
Abstract
The eye presents a very dynamic biomechanical environment, and thus ocular cells must be highly mechanosensitive and mechanoresponsive. Moreover, defects in mechanobiological pathways contribute to a number of sight-threatening ocular diseases, highlighting the importance of ocular mechanobiology. We here give a concise overview of the mechanobiology of ocular cells in the lens and cornea (and how mechanobiology plays a role in associated pathologies in these tissues), before providing a detailed review of the mechanobiology of the common blinding disease, glaucoma. Mechanical stimuli are intimately linked with the pathology of glaucoma, both in terms of altered homeostasis of the eye’s internal pressure control system and in the response of neural cells to elevated pressure in the eye. A complex array of mechanosensory elements (stretch-activated ion channels, integrins, G protein-coupled receptors) work together with intersecting networks of mechanotransducing pathways in cells of both the posterior and anterior eye in glaucoma. Despite intense research efforts over the past decades, much remains unknown about the mechanobiology of glaucoma. Continued investigation of glaucomatous mechanobiology is important, as it may reveal novel targets for treating this challenging disease.
Similar content being viewed by others


Introduction
The eye is a remarkably complex organ that permits sensing of visual information, an ability which is hugely advantageous for almost all higher organisms, including humans. Indeed, vision loss is consistently ranked as one of the most feared disabilities with significant impact on quality of life1,2, and thus understanding the function of the eye has long been of great scientific and clinical interest.
Our goal in this work is to provide a concise survey of mechanobiology in vision. To those not familiar with the field, it may be initially surprising that mechanobiology is critically important in ocular physiology and pathology. However, the mammalian eye is a very biomechanically active environment: it is endowed with external (extraocular) and internal muscles, with a fluid circulatory system that internally pressurizes the eye globe, and with a rich vascular network. Further, the posterior eye is acted upon by cerebrospinal fluid pressure. It follows that ocular cells and tissues must be able to sense, transduce and respond to a variety of biomechanical stimuli.
Terminology
For the avoidance of doubt, we here explicitly define the terminology we will use throughout this review. We define mechanosensing as the ability of a cell to sense mechanical cues from its microenvironment3, whereas we define mechanotransduction as the cellular processes that translate mechanical cues into biochemical signals4. Collectively, we refer to these processes, as well as any downstream signaling triggered by mechanotransduction, as mechanobiology.
Brief overview of two ocular pathologies involving mechanobiology
Mechanobiological processes often become particularly evident in pathological states. Here we touch on two such states in the eye to give readers a sense of the scope of ocular mechanobiology: posterior capsule opacification (PCO) and keratoconus. We note that the brief treatment of these two topics is not a reflection of them being unimportant; quite the contrary, they are very important, but are somewhat under-studied, and thus present excellent opportunities for novel research. We further recognize that there are other important ophthalmic diseases that likely involve abnormal biomechanics, including myopia5,6, age-related macular degeneration7, and diabetic retinopathy8,9. However, even though biomechanics and mechanobiology play roles in these conditions, the evidence for aberrant mechanobiological processes being critical initiators of pathology in these conditions is much less robust. Due to space constraints, we thus do not further discuss these conditions in this work, and note that further research in these areas is needed.
The lens and posterior capsular opacification
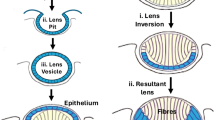
The lens helps focus light on the retina and its form and function are complex. Accommodation is the process by which the eye changes its focal length, and involves the action of the ciliary muscles so as to change the shape of the lens10. Anatomically, moving from the center of the lens to its outer surface, the lens is formed by lens fibers; the lens epithelium whose apical surface is oriented towards the center of the lens; and the lens capsule, the basement membrane of the epithelium (Fig. 1)11. This capsule is rather thick (4–15 µm in humans, depending on age and location12) and demonstrates heterogeneous biomechanical properties that are predicted to lead to a spatially equibiaxial tensile stress field in the capsule13,14, strongly suggesting the existence of a homeostatic mechano-feedback loop mediated by lens epithelial cells. This homeostatic environment is disrupted in patients receiving cataract surgery, in which a portion of the anterior lens capsule is removed; this disruption leads to PCO in c. 30% patients 5 years after surgery15, with significant negative effects on vision. The pathophysiology of PCO is complex but in some forms is thought to involve aberrant migration and extracellular matrix (ECM) production by lens epithelial cells after capsular disruption16. It is thus thought that lens epithelial cell mechanobiology may be important in PCO17, yet surprisingly little is known about relevant pathways. Cheng and colleagues have shown that Eph-ephrin signaling is important in lens biomechanical signaling18,19, yet much more remains to be discovered about mechanobiology in this intriguing and clinically important area.

. The inset at top right (red box) shows a magnified schematic view of the cornea, including (from anterior to posterior): the corneal epithelium (Epi), Bowman’s Layer (BL), the corneal stroma with resident keratocytes (dark blue), Descemet’s membrane (DM), and the corneal endothelium (Endo). The insets at top left show two magnified views of the conventional outflow pathway, located where the cornea, iris and sclera join. The purple boxed inset gives a schematic overview, while the green boxed image is a scanning electron micrograph from a human eye, in which Schlemm’s canal (SC), the trabecular meshwork (TM), the cornea (C) and the sclera (S) are identified. At lower right (orange box), the lens is shown, with details of its internal structure. Finally, at lower left (blue box), a schematic view of the optic nerve and surrounding structures is shown, with blood vessels and other structures identified, including the posterior ciliary arteries (PCA), the retina, the sclera (S), the central retinal artery and vein (CRA), the subarachnoid space, the optic nerve, the pia mater, the dural sheath, and the circle of Zinn and Haller (CZ). Immediately to its left (purple box) is an en face view of the optic nerve head region, in which a digestion process has been carried out to leave only the major connective tissues, namely the sclera and lamina cribrosa (LC). The schematic image of the cornea was created in Biorender.com and is based on ref. 284, the image of the lens is taken from ref. 285, the overview of the conventional outflow pathway is modified from ref. 286, the scanning electron micrograph of the outflow pathway is modified from ref. 159, the overview of the optic nerve head is modified from ref. 287, and the en face view of the optic nerve head is modified from ref. 288.
The cornea and keratoconus
The cornea is the clear tissue at the front of the eye which must transmit light efficiently for good visual acuity (Fig. 1). It also has an important structural role, which conflicts somewhat with the requirement for transparency. The cornea has therefore evolved to have a remarkable structure: a central stroma is located between anterior epithelial and posterior endothelial cells, each of which has a basement membrane, resulting in a five layer “sandwich”.
Considering first the anterior-most aspect of the cornea, corneal epithelial cells are endowed with a number of known mechanosensors, including several TRPV-family stretch-activated ion channels20,21,22,23,24, focal adhesion kinase (FAK) and paxillin25, and integrins26 including α6β4 and α3β1. Further, the corneal epithelium is exposed to a variety of external stresses, notably shear stresses including those due to tear film motion and blinking, as well as more complex stresses due to eye rubbing and contact lens wear27. These stresses, as well as the effects of substrate stiffness and topography, have been shown to affect a number of important aspects of corneal epithelial phenotype, including cytoskeletal architecture, junctional architecture, migration, apoptosis, and stemness, as reviewed in refs. 27,28.
We next consider the stroma, where stromal keratocytes are embedded in a dense connective tissue notable for its highly ordered collagen bundle structure and regular spacing, necessary for tissue optical clarity. Keratocytes are responsible for maintaining stromal ECM structure in the face of mechanical loading due to intraocular pressure (IOP), as well as for wound repair. Stromal keratocytes are sensitive to substrate stiffness and topography in a complex manner that depends on the local signaling environment, e.g., responses are modulated by levels of transforming growth factor-β1 (TGF-β1), interleukin-1β and tumor necrosis factor-α. Interested readers are referred to the excellent review by Thomasy et al. of this topic28. These cells are also sensitive to stretch29,30,31, important in the development of the common pathology known as keratoconus, in which the cornea experiences significant abnormal deformation32. Mechanosensing and mechanotransduction in corneal keratocytes are intriguing: these cells form a syncytium and it has been shown that transient cell membrane disruptions (TPMDs; essentially transient micro-tears in the cell membrane) induced by ostensibly benign events such as eye rubbing, led to calcium increases in the affected cell, which in turn caused calcium waves in neighboring cells33. Focal adhesions have also been implicated as an important mechanosensory in corneal keratocytes34, with downstream signaling involving YAP/TAZ28.
Finally, we consider the corneal endothelium, which actively pumps ions, and hence fluid, to de-swell the cornea and thereby control stromal hydration and maintain optical clarity. These endothelial cells lay down a specialized basement membrane (Descemet’s membrane; DM) and changes in the stiffness or topography of DM area strongly associated with corneal endothelial pathologies35. A number of studies have identified YAP and TAZ as key mechanotransducers in corneal endothelial cells (carefully reviewed in28), with changes in YAP/TAZ nuclear translocation being driven by changes in DM stiffness. YAP/TAZ mechanotransduction is modulated by TGF-β1, TGF-β2 and Wnt signaling, and these pathways are the subject of multiple studies in corneal tissue engineering as treatment for corneal dystrophies28. In brief, increased nuclear translocation of YAP led to more endothelial cell proliferation; conversely, several models in which YAP or TAZ nuclear translocation was reduced demonstrated reduced endothelial cell numbers, abnormal endothelial morphology and reduced endothelial regeneration after wounding, which are also features of corneal dystrophies. These data implicate YAP/TAZ signaling as being important in corneal dystrophies, although specific downstream signaling events from altered YAP/TAZ activation in these pathologies are currently unknown.
Glaucoma
Glaucoma is a complex family of pathologies of diverse etiology whose members share certain common clinical features, including structural change in the optic nerve head (ONH; Fig. 1) and progressive, irreversible damage to retinal ganglion cell (RGC) axons36,37,38. The RGCs are responsible for transmission of visual information from the retina to the brain, and damage to these cells is the sight-threatening event in glaucoma and cause of visual field deficits39,40,41,42. This disease affects nearly 80 million people worldwide, is the second leading cause of blindness in the modern world, costs $3+ billion annually in the US alone43, and will increase in prevalence as the population ages44.
Relevant to this article, the pathophysiology of glaucoma is closely intertwined with mechanics and mechanobiology. Many (although not all) glaucoma patients demonstrate elevated IOP; however, no matter the starting IOP, significant, sustained IOP reduction benefits patients45,46,47,48. Consequently, all current therapeutic approaches seek to reduce IOP, indicating a key role for mechanobiology in this disease49,50. Thus, and perhaps not surprisingly, glaucoma is the ophthalmic condition in which mechanobiology has been best studied, as will be seen below. Specifically, the study of why IOP is dysregulated in some glaucoma patients, and why elevated IOP causes RGC damage, are two topics of intense study that directly involve mechanobiology. We consider each of these in turn below.
Mechanobiology of the outflow pathway in glaucoma
IOP is determined and maintained by the constant secretion of aqueous humor, a clear fluid that fills the anterior eye, and by the resistance to its outflow from the anterior chamber. Outflow passes through the trabecular meshwork (TM) and Schlemm’s canal (SC) inner wall endothelium with its basal lamina51,52,53, the principal tissues of the conventional outflow pathway (Fig. 2A). Pathological alterations in the outflow tissues cause the characteristically increased outflow resistance in high-IOP glaucoma52,54. Strong evidence supports the existence of multiple mechanobiological homeostatic feedback loops regulating IOP55; generally, IOP elevation causes increased mechanical stimulation (stretch on TM cells and shear stress acting on SC cells), which in turn leads to phenotypic changes in outflow pathway tissues predicted to renormalize IOP, including matrix metalloproteinase secretion in response to TM stretch56,57,58, increased nitric oxide production in response to shear stress on SC cells59,60, and formation of pores in SC cells61,62. Thus, impairment of the ability of TM and/or SC cells to sense or transduce mechanobiological stimuli are expected to impair IOP homeostasis; as we will see below, there are a number of molecular players in these homeostatic pathways.

