthspan in a model of chronological aging. Aging Cell 21, e13589 (2022).
Kellenberger, E., Kuhn, I., Schuber, F. & Muller-Steffner, H. Flavonoids as inhibitors of human CD38. Bioorg. Med. Chem. Lett. 21, 3939–3942 (2011).
Wang, H., Li, S., Zhang, G., Wu, H. & Chang, X. Potential therapeutic effects of cyanidin-3-O-glucoside on rheumatoid arthritis by relieving inhibition of CD38+ NK cells on Treg cell differentiation. Arthritis Res. Ther. 21, 220 (2019).
Tang, D., Chen, K., Huang, L. & Li, J. Pharmacokinetic properties and drug interactions of apigenin, a natural flavone. Expert Opin. Drug Metab. Toxicol. 13, 323–330 (2017).
Escande, C. et al. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 62, 1084–1093 (2013).
Ogura, Y., Kitada, M., Xu, J., Monno, I. & Koya, D. CD38 inhibition by apigenin ameliorates mitochondrial oxidative stress through restoration of the intracellular NAD+/NADH ratio and Sirt3 activity in renal tubular cells in diabetic rats. Aging 12, 11325–11336(2020).
Covarrubias, A. J. et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat. Metab. 2, 1265–1283 (2020).
Adamia, S. et al. Combination therapy targeting Erk1/2 and CDK4/6i in relapsed refractory multiple myeloma. Leukemia 36, 1088–1101 (2022).
Gozzetti, A. et al. Anti CD38 monoclonal antibodies for multiple myeloma treatment. Hum. Vaccin Immunother. 18, 2052658 (2022).
Sanchez, L., Wang, Y., Siegel, D. S. & Wang, M. L. Daratumumab: a first-in-class CD38 monoclonal antibody for the treatment of multiple myeloma. J. Hematol. Oncol. 9, 51 (2016).
van de Donk, N. W. C. J. et al. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol. Rev. 270, 95–112 (2016).
Abadier, M., Estevam, J., Berg, D. & Fedyk, E. R. Mezagitamab induces immunomodulatory effect in patients with relapsed/refractory multiple myeloma (RRMM). Blood 136, 9–9 (2020).
Dwivedi, S., Rendón-Huerta, E. P., Ortiz-Navarrete, V. & Montaño, L. F. CD38 and regulation of the immune response cells in cancer. J. Oncol. 2021, 6630295 (2021).
Raab, M. S. et al. MOR202, a novel anti-CD38 monoclonal antibody, in patients with relapsed or refractory multiple myeloma: a first-in-human, multicentre, phase 1-2a trial. Lancet Haematol. 7, e381–e394 (2020).
Romano, A. et al. Mechanisms of action of the new antibodies in use in multiple myeloma. Front. Oncol. 11, 684561 (2021).
Franssen, L. E., Stege, C. A. M., Zweegman, S., van de Donk, N. W. C. J. & Nijhof, I. S. Resistance mechanisms towards CD38-Directed antibody therapy in multiple myeloma. J. Clin. Med. 9, 1195 (2020).
Jiao, Y. et al. CD38: targeted therapy in multiple myeloma and therapeutic potential for solid cancers. Expert Opin. Investig. Drugs 29, 1295–1308 (2020).
Ugamraj, H. S. et al. TNB-738, a biparatopic antibody, boosts intracellular NAD+ by inhibiting CD38 ecto-enzyme activity. MAbs 14, 2095949 (2022).
Qian, M. et al. Boosting ATM activity alleviates aging and extends lifespan in a mouse model of progeria. eLife 7, e34836 (2018).
Hu, Y. et al. Aβ promotes CD38 expression in senescent microglia in Alzheimer’s disease. Biol. Res. 55, 10 (2022).
Zha, S., Li, Z., Cao, Q., Wang, F. & Liu, F. PARP1 inhibitor (PJ34) improves the function of aging-induced endothelial progenitor cells by preserving intracellular NAD+ levels and increasing SIRT1 activity. Stem Cell Res. Ther. 9, 224 (2018).
Min, A. & Im, S.-A. PARP inhibitors as therapeutics: beyond modulation of PARylation. Cancers12, 394 (2020).
Singh, N., Pay, S. L., Bhandare, S. B., Arimpur, U. & Motea, E. A. Therapeutic strategies and biomarkers to modulate PARP activity for targeted cancer therapy. Cancers12, 972 (2020).
Paldino, E. et al. Modulation of inflammasome and pyroptosis by olaparib, a PARP-1 inhibitor, in the R6/2 mouse model of Huntington’s disease. Cells 9, 2286 (2020).
Kovacs, K. et al. PARP inhibitor protects against chronic hypoxia/reoxygenation-induced retinal injury by regulation of MAPKs, HIF1α, Nrf2, and NFκB. Invest. Ophthalmol. Vis. Sci. 60, 1478–1490 (2019).
Gariani, K. et al. Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease. J. Hepatol. 66, 132–141 (2017).
Bridges, K. A. et al. Niraparib (MK-4827), a novel poly(ADP-Ribose) polymerase inhibitor, radiosensitizes human lung and breast cancer cells. Oncotarget 5, 5076–5086 (2014).
Mogol, A. N., Kaminsky, A. Z., Dutton, D. J. & Madak Erdogan, Z. Targeting NAD+ metabolism: preclinical insights into potential cancer therapy strategies. Endocrinology 165, bqae043 (2024).
