
Abstract
The neurovascular unit includes multiple cell types that communicate with each other on a second-by-second basis using traditional neurotransmitters and other signaling molecules, however the function of each cell or the mechanisms by which homeostasis is maintained are still unclear. Here, we review the important elements of the astro- and neurovascular unit and the modulators that contribute to the orchestration of functional hyperemia in health and disease.
Similar content being viewed by others


Introduction
The bioenergetic processes of the brain are complex, well-integrated, and vary across regions and substructures. The maintenance of these processes is key for preserving cell-to-cell communication, where at the nexus of this homeostasis exists the physical and biochemical interactions between the vascular (e.g. vessels and capillaries) and cellular components (e.g. neurons, astrocytes, microglia, and pericytes). Necessary substrates such as glucose and molecular oxygen are delivered to the brain and subsequently transported to areas of high activity/energetic demand; the resulting products and byproducts of these reactions contribute to a rich network of negative and positive feedback loops to control cellular communication, vasoreactivity, as well as astro- and neurovascular coupling1,2.
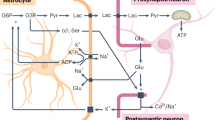
Several cell types comprise and surround the angioarchitecture of the brain1,3,4,5,6,7,8 (Fig. 1). A single layer of endothelial cells forms the boundary between the vessel lumen and the basement membrane. In arteries/arterioles and veins/venules, contractile vascular smooth muscle cells (VSMCs) surround the endothelium. Arteries are comprised of more VSMCs than veins and are structurally distinct from the less contractile venous VSMCs, reflecting the discrete functional roles of arteries and veins9,10. In smaller capillaries, VSMCs are absent, and instead pericytes engage with the vessel11. Astrocyte endfeet wrap around capillaries and penetrating vessels and are thought to play a key role in vasoreactivity5. Neurons and interneurons are known to engage with this complex to either directly or indirectly integrate local neurovascular coupling12,13. Finally, microglia mediate neuroinflammation, contribute to neuronal signaling, and modulate cerebral blood flow (CBF) and astro-neurovascular coupling (ANVC)14,15,16. Penetrating arteries branch into arterioles, which divert blood into capillary transitions, then into the capillary bed. Blood flows from the capillaries into the venules and ultimately to the veins. Smaller capillary structures function to remove waste (i.e. CO2) and exchange lymphatic fluids.

Pial vessels branch off into terminal arterioles and venules. From this, dense networks of capillaries form the basis of the capillary bed. Cellular components of the ANVU include contractile pericytes that interface with the endothelial cells of capillaries, astrocytes that project endfeet onto the vasculature, and the neurons that interface with these complexes. Tripartite synapses are composed of pre- and post-synaptic neurons and specialized astrocyte processes.
Despite the large body of work dedicated to evaluating the function and the cell types that participate in ANVC, the subtleties of the associations between them, including the direction (i.e., feedforward or feedback) or the degree of the response, as well as mediators involved, are all under deep scientific scrutiny. To date, there are many controversies regarding the sources and modulators of cellular signals that control vascular tone. Therefore, here, we review key cellular components physically associated with blood vessels, specific modulators responsible for functional vascular coupling, and the temporal associations between vascular elements both in healthy and diseased states. We also emphasize the role of chronic insulin signaling in functional hyperemia and ANVC. Cell-specific vasomodulators discussed in this manuscript are summarized in a table format (Table 1). While here focused on the major contributors (i.e., neurons, astrocytes, and pericytes), we recognize the additional critical roles of VSMCs and endothelial cells as key mediators of vascular tone and have highlighted their signaling pathways specifically associated with these three cell types within Table 1.
Anatomy, function, and disease
Neurons
Neurons are considered the fundamental functional unit of the nervous system, despite being largely outnumbered by glia17. These cells are excitable, propagate information, and contribute to local hemodynamic responses, ultimately participating in the control of cerebral blood flow (CBF). Unsurprisingly, measures of CBF reflect neuronal activity, termed ‘functional hyperemia’, which is the underlying principle behind BOLD, fMRI, and PET1. However, the degree to which neuronal signaling participates in modulating vascular tone is still unclear. Classically, neuronal control of vascular function was thought to be mediated through a feedback mechanism, whereby, inherent to increases in neuronal activity, an increased need for energy supply signals for changes in vasomotor tone (i.e. dilation)1. Contrastingly, based on evidence of the abundant energy supply available in the brain1,18, the feedforward mechanism, which augments vessel size via neuronal activation and byproduct production, has been postulated to exist and function apart from bioenergetic demands. Some groups also consider a combination of the two mechanisms to work in tandem4, and others have postulated hypotheses that may function independently of neuronal activity, including responses to prevent capillary stalls or control temperature fluctuations and waste removal19. Regardless of the direction of these signaling events, several neuronally derived vasoactive compounds have been identified. Compounds that cause vasodilation are primarily the most well described; however, neuronal control of vascular tone exists across a spectrum of both constrictive and dilatory effects, where the degree of the resulting response can vary widely across modulators and the local microenvironment (see “Neuron-Specific Modulators of Vascular Function”).
Given the integral role of these cells, it is not surprising that neurodegenerative diseases directly and indirectly negatively impact ANVC. Alzheimer’s disease (AD) and related dementias (ADRDs) are often underscored by hypoperfusion, impaired BBB function, and deteriorations in cell-to-cell communication20, and it is well described that vascular contributions to dementia (VCIDs) are numerous and have a clear impact on ANVC21,22,23,24,25. Neurometabolic processes are known to influence hemodynamics and are, in turn, also likely modulated by signals received from the vasculature. In cerebral cortical and hippocampal brain slices obtained from aged 5xFAD animals, disrupted glucose metabolism has been detected across neurons and astrocytes26. Indeed, many metabolic processes, such as glucose utilization27, mitochondrial function28, norepinephrine (NE) signaling29, and insulin signaling30 are disrupted with disease. In neuronal culture, insulin and insulin sensitizers (i.e. rosiglitazone and pioglitazone) have been shown to alter calcium-dependent processes31,32. Calcium dysregulation, often hyperexcitability, is another major characteristic of ADRDs that extends across cell types (e.g. neurons22), astrocytes23,24, and pericytes25). This might suggest that perhaps insulin resistance contributes to the vascular phenotype observed in ADRDs. Work from our laboratory has also shown that intranasal insulin (INI) increases neuronal Ca2+ measures of network synchrony in aged rats33.
Astrocytes
Astrocytes are stellate, spongiform cells defined by processes that wrap around the vasculature or interface with pericytes, VSMCs (i.e. endfeet), and neurons (i.e. tripartite synapses)34. Although astrocytes require considerably less energy than neurons overall (~5–15%), they are more abundant in the brain17. Astrocytes do not produce action potentials but do signal extensively using Ca2+ and K+ gradients. Astrocytes may also engage in activity-dependent release of gliotransmitters such as glutamate, GABA, ATP, NE35, and D-serine. This cell type also has been shown to display sensitivity to insulin36,37. Traditionally, these cells were thought to only function ancillary to neurons. A primary role of astrocytes was described in the astrocyte-neuron-lactate shuttle hypothesis (ANLSH), in which neuronally-derived glutamate induces glycolysis in astrocytes during activation. Specifically, co-transportation of Na+ with glutamate activates Na+/K+-ATPases, which then stimulates glycolysis where glucose is consumed and lactate is produced and exported to neurons as a source of energy38. However, this model has been challenged in recent years, with evidence indicating that the ANLSH is not well translated to in vivo settings or during stimulation39. Indeed, several groups, including ours, now report that neurons readily take up glucose, perhaps even more than astrocytes40,41,42,43,44. Furthermore, during activation, rates of O2 consumption appear to be more indicative of enhancements in neuronal glycolysis rather than oxidation of lactate, suggesting contributions of lactate as a major fuel source may be subtle45. In vivo, activated astrocytes appear to be engaged in more metabolic processes, including TCA, glycogenolysis, and pyruvate carboxylation, functioning beyond glycolysis alone45,46. Recent work highlights astrocytic involvement in a variety of roles including maintenance of the blood brain barrier (BBB), neurogenesis, regulation of myelination through gap junctions with oligodendrocytes and synaptic transmission and synapse turnover47, and plasticity48. Astrocyte morphology is exceptionally diverse across brain regions and species; nevertheless, these fundamental structural associations position astrocytes at the nexus of the astro-neurovascular unit (ANVU)47,49. Despite this, astrocytic contributions to ANVC are still unclear; recent work from our group and others have emphasized that astrocyte calcium responses follow changes in vessel size24,50,51,52. It has also been hypothesized that astrocytes may participate in vasoreactivity as only a tonic regulator of vessel size, whereas neuronal response is primarily a phasic controller53.