A Serial block-face scanning electron micrograph showing the interface between a Schlemm’s canal (SC) inner wall cell and a juxtacanalicular-trabecular meshwork (JCT-TM) cell, with direction of aqueous humor flow indicated by the yellow arrow (reproduced and adapted from ref. 289 with permission from the authors under a Creative Commons license). B Schematic overview of relevant mechanosensing machineries including mechanosensitive ion channels (TRPV4, Piezo1, TREK-1), integrin transmembrane receptors, and G protein-coupled receptors (angiotensin II type 1 (AT1R) receptor). C Schematic overview of relevant mechanotransduction machineries including focal adhesion complexes (talin, paxillin, kindlin, vinculin, FAK, α-actinin, zyxin, VASP), cytoskeleton networks (actin filaments, intermediate filaments, microtubules), and shuttling proteins (YAP/TAZ, Wnt/β-catenin, glucocorticoid (GC)/soluble GC receptor). Schematics in (B, C) created using BioRender.com.
In addition to direct alterations in mechanosensing and/or mechanotransduction in outflow pathway tissues, there are important secondary effects in glaucomatous eyes. Notably, compelling evidence supports the observation that the TM from glaucoma eyes is markedly stiffer compared to that from ostensibly normal eyes63,64,65,66, which in turn may negatively affect the tissue’s response to mechanical stretch even from normal IOP fluctuations67. Consequently, TM and SC inner wall cells in the glaucomatous outflow tract are exposed to altered biomechanical stresses61,66 that govern diverse mechanobiological adaptation processes involving cellular mechanosensing and mechanotransduction apparatuses. We discuss these separately in the following sections.
Mechanosensing
TM cells are equipped with several key mechanosensors such as mechanically-gated ion channels, integrin transmembrane receptors, and G protein-coupled receptors (GPCRs) (Fig. 2B).
Mechanosensitive ion channels
Mechanosensitive ion channels are directly and rapidly activated by stresses acting on the lipid bilayer or its associated non-membrane components68. Among the various types of channels found in the TM, TRPV469,70,71,72,73,74,75,76,77, Piezo178,79,80,81, and TREK-182,83,84,85 are the best understood. Interested readers are referred to recent reviews for further details on mechanosensitive ion channel biology86,87,88. TRPV4 channels in TM cells are activated by physiological pressure steps80,85 and strains69,70 to activate outflow-relevant downstream signaling components/mechanisms including Rho kinase, F-actin, paxillin and vinculin, reorganization of membrane lipids, and ECM release69,70,71. Recent evidence suggests that TRPV4 activity underpins increased outflow resistance under both physiological and pathological conditions75. The data that tonic TRPV4 activity is obligatory to maintain TM contractility and TGF-β2-induced ocular hypertension75 may help resolve contradictory conclusions from previous studies, which implicated TRPV4 signaling in both ocular hypertension and hypotension69,70,71,72,76,80,89. For instance, TRPV4 has been proposed to lower IOP through phosphoinositide signaling in primary cilia89, promote the release of polyunsaturated fatty acids72, and activate downstream Piezo1 mechanosensing90. However, TRPV4-regulated Ca2+ influx in TM cells is unaffected by primary cilia ablation69, polyunsaturated fatty acids stimulate rather than inhibit TRPV469, and TRPV4 signaling in TM cells is unaffected by pharmacological or genetic Piezo1 inhibition80. Furthermore, Piezo1 inhibition reduces outflow facility ex vivo and in vivo80,81, suggesting opposing functions for Piezo1 vs. TRPV4 activation in the context of IOP homeostasis. Taken together, ion channel-mediated mechanosensing plays a clear role in modulating TM cell behavior, with only little published information available on SC cells. It is conceivable that small-molecule TRPV4 antagonists could be explored for clinical IOP lowering strategies to protect retinal neurons; yet, additional studies will be required to refine the involvement and cross-regulation of different mechanosensitive ion channels in outflow tissue physiology and glaucoma onset/progression.
Integrin transmembrane receptors
Integrin transmembrane receptors link the ECM to the actomyosin cytoskeleton via a complex of adapter proteins91. There are 12 different integrins in the outflow pathway92. While most of them are expressed in both the TM and SC (with some differences in the levels of α2β1, α3β1, α6β1, αvβ3, αvβ5 integrins92), their activity is likely to vary and subject to modulation. Consequently, the mechanosensing function of integrins is thought to change dynamically in tandem with alterations in the biophysical properties of the outflow tract. Interested readers are referred to recent reviews for more details on the plethora of biological processes in the TM controlled by integrin signaling92,93,94,95. Of all integrins in the outflow pathway, the αvβ3 integrin is most likely to be a key player in glaucoma pathophysiology. Unlike other integrins, αvβ3 integrin has several ligands implicated in outflow tissue and IOP homeostasis. For instance, connective tissue growth factor96 and fluid shear stress97 were shown to increase αvβ3 integrin activity in TM cells, with further implications for SC inner wall cells97. The glaucoma-associated glucocorticoid dexamethasone also increases the expression and activation of αvβ3 integrin through a secondary effect involving the transcription factor NFATc198,99. By the same token, pro-fibrotic TGFβ2100 was shown to increase TM cell αvβ3 integrin expression101, with its activation further driving TGFβ2 expression via a potential feedback loop102. In addition, TGFβ2-induced thrombospondin-1 expression can activate αvβ3 integrin signaling103.
Another important feature of αvβ3 integrin in mechanosensing is its spatial localization; αvβ3 integrins are found in podosomes/invadopodia-/filopodia-like structures (including tunneling nanotubes) in TM cells104,105,106 and exhibit stable localization in focal adhesions driven by forces on the actomyosin network107,108. However, αvβ3 integrin has a weaker bond strength and faster binding/unbinding rates compared to other integrins such as α5β1 integrin within focal adhesions109. This makes αvβ3 integrin well suited to sense and modulate changes in the contractile properties of the TM/SC cytoskeleton (possibly acting as an on/off switch) involving force-induced changes in its interactions with ECM ligands110. Lastly, we consider known αvβ3 integrin crosstalk. Cooperative crosstalk between activated αvβ3 and β1 integrins in TM cells drives the formation of glaucoma-associated cross-linked actin networks111 in a Rac1-dependent manner99,103,112. In another Rac-1-mediated signaling event, activated αvβ3 integrin was shown to decrease phagocytosis by inhibiting αvβ5 integrin activity113,114 via transdominant crosstalk115. Cooperative signaling between αvβ3 and α5β1 integrins in focal adhesions facilitates RhoA-mediated fibronectin fibrillogenesis in TM cells; specifically, it increases the deposition of alternatively spliced fibronectin-EDA116 that is associated with ECM changes observed in ocular hypertensive glaucoma117. Consistent with these critical functions in TM cellular processes, αvβ3 integrin activation was shown to decrease outflow facility and increase IOP in mice, while its knockdown had the opposite effect and decreased IOP118. In summary, owing to their combined biochemical and mechanical properties, integrin receptor-mediated mechanosensing plays a central role in modulating outflow cell/tissue homeostasis and glaucoma pathophysiology. The wealth of data on αvβ3 integrin signaling highlights the importance of investigating mechanistic details of how integrins sense, respond to, and interact with ECM of varying properties with exquisite specificity. And while not without challenges, recent advances in other research fields119 suggest that clinical development of new small-molecule inhibitors targeting αvβ1 integrins may be a viable strategy for treating ocular hypertensive glaucoma given the crosstalk with αvβ3 integrin in aberrant cross-linked actin network formation.
G protein-coupled receptors
GPCRs comprise the largest class of cell surface receptors120. Emerging evidence suggests that mechanosensitive 7-transmembrane GPCRs facilitate cellular responses to biophysical cues, such as mechanical stretch and fluid shear stress121,122, in addition to light or chemical ligands121. This is thought to be a slower mechanism (seconds to minutes) compared to mechanically-gated ion channels (milliseconds)121. The angiotensin II type 1 (AT1R) receptor was among the first GPCRs for which ligand-independent activation by mechanical forces was shown123, with clear implications in modulating outflow function and IOP homeostasis. Interested readers are referred to recent reviews for additional details on the role of the renin-angiotensin system in outflow (patho)physiology124,125,126. Upon binding to AT1R, angiotensin II has been shown to induce TM cell proliferation and increase mRNA and protein expression of glaucoma-associated fibrotic markers (e.g., collagen type I, fibronectin, phospho-myosin light chain (p-MLC), α-smooth muscle actin (αSMA)), possibly via a NOX4/ROS axis in cooperation with the SMAD3/TGFβ pathway127,128,129. Other studies showed that angiotensin II decreases outflow facility in rabbits130 and monkeys131,132, with additional evidence that AT1R activation may modulate uveoscleral outflow and elevate IOP133,134,135. By contrast, angiotensin (1–7)—the product of angiotensin II conversion by ACE2136—was shown to oppose the molecular and cellular effects of Ang II and lower IOP130,137, with potential benefits for glaucoma therapy138. Taken together, GPCR-mediated mechanosensing contributes to modulating aqueous humor dynamics and IOP homeostasis. Of note, there are presently no studies that directly interrogated mechanosensitive GPCRs in SC cell/tissue function despite the known regulatory role of shear stress139. This opens the door for nuanced future investigations to refine the involvement and regulation (incl. crosstalk) of the renin-angiotensin system in outflow tissue physiology and pathophysiology in glaucoma.
We recognize that there are additional mechanosensing structures present throughout the outflow pathway that may mediate cellular responses to mechanical stretch and fluid shear stress. These include the somewhat lesser studied (yet no less important) putative mechanosensitive GPCRs140 as well as receptor tyrosine kinases, glycocalyx proteins, and junctional proteins3. Unfortunately, their inclusion is outside the scope of this work and readers are referred to the primary citations above.
Mechanotransduction
TM cells are endowed with key mechanotransduction machineries, including focal adhesion complexes, cytoskeleton networks, and shuttling proteins (Fig. 2C).
Focal adhesion complexes
Focal adhesion complexes transfer mechanical cues from the ECM to the actomyosin cytoskeleton. To execute this task, focal adhesions have two distinct compartments: an integrin transmembrane receptor91 (discussed above) and an intracellular linkage complex composed of the large adapter protein talin141 and numerous additional proteins such as kindlin, vinculin, paxillin, zyxin, and vasodilator-stimulated phosphoprotein (VASP)91 that jointly form the interface with actin filaments. Another critical component of many cell-matrix junctions is FAK, serving important functions in downstream signaling142. The molecular composition of focal adhesions is highly variable, dynamic, and sensitive to ECM composition and mechanics143; this has direct implications for outflow tissue function and IOP regulation in health and disease. For example, ECM rigidity/stiffness has been shown to modulate focal adhesion size in TM cells. Vinculin puncta were larger and more frequent on stiff compared to soft matrix, independent of the culture substrate type (i.e., glass, synthetic polyacrylamide gel, natural ECM hydrogel)144,145. Of note, comparable observations were made in SC cells, showing more prominent vinculin-containing focal adhesions with increasing substrate stiffness61. In TM cells, FAK activation via phosphorylation increased with increased substrate rigidity144. By the same token, mechanical stretch was found to increase colocalization between vinculin and phospho-FAK, along with paxillin, in a TRPV4- and Rho-associated kinase (ROCK)-dependent manner70. As with other focal adhesion components, mechanical stretch augmented zyxin phosphorylation and translocation toward actin stress fibers in TM cells70. Lastly, as in indirect link to glaucoma pathophysiology, VASP was detected in the aqueous humor of glaucoma patients, suggesting that the outflow tissue endothelial barrier is altered in glaucoma146. Taken together, focal adhesion-mediated mechanotransduction processes play an integral role in TM/SC cell (patho)biology. Yet, there is no data on functional modulation of aqueous humor outflow facility and IOP homeostasis.
Cytoskeleton networks
The propagation of extracellular and cell-generated forces is mediated by the regulation of cytoskeleton tension147. The cytoskeleton networks are dynamic structures composed of filamentous and crosslinking proteins that provide mechanical support to cells and control their physiological functions148. There are three principal cytoskeleton networks that differ in composition: actin filaments, microtubules, and intermediate filaments—each displaying a highly organized structure. Filamentous (F)-actin and non-muscle myosin II form the contractile actomyosin system that is present in essentially all cells149. Multiple globular (G)-actin subunits polymerize to form linear F-actin, and several F-actin fibers arrange into actin bundles. The small Rho GTPase RhoA directly promotes the assembly of contractile actin stress fiber bundles by activating the downstream effectors ROCK and the formin mDia1/2150,151. The actomyosin network in TM cells, closely resembling that of smooth muscle cells, was first described in the late 1970s152,153. Since then, hundreds of research reports have focused on and refined the critical role of the actomyosin system in outflow tissue homeostasis as well as in the development of ocular hypertensive glaucoma. Interested readers are referred to recent reviews for further details154,155,156.