Jones, P., Wilcoxen, K., Rowley, M. & Toniatti, C. Niraparib: a poly(ADP-ribose) polymerase (PARP) inhibitor for the treatment of tumors with defective homologous recombination. J. Med. Chem. 58, 3302–3314 (2015).
Martin-Oliva, D. et al. Inhibition of poly(ADP-ribose) polymerase modulates tumor-related gene expression, including hypoxia-inducible factor-1 activation, during skin carcinogenesis. Cancer Res. 66, 5744–5756 (2006).
Wang, G. et al. PARP-1 inhibitor, DPQ, attenuates LPS-induced acute lung injury through inhibiting NF-κB-mediated inflammatory response. PLoS ONE 8, e79757 (2013).
Damiani, R. M. et al. Influence of PARP-1 inhibition in the cardiotoxicity of the topoisomerase 2 inhibitors doxorubicin and mitoxantrone. Toxicol. Vitr. 52, 203–213 (2018).
Caldini, R. et al. Low doses of 3-aminobenzamide, a poly(ADP-ribose) polymerase inhibitor, stimulate angiogenesis by regulating expression of urokinase type plasminogen activator and matrix metalloprotease 2. Vasc. Cell 3, 12 (2011).
Woodhouse, B. C. & Dianov, G. L. Poly ADP-ribose polymerase-1: an international molecule of mystery. DNA Repair7, 1077–1086 (2008).
Lo, E. H., Bosque-Hamilton, P. & Meng, W. Inhibition of poly(ADP-ribose) polymerase: reduction of ischemic injury and attenuation of N-methyl-D-aspartate-induced neurotransmitter dysregulation. Stroke 29, 830–836 (1998).
Liaudet, L. et al. Suppression of poly (ADP-ribose) polymerase activation by 3-aminobenzamide in a rat model of myocardial infarction: long-term morphological and functional consequences. Br. J. Pharmacol. 133, 1424–1430 (2001).
Krishnan Muthaiah, V. P., Kaliyappan, K. & Mahajan, S. D. Poly ADP-ribose polymerase-1 inhibition by 3-aminobenzamide recuperates HEI-OC1 auditory hair cells from blast overpressure-induced cell death. Front. Cell Dev. Biol. 11, 1047308 (2023).
Ohmoto, A. & Yachida, S. Current status of poly(ADP-ribose) polymerase inhibitors and future directions. Onco Targets Ther. 10, 5195–5208 (2017).
Kaci, F. N. & Daglioglu, C. 3-Aminobenzamide-linked multifunctional nanoparticles enhances anticancer activity of low-dose cisplatin chemotherapy in lung adenocarcinoma. J. Drug Deliv. Sci. Technol. 100, 106038 (2024).
Chung, S. et al. Regulation of SIRT1 in cellular functions: role of polyphenols. Arch. Biochem. Biophys. 501, 79–90 (2010).
Iside, C., Scafuro, M., Nebbioso, A. & Altucci, L. SIRT1 activation by natural phytochemicals: an overview. Front. Pharmacol. 11, 1225 (2020).
Łanoszka, K. & Vlčková, N. Natural Sirtuin1 activators and atherosclerosis: an overview. Curr. Atheroscler. Rep. 25, 979–994 (2023).
Kim, J. E. et al. Mitochondrial SIRT3 as a protective factor against cyclosporine A-induced nephrotoxicity. Sci. Rep. 14, 10143 (2024).
Trinh, D., Al Halabi, L., Brar, H., Kametani, M. & Nash, J. E. The role of SIRT3 in homeostasis and cellular health. Front. Cell Neurosci. 18, 1434459 (2024).
Harris, P. S., Gomez, J. D., Backos, D. S. & Fritz, K. S. Characterizing Sirtuin 3 deacetylase affinity for aldehyde dehydrogenase 2. Chem. Res. Toxicol. 30, 785–793 (2017).
Li, Y. et al. Role of SIRT3 in neurological diseases and rehabilitation training. Metab. Brain Dis. 38, 69–89 (2023).
Ma, S., Fan, L. & Cao, F. Combating cellular senescence by sirtuins: implications for atherosclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 1822–1830 (2019).
Troelsen, K. S. et al. Mitochondria-targeted inhibitors of the human SIRT3 lysine deacetylase. RSC Chem. Biol. 2, 627–635 (2021).
Pillai, V. B. et al. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget 8, 34082–34098 (2017).
Peng, F. et al. 2-APQC, a small-molecule activator of Sirtuin-3 (SIRT3), alleviates myocardial hypertrophy and fibrosis by regulating mitochondrial homeostasis. Signal Transduct. Target Ther. 9, 133 (2024).
Lee, J. H., Yun, C. W., Hur, J. & Lee, S. H. Fucoidan rescues p-cresol-induced cellular senescence in mesenchymal stem cells via FAK-Akt-TWIST axis. Mar. Drugs 16, 121 (2018).
Lin, Y. et al. The anti-cancer effects of fucoidan: a review of both in vivo and in vitro investigations. Cancer Cell Int. 20, 154 (2020).
Li, J.-J. et al. Nicotinamide N-methyltransferase (NNMT): a new hope for treating aging and age-related conditions. Metabolites 14, 343 (2024).
Rahnasto-Rilla, M. K. et al. The identification of a SIRT6 activator from brown algae Fucus distichus. Mar. Drugs 15, 190 (2017).