As with neurons, populations of hyperactive astrocytes have been identified in ADRDs. This hyperactivity appears to be a key functional phenotype of astrocyte reactivity found in aging and most forms of neurodegeneration. Signs of hyperactivity in reactive astrocytes include elevated resting calcium levels, increased spontaneous calcium activity, and/or increased calcium transient amplitudes. The calcium dysregulation in reactive astrocytes is likely to give rise to aberrant activation and processing of many different downstream calcium-dependent signaling mediators implicated in disease. For instance, the protein phosphatase calcineurin undergoes extensive calcium-dependent proteolysis in astrocytes associated with AD and vascular pathologies (in both humans and rodent models), leading to hyperactivation of inflammatory transcription factors like NFβB and NFAT. The transformation of the reactive astrocyte transcriptome via aberrant calcineurin activity appears to be a fundamental mechanism linking astrocytic calcium dysregulation to neuroinflammation, glutamate uptake deficits, and synapse dysfunction. Interestingly, suppression of astrocytic calcineurin/NFAT signaling in the context of hyperhomocysteinemia (HHcy) and small cerebral vessel disease improved neurovascular coupling, capillary red blood cell velocity, and cerebral perfusion, suggesting that calcium dysregulation and astrocyte reactivity can compromise cerebrovascular function and disrupt the delivery of oxygen and energy substrates to the brain. Indeed, work from Sompol and colleagues shows that in a diet-induced model of HHcy, which recapitulates pathologies of vascular dementia, astrocyte calcium signaling was elevated with disease24. Glial cells participate in the formation of dense neuritic plaques and are engaged in the deposition of Aβ54. Recent work has shown that astrocyte signaling may be particularly important for the glymphatic clearance of Aβ out of the brain through the perivascular space. Activated astrocytes in aging mice and models recapitulating AD pathology have also been shown to have increased levels of calcineurin, which is known to modulate inflammatory responses55. Decreased glucose utilization27, reduced brain insulin receptors30, mitochondrial dysfunction28, and dysregulated Ca2+ signaling56,57 in the astrocytes also appear to contribute to disease progression58,59. Interestingly, NVC appears in the somatosensory cortex appears to be undisturbed in aged female 5xFAD mice, despite reductions in CBF. However, these reductions may be explained by the reductions in the baseline diameter of the penetrating arterioles60.
Pericytes
Though understudied relative to neurons and glial cells, pericytes are also a critical component of the ANVU where they regulate CBF, maintain the BBB, provide microvascular stability, and shape vascular remodeling61. Because of this functional and structural diversity, several subcategories of pericytes have been classified based upon morphology and location in the capillary bed. However, due to the difficulty in distinguishing pericytes from other mural cells (i.e. VSMCs) and inconsistent nomenclature usage across groups (i.e. whether or not all pericytes are contractile), it is often difficult to resolve findings across studies11. Capillaries dilate prior to arterioles and have been reported to cause an ~84% increase in blood flow with sensory input62. Studies of pericytes have also shown that these cells are contributors to ADRDs. The inhibition of voltage-gated calcium channels with nimodipine, an L-type voltage-gated calcium channel blocker (LVGCCs), to lower resting pericyte calcium in a rodent model of amyloidosis has been shown to alter elevations in measures of CBF (i.e. vasodilation)25. Similar results have been found in healthy animals treated with systemic nimodipine, where alterations in pericyte calcium levels modulate CBF63; note, however, that systemic applications of LVGCC blockers will likely impact multiple cell types, including VSMCs. Additionally, pericyte loss leaves neurons vulnerable to ischemic and excitotoxic damage, loss of BBB integrity, and reduced measures of CBF64,65. Pericyte-deficient mice display neurovascular uncoupling and reduced O2 supply in the brain; delated capillary dilation and poor neuronal excitability were observed in these animals, with no alterations in arteriolar and endothelium-dependent vasodilation66. Pericytes have been shown to participate in the clearance of Aβ and pericyte loss appears to contribute to Aβ accumulation and further pericyte loss67. AD is also associated with pericyte constriction, perhaps due to aberrant calcium levels25, which could underlie hypoperfusion seen in AD patients. Likewise, pericyte death and constriction have been associated with ischemia and stroke62.
Neuron-specific modulators of vascular function
eNOS is largely considered the primary mediator of NO-derived vasodilation, where endothelium-derived NO leads to the formation of cGMP and relaxation of VSMCs. Interestingly, recent work has demonstrated that nNOS (primarily neuronally derived) also contributes to mediating vascular tone. Indeed, nNOS activity is initiated by an increase in intracellular calcium during depolarization, ultimately causing the release of NO and other byproducts68. Furthermore, NO is known to mediate increases in cGMP levels resulting from glutamatergic signaling69. The role of NO is known to be a key mediator of both acute and prolonged vasomotor responses70. Surprisingly, mice lacking either eNOS71 or nNOS69,71 display normal vasoactivity in response to ACh, suggesting that acute vasomotor response is unchanged in these animals and can occur independently from NO action. Perhaps in a feedforward mechanism, in the nucleus accumbens and ventral striatum, NO has also been shown to induce ACh release, which may mediate hyperemic responses through activation of muscarinic receptors72,73. This appears to be a generalized mechanism, as topical treatments of ACh onto penetrating arterioles of bovine and human tissues cause vasodilation74. It is well recognized that direct activation of muscarinic receptors via ACh mediates vasodilation; indeed, neuronal release of ACh also appears to facilitate NO release by binding to endothelial type 5 muscarinic receptors (M5)75. In fact, M5 receptor knockout mice exposed to ACh fail to display alterations in vascular tone, but do respond to ADP, another potent vasodilator76.
In pyramidal neurons from the mouse cortex, NMDA-induced vasodilation has been shown to be dependent on cyclooxygenase 2 (COX-2) and PGE2 receptor activation on endothelial cells77. Glutamate-mediated vasoreactivity, as with NO and ACh, is reliant on calcium as a second messenger to modulate vessel size. Neuronal intracellular calcium has been correlated to vasomotion in the anesthetized rat78. Independent of the vascular beds, cell types, or the excitatory modulators engaged, clearly, calcium is a critical mediator of vasoreactivity. For instance, the application of barium, known to depolarize membranes, onto the cerebral surface has been shown to constrict pial arterioles. This constriction was inhibited upon subsequent treatment with verapamil, a calcium channel blocker79. In contrast, the topical application of a calcium ionophore dilated pial arterioles80. Glutamate-mediated vasoreactivity, as with NO and ACh, is reliant on Ca2+ as a second messenger to modulate vessel size. Indeed, the topical application of a calcium ionophore (A-23187) has been shown to dilate pial arterioles via COX activation through an endothelium-dependent mechanism80. These data suggest that the Ca2+ signaling modulates vessel size through a variety of pathways beyond neuronal control.
Whether independent of neurons or dependent on the byproducts of neuronal respiration, CO2 is a powerful local vasodilator19,81. The increase in vessel size invoked by CO2 is thought to be mediated by changes in pH in the brain82, arterial pressure of CO283, capillary pO2, and hemoglobin saturation84. Thus, pH, CO2, or O2 contribute to local vasoreactivity85 through VSMC activation, opening K+ channels and altering Ca2+channel activity and gap junctions13. In contrast to CO2, local NE release activates adrenergic α1 receptors in the vasculature to increase blood pressure. Indeed, in the brain, early work shows topical treatment of NE onto pial vessels induces constriction86. Clearly, therefore, localized levels of modulators in the brain participate in ANVC and tone to drive vasoreactivity.