The F-actin cytoskeletal arrangement differs significantly in healthy compared to glaucomatous TM cells in vitro and in vivo157,158, with the former exhibiting an organized pattern with linear stress fibers and the latter exhibiting a disorganized network of tangled actin structures known as crosslinked actin networks. Actin fibers in both the TM and SC are affected by mechanical factors159,160 and they have been associated with increased cell/tissue stiffness, outflow resistance and IOP65,111, providing compelling evidence for the critical importance of the actomyosin cytoskeleton in aqueous humor outflow modulation128,159. Therefore, the outflow tissue actomyosin system has been the focus of several pharmacological approaches to improve outflow function and lower IOP in glaucoma. Strategies targeting the Rho-GTPase signaling pathway have proved most promising, given its crucial role in regulating aspects of cell shape, motility, proliferation, and apoptosis throughout the body161. Upon binding to GTP, Rho proteins stimulate the downstream effector ROCK. ROCK in turn promotes myosin II activity by activating/phosphorylating myosin light chain (MLC) and by inhibiting MLC phosphatase. In the conventional outflow pathway, ROCK regulates TM cell actomyosin contraction, adhesion, shape and stiffness, as well as ECM reorganization via this process—all involved in outflow resistance generation162. The specific ROCK inhibitor Y27632 was shown to induce potent but reversible changes in TM cell morphology and to decrease actin stress fibers, focal adhesions, and cell-cell junctions in vitro163,164 in a substrate-independent manner165. In vivo administration of Y27632 resulted in increased outflow facility and decreased IOP in different animal models as well as in enucleated eyes128,163,164,166,167. Consequently, ROCK inhibitors were pursued for therapeutic development. Ripasudil entered the commercial market in Japan in 2014167,168, while Netarsudil became available in the USA in 2018 followed by approval for use in Europe in 2019169,170. Taken together, cytoskeleton network-mediated mechanotransduction—and specifically the actomyosin system—contributes to modulating outflow cell/tissue homeostasis and glaucoma pathophysiology. The development of ROCK inhibitors for clinical management of ocular hypertensive glaucoma deserves to be considered a true “bench-to-bedside” success story that highlights the potential of targeted cytoskeleton-modulating therapies.
Shuttling proteins
The mechanical information arising from ECM modifications (perceived by focal adhesion complexes and propagated via cytoskeleton networks) impacts cytoplasmic proteins, inducing their structural modification and subsequent translocation to the nucleus4. Among the first proteins identified to shuttle between subcellular compartments was the focal adhesion adapter zyxin. Upon mechanical stretch, zyxin detaches from the membrane site and moves into the nucleus171. Several mechano-actuated shuttling proteins not physically connected to focal adhesions have also been described. The emerging picture is that bidirectional shuttling proteins are key factors in cellular mechanotransduction. Relevant to outflow tissue (patho)physiology are the YAP/TAZ172, Wnt/β-catenin173, and glucocorticoid174 pathways—all governed by protein nuclear translocation. Of note, all of these signaling pathways crosstalk with YAP/TAZ mechanotransduction in TM cells. Interested readers are referred to a recent comprehensive review by Ghosh and Herberg of this complex topic175.
YAP and its paralog TAZ172 are transcriptional coactivators and master regulators of cellular mechanotransduction176,177. YAP/TAZ activity is modulated by a broad network of input cues, serving as a signaling nexus and integrator of multiple pathways178. YAP/TAZ proteins constantly shuttle between the cytoplasm and nucleus. Upon nuclear translocation, they bind to cognate transcription factors, such as TEAD family proteins, as YAP/TAZ lack DNA-binding domains. Via this principal regulatory mechanism, YAP/TAZ drive the expression of known downstream effectors and glaucoma-related putative effectors (e.g., transglutaminase-2, fibronectin, αSMA). In the Hippo-independent pathway, YAP/TAZ activity is primarily regulated by tension of the F-actin cytoskeleton that is contingent on the rigidity of the cellular substrate. For instance, increased ECM stiffness strongly promotes YAP/TAZ nuclear translocation to drive stiff-responsive gene expression involving increased ROCK activity and actomyosin contractile force generation179,180. Substantial evidence supports a clear role of YAP/TAZ mechanotransduction in ocular development, homeostasis, and disease181,182. For example, YAP/TAZ were found to be activated by glaucoma-associated stressors in TM cells145,183,184,185,186,187,188,189,190,191,192,193,194,195 and SC cells196,197. A recent genome-wide association study further identified the gene YAP1 encoding for YAP—but not TAZ—among previously unknown glaucoma risk loci across different ancestries198, suggesting a potential causal link between abnormal YAP/TAZ mechanotransduction and outflow dysfunction in ocular hypertensive glaucoma.
The evolutionarily conserved Wnt signaling pathway regulates critical aspects of embryonic development and tissue homeostasis173. Extracellular Wnt proteins stimulate diverse intracellular signal transduction cascades, including the canonical Wnt/β-catenin pathway and the alternative non-canonical pathway (i.e., Wnt/Ca2+ and Wnt planar cell polarity) that are subject to mutual regulation173,199. Without Wnt signaling, cytosolic β-catenin is recruited into the APC/Axin/GSK3/CK1 destruction complex, resulting in its phosphorylation followed by degradation. Binding of Wnt to its receptor complex composed of Frizzled and the low-density-lipoprotein-related protein5/6 triggers a series of events that disrupts the APC/Axin/GSK3 complex required for the targeted destruction of β-catenin. Via this mechanism, β-catenin triggers transcription by nuclear translocation and interaction with TCF/LEF transcription factors200. Wnt/β-catenin signaling has been studied extensively in numerous ocular diseases including glaucoma201,202. Wnt inhibitors such as sFRP1 and DKK1 are elevated in glaucoma, and their overexpression results in decreased aqueous humor outflow203. Furthermore, sFRP-1 levels are increased in TM cells cultured on stiff substrates195, and Wnt inhibition has been shown to cause cell stiffening204.
The pleiotropic actions of glucocorticoids are mediated by glucocorticoid signaling174. Glucocorticoids bind to intracellular glucocorticoid receptors, transcription factors of the nuclear receptor superfamily that are typically activated by ligands. These ligand-receptor complexes translocate into the nucleus where they interact with glucocorticoid response elements to modulate broad transcriptional activities (i.e., target genes comprise ~10–20% of the human genome) in what is considered the classical or “genomic” glucocorticoid pathway205. In contrast, glucocorticoids can also exert their actions in a more rapid “non-genomic” manner that affects several signaling pathways such as PKC, Ca2+/calmodulin protein kinase II, nitric oxide, mitogen-activated protein kinase, caveolin-1, RhoA/ROCK206; this mechanism does not require nuclear glucocorticoid receptor-mediated transcription or translation174. Sustained use of widely prescribed glucocorticoids, such as prednisone/prednisolone or dexamethasone, has long been known to induce ocular hypertension in susceptible individuals causing glucocorticoid-induced glaucoma (a type of secondary open-angle glaucoma)207,208,209,210,211. Exposure of TM cells to dexamethasone induces ECM deposition (e.g., collagens, fibronectin, laminin, glycosaminoglycans, elastin) and cell/ECM stiffening212,213. Furthermore, dexamethasone drives the formation of characteristic crosslinked actin networks111,214, induces ER stress215, and compromises TM cell phagocytic function216. Taken together, shuttling protein-mediated mechanotransduction plays a clear role in modulating TM cell behavior, with only little published information available on SC cells. The YAP/TAZ mechanotransduction pathway is by far the best understood. According to clinicaltrials.gov, there are several active clinical trials centered around blocking YAP/TAZ-TEAD interaction for the treatment of different cancers, with the potential to “cross-pollinate” and inform future glaucoma drug development approaches.
We recognize that there are additional cytoskeletal networks and shuttling protein-mediated mechanotransduction pathways relevant to the outflow pathway. These include the septin component of the cytoskeleton217 and the TGFβ218 and Notch219 signaling pathways. Due to space constraints, we are not able to discuss them in this work.
Mechanobiology of RGC damage
RGC damage in glaucoma is an incompletely understood process, for which many mechanisms have been postulated. Here, we focus on mechanical insult at the level of the ONH, and more specifically the connective tissue region within the ONH known as the lamina cribrosa (LC; Figs. 1 and 3), since significant data supports this as an important mechanism49,220. For example, RGC damage occurs within the LC221, where significant mechanical tissue deformation occurs222; patterns of vision loss in glaucoma are consistent with the heterogeneous physical structure of the LC223; and ONH cells are mechanoresponsive224,225,226,227.

Black and white background shows detailed anatomy in a human eye (R = retina; S = sclera; LC = lamina cribrosa; CRV = central retinal vessel). Lower left inset shows individually labeled optic nerve head astrocytes in a cross-sectional view through the optic nerve at the level of the lamina cribrosa. Individual cell types are overlain on the background image (RGC = retinal ganglion cell, including myelin sheath posterior to the lamina cribrosa; A = optic nerve head astrocytes; MG = optic nerve head microglial cells). Note that cells are not to scale. Inset at upper left show mechanobiologic machinery in these cell types. MSIC = mechanosensitive ion channel, including TRPV4 and Piezo1; MAPKs = mitogen-activated protein kinases, including ERK, c-JUN/JNK, p38 and p42/44. Background image from ref. 290 with permission; lower left inset from ref. 291, with permission. Cell overlays and mechanobiologic insets created using BioRender.com.
The ONH is an anatomically complex tissue containing multiple cell types, including astrocytes, RGCs, microglia, and vascular endothelia (Fig. 3); further, glaucoma is characterized by diverse cell and tissue-level changes, including impaired vascular function and changes in microglial activation and astrocyte reactivity, as elegantly reviewed in228. This complexity complicates understanding of the molecular and cellular pathways by which biomechanical insult culminates in RGC axonal dysfunction, and there are almost certainly multiple injury cascades228.
Among the many causes of RGC loss in glaucoma, significant evidence points to an important role for the mechanobiology of ONH astrocytes229,230,231,232,233,234,235. Astrocytes are the major glial cell type in the ONH (Fig. 3), and have many functions228,236,237. In response to pathological mechanical deformation, astrocytes transition from a quiescent phenotype to a more reactive, proliferative one characterized by increased expression of intermediate filaments (e.g., glial fibrillary acidic protein, vimentin and nestin) and s100β, and elevated release of pro-inflammatory cytokines such as TNF-α, among other changes229,238. Astrocyte reactivity was originally thought to be solely deleterious; e.g., reactive astrocytes remodel the ONH via synthesis of ECM proteins and matrix metalloproteinases229,239, which may contribute to RGC death by altering the normal biomechanical supportive functions of the ECM234,235,240. However, more nuanced recent work shows that astrocyte reactivity can also promote RGC survival228, e.g., astrocyte-specific knock out of the transcription factor STAT3 increased RGC loss241, and lipoxins secreted by reactive astrocytes are neuroprotective in experimental models of glaucoma242. More generally, it is now understood that astrocytes can exert both neurotoxic and neuroprotective effects after injury243.
Multiple studies have shown that astrocytes respond to mechanical insult, such as that induced by elevated IOP227,231,232,244,245,246, strongly suggesting that astrocyte mechanobiology is an important aspect of the pathogenesis of glaucoma. Which molecular pathway(s) are involved in ONH astrocyte mechanobiology, specifically in astrocytic response to stimuli such as those due to elevated IOP? Despite its importance, the answer to this question remains unknown, although evidence suggests that mechanosensitive ion channels are very important in this process247. Indeed, more than a dozen mechanosensitive ion channels have been reported to be expressed by astrocytes248, although not all of these are “primarily” mechano-activated, i.e., directly respond to mechanical stretch, rather than e.g., modulating the effects of other channels. More specifically, channels present in mouse ONH astrocytes, as determined by several mRNA assays248, include TRPC1-6, TRPM2, 4, 6 and 7, TRPV2 and 4, TRPP1 and 2, and Piezo1 and 2. However, which of these channels are most important in ONH astrocyte mechanosensation in vivo remains uncertain.
Downstream mechanotransduction likely involves several MAPKs, including ERK, c-JUN/JNK, p38249 and p42/44250. In this context, p38 is particularly interesting: pharmacologic inhibition of p38 MAPK by small molecules was protective against optic nerve degeneration in a rat microbead model of ocular hypertension, but surprisingly was not protective at longer time points in the DBA/2J mouse model or the squirrel monkey microbead model251,252. However, these results are hard to interpret mechanistically, due to the multiple cell types expressing p38 in the ONH and the fact they target all four p38 MAPK isoforms, each of which have distinctive dell type-dependent functions. Nonetheless, much interest remains in the potential of modulating p38 activity as a neuroprotective treatment in glaucoma.