Zhang, L. et al. Fucoidans are novel senotherapeutics that enhance sirt6 and DNA repair activity. Innov. Aging 6, 732–732 (2022).
Kim, S. et al. Sirtuin 7 inhibitor attenuates colonic mucosal immune activation in mice-potential therapeutic target in inflammatory bowel disease. Biomedicines 10, 2693 (2022).
Sun, W.-D. et al. Nicotinamide N-methyltransferase (NNMT): a novel therapeutic target for metabolic syndrome. Front. Pharmacol. 15, 1410479 (2024).
Liu, M. et al. Serum N(1)-methylnicotinamide is associated with obesity and diabetes in Chinese. J. Clin. Endocrinol. Metab. 100, 3112–3117 (2015).
Ruf, S. et al. Novel tricyclic small molecule inhibitors of Nicotinamide N-methyltransferase for the treatment of metabolic disorders. Sci. Rep. 12, 15440 (2022).
Kannt, A. et al. A small molecule inhibitor of Nicotinamide N-methyltransferase for the treatment of metabolic disorders. Sci. Rep. 8, 3660 (2018).
Chen, D. et al. Novel propargyl-linked bisubstrate analogues as tight-binding inhibitors for nicotinamide N-methyltransferase. J. Med. Chem. 62, 10783–10797 (2019).
Policarpo, R. L. et al. High-affinity alkynyl bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT). J. Med. Chem. 62, 9837–9873 (2019).
Meng, Y., Iyamu, I. D., Ahmed, N. A. M. & Huang, R. Comparative analysis of two NNMT bisubstrate inhibitors through chemoproteomic studies: uncovering the role of unconventional SAM analogue moiety for improved selectivity. ACS Chem. Biol. 19, 89–100 (2024).
Lee, H.-Y. et al. Covalent inhibitors of nicotinamide N-methyltransferase (NNMT) provide evidence for target engagement challenges in situ. Bioorg. Med. Chem. Lett. 28, 2682–2687 (2018).
Neelakantan, H. et al. Selective and membrane-permeablesmall molecule inhibitors of nicotinamide N-methyltransferase reverse high fat diet-induced obesity in mice. Biochem Pharmacol. 147, 141–152 (2018).
Neelakantan, H. et al. Small molecule nicotinamide N-methyltransferase inhibitor activates senescent muscle stem cells and improves regenerative capacity of aged skeletal muscle. Biochem. Pharmacol. 163, 481–492 (2019).
Dimet-Wiley, A. L. et al. Nicotinamide N-methyltransferase inhibition mimics and boosts exercise-mediated improvements in muscle function in aged mice. Sci. Rep. 14, 15554 (2024).
Ahmed-Belkacem, R., Debart, F. & Vasseur, J. Bisubstrate strategies to target methyltransferases. Eur. J. Org. Chem. 2022, e202101481 (2022).
Gao, Y., Martin, N. I. & Van Haren, M. J. Nicotinamide N-methyl transferase (NNMT): an emerging therapeutic target. Drug Discov. Today 26, 2699–2706 (2021).
Sampson, C. M. et al. Combined nicotinamide N-methyltransferase inhibition and reduced-calorie diet normalizes body composition and enhances metabolic benefits in obese mice. Sci. Rep. 11, 5637 (2021).
Xu, Q. et al. Mechanism research and treatment progress of NAD pathway related molecules in tumor immune microenvironment. Cancer Cell Int. 22, 242 (2022).
Zhu, Y. et al. From rate-limiting enzyme to therapeutic target: the promise of NAMPT in neurodegenerative diseases. Front. Pharmacol. 13, 920113 (2022).
Garten, A. et al. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 11, 535–546 (2015).
van der Veer, E. et al. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J. Biol. Chem. 282, 10841–10845 (2007).
Song, J. et al. Nicotinamide phosphoribosyltransferase is required for the calorie restriction-mediated improvements in oxidative stress, mitochondrial biogenesis, and metabolic adaptation. J. Gerontol. A Biol. Sci. Med. Sci. 69, 44–57 (2014).
Wei, Y., Xiang, H. & Zhang, W. Review of various NAMPT inhibitors for the treatment of cancer. Front. Pharmacol. 13, 970553 (2022).
Xu, T.-Y. et al. Discovery and characterization of novel small-molecule inhibitors targeting nicotinamide phosphoribosyltransferase. Sci. Rep. 5, 10043 (2015).
Zhang, S.-L. et al. Crystal structure-based comparison of two NAMPT inhibitors. Acta Pharm. Sin. 39, 294–301 (2018).
Ghanem, M. S. et al. Identification of NAPRT inhibitors with anti-cancer properties by in silico drug discovery. Pharmaceuticals 15, 848 (2022).
Parisotto, M. et al. The NAMPT inhibitor FK866 increases metformin sensitivity in pancreatic cancer cells. Cancers14, 5597 (2022).
Zeng, M. et al. Nicotinamide phosphoribosyltransferase inhibitor ameliorates mouse aging-induced cognitive impairment. J. Cereb. Blood Flow. Metab. 41, 2510–2523 (2021).
Holen, K., Saltz, L. B., Hollywood, E., Burk, K. & Hanauske, A.-R. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest. N. Drugs 26, 45–51 (2008).
Akiu, M. et al. Discovery of DS68702229 as a potent, orally available NAMPT (nicotinamide phosphoribosyltransferase) activator. Chem. Pharm. Bull.69, 1110–1122 (2021).