While neurons are engaged in ANVC, interneurons also participate either directly or indirectly in localized functional hyperemia. Specifically, GABA-releasing interneurons in the cerebellum expressing somatostatin (SOM) and NOS have been identified to also cause vasodilation, while other populations have been shown to induce vasoconstriction87. Importantly, the neuronal contribution to ANVC is a complex phenomenon that depends on several factors (known and unknown), modulators, and the local microenvironment (i.e. temperature, pH, pO2, pCO2, as well as Ca2+), and, while it is not yet clear which vasomodulators are the most critical, studies modeling neurodegeneration highlight the presence of disrupted signaling pathways between the neurons and vasculature.
Astrocyte-specific modulators of vascular function
Over the past twenty years, astrocyte-specific processes have emerged as key contributors of ANVC. Currently, astrocytes are recognized to have the capacity to initiate, tune, and transmit changes in vascular tone through a diverse variety of signaling pathways; however, the extent and sequence of how these mechanisms coordinate blood flow remains unclear. Several calcium-directed processes have been proposed to mediate astrocyte-driven vasoreactivity. Classical descriptions of ANVC often associate elevations in astrocytic calcium with phospholipase C (PLC)88, ryanodine receptor (RyR)89, or metabotropic glutamate receptor (mGluRs) activation90, driving the release of AA and its metabolites (i.e. EETs, PGE2, or 20-HETE) to elicit a vasomotor (in either direction) response through VSMCs or pericytes91,92,93,94. Early work using flash photolysis of caged Ca2+ in astrocyte endfeet demonstrated vasoconstrictions driven by AA metabolism into 20-HETE95, while another group produced vasodilations through a COX 1 metabolic pathway96, and, indeed, electrophysiological experiments have also linked elevated endfoot calcium to vessel dilation.97. Purinergic receptors such as P2X1R, an ATP receptor, are known to cause elevations in calcium levels via PGE2 and contribute to capillary dilations in cortical slices while inhibition reduced vasodilation90. Thus, the local signaling dynamics that control ANVC have been identified, although it is difficult to generalize the outcome of specific modulators, particularly when considering the structural elements across different vascular beds and regions of the brain.
Conversely, evidence from other groups shows a lack of associations between astrocyte calcium and ANVC. Several groups using the IP3R2KO mouse model (i.e., lacking a primary intracellular Ca2+release channel) report significant reductions in somatic and endfoot Ca2+with little impact on in vivo vasoreactivity48,51,98,99. However, experiments using neocortical slices from the same model suggest that IP3R2 is required for astrocyte mGluR-facilitated changes in vessel size90,100, and hippocampal slices show preserved GPCR-sensitive calcium excursions in astrocyte processes, indicating perhaps unrecognized pathways. It is important to note, however, that astrocytic mGluR5, key in Ca2+ signaling, is detected only during early development in rodents101 and, therefore, is unlikely to contribute to ANVC with age. Similarly, using two-photon microscopy and optogenetic astrocyte stimulation, elevations in calcium levels (soma and endfeet) did not induce changes in arteriole diameter102, nor did blocking them prevent arteriole dilation90. These studies and others argue that large somatic calcium transients may not be the primary drivers of ANVC and that small, localized, and perhaps undetectable calcium transients within astrocyte processes may host subtle triggers51,103,104.
Astrocytes may also modulate vascular tone through ion signal coupling. Astrocytic siphoning of K+ from neurons onto the arteriole wall has also been postulated to contribute to vasodilation105. The local microenvironment has also been described to influence the direction and magnitude of vascular responses. In hippocampal slices, elevated pO2 causes adenosine and lactate to dilate arterioles, whereas low pO2 evokes constrictions through adenosine106. Catecholamines such as NE are well established as potent modulators of vascular tone, and transient increases in NE levels have been linked to increased astrocyte Ca2+ activity107, which in turn modulates neuronal activity108. Although astrocytes express catecholaminergic enzymes involved in NE synthesis (i.e. tyrosine hydroxylase109 and L-amino acid decarboxylase110) as well as the machinery for neuromodulator release111, it is unknown if these cells actually secrete NE.
Emerging evidence suggests bidirectional communication between astrocytes and cerebral vessels provides feedback that regulates vascular tone and perfusion. In fact, it is thought that a conserved mechanism between astrocyte calcium and vasoreactivity may operate in specific contexts. Recently, various stimulation-evoked vascular-to-astrocyte signaling processes have been investigated. Astrocyte endfoot calcium transients are initiated by both glutamate and NO following stimulation50. Interestingly, mechanotransduction through transient receptor potential channels (TRPs) localized in endfeet has also been shown to activate calcium signaling during vasoconstriction; however, downstream COX-1 activation establishes a negative feedback mechanism that attenuates further vasoconstrictions112,113. Additionally, studies investigating vascular-astrocyte dynamics in awake mice found endfoot calcium signaling follows changes in vessel size upon stimulation24,50,114,115. It is plausible that astrocytic calcium maintains tonic control of CBF and is modulated by metabolic factors and neuronal signaling in response to stimulation53. Thus, endfoot calcium signaling may represent a critical checkpoint in functional hyperemia from which microvascular signaling may further tune vascular oscillations and perfusion rate.
Disruptions in energy metabolism or integrity of the ANVU as observed in ADRDs, may interrupt the balance of these regulatory mechanisms, and the timing of vascular and astrocytic signaling events. In an aged model of amyloidosis, some elements of hyperactive Ca2+ signaling was observed, along with the delayed onset and reduced magnitude of stimulation evoked endfoot Ca2+ signals following vasodilation115. In this context, Ca2+ dysregulation may disrupt downstream processes such as AA metabolite production, regulation of neuronal excitability, K+ shuttling and other ion gradient maintenance, and Ca2+-sensitive K+ (BK) and TRP channel function. Excessive Ca2+ signaling has also been shown to activate calcineurin (CN) signaling, which in the presence of Aβ42, further elevates calcium levels116,117,118. Finally, CN signaling is implicated in neuroinflammatory processes that result in damage of the blood brain barrier, further exasperating vascular functional impairments119. For these reasons, it is predicted that targeting astrocyte reactive processes in disease states may alleviate cerebrovascular dysfunction and restore metabolic homeostasis.
Pericyte-specific mediators of vascular function
Whereas VSMCs are present on larger vessels, pericytes are uniquely located on the surface of capillaries and the transitional zones between the capillary bed and arterioles and venules. Thus, these contractile cells appear integral for controlling local vasoreactivity, particularly the microvasculature, although it is clear that not all pericytes are contractile9,120,121. Despite the rich history characterizing pericytes in the brain, it is still difficult to assess pericyte function in vivo and the number of pericyte types, inconsistent nomenclature used throughout the literature, and difficultly targeting these cells contribute to the lack of consensusa11. While it has been demonstrated extensively that pericytes are juxtaposed to the vasculature, particularly the capillary beds, it appears that pericytes are particularly well suited to controlling local vascular flow, and, indeed, in vivo loss of pericytes in aging animals is associated with poorer control of localized capillary blood flow122. Furthermore, recent work has highlighted the novel role of pericytes in controlling angiogenesis in conditions associated with myelin repair, hypoxia, and aging122,123,124.
Whether or not an intermediary step exists between pericytes and capillaries to control the local microvascular tone (i.e. transducing a signal originating from another cell type vs. direct microvascular control), recent evidence demonstrates that loss of pericytes is associated with neurovascular dysfunction, suggesting that these cells may be active participants in ANVC. However, other studies have shown that pericyte density does not change with aging, despite presenting impaired ANVC in response to hypercapnia125.
Both cholinergic and adrenergic receptors have been identified on pericytes in vitro. Additionally, pericytes have been shown to express receptors for vasopressin, vasoactive intestinal peptide (VIP), endothelin 1 (ET-1), and angiotensin II (ANG II)126. Much of the existing work describing pericytic modulators of vascular function primarily details metabolites derived from other cell types. Indeed, the 20-HETE metabolite in the pericyte is derived from AA released by astrocytes and induces vasoconstriction11. Likewise, PGE2 release from astrocytes is thought to target the EP4 receptor on pericytes, promoting vasorelaxation; however, this process can be inhibited by neuronally derived NO127. As in most vascular beds, treatment of cerebellar slices with NE also caused a constrictive response128,129; however, interestingly, NE release from neurons onto pericytes causes a contractile response that does not alter intracellular calcium levels130. In culture, ATP treatment induced elevations in pericytic calcium and invoked constriction129,131. Nevertheless, pericytic calcium is an important mediator of vessel tone and appears to be in part be dependent on potassium flux, perhaps driven by Katp channels132; stimulation of pericytes via Katp channel agonists have been shown to elevate capillary blood flow133. Moreover, ex vivo investigations of pericyte contributions to the ANVC show that pericytes can direct local blood flow to areas of high neuronal activity according to large fluctuations of K+134.