Downstream of mechanotransduction, mechanical strain causes many phenotypic changes in astrocytes consistent with glaucomatous pathophysiology, including migration and remodeling of the actin cytokeleton231,232,245,246, changes in ECM turnover100,227,233,244,253, and alterations in gap junctions254. There is also evidence of transient cell membrane damage when cells are exposed to pathological levels of strain, which can be repaired by Annexin4 in healthy ONH astrocytes255.
Of course, astrocytes are not the only glial cell type in the ONH - microglia are also present. The mechanobiology of these cell types is somewhat less well-studied in the ONH. Microglia have been implicated as having an important role in neuroinflammatory processes involved in glaucoma256,257,258. However, their role is certainly complex: for example, microglial depletion in mice with ocular hypertension had no demonstrable effect on ONH pathology or RGC loss, suggesting that astrocytic changes may be more important in driving glaucomatous optic neuropathy than microglial changes259. The mechanobiology of the microglia within the ONH shares features with brain microglia260, yet mechanobiological pathways in these cells are understudied.
Finally, it is now appreciated that RGCs are also mechanosensitive and may autonomously play a role in their own dysfunction in the glaucomatous eye261. Indeed, it has long been appreciated that macroscopic mechanical insult to RGC axons might play a role in RGC dysfunction, as RGC axons are “kinked” as they pass through the tortuous passages of the lamina cribrosa36,262,263. However, more recently we have learned that RGCs are directly mechanosensitive256; for example, they express TRPV4264, the pannexin hemichannel Panx1265 and the P2X7 receptor266, and Piezo1 and Piezo2267,268. The distribution of these mechanosensory elements is spatially non-uniform in RGCs; for example, Piezo1 is reported to be expressed in soma and axons of RGCs, but not dendrites, which may be relevant to glaucoma since changes to RGCs occur at different times in different cell compartments (axons vs. soma, etc.)269,270. The Panx1/P2X7 system is particularly interesting: Mitchell and colleagues have shown, over a series of papers, that ATP release by Panx1 hemichannels acts as an autocrine signal via the P2X7 receptor that promotes neurodegeneration in RGCs80,81,82,83 and ONH astrocytes271,272. However, there remains a major knowledge gap about how (and whether) direct mechanostimulation can cause RGC death, in part due to the many mechanisms that contribute to glaucomatous RGC death273,274 and in part due to the complex interactions between RGCs and glial cells in the optic nerve head. We suggest that elucidating mechanistic pathways between astrocytic and RGC mechanosensation and RGC cell death is an important, understudied research area.
Conclusions and open questions
In this review, we have provided an overview of the mechanobiological landscape in the eye, with an initial focus on cornea and lens before diving deeper into anterior and posterior tissues involved in glaucoma. We define mechanosensation and mechanotransduction mechanisms, and unpack the respective key players that different ocular cells use to adapt to the complex and dynamically changing microenvironment within their tissue niches.
Unsurprisingly, many open questions remain, a few of which are listed below. We note that many of these questions, although framed in the context of the eye, are quite general and addressing these questions would have broad implications for our understanding of normal physiology and of pathophysiology.
-
Do age-related changes in mechanosensing/mechanotransduction processes contribute to ocular pathologies? Age is a major risk factor for glaucoma and other ocular pathologies; yet, it is unclear how and to what extent the cellular programs and machineries in relevant tissues undergo pathological alterations over time.
-
How do mechanobiological pathways adapt to varying biomechanical loads? Ocular tissues are subject to remarkably dynamic changes in their biophysical environment, with time scales ranging from seconds (ocular pulse) to many hours (diurnal variations)275,276. However, mechanistic details on how cells integrate this variable “mechanical dosing” in health and disease are scarce.
-
What role does the nuclear envelope play in ocular mechanotransduction? Nuclear-cytoskeletal coupling is critical for force transmission to the nucleus and for the subsequent biological responses277. Studies in other fields have pointed to the nuclear envelope as a regulator of biochemical and biophysical interactions between the nucleus and cytoskeleton; yet, very little is known about the regulation of (epi) genome organization and gene expression by “nuclear mechanotransduction” in ocular cells.
-
In glaucoma, how do inflammation, metabolic stress and immune function in both anterior and posterior tissues interact with aberrant mechanosensing/mechanotransduction to potentiate the effects of IOP in RGC loss? This has become a critical question in light of recent findings indicating an important, yet poorly understood, role for these systemic factors in glaucoma278,279,280,281,282.
-
How much RGC damage in glaucoma is directly attributable to RGC mechanosensation vs. indirect effects of ONH glial cell mechanobiology? It seems likely that both mechanisms are involved, but direct evidence about the relative importance of the two mechanisms is scarce and may be dependent on disease state. Relatedly, what mechanobiology-driven interaction(s) between astrocytes and microglia contribute to RGC loss; both cell types, which are the major glial cells of the optic nerve, have been implicated in RGC loss yet the cross-talk between the two glial cell types has not been extensively studied259,283.
These are exciting areas for future mechanobiology research that hopefully will help identify the causal mechanism(s) underlying glaucoma and other sight-threatening pathologies, and to facilitate the screening and development of novel treatments.
Data availability
No datasets were generated or analyzed during the current study.
References
Brown, M. M., Brown, G. C., Sharma, S. & Busbee, B. Quality of life associated with visual loss: a time tradeoff utility analysis comparison with medical health states. Ophthalmology 110, 1076–1081 (2003).
Vu, H. T., Keeffe, J. E., McCarty, C. A. & Taylor, H. R. Impact of unilateral and bilateral vision loss on quality of life. Br. J. Ophthalmol. 89, 360–363 (2005).
Chen, Y., Ju, L., Rushdi, M., Ge, C. & Zhu, C. Receptor-mediated cell mechanosensing. Mol. Biol. Cell 28, 3134–3155 (2017).
Martino, F., Perestrelo, A. R., Vinarský, V., Pagliari, S. & Forte, G. Cellular Mechanotransduction: from tension to function. Front. Physiol. 9, 824 (2018).
Grytz, R., Yang, H., Hua, Y., Samuels, B. C. & Sigal, I. A. Connective tissue remodeling in myopia and its potential role in increasing risk of glaucoma. Curr. Opin. Biomed. Eng. 15, 40–50 (2020).
Boote, C. et al. Scleral structure and biomechanics. Prog. Retin. Eye Res. 74, 100773 (2020).
Piskova, T., Kozyrina, A. N. & Di Russo, J. Mechanobiological implications of age-related remodelling in the outer retina. Biomater. Adv. 147, 213343 (2023).
Cheng, Y., Ren, T. & Wang, N. Biomechanical homeostasis in ocular diseases: a mini-review. Front. Public Health 11, 1106728 (2023).
Križaj, D., Cordeiro, S. & Strauß, O. Retinal TRP channels: Cell-type-specific regulators of retinal homeostasis and multimodal integration. Prog. Retin. Eye Res. 92, 101114 (2023).
Croft, M. A. & Kaufman, P. L. Accommodation and presbyopia: the ciliary neuromuscular view. Ophthalmol. Clin. N. Am. 19, 13–24 (2006). v.
Cheng, C. Lens Structure. in Reference Module in Neuroscience and Biobehavioral Psychology. https://doi.org/10.1016/B978-0-443-13820-1.00142-0 (Elsevier, 2024).
Barraquer, R. I., Michael, R., Abreu, R., Lamarca, J. & Tresserra, F. Human lens capsule thickness as a function of age and location along the sagittal lens perimeter. Investig. Ophthalmol. Vis. Sci. 47, 2053–2060 (2006).
Pedrigi, R. M., Dziezyc, J. & Humphrey, J. D. Altered mechanical behavior and properties of the human anterior lens capsule after cataract surgery. Exp. Eye Res. 89, 575–580 (2009).
Pedrigi, R. M., David, G., Dziezyc, J. & Humphrey, J. D. Regional mechanical properties and stress analysis of the human anterior lens capsule. Vis. Res. 47, 1781–1789 (2007).
Schaumberg, D. A., Dana, M. R., Christen, W. G. & Glynn, R. J. A systematic overview of the incidence of posterior capsule opacification. Ophthalmology 105, 1213–1221 (1998).
Wormstone, I. M., Wormstone, Y. M., Smith, A. J. O. & Eldred, J. A. Posterior capsule opacification: What’s in the bag?. Prog. Retin. Eye Res. 82, 100905 (2021).
Ameku, K. A., Berggren, C. C. & Pedrigi, R. M. Implantation of a capsular tension ring during cataract surgery attenuates predicted remodeling of the post-surgical lens capsule along the visual axis. Front. Bioeng. Biotechnol. 11, 1300830 (2024).
Murugan, S. & Cheng, C. Roles of Eph-ephrin signaling in the eye lens cataractogenesis, biomechanics, and homeostasis. Front. Cell Dev. Biol. 10, 852236 (2022).
Cheng, C. Tissue, cellular, and molecular level determinants for eye lens stiffness and elasticity. Front. Ophthalmol. 4, 1456474 (2024).
Yamada, T. et al. Functional expression of transient receptor potential vanilloid 3 (TRPV3) in corneal epithelial cells: involvement in thermosensation and wound healing. Exp. Eye Res. 90, 121–129 (2010).
Yang, H. et al. EGF stimulates growth by enhancing capacitative calcium entry in corneal epithelial cells. J. Membr. Biol. 194, 47–58 (2003).
Yang, Y. et al. Wakayama symposium: dependence of corneal epithelial homeostasis on transient receptor potential function. Ocul. Surf. 11, 8–11 (2013).
Zhang, F. et al. Transient receptor potential vanilloid 1 activation induces inflammatory cytokine release in corneal epithelium through MAPK signaling. J. Cell. Physiol. 213, 730–739 (2007).
Mergler, S. et al. Thermosensitive transient receptor potential channels in human corneal epithelial cells. J. Cell. Physiol. 226, 1828–1842 (2011).
Teranishi, S., Kimura, K. & Nishida, T. Role of formation of an ERK-FAK-paxillin complex in migration of human corneal epithelial cells during wound closure in vitro. Investig. Ophthalmol. Vis. Sci. 50, 5646–5652 (2009).
McKay, T. B., Schlötzer-Schrehardt, U., Pal-Ghosh, S. & Stepp, M. A. Integrin: basement membrane adhesion by corneal epithelial and endothelial cells. Exp. Eye Res. 198, 108138 (2020).
Masterton, S. & Ahearne, M. Mechanobiology of the corneal epithelium. Exp. Eye Res. 177, 122 (2018).
Thomasy, S. M., Leonard, B. C., Greiner, M. A., Skeie, J. M. & Raghunathan, V. K. Squishy matters—Corneal mechanobiology in health and disease. Prog. Retin. Eye Res. 99, 101234 (2024).
Lan, W. et al. Gene expression of down-regulation of lysyl oxidases family in keratoconus corneal fibroblasts induced by combination of mechanical stretching and prostaglandin E 2]. Sheng Wu Yi Xue Gong. Cheng Xue Za Zhi34, 239–245 (2017).
Du, G.-L., Chen, W.-Y., Li, X.-N., He, R. & Feng, P.-F. Induction of MMP‑1 and ‑3 by cyclical mechanical stretch is mediated by IL‑6 in cultured fibroblasts of keratoconus. Mol. Med. Rep. 15, 3885–3892 (2017).
Akoto, T. et al. Unravelling the impact of cyclic mechanical stretch in keratoconus—a transcriptomic profiling study. Int. J. Mol. Sci. 24, 7437 (2023).
Santodomingo-Rubido, J. et al. Keratoconus: an updated review. Cont. Lens Anterior Eye 45, 101559 (2022).
Chen, Z., Lu, X., McGee-Lawrence, M. E. & Watsky, M. A. Transient cell membrane disruptions induce calcium waves in corneal keratocytes. Sci. Rep. 10, 2840 (2020).
Maruri, D. P., Iyer, K. S., Schmidtke, D. W., Petroll, W. M. & Varner, V. D. Signaling downstream of focal adhesions regulates stiffness-dependent differences in the TGF-β1-mediated myofibroblast differentiation of corneal keratocytes. Front. Cell Dev. Biol. 10, 886759 (2022).
Ali, M., Raghunathan, V., Li, J. Y., Murphy, C. J. & Thomasy, S. M. Biomechanical relationships between the corneal endothelium and Descemet’s membrane. Exp. Eye Res. 152, 57–70 (2016).