Pinkerton, A. B. et al. Optimization of a urea-containing series of nicotinamide phosphoribosyltransferase (NAMPT) activators. Bioorg. Med. Chem. Lett. 41, 128007 (2021).
Tang, S. et al. Chemistry-led investigations into the mode of action of NAMPT activators, resulting in the discovery of non-pyridyl class NAMPT activators. Acta Pharm. Sin. B 13, 709–721 (2023).
Yao, H. et al. Discovery of small-molecule activators of nicotinamide phosphoribosyltransferase (NAMPT) and their preclinical neuroprotective activity. Cell Res. 32, 570–584 (2022).
Hong, W., Mo, F., Zhang, Z., Huang, M. & Wei, X. Nicotinamide mononucleotide: a promising molecule for therapy of diverse diseases by targeting NAD+ metabolism. Front. Cell Dev. Biol. 8, 246 (2020).
Liang, J. et al. Impact of NAD+ metabolism on ovarian aging. Immun. Ageing 20, 70 (2023).
Kim, M. et al. Discovery of a novel NAMPT inhibitor that selectively targets NAPRT-deficient EMT-subtype cancer cells and alleviates chemotherapy-induced peripheral neuropathy. Theranostics 13, 5075–5098 (2023).
Olesen, U. H., Thougaard, A. V., Jensen, P. B. & Sehested, M. A preclinical study on the rescue of normal tissue by nicotinic acid in high-dose treatment with APO866, a specific nicotinamide phosphoribosyltransferase inhibitor. Mol. Cancer Ther. 9, 1609–1617 (2010).
Piacente, F. et al. Nicotinic acid phosphoribosyltransferase regulates cancer cell metabolism, susceptibility to NAMPT inhibitors, and DNA repair. Cancer Res. 77, 3857–3869 (2017).
Franco, J. et al. Structure-based identification and biological characterization of new NAPRT inhibitors. Pharmaceuticals 15, 855 (2022).
Baldassarri, C. et al. Properly substituted benzimidazoles as a new promising class of nicotinate phosphoribosyltransferase (NAPRT) modulators. Pharmaceuticals 16, 189 (2023).
Sporny, M. et al. Structural basis for SARM1 inhibition and activation under energetic stress. eLife 9, e62021 (2020).
Chen, J. & Li, H. Characterization of novel SARM1 inhibitors for the treatment of chemotherapy-induced peripheral neuropathy. Biomedicines 12, 2123 (2024).
Yoshino, J. ACMSD: a novel target for modulating NAD+ homeostasis. Trends Endocrinol. Metab. 30, 229–232 (2019).
Katsyuba, E. et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 563, 354–359 (2018).
Cianci, M. et al. Structural Basis of Human Dimeric α-Amino-β-Carboxymuconate-ε-Semialdehyde Decarboxylase Inhibition With TES-1025. Front Mol Biosci. 9, 834700 (2022).
Yang, Y. et al. Diflunisal derivatives as modulators of ACMS decarboxylase targeting the tryptophan-kynurenine pathway. J. Med. Chem. 64, 797–811 (2021).
Balakrishnan, K. et al. Phase 2 and pharmacodynamic study of oral forodesine in patients with advanced, fludarabine-treated chronic lymphocytic leukemia. Blood 116, 886–892 (2010).
Dummer, R. et al. Final results of a multicenter phase II study of the purine nucleoside phosphorylase (PNP) inhibitor forodesine in patients with advanced cutaneous T-cell lymphomas (CTCL) (Mycosis fungoides and Sézary syndrome). Ann. Oncol. 25, 1807–1812 (2014).
Feuz, M. B., Meyer-Ficca, M. L. & Meyer, R. G. Beyond pellagra-research models and strategies addressing the enduring clinical relevance of NAD deficiency in aging and disease. Cells 12, 500 (2023).
Fang, E. F. et al. NAD+ in aging: molecular mechanisms and translational implications. Trends Mol. Med. 23, 899–916 (2017).
Gong, B. et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 34, 1581–1588 (2013).
Sorrentino, V. et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 552, 187–193 (2017).
Pirinen, E. et al. Niacin cures systemic NAD+ deficiency and improves muscle performance in adult-onset mitochondrial myopathy. Cell Metab. 31, 1078–1090.e5 (2020).
Ferrell, M. et al. A terminal metabolite of niacin promotes vascular inflammation and contributes to cardiovascular disease risk. Nat. Med. 30, 424–434 (2024).
Orr, M. E. et al. A randomized placebo-controlled trial of nicotinamide riboside in older adults with mild cognitive impairment. Geroscience 46, 665–682 (2024).
Schmeisser, K. et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 9, 693–700 (2013).
Beltrà, M. et al. NAD+ repletion with niacin counteracts cancer cachexia. Nat. Commun. 14, 1849 (2023).
Migliavacca, E. et al. Mitochondrial oxidative capacity and NAD+ biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 10, 5808 (2019).
Hiona, A. & Leeuwenburgh, C. The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Exp. Gerontol. 43, 24–33 (2008).
Askanas, V. & Engel, W. K. Sporadic inclusion-body myositis: conformational multifactorial ageing-related degenerative muscle disease associated with proteasomal and lysosomal inhibition, endoplasmic reticulum stress, and accumulation of amyloid-β42 oligomers and phosphorylated tau. Presse Med. 40, e219–e235 (2011).
Joshi, P. R. et al. Functional relevance of mitochondrial abnormalities in sporadic inclusion body myositis. J. Clin. Neurosci. 21, 1959–1963 (2014).