Beyond calcium as a mediator of vascular tone: insulin
In the brain, insulin plays a critical role in maintaining glucose metabolism135,136, inhibiting NE reuptake137, increasing glycine uptake138, and stimulating protein kinase C activity139 across multiple cell types. Insulin receptors (IRs) are widely expressed throughout the brain140,141, including in neurons137, glial cells such as astrocytes and microglia142,143, and endothelial cells144. IRs exist in two alternatively spliced isoforms: the B isoform (IR-B) and the A isoform (IR-A), the latter of which lacks exon 11. In vitro studies have detected IR-A exclusively in neurons. In vivo work demonstrates that IR-B is present in both neuronal and astrocytic populations but is predominantly expressed by astrocytes145,146. This isoform-specific distribution highlights fundamental differences in insulin signaling between astrocytes and neurons, leading to distinct functional consequences for glucose metabolism and insulin-mediated processes in the brain. The enrichment of IR-B in astrocytes suggests a specialized role in regulating brain glucose metabolism and maintaining energy homeostasis. Functionally, IR-A shows a higher affinity for insulin growth factor II (IGF-II)147 and is associated with mitogenic pathways such as phosphoinositide 3-kinase (PI3K)148,149, although its rapid internalization limits sustained signaling150,151. In contrast, IR-B has a higher affinity for insulin and acts as a more effective regulator of glucose metabolism, particularly in glucose uptake152,153,154. It internalizes more slowly than IR-A, allowing for prolonged metabolic signaling via endosomal pathways. These differences in ligand affinity, expression patterns, and downstream signaling underscore the distinct biological roles of IR-A and IR-B across different cell types. Under normal conditions, insulin binding triggers autophosphorylation of IRs, leading to the activation of two primary signaling pathways: phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) and Ras-Mitogen-activated protein kinase (MAPK)143,155. Through insulin receptor substrate 1 (IRS-1) and IRS-2 activation, the canonical PI3K/AKT pathway regulates glucose transporter type 4 (GLUT4) translocation and glucose uptake156.
Cortical insulin levels decline with aging and in Alzheimer’s disease (AD), accompanied by a reduction in insulin receptor density157,158. While IR ligand affinities remain largely unchanged in AD compared to young individuals, they decrease with advancing age157. Impaired glucose metabolism, potentially resulting from altered CNS IR function or downstream signaling defects, has been implicated in AD onset157. These deficits in insulin signaling contribute to various insulin resistance phenotypes.
Mechanisms of brain insulin resistance
Insulin resistance in the brain can arise from multiple mechanisms, including decreased IR expression, impaired ligand binding, and reduced tyrosine kinase activity159,160. Additionally, impaired insulin transport across the blood–brain barrier (BBB) can exacerbate insulin resistance161. The question of whether the brain is insulin-sensitive has been debated for decades162. However, the widespread distribution of IRs and extensive research demonstrating insulin’s effects on neuronal and glial function strongly support the notion that the brain is an insulin-sensitive organ. Given the differences between peripheral and central insulin action, including variations in IR isoforms, substrate affinity, duration of signaling, and glucose uptake mechanisms, brain insulin resistance cannot be solely defined by peripheral metabolic criteria148, highlighting the importance of central insulin signaling across multiple brain regions (Fig. 2).

Insulin resistance contributes significantly to vascular dysfunction and ANVU dysfunction, through impaired glucose metabolism, oxidative stress, and inflammation. This leads to microvascular rarefaction and vasodilator dysfunction, reducing cerebral perfusion and increasing microinfarcts, microbleeds, and BBB dysregulation. Additionally, insulin resistance promotes amyloid-β accumulation and tau hyperphosphorylation via disrupted insulin signaling, while hypertension further impairs perivascular and glymphatic clearance. Together, these mechanisms accelerate Alzheimer’s-associated pathology and cognitive impairment.
Under conditions of insulin resistance, neurons and glial cells fail to respond effectively to insulin, leading to impaired glucose metabolism, mitochondrial dysfunction, and chronic neuroinflammation163. Dysregulation of the PI3K/AKT and MAPK pathways, essential for neuronal survival and synaptic plasticity, further exacerbates neurodegeneration164. Brain insulin resistance has been directly linked to increased tau hyperphosphorylation and amyloid-β accumulation, the two hallmark pathologies of AD165. Additionally, insulin-degrading enzyme (IDE), which plays a crucial role in Aβ clearance, exhibits decreased activity in AD166,167, a proposal that is not consistent with reductions in central insulin levels, where less insulin would be metabolized, thus providing enhanced opportunities for IDE to metabolize Aβ. Chronic metabolic stress, obesity, and type 2 diabetes further amplify these disruptions, accelerating cognitive decline168,169. Furthermore, induced hyperinsulinemia or exogenous administration of insulin has been shown to improve cognition in AD patients and aged individuals170,171.
Intranasal insulin as evidence for brain insulin sensitivity
Despite ongoing debates regarding central insulin signaling or mechanisms underlying the changes in insulin sensitivity, the success of intranasal insulin in humans in enhancing cognitive function, regulating brain metabolism, and modulating neuroplasticity provides compelling evidence that the brain is an insulin-sensitive organ. Clinical studies have demonstrated that intranasal insulin administration improves declarative memory in healthy individuals172, obese patients173 and patients with mild cognitive impairment (MCI) or AD171,174. Our lab’s findings further support the notion that insulin remains functionally relevant in AD, reinforcing the importance of targeting insulin pathways as a potential therapeutic strategy32,175,176,177. In rats and mice, INI administration shifts astrocytic energy synthesis to fatty acids via fatty acid oxidation, perhaps to produce additional lactate for neuronal use177,178.
The impact of insulin on vascular function
Insulin is known to modulate vasomotor tone both in the periphery and in the CNS. The activation of IRs on the vascular endothelium is known to elicit vasodilation or vasoconstriction through two different pathways (i.e. PI-3K/eNOS and MAPK, respectively)179. Indeed, insulin action regulates generation of NO through PI-3K stimulation180. Central administration of insulin in vivo is associated with increases in heart rate and blood flow and is thought to be mediated in part by eNOS181. When IR is knocked out from endothelial cells, animals display impaired BBB function182. Likewise, enhanced insulin signaling appears to attenuate angiogenesis; increased vascularity has been noted in global IR-knock out mice, which is lost with age183. Thus, results are beginning to suggest the importance of multiple cell types and associated signaling pathways in controlling the structure of the angioarchitecture. Finally, in the clinic, INI treatment has been shown to elevate CBF measures179,184,185. Central hyperinsulinemia, as seen in T2DM and metabolic syndrome, is associated with hypoperfusion, increased risk of cardiovascular disease, and neurovascular uncoupling186,187. In AD, insulin receptors associated with the vasculature display impaired function, suggesting insulin resistance at the level of the BBB188,189. Thus, insulin signaling in the brain appears capable of targeting multiple cytometabolic elements of the ANVU (e.g. neurons, astrocytes, pericytes, endothelial cells, etc.).
Conclusions
Although some of the signaling modulators of ANVC are well defined, it is key to consider both the spatial and temporal associations between vasoactive factors. Recent reports investigating the timing of vasoreactivity place vessel responses before astrocytic calcium flux, despite many early studies describing the opposite1, perhaps due to methodological differences (i.e. conducted in vitro or in anesthetized animals)1,4,190. With the increasing accessibility of 2-photon microscopy and ability to image awake animals, more evidence is emerging supporting this sequence of events. Indeed, data from our group in awake mice and others in anesthetized mice have shown that vasodilation precedes the influx of astrocytic calcium24,50. Neuronal Ca2+ events have also been shown to precede astrocyte response by a few seconds, however it is important to note that a subset of astrocytes (estimated at ~5%) can respond as quickly as these neurons191. Neuronal measures of activity have now been shown to rapidly ( < 2 s) follow vasodilation192, suggesting that neurons may not be the originators of the dilatory response. Given the nature of the length of astrocytic (tens of seconds) and neuronal ( ~ 1 s) Ca2+ responses, the duration of sensory stimulation likely dictates to what extent astrocytes and neurons in the somatosensory cortex contribute to functional hyperemia under tonic and phasic conditions. Despite this, many recent studies utilizing 2-photon imaging are still limited by the thin imaging plane and may not be able to identify vascular reactivity in planes above or below the focal plane where Ca2+ is measured at astrocytic foci. Therefore, future studies should investigate the bioenergetic responses of cells in a larger plane (i.e. 3D 2-photon imaging) to fully characterize ANVC.