Burgoyne, C. F. et al. The optic nerve head as a biomechanical structure: a new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog. Retin. Eye Res. 24, 39–73 (2005).
Weinreb, R. N. & Khaw, P. T. Primary open-angle glaucoma. Lancet 363, 1711–1720 (2004).
Epstein, D. L. Chandler and Grant’s Glaucoma (eds Epstein, D. L., Allingham, R. R. & Schuman, J. S.) (Lippincott Williams & Wilkins, 1997).
Tamm, E. R. et al. Biological aspects of axonal damage in glaucoma: a brief review. Exp. Eye Res. 157, 5–12 (2017).
Stowell, C. et al. Biomechanical aspects of axonal damage in glaucoma: a brief review. Exp. Eye Res. 157, 13–19 (2017).
Jayaram, H., Kolko, M., Friedman, D. S. & Gazzard, G. Glaucoma: now and beyond. Lancet 402, 1788–1801 (2023).
Quigley, H. A. Neuronal death in glaucoma. Prog. Retin. Eye Res. 18, 39–57 (1999).
Varma, R., Lee, P. P., Goldberg, I. & Kotak, S. An assessment of the health and economic burdens of glaucoma. Am. J. Ophthalmol. 152, 515–522 (2011).
Quigley, H. A. & Broman, A. T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 90, 262–267 (2006).
Heijl, A., Leske, M. C., Bengtsson, B., Hyman, L. & Hussein, M. Reduction of intraocular pressure and glaucoma progression: results from the Early Manifest Glaucoma trial. Arch. Ophthalmol. 120, 1268–1279 (2002).
Leske, M. C. et al. Factors for glaucoma progression and the effect of treatment: the early manifest glaucoma trial. Arch. Ophthalmol. 121, 48–56 (2003).
Anderson, D. R., Drance, S. M. & Schulzer, M. Natural history of normal-tension glaucoma. Ophthalmology 108, 247–253 (2001).
Gaasterland, D. E. et al. The Advanced Glaucoma Intervention Study (AGIS): 7. The relationship between control of intraocular pressure and visual field deterioration. Am. J. Ophthalmol. 130, 429–440 (2000).
Burgoyne, C. F., Downs, J. C., Bellezza, A. J., Suh, J.-K. F. & Hart, R. T. The optic nerve head as a biomechanical structure: a new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog. Retin. Eye Res. 24, 39 (2005).
Sigal, I. A. & Ethier, C. R. Biomechanics of the optic nerve head. Exp. Eye Res. 88, 799–807 (2009).
Tamm, E. R. The trabecular meshwork outflow pathways: structural and functional aspects. Exp. Eye Res. 88, 648–655 (2009).
Tamm, E. R., Braunger, B. M. & Fuchshofer, R. Intraocular pressure and the mechanisms involved in resistance of the aqueous humor flow in the trabecular meshwork outflow pathways. Prog. Mol. Biol. Transl. Sci. 134, 301–314 (2015).
Overby, D. R., Stamer, W. D. & Johnson, M. The changing paradigm of outflow resistance generation: towards synergistic models of the JCT and inner wall endothelium. Exp. Eye Res. 88, 656–670 (2009).
Kwon, Y. H., Fingert, J. H., Kuehn, M. H. & Alward, W. L. M. Primary open-angle glaucoma. N. Engl. J. Med. 360, 1113–1124 (2009).
Acott, T. S. et al. Intraocular pressure homeostasis: maintaining balance in a high-pressure environment. J. Ocul. Pharmacol. Ther. 00, 1–8 (2014).
Bradley, J. M. B. et al. Effect of matrix metalloproteinases activity on outflow in perfused human organ culture. Investig. Ophthalmol. Vis. Sci. 39, 2649–2658 (1998).
Bradley, J. M. B. et al. Effects of mechanical stretching on trabecular matrix metalloproteinases. Investig. Ophthalmol. Vis. Sci. 42, 1505–1513 (2001).
Bradley, J. M. B. & Acott, T. S. Trabecular cell stretch/distortion increases gelatinase A expression [ARVO abstract]. Investig. Ophthalmol. Vis. Sci. 39, S484 (1998).
Reina-Torres, E. et al. The vital role for nitric oxide in intraocular pressure homeostasis. Prog. Retin. Eye Res. 83, 100922 (2021).
Overby, D. R. et al. The factors affecting the stability of IOP homeostasis. Investig. Ophthalmol. Vis. Sci. 65, 4 (2024).
Overby, D. R. et al. Altered mechanobiology of Schlemm’s canal endothelial cells in glaucoma. Proc. Natl. Acad. Sci. USA 111, 13876–13881 (2014).
Braakman, S. T. et al. Biomechanical strain as a trigger for pore formation in Schlemm’s canal endothelial cells. Exp. Eye Res. 127, 224–235 (2014).
Last, J. A. et al. Elastic modulus determination of normal and glaucomatous human trabecular meshwork. Investig. Ophthalmol. Vis. Sci. 52, 2147–2152 (2011).
Wang, K. et al. Estimating human trabecular meshwork stiffness by numerical modeling and advanced OCT imaging. Investig. Ophthalmol. Vis. Sci. 58, 4809–4817 (2017).
Wang, K., Read, A. T., Sulchek, T. & Ethier, C. R. Trabecular meshwork stiffness in glaucoma. Exp. Eye Res. 158, 3–12 (2017).
Vahabikashi, A. et al. Increased stiffness and flow resistance of the inner wall of Schlemm’s canal in glaucomatous human eyes. Proc. Natl. Acad. Sci. USA 116, 26555–26563 (2019).
Liton, P. B. & Gonzalez, P. Stress response of the trabecular meshwork. J. Glaucoma 17, 378–385 (2008).
Martinac, B. Mechanosensitive ion channels: molecules of mechanotransduction. J. Cell Sci. 117, 2449–2460 (2004).
Ryskamp, D. A. et al. TRPV4 regulates calcium homeostasis, cytoskeletal remodeling, conventional outflow and intraocular pressure in the mammalian eye. Sci. Rep. 6, 30583 (2016).
Lakk, M. & Križaj, D. TRPV4-Rho signaling drives cytoskeletal and focal adhesion remodeling in trabecular meshwork cells. Am. J. Physiol. Cell Physiol. 320, C1013–C1030 (2021).
Lakk, M. et al. Membrane cholesterol regulates TRPV4 function, cytoskeletal expression, and the cellular response to tension. J. Lipid Res. 62, 100145 (2021).
Uchida, T. et al. TRPV4 is activated by mechanical stimulation to induce prostaglandins release in trabecular meshwork, lowering intraocular pressure. PloS ONE 16, e0258911 (2021).
Yarishkin, O. et al. Emergent temporal signaling in human trabecular meshwork cells: role of TRPV4-TRPM4 interactions. Front. Immunol. 13, 805076 (2022).
Baumann, J. M. et al. TRPV4 and chloride channels mediate volume sensing in trabecular meshwork cells. Am. J. Physiol. Cell Physiol. 327, C403–C414 (2024).
Rudzitis, C. N. et al. TRPV4 overactivation enhances cellular contractility and drives ocular hypertension in TGFβ2 overexpressing eyes. BioRxiv Prepr. Serv. Biol. 2024.11.05.622187. https://doi.org/10.1101/2024.11.05.622187 (2024).
Redmon, S. N. et al. TRPV4 subserves physiological and pathological elevations in intraocular pressure. Res. Sq. rs.3.rs-4714050. https://doi.org/10.21203/rs.3.rs-4714050/v1 (2024).
Patel, P. D. et al. Impaired TRPV4-eNOS signaling in trabecular meshwork elevates intraocular pressure in glaucoma. Proc. Natl. Acad. Sci. USA 118, e2022461118 (2021).
Morozumi, W. et al. Piezo1 activation induces fibronectin reduction and PGF2α secretion via arachidonic acid cascade. Exp. Eye Res. 215, 108917 (2022).
Uchida, T. et al. Mechanical stretch induces Ca2+ influx and extracellular release of PGE2 through Piezo1 activation in trabecular meshwork cells. Sci. Rep. 11, 4044 (2021).
Yarishkin, O. et al. Piezo1 channels mediate trabecular meshwork mechanotransduction and promote aqueous fluid outflow. J. Physiol. 599, 571–592 (2021).
Zhu, W. et al. The role of Piezo1 in conventional aqueous humor outflow dynamics. iScience 24, 102042 (2021).
Yarishkin, O., Baumann, J. M. & Križaj, D. Mechano-electrical transduction in trabecular meshwork involves parallel activation of TRPV4 and TREK-1 channels. Channels 13, 168–171 (2019).
Yarishkin, O., Phuong, T. T. T. & Križaj, D. Trabecular meshwork TREK-1 channels function as polymodal integrators of pressure and pH. Invest. Ophthalmol. Vis. Sci. 60, 2294–2303 (2019).
Carreon, T. A., Castellanos, A., Gasull, X. & Bhattacharya, S. K. Interaction of cochlin and mechanosensitive channel TREK-1 in trabecular meshwork cells influences the regulation of intraocular pressure. Sci. Rep. 7, 452 (2017).
Yarishkin, O. et al. TREK-1 channels regulate pressure sensitivity and calcium signaling in trabecular meshwork cells. J. Gen. Physiol. 150, 1660–1675 (2018).
Garcia-Sanchez, J., Lin, D. & Liu, W. W. Mechanosensitive ion channels in glaucoma pathophysiology. Vis. Res. 223, 108473 (2024).
Zhang, X., Wang, F. & Su, Y. TRPV: an emerging target in glaucoma and optic nerve damage. Exp. Eye Res. 239, 109784 (2024).
Chen, Y., Su, Y. & Wang, F. The Piezo1 ion channel in glaucoma: a new perspective on mechanical stress. Hum. Cell 35, 1307–1322 (2022).
Luo, N. N. et al. Primary cilia signaling mediates intraocular pressure sensation. Proc. Natl. Acad. Sci. USA 111, 12871–12876 (2014).
Jing, L., Liu, K., Wang, F. & Su, Y. Role of mechanically-sensitive cation channels Piezo1 and TRPV4 in trabecular meshwork cell mechanotransduction. Hum. Cell 37, 394–407 (2024).
Kechagia, J. Z., Ivaska, J. & Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 20, 457–473 (2019).
Faralli, J. A., Filla, M. S. & Peters, D. M. Integrin crosstalk and its effect on the biological functions of the trabecular meshwork/schlemm’s canal. Front. Cell Dev. Biol. 10, 886702 (2022).
Gagen, D., Faralli, J. A., Filla, M. S. & Peters, D. M. The role of integrins in the trabecular meshwork. J. Ocul. Pharmacol. Ther. 30, 110–120 (2014).
Filla, M. S., Faralli, J. A., Peotter, J. L. & Peters, D. M. The role of integrins in glaucoma. Exp. Eye Res. 158, 124–136 (2017).
Faralli, J. A., Filla, M. S. & Peters, D. M. Role of integrins in the development of fibrosis in the trabecular meshwork. Front. Ophthalmol. 3, 1274797 (2023).
Junglas, B., Yu, A. H. L., Welge-Lüssen, U., Tamm, E. R. & Fuchshofer, R. Connective tissue growth factor induces extracellular matrix deposition in human trabecular meshwork cells. Exp. Eye Res. 88, 1065–1075 (2009).
Johnstone, M. et al. Aqueous outflow regulation—21st century concepts. Prog. Retin. Eye Res. 83, 100917 (2021).
Faralli, J. A., Gagen, D., Filla, M. S., Crotti, T. N. & Peters, D. M. Dexamethasone increases αvβ3 integrin expression and affinity through a calcineurin/NFAT pathway. Biochim. Biophys. Acta 1833, 3306–3313 (2013).
Filla, M. S., Schwinn, M. K., Nosie, A. K., Clark, R. W. & Peters, D. M. Dexamethasone-associated cross-linked actin network formation in human trabecular meshwork cells involves β3 integrin signaling. Invest. Ophthalmol. Vis. Sci. 52, 2952–2959 (2011).
Fuchshofer, R. & Tamm, E. R. The role of TGF-β in the pathogenesis of primary open-angle glaucoma. Cell Tissue Res. 347, 279–290 (2012).
Tsukamoto, T., Kajiwara, K., Nada, S. & Okada, M. Src mediates TGF-β-induced intraocular pressure elevation in glaucoma. J. Cell. Physiol. 234, 1730–1744 (2019).