Aguennouz, M. et al. Telomere shortening is associated to TRF1 and PARP1 overexpression in Duchenne muscular dystrophy. Neurobiol. Aging 32, 2190–2197 (2011).
Ryu, D. et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl. Med. 8, 361ra139 (2016).
Frederick, D. W. et al. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. 24, 269–282 (2016).
Das, A. et al. Impairment of an endothelial NAD+-H2S signaling network is a reversible cause of vascular aging. Cell 173, 74–89.e20 (2018).
Pirinen, E. et al. Pharmacological inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 19, 1034–1041 (2014).
Argilés, J. M., Busquets, S., Stemmler, B. & López-Soriano, F. J. Cancer cachexia: understanding the molecular basis. Nat. Rev. Cancer 14, 754–762 (2014).
Park, J. M., Han, Y. M., Lee, H. J., Park, Y. J. & Hahm, K. B. Nicotinamide riboside vitamin B3 mitigated C26 adenocarcinoma-induced cancer cachexia. Front. Pharmacol. 12, 665493 (2021).
Campelj, D. G. et al. The paradoxical effect of PARP inhibitor BGP-15 on irinotecan-induced cachexia and skeletal muscle dysfunction. Cancers12, 3810 (2020).
Sorensen, J. C. et al. BGP-15 protects against oxaliplatin-induced skeletal myopathy and mitochondrial reactive oxygen species production in mice. Front. Pharmacol. 8, 137 (2017).
Basse, A. L. et al. Nampt controls skeletal muscle development by maintaining Ca2+ homeostasis and mitochondrial integrity. Mol. Metab. 53, 101271 (2021).
Kim, H.-J., Jung, D.-W. & Williams, D. R. Age is just a number: progress and obstacles in the discovery of new candidate drugs for sarcopenia. Cells 12, 2608 (2023).
Tsai, S.-Y. Lost in translation: challenges of current pharmacotherapy for sarcopenia. Trends Mol. Med. 30, 1047–1060 (2024).
Duan, D., Goemans, N., Takeda, S., Mercuri, E. & Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 7, 13 (2021).
Cardoso, D., Barthélémy, I., Blot, S. & Muchir, A. Replenishing NAD+ content reduces aspects of striated muscle disease in a dog model of Duchenne muscular dystrophy. Skelet. Muscle 13, 20 (2023).
Frederick, D. W. et al. Complementary NAD+ replacement strategies fail to functionally protect dystrophin-deficient muscle. Skelet. Muscle 10, 30 (2020).
Amjad, S. et al. Role of NAD+ in regulating cellular and metabolic signaling pathways. Mol. Metab. 49, 101195 (2021).
Bartman, S., Coppotelli, G. & Ross, J. M. Mitochondrial dysfunction: a key player in brain aging and diseases. Curr. Issues Mol. Biol. 46, 1987–2026 (2024).
Lautrup, S., Sinclair, D. A., Mattson, M. P. & Fang, E. F. NAD+ in brain aging and neurodegenerative disorders. Cell Metab. 30, 630–655 (2019).
Wang, X. et al. NAD+ in Alzheimer’s disease: molecular mechanisms and systematic therapeutic evidence obtained in vivo. Front. Cell Dev. Biol. 9, 668491 (2021).
Yu, M., Sporns, O. & Saykin, A. J. The human connectome in Alzheimer disease - relationship to biomarkers and genetics. Nat. Rev. Neurol. 17, 545–563 (2021).
Yao, Z., Yang, W., Gao, Z. & Jia, P. Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci. Lett. 647, 133–140 (2017).
Blacher, E. et al. Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. Ann. Neurol. 78, 88–103 (2015).
Maggiore, A. et al. Neuroprotective effects of PARP inhibitors in Drosophila models of Alzheimer’s disease. Cells 11, 1284 (2022).
Fathi, M. et al. Dynamic changes in metabolites of the kynurenine pathway in Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: a systematic Review and meta-analysis. Front. Immunol. 13, 997240 (2022).
Gabrawy, M. M. et al. Dual treatment with kynurenine pathway inhibitors and NAD+ precursors synergistically extends life span in Drosophila. Aging Cell 23, e14102 (2024).
Schöndorf, D. C. et al. The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson’s disease. Cell Rep. 23, 2976–2988 (2018).
Flønes, I. H. et al. Mitochondrial complex I deficiency stratifies idiopathic Parkinson’s disease. Nat. Commun. 15, 3631 (2024).
González-Rodríguez, P. et al. Disruption of mitochondrial complex I induces progressive Parkinsonism. Nature 599, 650–656 (2021).
Jia, H. et al. High doses of nicotinamide prevent oxidative mitochondrial dysfunction in a cellular model and improve motor deficit in a Drosophila model of Parkinson’s disease. J. Neurosci. Res. 86, 2083–2090 (2008).
Dölle, C. et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 7, 13548 (2016).
Lehmann, S., Costa, A. C., Celardo, I., Loh, S. H. Y. & Martins, L. M. Parp mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson’s disease. Cell Death Dis. 7, e2166 (2016).
Lehmann, S., Loh, S. H. Y. & Martins, L. M. Enhancing NAD+ salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 6, 141–147 (2017).
Caito, S. W. & Aschner, M. NAD+ Supplementation attenuates methylmercury dopaminergic and mitochondrial toxicity in Caenorhabditis elegans. Toxicol. Sci. 151, 139–149 (2016).