Furthermore, given the role of NE in mediating vasoconstriction, understanding how metabolic dysregulation impacts its release could potentially reveal novel therapeutic targets. Indeed, literature suggests that NE modulates astrocyte activity. In vivo light stimulation paired with NE release has been shown to enhance astrocytic Ca2+ signaling193, and is perhaps associated with vasoreactivity. Importantly, astrocyte Ca2+ signaling differs between awake and asleep mice194, perhaps mediated by NE, as the release of NE is thought to propagate Ca2+ signaling in the cortex of awake mice, primarily through α1 receptors195. NE signaling onto astrocytes is also thought to induce the release of ATP for the regulation of postsynaptic efficacy196. Therefore, future work should assess the impact of neuronal and astrocyte photoactivation on α1 adrenergic receptors on vascular smooth muscle cells in a model of diabetes and obesity to investigate the role of NE in ANVC and to test the hypothesis that astrocytes modulate NE release onto the vasculature.
Data availability
No datasets were generated or analysed during the current study.
References
Iadecola, C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 96, 17–42 (2017).
Sokoloff, L. Local cerebral energy metabolism: its relationships to local functional activity and blood flow. Ciba Found Symp. 171–197 (1978).
Iadecola, C. et al. The neurovasculome: key roles in brain health and cognitive impairment: a scientific statement from the American Heart Association/American Stroke Association. Stroke 54, e251–e271 (2023).
Attwell, D. et al. Glial and neuronal control of brain blood flow. Nature 468, 232–243 (2010).
Koehler, R. C., Gebremedhin, D. & Harder, D. R. Role of astrocytes in cerebrovascular regulation. J. Appl. Physiol. 100, 307–317 (2006).
Lecrux, C. & Hamel, E. The neurovascular unit in brain function and disease. Acta Physiol. 203, 47–59 (2011).
Abbott, N. J., Rönnbäck, L. & Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53 (2006).
Iadecola, C. & Nedergaard, M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 10, 1369–1376 (2007).
Hill, R. A. et al. Regional blood flow in the normal and ischemic brain is controlled by arteriolar smooth muscle cell contractility and not by capillary pericytes. Neuron 87, 95–110 (2015).
Ross, J. M. et al. The expanding cell diversity of the brain vasculature. Front. Physiol. 11, 600767 (2020).
Hartmann, D. A., Coelho-Santos, V. & Shih, A. Y. Pericyte control of blood flow across microvascular zones in the central nervous system. Annu Rev. Physiol. 84, 331–354 (2022).
Cauli, B. et al. Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J. Neurosci. 24, 8940–8949 (2004).
Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360 (2004).
Ding, Z. et al. Emerging roles of microglia in neuro-vascular unit: implications of microglia-neurons interactions. Front. Cell Neurosci. 15, 706025 (2021).
Csaszar, E. et al. Microglia modulate blood flow, neurovascular coupling, and hypoperfusion via purinergic actions. J. Exp. Med. 219, e20211071 (2022).
Zhao, X. et al. Microglial interactions with the neurovascular system in physiology and pathology. Dev. Neurobiol. 78, 604–617 (2018).
Nedergaard, M., Ransom, B. & Goldman, S. A. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 26, 523–530 (2003).
Raichle, M. E. & Mintun, M. A. Brain work and brain imaging. Annu Rev. Neurosci. 29, 449–476 (2006).
Drew, P. J. Neurovascular coupling: motive unknown. Trends Neurosci. 45, 809–819 (2022).
Korte, N., Nortley, R. & Attwell, D. Cerebral blood flow decrease as an early pathological mechanism in Alzheimer’s disease. Acta Neuropathol. 140, 793–810 (2020).
Snyder, H. M. et al. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimers Dement 11, 710–717 (2015).
Khachaturian, Z. S. The role of calcium regulation in brain aging: reexamination of a hypothesis. Aging 1, 17–34 (1989).
Shah, D. et al. Astrocyte calcium dysfunction causes early network hyperactivity in Alzheimer’s disease. Cell Rep. 40, 111280 (2022).
Sompol, P. et al. Targeting astrocyte signaling alleviates cerebrovascular and synaptic function deficits in a diet-based mouse model of small cerebral vessel disease. J. Neurosci. 43, 1797–1813 (2023).
Korte, N. et al. Inhibiting Ca(2+) channels in Alzheimer’s disease model mice relaxes pericytes, improves cerebral blood flow and reduces immune cell stalling and hypoxia. Nat. Neurosci. 27, 2086–2100 (2024).
Andersen, J. V. et al. Hippocampal disruptions of synaptic and astrocyte metabolism are primary events of early amyloid pathology in the 5xFAD mouse model of Alzheimer’s disease. Cell Death Dis. 12, 954 (2021).
Mosconi, L. et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med Mol. Imaging 36, 811–822 (2009).
Brooks, W. M. et al. Gene expression profiles of metabolic enzyme transcripts in Alzheimer’s disease. Brain Res. 1127, 127–135 (2007).
Gannon, M. & Wang, Q. Complex noradrenergic dysfunction in Alzheimer’s disease: Low norepinephrine input is not always to blame. Brain Res. 1702, 12–16 (2019).
Frolich, L. et al. A disturbance in the neuronal insulin receptor signal transduction in sporadic Alzheimer’s disease. Ann. N. Y Acad. Sci. 893, 290–293 (1999).
Pancani, T. et al. Distinct modulation of voltage-gated and ligand-gated Ca2+ currents by PPAR-gamma agonists in cultured hippocampal neurons. J. Neurochem. 109, 1800–1811 (2009).
Maimaiti, S. et al. Novel calcium-related targets of insulin in hippocampal neurons. Neuroscience 364, 130–142 (2017).
Lin, R. L. et al. Sensitivity of the S1 neuronal calcium network to insulin and Bay-K 8644 in vivo: relationship to gait, motivation, and aging processes. Aging Cell 21, e13661 (2022).
Foo, L. C. et al. Development of a method for the purification and culture of rodent astrocytes. Neuron 71, 799–811 (2011).
Renden, R. B. et al. Modulatory effects of noradrenergic and serotonergic signaling pathway on neurovascular coupling. Commun. Biol. 7, 287 (2024).
Sofroniew, M. V. & Vinters, H. V. Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35 (2010).
Cai, W. et al. Insulin regulates astrocyte gliotransmission and modulates behavior. J. Clin. Investig. 128, 2914–2926 (2018).
Pellerin, L. & Magistretti, P. J. Glutamate uptake into astrocytes stimulates aerobic glycolysis—a mechanism coupling neuronal-activity to glucose-utilization. Proc. Natl. Acad. Sci. USA 91, 10625–10629 (1994).
Yellen, G. Fueling thought: Management of glycolysis and oxidative phosphorylation in neuronal metabolism. J. Cell Biol. 217, 2235–2246 (2018).
Frazier, H. N. et al. Elevating insulin signaling using a constitutively active insulin receptor increases glucose metabolism and expression of GLUT3 in hippocampal neurons. Front. Neurosci. 14, 668 (2020).
Pancani, T. et al. Imaging of a glucose analog, calcium and NADH in neurons and astrocytes: Dynamic responses to depolarization and sensitivity to pioglitazone. Cell Calcium 50, 548–558 (2011).
Diaz-Garcia, C. M. et al. Neuronal Stimulation Triggers Neuronal Glycolysis and Not Lactate Uptake. Cell Metab. 26, 361–374 e4 (2017).
Bak, L. K. et al. Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 109, 87–93 (2009).
Lundgaard, I. et al. Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat. Commun. 6, 6807 (2015).
Dienel, G. A. Brain lactate metabolism: the discoveries and the controversies. J. Cereb. Blood Flow. Metab. 32, 1107–1138 (2012).
Hertz, L., Peng, L. & Dienel, G. A. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow. Metab. 27, 219–249 (2007).