Filla, M. S., Meyer, K. K., Faralli, J. A. & Peters, D. M. Overexpression and activation of αvβ3 integrin differentially affects TGFβ2 signaling in human trabecular meshwork cells. Cells 10, 1923 (2021).
Filla, M. S., Schwinn, M. K., Sheibani, N., Kaufman, P. L. & Peters, D. M. Regulation of cross-linked actin network (CLAN) formation in human trabecular meshwork (HTM) cells by convergence of distinct beta1 and beta3 integrin pathways. Investig. Ophthalmol. Vis. Sci. 50, 5723–5731 (2009).
Sun, Y. Y., Bradley, J. M. & Keller, K. E. Phenotypic and functional alterations in tunneling nanotubes formed by glaucomatous trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 60, 4583–4595 (2019).
Keller, K. E., Bradley, J. M., Sun, Y. Y., Yang, Y.-F. & Acott, T. S. Tunneling nanotubes are novel cellular structures that communicate signals between trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 58, 5298–5307 (2017).
Aga, M., Bradley, J. M., Keller, K. E., Kelley, M. J. & Acott, T. S. Specialized podosome- or invadopodia-like structures (PILS) for focal trabecular meshwork extracellular matrix turnover. Investig. Ophthalmol. Vis. Sci. 49, 5353–5365 (2008).
Morgan, M. R. et al. Syndecan-4 phosphorylation is a control point for integrin recycling. Dev. Cell 24, 472–485 (2013).
Wang, X. & Ha, T. Defining single molecular forces required to activate integrin and notch signaling. Science 340, 991–994 (2013).
Roca-Cusachs, P., Gauthier, N. C., Del Rio, A. & Sheetz, M. P. Clustering of alpha(5)beta(1) integrins determines adhesion strength whereas alpha(v)beta(3) and talin enable mechanotransduction. Proc. Natl. Acad. Sci. USA 106, 16245–16250 (2009).
Seetharaman, S. & Etienne-Manneville, S. Integrin diversity brings specificity in mechanotransduction. Biol. Cell 110, 49–64 (2018).
Bermudez, J. Y., Montecchi-Palmer, M., Mao, W. & Clark, A. F. Cross-linked actin networks (CLANs) in glaucoma. Exp. Eye Res. 159, 16–22 (2017).
Filla, M. S., Woods, A., Kaufman, P. L. & Peters, D. M. Beta1 and beta3 integrins cooperate to induce syndecan-4-containing cross-linked actin networks in human trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 47, 1956–1967 (2006).
Peotter, J. L. et al. Involvement of Tiam1, RhoG and ELMO2/ILK in Rac1-mediated phagocytosis in human trabecular meshwork cells. Exp. Cell Res. 347, 301–311 (2016).
Gagen, D., Filla, M. S., Clark, R., Liton, P. & Peters, D. M. Activated αvβ3 integrin regulates αvβ5 integrin-mediated phagocytosis in trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 54, 5000–5011 (2013).
Gonzalez, A. M., Bhattacharya, R., deHart, G. W. & Jones, J. C. R. Transdominant regulation of integrin function: mechanisms of crosstalk. Cell. Signal. 22, 578–583 (2010).
Filla, M. S. et al. Activation of αvβ3 Integrin Alters Fibronectin Fibril Formation in Human Trabecular Meshwork Cells in a ROCK-Independent Manner. Investig. Ophthalmol. Vis. Sci. 60, 3897–3913 (2019).
Roberts, A. L. et al. Fibronectin extra domain A (FN-EDA) elevates intraocular pressure through Toll-like receptor 4 signaling. Sci. Rep. 10, 9815 (2020).
Faralli, J. A., Filla, M. S. & Peters, D. M. Effect of αvβ3 integrin expression and activity on intraocular pressure. Investig. Ophthalmol. Vis. Sci. 60, 1776–1788 (2019).
Slack, R. J., Macdonald, S. J. F., Roper, J. A., Jenkins, R. G. & Hatley, R. J. D. Emerging therapeutic opportunities for integrin inhibitors. Nat. Rev. Drug Discov. 21, 60–78 (2022).
Rosenbaum, D. M., Rasmussen, S. G. F. & Kobilka, B. K. The structure and function of G-protein-coupled receptors. Nature 459, 356–363 (2009).
Chachisvilis, M., Zhang, Y.-L. & Frangos, J. A. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc. Natl. Acad. Sci. USA 103, 15463–15468 (2006).
Erdogmus, S. et al. Helix 8 is the essential structural motif of mechanosensitive GPCRs. Nat. Commun. 10, 5784 (2019).
Zou, Y. et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat. Cell Biol. 6, 499–506 (2004).
Agarwal, R. & Iezhitsa, I. Advances in targeting the extracellular matrix for glaucoma therapy: current updates. Expert Opin. Ther. Targets 27, 1217–1229 (2023).
Buonfiglio, F., Pfeiffer, N. & Gericke, A. Glaucoma and the ocular renin-angiotensin-aldosterone system: update on molecular signalling and treatment perspectives. Cell. Signal. 122, 111343 (2024).
Holappa, M., Vapaatalo, H. & Vaajanen, A. Many faces of renin-angiotensin system—focus on eye. Open Ophthalmol. J. 11, 122–142 (2017).
Li, H. et al. Elevated angiotensin-II levels contribute to the pathogenesis of open-angle glaucoma via inducing the expression of fibrosis-related genes in trabecular meshwork cells through a ROS/NOX4/SMAD3 axis. Cell Transpl. 32, 9636897231162526 (2023).
Rao, P. V., Deng, P., Sasaki, Y. & Epstein, D. L. Regulation of myosin light chain phosphorylation in the trabecular meshwork: role in aqueous humour outflow facility. Exp. Eye Res. 80, 197–206 (2005).
Shen, F., Zhang, L. & Liu, T. Effects of angiotensin II on the 3H-TdR incorporation and synthesis of collagen in cultured bovine trabecular meshwork cells. Yan Ke Xue Bao 17, 209–212 (2001).
Vaajanen, A., Vapaatalo, H., Kautiainen, H. & Oksala, O. Angiotensin (1-7) reduces intraocular pressure in the normotensive rabbit eye. Investig. Ophthalmol. Vis. Sci. 49, 2557–2562 (2008).
Kaufman, P. L. & Bárány, E. H. Adrenergic drug effects on aqueous outflow facility following ciliary muscle retrodisplacement in the cynomolgus monkey. Investig. Ophthalmol. Vis. Sci. 20, 644–651 (1981).
Kaufman, P. L. & Rentzhog, L. Effect of total iridectomy on outflow facility responses to adrenergic drugs in cynomolgus monkeys. Exp. Eye Res. 33, 65–74 (1981).
Costagliola, C. et al. Effect of oral losartan potassium administration on intraocular pressure in normotensive and glaucomatous human subjects. Exp. Eye Res. 71, 167–171 (2000).
Wang, R.-F., Podos, S. M., Mittag, T. W. & Yokoyoma, T. Effect of CS-088, an angiotensin AT1 receptor antagonist, on intraocular pressure in glaucomatous monkey eyes. Exp. Eye Res. 80, 629–632 (2005).
Shah, G. B., Sharma, S., Mehta, A. A. & Goyal, R. K. Oculohypotensive effect of angiotensin-converting enzyme inhibitors in acute and chronic models of glaucoma. J. Cardiovasc. Pharmacol. 36, 169–175 (2000).
Jiang, F. et al. Angiotensin-converting enzyme 2 and angiotensin 1-7: novel therapeutic targets. Nat. Rev. Cardiol. 11, 413–426 (2014).
Foureaux, G. et al. Antiglaucomatous effects of the activation of intrinsic Angiotensin-converting enzyme 2. Investig. Ophthalmol. Vis. Sci. 54, 4296–4306 (2013).
Sharif, N. A. Novel potential treatment modalities for ocular hypertension: focus on angiotensin and bradykinin system axes. J. Ocul. Pharmacol. Ther. 31, 131–145 (2015).
McDonnell, F. et al. Shear stress in Schlemm’s canal as a sensor of intraocular pressure. Sci. Rep. 10, 5804 (2020).
Wilde, C., Mitgau, J., Suchý, T., Schöneberg, T. & Liebscher, I. Translating the force-mechano-sensing GPCRs. Am. J. Physiol. Cell Physiol. 322, C1047–C1060 (2022).
Klapholz, B. & Brown, N. H. Talin—the master of integrin adhesions. J. Cell Sci. 130, 2435–2446 (2017).
Mitra, S. K., Hanson, D. A. & Schlaepfer, D. D. Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 (2005).
Schiller, H. B. & Fässler, R. Mechanosensitivity and compositional dynamics of cell-matrix adhesions. EMBO Rep. 14, 509–519 (2013).
Schlunck, G. et al. Substrate rigidity modulates cell matrix interactions and protein expression in human trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 49, 262–269 (2008).
Li, H., Raghunathan, V., Stamer, W. D., Ganapathy, P. S. & Herberg, S. Extracellular matrix stiffness and TGFβ2 regulate YAP/TAZ activity in human trabecular meshwork cells. Front. Cell Dev. Biol. 10, 844342 (2022).
Saccà, S. C., Centofanti, M. & Izzotti, A. New proteins as vascular biomarkers in primary open angle glaucomatous aqueous humor. Investig. Ophthalmol. Vis. Sci. 53, 4242–4253 (2012).
Discher, D. E., Janmey, P. & Wang, Y.-L. Tissue cells feel and respond to the stiffness of their substrate. Science 310, 1139–1143 (2005).
Fletcher, D. A. & Mullins, R. D. Cell mechanics and the cytoskeleton. Nature 463, 485–492 (2010).
Tojkander, S., Gateva, G. & Lappalainen, P. Actin stress fibers–assembly, dynamics and biological roles. J. Cell Sci. 125, 1855–1864 (2012).
Watanabe, N. et al. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 16, 3044–3056 (1997).
Leung, T., Chen, X. Q., Manser, E. & Lim, L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell. Biol. 16, 5313–5327 (1996).
Grierson, I. & Rahi, A. H. Microfilaments in the cells of the human trabecular meshwork. Br. J. Ophthalmol. 63, 3–8 (1979).
Gipson, I. K. & Anderson, R. A. Actin filaments in cells of human trabecular meshwork and Schlemm’s canal. Investig. Ophthalmol. Vis. Sci. 18, 547–561 (1979).
Tan, J. C. H., Peters, D. M. & Kaufman, P. L. Recent developments in understanding the pathophysiology of elevated intraocular pressure. Curr. Opin. Ophthalmol. 17, 168–174 (2006).
Tian, B., Gabelt, B. T., Geiger, B. & Kaufman, P. L. The role of the actomyosin system in regulating trabecular fluid outflow. Exp. Eye Res. 88, 713–717 (2009).
Kaufman, P. L. Deconstructing aqueous humor outflow—the last 50 years. Exp. Eye Res. 197, 108105 (2020).
Hoare, M.-J. et al. Cross-linked actin networks (CLANs) in the trabecular meshwork of the normal and glaucomatous human eye in situ. Investig. Ophthalmol. Vis. Sci. 50, 1255–1263 (2009).
Clark, A. F., Miggans, S. T., Wilson, K., Browder, S. & McCartney, M. D. Cytoskeletal changes in cultured human glaucoma trabecular meshwork cells. J. Glaucoma 4, 183–188 (1995).
Ethier, C. R., Read, A. T. & Chan, D. Biomechanics of Schlemm’s canal endothelial cells: influence on F-actin architecture. Biophys. J. 87, 2828 (2004).
Tumminia, S. J. et al. Mechanical stretch alters the actin cytoskeletal network and signal transduction in human trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 39, 1361–1371 (1998).
Riento, K. & Ridley, A. J. Rocks: multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 4, 446–456 (2003).
Rao, P. V., Pattabiraman, P. P. & Kopczynski, C. Role of the Rho GTPase/Rho kinase signaling pathway in pathogenesis and treatment of glaucoma: Bench to bedside research. Exp. Eye Res. 158, 23–32 (2017).
Rao, P. V., Deng, P. F., Kumar, J. & Epstein, D. L. Modulation of aqueous humor outflow facility by the Rho kinase-specific inhibitor Y-27632. Investig. Ophthalmol. Vis. Sci. 42, 1029 (2001).
Honjo, M. et al. Effects of rho-associated protein kinase inhibitor Y-27632 on intraocular pressure and outflow facility. Investig. Ophthalmol. Vis. Sci. 42, 137 (2001).