Shan, C. et al. Protective effects of β- nicotinamide adenine dinucleotide against motor deficits and dopaminergic neuronal damage in a mouse model of Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 94, 109670 (2019).
Turconi, G. et al. Nicotinamide riboside first alleviates symptoms but later downregulates dopamine metabolism in proteasome inhibition mouse model of Parkinson’s disease. Heliyon 10, e34355 (2024).
Yang, B. et al. NAD+ supplementation prevents STING-induced senescence in ataxia telangiectasia by improving mitophagy. Aging Cell 20, e13329 (2021).
Hughes, R. O. et al. Small molecule SARM1 inhibitors recapitulate the SARM1-/- phenotype and allow recovery of a metastable pool of axons fated to degenerate. Cell Rep. 34, 108588 (2021).
Alexandris, A. S. et al. Protective effects of NAMPT or MAPK inhibitors and NaR on Wallerian degeneration of mammalian axons. Neurobiol. Dis. 171, 105808 (2022).
Takaso, Y. et al. Deletion of CD38 and supplementation of NAD+ attenuate axon degeneration in a mouse facial nerve axotomy model. Sci. Rep. 10, 17795 (2020).
Chanvillard, L., Tammaro, A. & Sorrentino, V. NAD+ metabolism and interventions in premature renal aging and chronic kidney disease. Cells 12, 21 (2022).
Cohen, J. J. Relationship between energy requirements for Na+ reabsorption and other renal functions. Kidney Int. 29, 32–40 (1986).
van der Rijt, S., Leemans, J. C., Florquin, S., Houtkooper, R. H. & Tammaro, A. Immunometabolic rewiring of tubular epithelial cells in kidney disease. Nat. Rev. Nephrol. 18, 588–603 (2022).
Takahashi, R. et al. The significance of NAD + metabolites and nicotinamide N-methyltransferase in chronic kidney disease. Sci. Rep. 12, 1–19 (2022).
Guan, Y. et al. Nicotinamide mononucleotide, an NAD+ precursor, rescues age-associated susceptibility to AKI in a sirtuin 1-dependent manner. J. Am. Soc. Nephrol. 28, 2337–2352 (2017).
Yasuda, I. et al. Pre-emptive short-term nicotinamide mononucleotide treatment in a mouse model of diabetic nephropathy. J. Am. Soc. Nephrol.32, 1355–1370 (2021).
Cho, K., Kim, H., Rodriguez-Iturbe, B. & Vaziri, N. D. Niacin ameliorates oxidative stress, inflammation, proteinuria, and hypertension in rats with chronic renal failure. Am. J. Physiol. Ren. Physiol. 297, F106–F113 (2009).
Zhen, X. et al. Nicotinamide supplementation attenuates renal interstitial fibrosis via boosting the activity of sirtuins. Kidney Dis.7, 186–199 (2021).
Marjot, T., Moolla, A., Cobbold, J. F., Hodson, L. & Tomlinson, J. W. Nonalcoholic fatty liver disease in adults: current concepts in etiology, outcomes, and management. Endocr. Rev. 41, bnz009 (2020).
Liu, Y. J. et al. ACMSD inhibition corrects fibrosis, inflammation, and DNA damage in MASLD/MASH. J. Hepatol. 82, 174–188 (2025).
Koltai, E. et al. Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech. Ageing Dev. 131, 21–28 (2010).
Barth, P. G. et al. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62, 327–355 (1983).
Damschroder, D. et al. Stimulating the sir2-spargel axis rescues exercise capacity and mitochondrial respiration in a Drosophila model of Barth syndrome. Dis. Model Mech. 15, dmm049279 (2022).
Ji, J. et al. NAD supplementation improves mitochondrial performance of cardiolipin mutants. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1867, 159094 (2022).
Zhang, H., Zhang, Y., Zhang, J. & Jia, D. Exercise alleviates cardiovascular diseases by improving mitochondrial homeostasis. J. Am. Heart Assoc. 13, e036555 (2024).
Caldwell, C. C., Petzinger, G. M., Jakowec, M. W. & Cadenas, E. Treadmill exercise rescues mitochondrial function and motor behavior in the CAG140 knock-in mouse model of Huntington’s disease. Chem. Biol. Interact. 315, 108907 (2020).
Hokari, F. et al. Muscle contractile activity regulates Sirt3 protein expression in rat skeletal muscles. J. Appl. Physiol.109, 332–340 (2010).
Cantó, C. et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 11, 213–219 (2010).
Dollerup, O. L. et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Am. J. Clin. Nutr. 108, 343–353 (2018).
Airhart, S. E. et al. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 12, e0186459 (2017).
Lapatto, H. A. K. et al. Nicotinamide riboside improves muscle mitochondrial biogenesis, satellite cell differentiation, and gut microbiota in a twin study. Sci. Adv. 9, eadd5163 (2023).
Berven, H. et al. NR-SAFE: a randomized, double-blind safety trial of high dose nicotinamide riboside in Parkinson’s disease. Nat. Commun. 14, 7793 (2023).
Elhassan, Y. S. et al. Nicotinamide riboside augments the aged human skeletal muscle NAD+ metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep. 28, 1717–1728.e6 (2019).
Ahmadi, A. et al. Randomized crossover clinical trial of coenzyme Q10 and nicotinamide riboside in chronic kidney disease. JCI Insight 8, e167274 (2023).