Verkhratsky, A. et al. Astrocytes in human central nervous system diseases: a frontier for new therapies. Signal Transduct. Target Ther. 8, 396 (2023).
Takata, N. et al. Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J. Neurosci. 31, 18155–18165 (2011).
Haydon, P. G. & Carmignoto, G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 86, 1009–1031 (2006).
Tran, C. H. T., Peringod, G. & Gordon, G. R. Astrocytes integrate behavioral state and vascular signals during functional hyperemia. Neuron 100, 1133–1148 e3 (2018).
Del Franco, A. P., Chiang, P. P. & Newman, E. A. Dilation of cortical capillaries is not related to astrocyte calcium signaling. Glia 70, 508–521 (2022).
Institoris, Á, Rosenegger, D. G. & Gordon, G. R. Arteriole dilation to synaptic activation that is sub-threshold to astrocyte endfoot Ca2+ transients. J. Cereb. Blood Flow. Metab. 35, 1411–1415 (2015).
Rosenegger, D. G. et al. Tonic local brain blood flow control by astrocytes independent of phasic neurovascular coupling. J. Neurosci. 35, 13463–13474 (2015).
Nagele, R. G. et al. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 25, 663–674 (2004).
Norris, C. M. et al. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J. Neurosci. 25, 4649–4658 (2005).
Huffels, C. F. M. et al. Calcium signaling in individual APP/PS1 mouse dentate gyrus astrocytes increases ex vivo with Abeta pathology and age without affecting astrocyte network activity. J. Neurosci. Res. 100, 1281–1295 (2022).
Mitroshina, E. V. et al. Novel algorithm of network calcium dynamics analysis for studying the role of astrocytes in neuronal activity in Alzheimer’s disease models. Int. J. Mol. Sci. 23, 15928 (2022).
Verkhratsky, A. et al. Astrocytes in Alzheimer’s disease. Neurotherapeutics 7, 399–412 (2010).
Olabarria, M. et al. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58, 831–838 (2010).
Zhukov, O. et al. Preserved blood-brain barrier and neurovascular coupling in female 5xFAD model of Alzheimer’s disease. Front. Aging Neurosci. 15, 1089005 (2023).
Krueger, M. & Bechmann, I. CNS pericytes: concepts, misconceptions, and a way out. Glia 58, 1–10 (2010).
Hall, C. N. et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508, 55–60 (2014).
Meza-Resillas, J. et al. Systemic nimodipine affects pericyte calcium signaling, resting hemodynamics and neurovascular coupling in healthy mouse brain. Neurotherapeutics. 22, e00614 (2025).
Nikolakopoulou, A. M. et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat. Neurosci. 22, 1089–1098 (2019).
Stobart, J. L. et al. Altered hemodynamics and vascular reactivity in a mouse model with severe pericyte deficiency. J. Cereb. Blood Flow. Metab. 43, 763–777 (2023).
Kisler, K. et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat. Neurosci. 20, 406–416 (2017).
Li, P. & Fan, H. Pericyte loss in diseases. Cells. 12 (2023).
Costa, E. D. et al. Neuronal nitric oxide synthase in vascular physiology and diseases. Front Physiol. 7, 206 (2016).
Irikura, K. et al. Cerebrovascular alterations in mice lacking neuronal nitric oxide synthase gene expression. Proc. Natl. Acad. Sci. USA 92, 6823–6827 (1995).
Hosford, P. S. & Gourine, A. V. What is the key mediator of the neurovascular coupling response?. Neurosci. Biobehav. Rev. 96, 174–181 (2019).
Meng, W. et al. ACh dilates pial arterioles in endothelial and neuronal NOS knockout mice by NO-dependent mechanisms. Am. J. Physiol. 271, H1145–H1150 (1996).
Prast, H. et al. Nitric oxide-induced release of acetylcholine in the nucleus accumbens: role of cyclic GMP, glutamate, and GABA. J. Neurochem. 71, 266–273 (1998).
Guevara-Guzman, R., Emson, P. C. & Kendrick, K. M. Modulation of in vivo striatal transmitter release by nitric oxide and cyclic GMP. J. Neurochem. 62, 807–810 (1994).
Elhusseiny, A. & Hamel, E. Muscarinic—but not nicotinic—acetylcholine receptors mediate a nitric oxide-dependent dilation in brain cortical arterioles: a possible role for the M5 receptor subtype. J. Cereb. Blood Flow. Metab. 20, 298–305 (2000).
Zhou, M. et al. Blood pressure partially mediated the association of insulin resistance and cerebral small vessel disease: a community-based study. J. Am. Heart Assoc. 13, e031723 (2024).
Yamada, M. et al. Cholinergic dilation of cerebral blood vessels is abolished in M(5) muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. USA 98, 14096–14101 (2001).
Lacroix, A. et al. COX-2-derived prostaglandin E2 produced by pyramidal neurons contributes to neurovascular coupling in the rodent cerebral cortex. J. Neurosci. 35, 11791–11810 (2015).
He, Y. et al. Ultra-slow single-vessel BOLD and CBV-based fMRI spatiotemporal dynamics and their correlation with neuronal intracellular calcium signals. Neuron 97, 925–939 e5 (2018).
Rosenblum, W. I. Inhibition of barium-induced constriction of cerebral surface arterioles by blockers of calcium channels. Blood Vessels 22, 139–144 (1985).
Rosenblum, W. I., McDonald, M. & Wormley, B. Calcium ionophore and acetylcholine dilate arterioles on the mouse brain by different mechanisms. Stroke 20, 1391–1395 (1989).
Rosenblum, W. I. Cerebral microcirculation: a review emphasizing the interrelationship of local blood flow and neuronal function. Angiology 16, 485–507 (1965).
Skinhoj, E. Regulation of cerebral blood flow as a single function of the interstitial pH in the brain. A hypothesis. Acta Neurol. Scand. 42, 604–607 (1966).
Caldwell, H. G. Blunted cerebrovascular CO(2) reactivity to satisfy the hungry heat stressed brain. J. Physiol. 599, 2513–2515 (2021).
Roche, M. et al. In vivo imaging with a water immersion objective affects brain temperature, blood flow and oxygenation. Elife. 8, e47324 (2019).
Iadecola, C. & Gottesman, R. F. Cerebrovascular alterations in Alzheimer disease. Circ. Res 123, 406–408 (2018).
Rosenblum, W. I. Contractile response of pial arterioles to norepinephrine. Effects in the mouse. Arch. Neurol. 31, 197–199 (1974).
Lauritzen, M. Reading vascular changes in brain imaging: is dendritic calcium the key. Nat. Rev. Neurosci. 6, 77–85 (2005).
Agulhon, C. et al. Calcium Signaling and Gliotransmission in Normal vs. Reactive Astrocytes. Front Pharm. 3, 139 (2012).
Lalo, U. & Pankratov, Y. Astrocyte ryanodine receptors facilitate gliotransmission and astroglial modulation of synaptic plasticity. Front Cell Neurosci. 18, 1382010 (2024).
Mishra, A. et al. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 19, 1619–1627 (2016).
Stella, N. et al. Glutamate induces the release of arachidonic acid by interacting with an atypical metabotropic receptor present on mouse brain astrocytes. Ren. Physiol. Biochem. 17, 153–156 (1994).
Metea, M. R. & Newman, E. A. Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J. Neurosci. 26, 2862–2870 (2006).
MacVicar, B. A. & Newman, E. A. Astrocyte regulation of blood flow in the brain. Cold Spring Harb. Perspect. Biol. 7, a020388 (2015).
Shi, Y. et al. Interaction of mechanisms involving epoxyeicosatrienoic acids, adenosine receptors, and metabotropic glutamate receptors in neurovascular coupling in rat whisker barrel cortex. J. Cereb. Blood Flow. Metab. 28, 111–125 (2007).
Mulligan, S. J. & MacVicar, B. A. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 431, 195–199 (2004).
Takano, T. et al. Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 9, 260–267 (2006).
Zonta, M. et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 6, 43–50 (2003).
Bonder, D. E. & McCarthy, K. D. Astrocytic Gq-GPCR-linked IP3R-dependent Ca2+ signaling does not mediate neurovascular coupling in mouse visual cortex in vivo. J. Neurosci. 34, 13139–13150 (2014).