Li, H., Henty-Ridilla, J. L., Bernstein, A. M., Ganapathy, P. S. & Herberg, S. TGFβ2 regulates human trabecular meshwork cell contractility via ERK and ROCK pathways with distinct signaling crosstalk dependent on the culture substrate. Curr. Eye Res. 47, 1165–1178 (2022).
Tian, B. & Kaufman, P. L. Effects of the Rho kinase inhibitor Y-27632 and the phosphatase inhibitor calyculin A on outflow facility in monkeys. Exp. Eye Res. 80, 215–225 (2005).
Inoue, T. & Tanihara, H. Rho-associated kinase inhibitors: a novel glaucoma therapy. Prog. Retin. Eye Res. 37, 1–12 (2013).
Inoue, T. & Tanihara, H. Ripasudil hydrochloride hydrate: targeting Rho kinase in the treatment of glaucoma. Expert Opin. Pharmacother. 18, 1669–1673 (2017).
Tanna, A. P. & Johnson, M. Rho kinase inhibitors as a novel treatment for glaucoma and ocular hypertension. Ophthalmology 125, 1741–1756 (2018).
Serle, J. B. et al. Two Phase 3 Clinical trials comparing the safety and efficacy of netarsudil to timolol in patients with elevated intraocular pressure: Rho kinase elevated IOP treatment trial 1 and 2 (ROCKET-1 and ROCKET-2). Am. J. Ophthalmol. 186, 116–127 (2018).
Nix, D. A. & Beckerle, M. C. Nuclear-cytoplasmic shuttling of the focal contact protein, zyxin: a potential mechanism for communication between sites of cell adhesion and the nucleus. J. Cell Biol. 138, 1139–1147 (1997).
Dupont, S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011).
Komiya, Y. & Habas, R. Wnt signal transduction pathways. Organogenesis 4, 68–75 (2008).
Kadmiel, M. & Cidlowski, J. A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 34, 518–530 (2013).
Ghosh, R. & Herberg, S. The role of YAP/TAZ mechanosignaling in trabecular meshwork and Schlemm’s canal cell dysfunction. Vis. Res. 224, 108477 (2024).
Totaro, A., Panciera, T. & Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 20, 888–899 (2018).
Piccolo, S., Dupont, S. & Cordenonsi, M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol. Rev. 94, 1287–1312 (2014).
Hansen, C. G., Moroishi, T. & Guan, K.-L. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 25, 499–513 (2015).
Halder, G., Dupont, S. & Piccolo, S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 13, 591–600 (2012).
Dupont, S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 343, 42–53 (2016).
Lee, M., Goraya, N., Kim, S. & Cho, S.-H. Hippo-yap signaling in ocular development and disease. Dev. Dyn. 247, 794–806 (2018).
Zhu, J.-Y., Lin, S. & Ye, J. YAP and TAZ, the conductors that orchestrate eye development, homeostasis, and disease. J. Cell. Physiol. 234, 246–258 (2018).
Bu, Q. et al. Targeting mechanics-induced trabecular meshwork dysfunction through YAP-TGFβ Ameliorates high myopia-induced ocular hypertension. Exp. Eye Res. 241, 109853 (2024).
Sung, M. S., Kim, S. Y., Eom, G. H. & Park, S. W. High VEGF concentrations accelerate human trabecular meshwork fibrosis in a TAZ-dependent manner. Int. J. Mol. Sci. 24, 9625 (2023).
Yoo, H. et al. Simvastatin attenuates glucocorticoid-induced human trabecular meshwork cell dysfunction via YAP/TAZ inactivation. Curr. Eye Res. 48, 736–749 (2023).
Wang, L., Tian, Y., Cao, Y., Ma, Q. & Zhao, S. MiR-137 promotes cell growth and inhibits extracellular matrix protein expression in H2O2-induced human trabecular meshwork cells by targeting Src. Neurosci. Lett. 755, 135902 (2021).
Liu, Z. et al. RhoA/ROCK-YAP/TAZ axis regulates the fibrotic activity in dexamethasone-treated human trabecular meshwork cells. Front. Mol. Biosci. 8, 728932 (2021).
Yemanyi, F. & Raghunathan, V. Lysophosphatidic Acid and IL-6 Trans-signaling Interact via YAP/TAZ and STAT3 signaling pathways in human trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 61, 29 (2020).
Dhamodaran, K., Baidouri, H., Sandoval, L. & Raghunathan, V. Wnt activation after inhibition restores trabecular meshwork cells toward a normal phenotype. Investig. Ophthalmol. Vis. Sci. 61, 30 (2020).
Yemanyi, F., Vranka, J. & Raghunathan, V. K. Crosslinked extracellular matrix stiffens human trabecular meshwork cells via dysregulating β-catenin and YAP/TAZ signaling pathways. Investig. Ophthalmol. Vis. Sci. 61, 41 (2020).
Ho, L. T. Y., Skiba, N., Ullmer, C. & Rao, P. V. Lysophosphatidic acid induces ECM production via activation of the mechanosensitive YAP/TAZ transcriptional pathway in trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 59, 1969–1984 (2018).
Peng, J. et al. YAP and TAZ mediate steroid-induced alterations in the trabecular meshwork cytoskeleton in human trabecular meshwork cells. Int. J. Mol. Med. 41, 164–172 (2018).
Chen, W.-S., Cao, Z., Krishnan, C. & Panjwani, N. Verteporfin without light stimulation inhibits YAP activation in trabecular meshwork cells: Implications for glaucoma treatment. Biochem. Biophys. Res. Commun. 466, 221–225 (2015).
Thomasy, S. M., Morgan, J. T., Wood, J. A., Murphy, C. J. & Russell, P. Substratum stiffness and latrunculin B modulate the gene expression of the mechanotransducers YAP and TAZ in human trabecular meshwork cells. Exp. Eye Res. 113, 66–73 (2013).
Raghunathan, V. K. et al. Role of substratum stiffness in modulating genes associated with extracellular matrix and mechanotransducers YAP and TAZ. Investig. Ophthalmol. Vis. Sci. 54, 378–386 (2013).
Li, H. et al. YAP/TAZ mediate TGFβ2-induced Schlemm’s canal cell dysfunction. Investig. Ophthalmol. Vis. Sci. 63, 15 (2022).
Li, H. et al. Targeting YAP/TAZ mechanosignaling to ameliorate stiffness-induced Schlemm’s canal cell pathobiology. Am. J. Physiol. Cell Physiol. 326, C513–C528 (2024).
Gharahkhani, P. et al. Genome-wide meta-analysis identifies 127 open-angle glaucoma loci with consistent effect across ancestries. Nat. Commun. 12, 1258 (2021).
van Amerongen, R., Mikels, A. & Nusse, R. Alternative Wnt signaling is initiated by distinct receptors. Sci. Signal. 1, re9 (2008).
Liu, J. et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 7, 3 (2022).
Webber, H. C., Bermudez, J. Y., Sethi, A., Clark, A. F. & Mao, W. Crosstalk between TGFβ and Wnt signaling pathways in the human trabecular meshwork. Exp. Eye Res. 148, 97–102 (2016).
Wang, W.-H. et al. Increased expression of the WNT antagonist sFRP-1 in glaucoma elevates intraocular pressure. Investig. Ophthalmol. Vis. Sci. 118, 1056–1064 (2008).
Mao, W. et al. Existence of the canonical Wnt signaling pathway in the human trabecular meshwork. Investig. Ophthalmol. Vis. Sci. 53, 7043–7051 (2012).
Morgan, J. T., Raghunathan, V. K., Chang, Y.-R., Murphy, C. J. & Russell, P. Wnt inhibition induces persistent increases in intrinsic stiffness of human trabecular meshwork cells. Exp. Eye Res. 132, 174–178 (2015).
Oakley, R. H. & Cidlowski, J. A. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 132, 1033–1044 (2013).
Panettieri, R. A. et al. Non-genomic effects of glucocorticoids: an updated view. Trends Pharmacol. Sci. 40, 38–49 (2019).
Bernstein, H. N., Mills, D. W. & Becker, B. Steroid-induced elevation of intraocular pressure. Arch. Ophthalmol.70, 15–18 (1963).
Tripathi, R. C., Parapuram, S. K., Tripathi, B. J., Zhong, Y. & Chalam, K. V. Corticosteroids and glaucoma risk. Drugs Aging 15, 439–450 (1999).
Jones, R. & Rhee, D. J. Corticosteroid-induced ocular hypertension and glaucoma: a brief review and update of the literature. Curr. Opin. Ophthalmol. 17, 163–167 (2006).
Kersey, J. P. & Broadway, D. C. Corticosteroid-induced glaucoma: a review of the literature. Eye Lond. Engl. 20, 407–416 (2006).
Phulke, S., Kaushik, S., Kaur, S. & Pandav, S. S. Steroid-induced glaucoma: an avoidable irreversible blindness. J. Curr. Glaucoma Pract. 11, 67–72 (2017).
Clark, A. F. & Wordinger, R. J. The role of steroids in outflow resistance. Exp. Eye Res. 88, 752–759 (2009).
Raghunathan, V. K. et al. Dexamethasone stiffens trabecular meshwork, trabecular meshwork cells, and matrix. Investig. Ophthalmol. Vis. Sci. 56, 4447–4459 (2015).
Clark, A. F. et al. Glucocorticoid-induced formation of cross-linked actin networks in cultured human trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 35, 281 (1994).
Kasetti, R. B., Maddineni, P., Millar, J. C., Clark, A. F. & Zode, G. S. Increased synthesis and deposition of extracellular matrix proteins leads to endoplasmic reticulum stress in the trabecular meshwork. Sci. Rep. 7, 14951 (2017).
Zhang, X., Ognibene, C. M., Clark, A. F. & Yorio, T. Dexamethasone inhibition of trabecular meshwork cell phagocytosis and its modulation by glucocorticoid receptor beta. Exp. Eye Res. 84, 275–284 (2007).
Das, A. & Kunwar, A. Septins: structural insights, functional dynamics, and implications in health and disease. J. Cell. Biochem. e30660. https://doi.org/10.1002/jcb.30660 (2024).
Itoh, S., Itoh, F., Goumans, M. J. & Ten Dijke, P. Signaling of transforming growth factor-beta family members through Smad proteins. Eur. J. Biochem. 267, 6954–6967 (2000).
Zhou, B. et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 7, 95 (2022).
Pitha, I., Du, L., Nguyen, T. D. & Quigley, H. IOP and glaucoma damage: the essential role of optic nerve head and retinal mechanosensors. Prog. Retin. Eye Res. 99, 101232 (2024).
Anderson, D. R. & Hendrickson, A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Investig. Ophthalmol. Vis. Sci. 13, 771 (1974).
Sigal, I. A., Flanagan, J. G., Tertinegg, I. & Ethier, C. R. Predicted extension, compression and shearing of optic nerve head tissues. Exp. Eye Res. 85, 312–322 (2007).
Quigley, H. A. & Addicks, E. M. Regional differences in the structure of the lamina cribrosa and their relation to glaucomatous optic-nerve damage. Arch. Ophthalmol. 99, 137–143 (1981).
Kirwan, R. P. et al. Influence of cyclical mechanical strain on extracellular matrix gene expression in human lamina cribrosa cells in vitro. Mol. Vis. 11, 798–810 (2005).
Kirwan, R. P., Crean, J. K., Fenerty, C. H., Clark, A. F. & O’Brien, C. J. Effect of cyclical mechanical stretch and exogenous transforming growth factor-beta1 on matrix metalloproteinase-2 activity in lamina cribrosa cells from the human optic nerve head. J. Glaucoma 13, 327 (2004).
Rogers, R. S., Dharsee, M., Ackloo, S. & Flanagan, J. G. Proteomics analyses of activated human optic nerve head lamina cribrosa cells following biomechanical strain. Investig. Ophthalmol. Vis. Sci. 53, 3806–3816 (2012).
Rogers, R. S., Dharsee, M., Ackloo, S., Sivak, J. M. & Flanagan, J. G. Proteomics analyses of human optic nerve head astrocytes following biomechanical strain. Mol. Cell. Proteom. MCP 11, M111.012302 (2012).
Alqawlaq, S., Flanagan, J. G. & Sivak, J. M. All roads lead to glaucoma: induced retinal injury cascades contribute to a common neurodegenerative outcome. Exp. Eye Res. 183, 88–97 (2019).
Hernandez, M. R. The optic nerve head in glaucoma: role of astrocytes in tissue remodeling. Prog. Retin. Eye Res. 19, 297–321 (2000).