Dollerup, O. L. et al. Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. J. Physiol. 598, 731–754 (2020).
Zhou, B. et al. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J. Clin. Invest. 130, 6054–6063 (2020).
Wang, D. D. et al. Safety and tolerability of nicotinamide riboside in heart failure with reduced ejection fraction. JACC Basic Transl. Sci. 7, 1183–1196 (2022).
Brakedal, B. et al. The NADPARK study: a randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 34, 396–407.e6 (2022).
Presterud, R. et al. Long-term nicotinamide riboside use improves coordination and eye movements in ataxia telangiectasia. Mov. Disord. 39, 360–369 (2024).
Connell, N. et al. NAD+-precursor supplementation with L-tryptophan, nicotinic acid, and nicotinamide does not affect mitochondrial function or skeletal muscle function in physically compromised older adults. J. Nutr. 151, 2917–2931 (2021).
Kuerec, A. H. et al. Towards personalized nicotinamide mononucleotide (NMN) supplementation: nicotinamide adenine dinucleotide (NAD) concentration. Mech. Ageing Dev. 218, 111917 (2024).
Yi, L. et al. The efficacy and safety of β-nicotinamide mononucleotide (NMN) supplementation in healthy middle-aged adults: a randomized, multicenter, double-blind, placebo-controlled, parallel-group, dose-dependent clinical trial. GeroScience 45, 29–43 (2023).
Yoshino, M. et al. Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science 372, 1224–1229 (2021).
Igarashi, M. et al. Chronic nicotinamide mononucleotide supplementation elevates blood nicotinamide adenine dinucleotide levels and alters muscle function in healthy older men. NPJ Aging 8, 5 (2022).
Walzik, D. et al. Acute exercise boosts NAD+ metabolism of human peripheral blood mononuclear cells. Brain Behav. Immun. 123, 1011–1023 (2025).
de Guia, R. M. et al. Aerobic and resistance exercise training reverses age-dependent decline in NAD+ salvage capacity in human skeletal muscle. Physiol. Rep. 7, e14139 (2019).
Olszanecka-Glinianowicz, M. et al. Relationship between circulating visfatin/NAMPT, nutritional status and insulin resistance in an elderly population - results from the PolSenior substudy. Metabolism 63, 1409–1418 (2014).
Lamb, D. A. et al. Resistance training increases muscle NAD+ and NADH concentrations as well as NAMPT protein levels and global sirtuin activity in middle-aged, overweight, untrained individuals. Aging12, 9447–9460 (2020).
Costford, S. R. et al. Skeletal muscle NAMPT is induced by exercise in humans. Am. J. Physiol. Endocrinol. Metab. 298, E117–E126 (2010).
Janssens, G. E. et al. Healthy aging and muscle function are positively associated with NAD+ abundance in humans. Nat. Aging 2, 254–263 (2022).
Benyó, Z. et al. GPR109A (PUMA-G/HM74A) mediates nicotinic acid–induced flushing. J. Clin. Invest. 115, 3634–3640 (2005).
Poynten, A. M. et al. Nicotinic acid-induced insulin resistance is related to increased circulating fatty acids and fat oxidation but not muscle lipid content. Metabolism 52, 699–704 (2003).
Chedere, A., Mishra, M., Kulkarni, O., Sriraman, S. & Chandra, N. Personalized quantitative models of NAD metabolism in hepatocellular carcinoma identify a subgroup with poor prognosis. Front. Oncol. 12, 954512 (2022).
Remie, C. M. E. et al. Nicotinamide riboside supplementation alters body composition and skeletal muscle acetylcarnitine concentrations in healthy obese humans. Am. J. Clin. Nutr. 112, 413–426 (2020).
Moore, M. P. & Mucinski, J. M. Impact of nicotinamide riboside supplementation on skeletal muscle mitochondria and whole-body glucose homeostasis: challenging the current hypothesis. J. Physiol. 598, 3327–3328 (2020).
Bird, J. G. et al. Highly efficient 5’ capping of mitochondrial RNA with NAD+ and NADH by yeast and human mitochondrial RNA polymerase. eLife 7, e42179 (2018).
Grudzien-Nogalska, E., Bird, J. G., Nickels, B. E. & Kiledjian, M. NAD-capQ’ detection and quantitation of NAD caps. RNA 24, 1418–1425 (2018).
Wolfram-Schauerte, M. & Höfer, K. NAD-capped RNAs - a redox cofactor meets RNA. Trends Biochem. Sci. 48, 142–155 (2023).
Melkonian, E. A. & Schury, M. P. Biochemistry, anaerobic glycolysis. In StatPearls (StatPearls Publishing, Treasure Island, FL, 2025).
Luengo, A. et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell 81, 691–707.e6 (2021).
Cha, Y. et al. Metabolic control of primed human pluripotent stem cell fate and function by the miR-200c-SIRT2 axis. Nat. Cell Biol. 19, 445–456 (2017).
Ma, R. et al. SIRT3 suppression resulting from the enhanced β-catenin signaling drives glycolysis and promotes hypoxia-induced cell growth in hepatocellular carcinoma cells. Cell Cycle 23, 435–447 (2024).
Finley, L. W. S. et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 19, 416–428 (2011).
Guo, J. et al. The role of NAD-dependent deacetylase sirtuin-2 in liver metabolic stress through regulating pyruvate kinase M2 ubiquitination. J. Transl. Med. 22, 656 (2024).