Nizar, K. et al. In vivo stimulus-induced vasodilation occurs without IP3 receptor activation and may precede astrocytic calcium increase. J. Neurosci. 33, 8411–8422 (2013).
He, L., Linden, D. J. & Sapirstein, A. Astrocyte inositol triphosphate receptor type 2 and cytosolic phospholipase A2 alpha regulate arteriole responses in mouse neocortical brain slices. PLoS One 7, e42194 (2012).
De Leo, C., Eftimiadi, C. & Schito, G. C. Rapid disappearance from the intestinal tract of bacteria resistant to rifaximin. Drugs Exp. Clin. Res 12, 979–981 (1986).
Ozawa, K. et al. Astrocytic GPCR-induced Ca(2+) signaling is not causally related to local cerebral blood flow changes. Int. J. Mol. Sci. 24 (2023).
Srinivasan, R. et al. Ca(2+) signaling in astrocytes from Ip3r2(-/-) mice in brain slices and during startle responses in vivo. Nat. Neurosci. 18, 708–717 (2015).
Stobart, J. L. et al. Cortical circuit activity evokes rapid astrocyte calcium signals on a similar timescale to neurons. Neuron 98, 726–735 e4 (2018).
Paulson, O. B. & Newman, E. A. Does the release of potassium from astrocyte endfeet regulate cerebral blood flow?. Science 237, 896–898 (1987).
Gordon, G. R. et al. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 456, 745–749 (2008).
Oe, Y. et al. Distinct temporal integration of noradrenaline signaling by astrocytic second messengers during vigilance. Nat. Commun. 11, 471 (2020).
Reitman, M. E. et al. Norepinephrine links astrocytic activity to regulation of cortical state. Nat. Neurosci. 26, 579–593 (2023).
Wang, Z. H. et al. Therapeutic effects of astrocytes expressing both tyrosine hydroxylase and brain-derived neurotrophic factor on a rat model of Parkinson’s disease. Neuroscience 113, 629–640 (2002).
Li, X. M. et al. Gene expression of aromatic L-amino acid decarboxylase in cultured rat glial cells. J. Neurochem 59, 1172–1175 (1992).
Vardjan, N., Verkhratsky, A. & Zorec, R. Pathologic potential of astrocytic vesicle traffic: new targets to treat neurologic diseases?. Cell Transplant. 24, 599–612 (2015).
Filosa, J. A., Bonev, A. D. & Nelson, M. T. Calcium dynamics in cortical astrocytes and arterioles during neurovascular coupling. Circ. Res. 95, e73–e81 (2004).
Haidey, J. N. et al. Astrocytes regulate ultra-slow arteriole oscillations via stretch-mediated TRPV4-COX-1 feedback. Cell Rep. 36, 109405 (2021).
Institoris, A. et al. Astrocytes amplify neurovascular coupling to sustained activation of neocortex in awake mice. Nat. Commun. 13, 7872 (2022).
Weiss, B. E. et al. Disrupted calcium dynamics in reactive astrocytes occur with endfeet-arteriole decoupling in an amyloid mouse model of Alzheimer’s disease. bioRxiv. p. 2025.01.24.634584 (2025).
Lim, D. et al. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. (1098-1136 (Electronic)).
Ronco, V. et al. Differential deregulation of astrocytic calcium signalling by amyloid-β, TNFα, IL-1β and LPS. Cell Calcium, 55, 219–229 (2014).
Sanchez-Mico, M. V. et al. Soluble Abeta produces an increase in cytosolic and mitochondrial calcium in astrocytes in vivo, in part by direct effect in astroglial cells. Alzheimer’s. Dement. 19, e077883 (2023).
Kook, S. Y. et al. Aβ₁₋₄₂-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca²⁺-calcineurin signaling. J. Nneurosci. 32, 8845–8854 (2012).
Grutzendler, J. & Nedergaard, M. Cellular control of brain capillary blood flow: in vivo imaging veritas. Trends Neurosci. 42, 528–536 (2019).
Wei, H. S. et al. Erythrocytes are oxygen-sensing regulators of the cerebral microcirculation. Neuron 91, 851–862 (2016).
Berthiaume, A. A. et al. Pericyte remodeling is deficient in the aged brain and contributes to impaired capillary flow and structure. Nat. Commun. 13, 5912 (2022).
Nielson, C. D. & Shih, A. Y. In vivo single cell optical ablation of brain pericytes. Front. Neurosci. 16, 900761 (2022).
Coelho-Santos, V. & Shih, A. Y. Pericytes: unsung heroes in myelin repair after neonatal brain hypoxia. Neuron 112, 2081–2083 (2024).
Balbi, M. et al. Dysfunction of mouse cerebral arteries during early aging. J. Cereb. Blood Flow. Metab. 35, 1445–1453 (2015).
Rucker, H. K., Wynder, H. J. & Thomas, W. E. Cellular mechanisms of CNS pericytes. Brain Res. Bull. 51, 363–369 (2000).
Sweeney, M. D., Ayyadurai, S. & Zlokovic, B. V. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat. Neurosci. 19, 771–783 (2016).
Markhotina, N., Liu, G. J. & Martin, D. K. Contractility of retinal pericytes grown on silicone elastomer substrates is through a protein kinase A-mediated intracellular pathway in response to vasoactive peptides. IET Nanobiotechnol. 1, 44–51 (2007).
Peppiatt, C. M. et al. Bidirectional control of CNS capillary diameter by pericytes. Nature 443, 700–704 (2006).
Korte, N. et al. Noradrenaline released from locus coeruleus axons contracts cerebral capillary pericytes via alpha(2) adrenergic receptors. J. Cereb. Blood Flow. Metab. 43, 1142–1152 (2023).
Horlyck, S. et al. ATP induces contraction of cultured brain capillary pericytes via activation of P2Y-type purinergic receptors. Am. J. Physiol. Heart Circ. Physiol. 320, H699–H712 (2021).
Gluck, C. et al. Distinct signatures of calcium activity in brain mural cells. Elife. 10, e70591 (2021).
Hariharan, A. et al. Brain capillary pericytes are metabolic sentinels that control blood flow through a K(ATP) channel-dependent energy switch. Cell Rep. 41, 111872 (2022).
Gonzales, A. L. et al. Contractile pericytes determine the direction of blood flow at capillary junctions. Proc. Natl. Acad. Sci. USA 117, 27022–27033 (2020).
Pearson-Leary, J. et al. Insulin modulates hippocampally-mediated spatial working memory via glucose transporter-4. Behav. Brain Res. 338, 32–39 (2018).
Garcia-Caceres, C. et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 166, 867–880 (2016).
Boyd, F. T. Jr. et al. Insulin receptors and insulin modulation of norepinephrine uptake in neuronal cultures from rat brain. J. Biol. Chem. 260, 15880–15884 (1985).
Yorek, M. A., Dunlap, J. A. & Ginsberg, B. H. Amino acid and putative neurotransmitter transport in human Y79 retinoblastoma cells. Effect of insulin and insulin-like growth factor. J. Biol. Chem. 262, 10986–10993 (1987).
Heidenreich, K. A. et al. Insulin stimulates the activity of a novel protein kinase C, PKC-epsilon, in cultured fetal chick neurons. J. Biol. Chem. 265, 15076–15082 (1990).
Havrankova, J., Roth, J. & Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 272, 827–829 (1978).
Hopkins, D. F. & Williams, G. Insulin receptors are widely distributed in human brain and bind human and porcine insulin with equal affinity. Diabet. Med. 14, 1044–1050 (1997).
Clarke, D. W. et al. Insulin binds to specific receptors and stimulates 2-deoxy-D-glucose uptake in cultured glial cells from rat brain. J. Biol. Chem. 259, 11672–11675 (1984).
Kum, W. et al. Insulin binding and effects on pyrimidine nucleoside uptake and incorporation in cultured mouse astrocytes. J. Neurochem. 49, 1293–1300 (1987).
Bar, R. S., Hoak, J. C. & Peacock, M. L. Insulin receptors in human endothelial cells: identification and characterization. J. Clin. Endocrinol. Metab. 47, 699–702 (1978).
Garwood, C. J. et al. Insulin and IGF1 signalling pathways in human astrocytes in vitro and in vivo; characterisation, subcellular localisation and modulation of the receptors. Mol. Brain 8, 51 (2015).