Lye-Barthel, M., Sun, D. & Jakobs, T. C. Morphology of astrocytes in a glaucomatous optic nerve. Investig. Opthalmol. Vis. Sci. 54, 909 (2013).
Tehrani, S., Johnson, E. C., Cepurna, W. O. & Morrison, J. C. Astrocyte processes label for filamentous actin and reorient early within the optic nerve head in a rat glaucoma model. Investig. Ophthalmol. Vis. Sci. 55, 6945–6952 (2014).
Tehrani, S. et al. Astrocyte structural and molecular response to elevated intraocular pressure occurs rapidly and precedes axonal tubulin rearrangement within the optic nerve head in a rat model. PLoS ONE 11, e0167364 (2016).
Johnson, E. C. & Morrison, J. C. Friend or foe? Resolving the impact of glial responses in glaucoma. J. Glaucoma 18, 341–353 (2009).
Neufeld, A. H. & Liu, B. Glaucomatous optic neuropathy: when glia misbehave. Neuroscientist 9, 485–495 (2003).
Son, J. L. et al. Glaucomatous optic nerve injury involves early astrocyte reactivity and late oligodendrocyte loss. Glia 58, 780–789 (2010).
Chong, R. S. & Martin, K. R. Glial cell interactions and glaucoma. Curr. Opin. Ophthalmol. 26, 73–77 (2015).
de Hoz, R. et al. Retinal macroglial responses in health and disease. BioMed. Res. Int. 2016, 2954721 (2016).
Sofroniew, M. V. & Vinters, H. V. Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35 (2010).
Ridet, J. L., Malhotra, S. K., Privat, A. & Gage, F. H. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 20, 570–577 (1997).
Munemasa, Y. & Kitaoka, Y. Molecular mechanisms of retinal ganglion cell degeneration in glaucoma and future prospects for cell body and axonal protection. Front. Cell. Neurosci. 6, 60 (2012).
Sun, D., Moore, S. & Jakobs, T. C. Optic nerve astrocyte reactivity protects function in experimental glaucoma and other nerve injuries. J. Exp. Med. 214, 1411–1430 (2017).
Livne-Bar, I. et al. Astrocyte-derived lipoxins A4 and B4 promote neuroprotection from acute and chronic injury. Investig. Ophthalmol. Vis. Sci. 127, 4403–4414 (2017).
Zamanian, J. L. et al. Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410 (2012).
Mulvihill, J. J. E. et al. Development of a platform for studying 3D astrocyte mechanobiology: compression of astrocytes in collagen gels. Ann. Biomed. Eng. 46, 365–374 (2018).
Morrison, J. C. et al. A period of controlled elevation of IOP (CEI) produces the specific gene expression responses and focal injury pattern of experimental rat glaucoma. Investig. Ophthalmol. Vis. Sci. 57, 6700–6711 (2016).
Tehrani, S. et al. Optic nerve head astrocytes display axon-dependent and -independent reactivity in response to acutely elevated intraocular pressure. Investig. Opthalmol. Vis. Sci. 60, 312 (2019).
Kirschner, A. et al. Mechanosensitive channel inhibition attenuates TGFβ2-induced actin cytoskeletal remodeling and reactivity in mouse optic nerve head astrocytes. Exp Eye Res. 212, 108791 (2021).
Choi, H. J., Sun, D. & Jakobs, T. C. Astrocytes in the optic nerve head express putative mechanosensitive channels. Mol. Vis. 21, 749–766 (2015).
Tezel, G., Chauhan, B. C., LeBlanc, R. P. & Wax, M. B. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Investig. Ophthalmol. Vis. Sci. 44, 3025–3033 (2003).
Mammone, T., Chidlow, G., Casson, R. J. & Wood, J. P. M. Expression and activation of mitogen-activated protein kinases in the optic nerve head in a rat model of ocular hypertension. Mol. Cell. Neurosci. 88, 270–291 (2018).
Dapper, J. D., Crish, S. D., Pang, I.-H. & Calkins, D. J. Proximal inhibition of p38 MAPK stress signaling prevents distal axonopathy. Neurobiol. Dis. 59, 26–37 (2013).
Lambert, W. S. et al. Of mice and monkeys: neuroprotective efficacy of the p38 inhibitor BIRB 796 depends on model duration in experimental glaucoma. Sci. Rep. 10, 8535 (2020).
Johnson, E. C. et al. The effect of chronically elevated intraocular pressure on the rat optic nerve head extracellular matrix. Exp. Eye Res. 62, 663–674 (1996).
Malone, P., Miao, H., Parker, A., Juarez, S. & Hernandez, M. R. Pressure induces loss of gap junction communication and redistribution of connexin 43 in astrocytes. Glia 55, 1085–1098 (2007).
Vicic, N. et al. Evidence of an Annexin A4 mediated plasma membrane repair response to biomechanical strain associated with glaucoma pathogenesis. J. Cell. Physiol. 237, 3687–3702 (2022).
Križaj, D. What is glaucoma? in Webvision: The Organization of the Retina and Visual System (eds Kolb, H., Fernandez, E., Jones, B. & Nelson, R.) (University of Utah Health Sciences Center, 1995).
Seitz, R., Ohlmann, A. & Tamm, E. R. The role of Müller glia and microglia in glaucoma. Cell Tissue Res. 353, 339–345 (2013).
Williams, P. A., Marsh-Armstrong, N., Howell, G. R. & Lasker/IRRF Initiative on Astrocytes and Glaucomatous Neurodegeneration Participants Neuroinflammation in glaucoma: a new opportunity. Exp. Eye Res. 157, 20–27 (2017).
Tan, Z., Guo, Y., Shrestha, M., Gregory-Ksander, M. S. & Jakobs, T. C. Depletion of optic nerve microglia does not improve visual function in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 62, 2383 (2021).
Zong, B. et al. Mechanosensitive Piezo1 channel in physiology and pathophysiology of the central nervous system. Ageing Res. Rev. 90, 102026 (2023).
Križaj, D. et al. From mechanosensitivity to inflammatory responses: new players in the pathology of glaucoma. Curr. Eye Res. 39, 105–119 (2014).
Wang, B. et al. Tortuous pore path through the glaucomatous lamina cribrosa. Sci. Rep. 8, 7281 (2018).
Quigley, H. A. & Addicks, E. M. Chronic experimental glaucoma in primates. II. Effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Investig. Ophthalmol. Vis. Sci. 19, 137–152 (1980).
Ryskamp, D. et al. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J. Neurosci. 31, 7089–7101 (2011).
Dvoriantchikova, G. et al. Genetic ablation of pannexin1 protects retinal neurons from ischemic injury. PLoS ONE 7, e31991 (2012).
Wheeler-Schilling, T. H., Marquordt, K., Kohler, K., Guenther, E. & Jabs, R. Identification of purinergic receptors in retinal ganglion cells. Brain Res. Mol. Brain Res. 92, 177–180 (2001).
Morozumi, W. et al. Piezo channel plays a part in retinal ganglion cell damage. Exp. Eye Res. 191, 107900 (2020).
Sripinun, P. et al. Piezo1 and Piezo2 channels in retinal ganglion cells and the impact of Piezo1 stimulation on light-dependent neural activity. BioRxiv Prepr. Serv. Biol. 2024.06.25.599602. https://doi.org/10.1101/2024.06.25.599602 (2024).
Tribble, J. R. et al. Midget retinal ganglion cell dendritic and mitochondrial degeneration is an early feature of human glaucoma. Brain Commun. 1, fcz035 (2019).
Whitmore, A. V., Libby, R. T. & John, S. W. M. Glaucoma: thinking in new ways-a rôle for autonomous axonal self-destruction and other compartmentalised processes?. Prog. Retin. Eye Res. 24, 639–662 (2005).
Beckel, J. M. et al. Mechanosensitive release of adenosine 5’-triphosphate through pannexin channels and mechanosensitive upregulation of pannexin channels in optic nerve head astrocytes: a mechanism for purinergic involvement in chronic strain. Glia 62, 1486–1501 (2014).
Albalawi, F. et al. The P2X7 Receptor primes IL-1β and the NLRP3 inflammasome in astrocytes exposed to mechanical strain. Front. Cell. Neurosci. 11, 227 (2017).
Yang, Y. & Sun, X. Retinal ganglion cell death in glaucoma: advances and caveats. Curr. Eye Res. 48, 1–10 (2023).
Almasieh, M., Wilson, A. M., Morquette, B., Cueva Vargas, J. L. & Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 31, 152–181 (2012).
Coleman, D. J. & Trokel, S. Direct-recorded intraocular pressure variations in a human subject. Arch. Ophthalmol. 82, 637 (1969).
Downs, J. C. et al. 24-hour IOP telemetry in the nonhuman primate: implant system performance and initial characterization of IOP at multiple timescales. Investig. Ophthalmol. Vis. Sci. 52, 7365–7375 (2011).
Uhler, C. & Shivashankar, G. V. Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat. Rev. Mol. Cell Biol. 18, 717–727 (2017).
El Hajji, S. et al. Insulin restores retinal ganglion cell functional connectivity and promotes visual recovery in glaucoma. Sci. Adv. 10, eadl5722 (2024).
Liu, K. C. et al. Resident tissue macrophages govern intraocular pressure homeostasis. BioRxiv Prepr. Serv. Biol. 2025.01.25.634888. https://doi.org/10.1101/2025.01.25.634888 (2025).
Williams, P. A. et al. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 355, 756–760 (2017).
Hirt, J. & Liton, P. B. Autophagy and mechanotransduction in outflow pathway cells. Exp. Eye Res. 158, 146–153 (2017).
Alarcon-Martinez, L. et al. Neurovascular dysfunction in glaucoma. Prog. Retin. Eye Res. 97, 101217 (2023).
Diemler, C. A. et al. Microglia depletion leads to increased susceptibility to ocular hypertension-dependent glaucoma. Front. Aging Neurosci. 16, 1396443 (2024).
Hammond, G., Young, R., Muir, D. & Quantock, A. The microanatomy of Bowman’s layer in the cornea of the pig: changes in collagen fibril architecture at the corneoscleral limbus. Eur. J. Anat. 24, 399–406 (2020).
Aliò, J. L., Anania, A. & Sagnelli, P. The aging of the human lens. in Age-Related Changes of the Human Eye (eds Cavallotti, C. A. P. & Cerulli, L.) 61–131. https://doi.org/10.1007/978-1-59745-507-7_5 (Humana Press, 2008).
Costagliola, C. et al. How many aqueous humor outflow pathways are there?. Surv. Ophthalmol. 65, 144–170 (2020).
Hayreh, S. S. Ischemic optic neuropathy. Prog. Retin. Eye Res. 28, 34–62 (2009).
Minckler, D. S. Histology of optic nerve damage in ocular hypertension and early glaucoma. Surv. Ophthalmol. 33, 401 (1989).
Lai, J. et al. The role of Schlemm’s canal endothelium cellular connectivity in giant vacuole formation: a 3D electron microscopy study. Investig. Opthalmol. Vis. Sci. 60, 1630 (2019).
Krey, H. F. & Bräuer, H. Chibret Augenatlas: Eine Repetition Für Ärtze Mit Zeigetafeln Für Patienten (Chibret Medical Service, 1998).
Waxman, S. et al. Individual astrocyte morphology in the collagenous lamina cribrosa revealed by multicolor DiOlistic labeling. Exp. Eye Res. 230, 109458 (2023).
Acknowledgements
We acknowledge Professor Levi Wood (Georgia Tech) for fruitful discussions about glial mechanobiology. We thank Dr. Haiyan Li for her help in generating certain elements of Figure 3. We gratefully acknowledge grant support from the NIH (R21EY033142 (C.R.E.), R01EY034096 (S.H.), R21EY036189 (S.H.)), the BrightFocus Foundation (CG2020001 (C.R.E.), G2024006S (S.H.)), unrestricted funds from the Georgia Research Alliance (C.R.E.), and unrestricted grants from Research to Prevent Blindness and Lions Region 20-Y1 to SUNY Upstate Medical University Department of Ophthalmology and Visual Sciences (S.H.).
Author information
Authors and Affiliations
Contributions
C.R.E. and S.H. wrote the main manuscript text. C.R.E. prepared Figures 1 and 3. S.H. prepared Figure 2. Both authors reviewed and edited the manuscript, including figures.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ethier, C.R., Herberg, S. Mechanobiology in the eye. npj Biol. Phys. Mech. 2, 18 (2025). https://doi.org/10.1038/s44341-025-00022-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44341-025-00022-6