Nishida, Y. et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol Cell 59, 321–332 (2015).
Yang, L. et al. Serine catabolism feeds NADH when respiration is impaired. Cell Metab. 31, 809–821.e6 (2020).
Khan, N. A. et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 6, 721–731 (2014).
Myakala, K. et al. NAD metabolism modulates inflammation and mitochondria function in diabetic kidney disease. J. Biol. Chem. 299, 104975 (2023).
de Castro, J. M., Stein, D. J., Medeiros, H. R., de Oliveira, C. & Torres, I. L. S. Nicotinamide riboside neutralizes hypothalamic inflammation and increases weight loss without altering muscle mass in obese rats under calorie restriction: a preliminary investigation. Front. Nutr. 8, 648893 (2021).
Kim, M.-B. et al. Nicotinamide riboside supplementation exerts an anti-obesity effect and prevents inflammation and fibrosis in white adipose tissue of female diet-induced obesity mice. J. Nutr. Biochem. 107, 109058 (2022).
Wang, L., Chen, C., Zhou, H., Tao, L. & Xu, E. Nicotinamide riboside-driven modulation of SIRT3/mtROS/JNK signaling pathways alleviates myocardial ischemia-reperfusion injury. Int. J. Med. Sci. 21, 2139–2148 (2024).
Abdellatif, M. et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci. Transl. Med. 13, eabd7064 (2021).
Xiong, X. et al. NAD+-boosting agent nicotinamide mononucleotide potently improves mitochondria stress response in Alzheimer’s disease via ATF4-dependent mitochondrial UPR. Cell Death Dis. 15, 744 (2024).
Hasegawa, K., Sakamaki, Y., Tamaki, M. & Wakino, S. Nicotinamide mononucleotide ameliorates adriamycin-induced renal damage by epigenetically suppressing the NMN/NAD consumers mediated by Twist2. Sci. Rep. 12, 13712 (2022).
Chandrasekaran, K. et al. NAD+ precursors repair mitochondrial function in diabetes and prevent experimental diabetic neuropathy. Int. J. Mol. Sci. 23, 4887 (2022).
Fang, E. F. et al. NAD+ augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 10, 5284 (2019).
de Zélicourt, A. et al. CD38-NADase is a new major contributor to Duchenne muscular dystrophic phenotype. EMBO Mol. Med. 14, e12860 (2022).
Gong, M. et al. Trigonelline inhibits tubular epithelial-mesenchymal transformation in diabetic kidney disease via targeting Smad7. Biomed. Pharmacother. 168, 115747 (2023).
Faivre, A. et al. Differential role of nicotinamide adenine dinucleotide deficiency in acute and chronic kidney disease. Nephrol. Dial. Transpl. 36, 60–68 (2021).
Dollerup, O. L. et al. Effects of nicotinamide riboside on endocrine pancreatic function and incretin hormones in nondiabetic men with obesity. J. Clin. Endocrinol. Metab. 104, 5703–5714 (2019).
Norheim, K. L. et al. Effect of nicotinamide riboside on airway inflammation in COPD: a randomized, placebo-controlled trial. Nat. Aging 4, 1772–1781 (2024).
Simic, P. et al. Nicotinamide riboside with pterostilbene (NRPT) increases NAD+ in patients with acute kidney injury (AKI): a randomized, double-blind, placebo-controlled, stepwise safety study of escalating doses of NRPT in patients with AKI. BMC Nephrol. 21, 342 (2020).
Dellinger, R. W. et al. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD+ levels in humans safely and sustainably: a randomized, double-blind, placebo-controlled study. NPJ Aging Mech. Dis. 3, 17 (2017).
van Dijk, A. E. et al. Acute effects of decaffeinated coffee and the major coffee components chlorogenic acid and trigonelline on glucose tolerance. Diab. Care 32, 1023–1025 (2009).
Acknowledgements
V.S. is supported by the NUHS Internal Grant Funding under NUS Start-up grant NUHSRO/2022/047/Startup/11, an MOE Tier 2 grant T2EP30124-0014, and a Vidi grant from the Netherlands Organization for Scientific Research (NWO; 09150172110059).
Author information
Authors and Affiliations
Contributions
K.Y., K.S.V., S.J., T.C.M.T., and V.S.: resourcing, table creation, and writing the original draft. K.Y., K.S.V., S.J., and T.C.M.T.: figure drawing. V.S.: conceptualization. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yusri, K., Jose, S., Vermeulen, K.S. et al. The role of NAD+ metabolism and its modulation of mitochondria in aging and disease. npj Metab Health Dis 3, 26 (2025). https://doi.org/10.1038/s44324-025-00067-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44324-025-00067-0
This article is cited by
-
Hepatic tryptophan metabolism links chronic stress to liver cancer
Nature Metabolism (2026)
-
Microglia Mitochondrial Metabolism in Neurological Diseases
Molecular Neurobiology (2026)
-
Mitochondrial immunometabolism in sepsis: orchestrating macrophage polarization and dysfunction
European Journal of Medical Research (2025)
-
Hypoxia-Inducible Factor 1-alpha (HIF1α), Nicotinamide Adenine Dinucleotide (NAD+, NADH), and Nitric Oxide (NO) interplay in critically ill patients, with implications for patient safety and targeted therapies: a review
Patient Safety in Surgery (2025)
-
Tunneling nanotubes between glioblastoma cells and T cells and hijack of mitochondria
Journal of Neuro-Oncology (2025)