Spencer, B. et al. Identification of insulin receptor splice variant B in neurons by in situ detection in human brain samples. Sci. Rep. 8, 4070 (2018).
Andersen, M. et al. IGF1 and IGF2 specificities to the two insulin receptor isoforms are determined by insulin receptor amino acid 718. Plos One 12, e0178885 (2017).
Belfiore, A. et al. Insulin receptor isoforms in physiology and disease: an updated view. Endocr. Rev. 38, 379–431 (2017).
Malakar, P. et al. Insulin receptor alternative splicing is regulated by insulin signaling and modulates beta cell survival. Sci. Rep. 6, 31222 (2016).
Vogt, B. et al. The two isotypes of the human insulin receptor (HIR-A and HIR-B) follow different internalization kinetics. Biochem. Biophys. Res. Commun. 177, 1013–1018 (1991).
Yamaguchi, Y. et al. Functional properties of two naturally occurring isoforms of the human insulin receptor in Chinese hamster ovary cells. Endocrinology 129, 2058–2066 (1991).
Belfiore, A. et al. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 30, 586–623 (2009).
Frasca, F. et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 19, 3278–3288 (1999).
Vella, V. et al. Novel mechanisms of tumor promotion by the insulin receptor isoform A in triple-negative breast cancer cells. Cells. 10 (2021).
White, M. F. & Kahn, C. R. The insulin signaling system. J. Biol. Chem. 269, 1-4 (1994).
Arnold, S. E. et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat. Rev. Neurol. 14, 168–181 (2018).
Frolich, L. et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. 105, 423–438 (1998).
Stanley, M., Macauley, S. L. & Holtzman, D. M. Changes in insulin and insulin signaling in Alzheimer’s disease: cause or consequence?. J. Exp. Med. 213, 1375–1385 (2016).
Pessin, J. E. & Saltiel, A. R. Signaling pathways in insulin action: molecular targets of insulin resistance. J. Clin. Investig. 106, 165–169 (2000).
Frazier, H. N. et al. Broadening the definition of brain insulin resistance in aging and Alzheimer’s disease. Exp. Neurol. 313, 79–87 (2019).
Banks, W. A., Owen, J. B. & Erickson, M. A. Insulin in the brain: there and back again. Pharm. Ther. 136, 82–93 (2012).
Milstein, J. L. & Ferris, H. A. The brain as an insulin-sensitive metabolic organ. Mol. Metab. 52, 101234 (2021).
de la Monte, S. M. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr. Alzheimer Res. 9, 35–66 (2012).
Razani, E. et al. The PI3K/Akt signaling axis in Alzheimer’s disease: a valuable target to stimulate or suppress?. Cell Stress Chaperones 26, 871–887 (2021).
Freude, S., Schilbach, K. & Schubert, M. The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer’s disease: from model organisms to human disease. Curr. Alzheimer Res. 6, 213–223 (2009).
Talbot, K. et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 122, 1316–1338 (2012).
Tian, Y., Jing, G. & Zhang, M. Insulin-degrading enzyme: roles and pathways in ameliorating cognitive impairment associated with Alzheimer’s disease and diabetes. Ageing Res. Rev. 90, 101999 (2023).
Soleimanzad, H. et al. Obesity in midlife hampers resting and sensory-evoked cerebral blood flow in mice. Obesity 29, 150–158 (2021).
Li, W. et al. Early effects of high-fat diet on neurovascular function and focal ischemic brain injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R1001–R1008 (2013).
Craft, S. et al. Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiol. Aging 17, 123–130 (1996).
Reger, M. A. et al. Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol. Aging 27, 451–458 (2006).
Benedict, C. et al. Intranasal insulin improves memory in humans: superiority of insulin aspart. Neuropsychopharmacology 32, 239–243 (2007).
Hallschmid, M. et al. Obese men respond to cognitive but not to catabolic brain insulin signaling. Int J. Obes. 32, 275–282 (2008).
Reger, M. A. et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimers Dis. 13, 323–331 (2008).
Maimaiti, S. et al. Intranasal insulin improves age-related cognitive deficits and reverses electrophysiological correlates of brain aging. J. Gerontol. A Biol. Sci. Med. Sci. 71, 30–39 (2016).
Pancani, T. et al. Effect of high-fat diet on metabolic indices, cognition, and neuronal physiology in aging F344 rats. Neurobiol. Aging 34, 1977–1987 (2013).
Anderson, K. L. et al. Impact of single or repeated dose intranasal zinc-free insulin in young and aged F344 rats on cognition, signaling, and brain metabolism. J. Gerontol. A Biol. Sci. Med. Sci. 72, 189–197 (2017).
Taib, B. et al. Insulin acts on astrocytes to shift their substrate preference to fatty acids. iScience 28, 111642 (2025).
Hughes, T. M. & Craft, S. The role of insulin in the vascular contributions to age-related dementia. Biochim. Biophys. Acta 1862, 983–991 (2016).
Potenza, M. A. et al. Treatment of spontaneously hypertensive rats with rosiglitazone and/or enalapril restores balance between vasodilator and vasoconstrictor actions of insulin with simultaneous improvement in hypertension and insulin resistance. Diabetes 55, 3594–3603 (2006).
Cabou, C. et al. Central insulin regulates heart rate and arterial blood flow: an endothelial nitric oxide synthase-dependent mechanism altered during. Diab. Diab. 56, 2872–2877 (2007).
Konishi, M. et al. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc. Natl. Acad. Sci. USA 114, E8478-E8487 (2017).
Fernandez, A. M. et al. Insulin regulates neurovascular coupling through astrocytes. Proc. Natl. Acad. Sci. USA 119, e2204527119 (2022).
Kullmann, S. et al. Insulin action in the human brain: evidence from neuroimaging studies. J. Neuroendocrinol. 27, 419–423 (2015).
Akintola, A. A. et al. Effect of intranasally administered insulin on cerebral blood flow and perfusion; a randomized experiment in young and older adults. Aging 9, 790–802 (2017).
Safar, M. E. et al. Hypertension and vascular dynamics in men and women with metabolic syndrome. J. Am. Coll. Cardiol. 61, 12–19 (2013).
Ausk, K. J., Boyko, E. J. & Ioannou, G. N. Insulin resistance predicts mortality in nondiabetic individuals in the U.S. Diab. Care 33, 1179–1185 (2010).
Leclerc, M. et al. Cerebrovascular insulin receptors are defective in Alzheimer’s disease. Brain 146, 75–90 (2023).
Case, S. L. et al. Falling short: the contribution of central insulin receptors to gait dysregulation in brain aging. Biomedicines. 10 (2022).
Chaigneau, E. et al. The relationship between blood flow and neuronal activity in the rodent olfactory bulb. J. Neurosci. 27, 6452–6460 (2007).
Winship, I. R., Plaa, N. & Murphy, T. H. Rapid astrocyte calcium signals correlate with neuronal activity and onset of the hemodynamic response in vivo. J. Neurosci. 27, 6268–6272 (2007).
Filosa, J. A. et al. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat. Neurosci. 9, 1397–1403 (2006).
Paukert, M. et al. Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 82, 1263–1270 (2014).
Bojarskaite, L. et al. Astrocytic Ca(2+) signaling is reduced during sleep and is involved in the regulation of slow wave sleep. Nat. Commun. 11, 3240 (2020).
Ding, F. et al. alpha1-Adrenergic receptors mediate coordinated Ca2+ signaling of cortical astrocytes in awake, behaving mice. Cell Calcium 54, 387–394 (2013).
Gordon, G. R. & Bains, J. S. Noradrenaline triggers multivesicular release at glutamatergic synapses in the hypothalamus. J. Neurosci. 25, 11385–11395 (2005).
Acknowledgements
The authors would like to thank Kristopher Gumbo for their illustrative help on Fig. 1.
Author information
Authors and Affiliations
Contributions
S.S. and T.H.L. contributed equally to the work and helped frame the conceptualization and literature review, as well as the graphics for the work. O.T., C.N., B.W., R.L.L., L.G. and N.W. helped edit and write sections of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sims, S.L., Lu, TH., Weiss, B.E. et al. Central cytometabolic functional vascular coupling in health and disease. npj Metab Health Dis 3, 47 (2025). https://doi.org/10.1038/s44324-025-00090-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44324-025-00090-1
