
Abstract
Cancer immunotherapies, including immune checkpoint blockade and engineered T cell therapies, have revolutionized cancer treatment but face challenges from cancer-specific heterogeneity, tumor-immune interaction complexity, age-associated immune alteration and interspecies limitations. Advances in organoid technologies, using the patient-derived, self-organizing three-dimensional tissues, provide physiologically relevant platforms to model tumor-immune interactions and evaluate therapies in a cancer- and patient-specific manner. This platform has significantly enhanced the predictive power of preclinical research and facilitated more effective immunotherapy development.
Similar content being viewed by others
Introduction
Cancer immunotherapy is rapidly emerging as a promising approach that harnesses and modulates the patient’s-own immune system to selectively eliminate tumor cells. It has demonstrated significant clinical success and is now widely recognized as an essential component of cancer treatment. Strategies like immune checkpoint blockade (ICB) and cellular engineering approaches have become mainstream treatments for certain cancer types due to their high specificity and effectiveness. ICB works by blocking inhibitory pathways that tumor often exploit, such as cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and program death-1 (PD-1)/PD-L1, to evade immune attack. Therapeutic antibodies to block these inhibitory receptors or their ligands can restore the T cell activity and promote a robust anti-tumor immune response. Cellular engineering approaches, especially adoptive T-cell therapies like chimeric antigen receptor (CAR)-T cell therapy, involving modifying patient-derived immune cells to express synthetic receptors that enhance their capabilities to target and kill tumor cells, have shown remarkable clinical success in hematological malignancies1,2,3,4. Similarly, T cell receptor-engineered T cell (TCR-T) therapy, which employs engineered αβ heterodimers to recognize tumor-specific peptides presented by MHC molecules, has demonstrated promising efficacy in a growing number of solid cancers5,6,7.
Despite remarkable advances in cancer immunotherapy, its clinical effectiveness often falls short of expectations. Many approaches that demonstrate promise in preclinical studies encounter significant challenges during clinical translation, largely due to fundamental interspecies differences8,9 that limit the predictive power of animal models. Even when immunotherapies are effective, their success is often restricted to specific cancer types10 or specific patient subgroups11,12,13, limiting their broad applicability. Moreover, these challenges are compounded by the intrinsic complexity of the immune system. The immune responses to cancer are highly dynamic and context dependent. This mechanistic uncertainty contributes to heterogeneity in patient responses and complicates the development of predictive biomarkers. Lacking the robust pathological or immunological parameters, treatment decisions are sometimes guided by retrospective and empirical analyses rather than prospective and personalized strategies, which creates substantial burdens for both patients and healthcare systems.
To address these challenges, it is critical to clearly define the precise indications and mechanisms of existing immunotherapies, develop more broadly effective treatment strategies, and establish reliable methods to predict clinical outcomes and guide therapeutic decision-making. Animal models have been widely used as a robust and established model for mechanistic investigation and therapeutic evaluation. In vitro approaches have advanced rapidly in recent years, with the development of various 3D spheroids14,15, organ-on-a-chip systems16,17, and organoids18,19,20,21,22 to address medically challenging questions and validate treatments. Achieving these goals requires a controllable, physiologically relevant in vitro platform that complements existing in vivo models and faithfully recapitulates the complex interactions between tumor and the human immune system.
Among the various in vitro platforms developed to date, organoids stand out as self-organizing, three-dimensional (3D) tissues that closely mimic the structural, functional, and biological complexities of native organs, offering significant potential to address current unmet research and clinical needs19,20,22. Organoids can be derived from pluripotent stem cells, adult tissue-resident stem or progenitor cells, or directly from differentiated and diseased tissues. Notably, patient-derived tumor organoids (PDOs) generated from primary cancer tissues preserve key histological, genetic, and phenotypic features of native tumors, including their proliferative potential and heterogeneity20,23. The human origin and self-organized cytoarchitecture of organoids offer unique advantages over conventional two-dimensional (2D) or xenograft models, enabling mechanistic studies that faithfully reflect human biology and disease progression. Beyond cancer, organoids have been widely adopted across multiple disciplines. In pharmacology and toxicology, organoid-based assays can outperform 2D cultures in predicting human-specific responses with more accuracy, as their 3D architecture and multicellular composition better capture tissue physiology24. In regenerative medicine, tissue-specific organoids derived from intestinal, neural, and renal tissues are being explored for transplantation and tissue repair25,26. Furthermore, organoids have enabled modeling of complex diseases such as cystic fibrosis27, neurodegeneration28, and viral infections29,30, demonstrating their broad biomedical relevance.
Organoids can be fabricated with different methods, depending on the desired biological fidelity and experimental throughput. Some commonly employed strategies for microphysiological system design include bioprinting, microfluidics, microwells and hanging drops, enabling either spontaneous self-assembly or guided organization under biological, physical and geometric constraints. These fabrication approaches have been thoroughly reviewed before19,31,32 and thus won’t be extensively discussed in this review. The continued evolution of organoid research has been tightly coupled with innovations in material science, gene editing and computational analysis. Functional biomaterials have transformed static organoid matrices into dynamic microenvironments that support vascularization and mechanical tuning. CRISPR-Cas9-based editing approach allows lineage tracing and precise interrogation of biological pathways. The vast, information-rich datasets generated from organoids, in combination with artificial intelligence (AI), facilitate multidimensional data integration and accelerate discovery. Together, these advances are transforming organoids into a powerful, integrative platform for both fundamental research and translational applications, particularly in cancer immunotherapy.
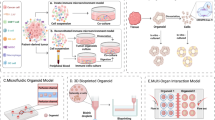
In this review, we first summarize major challenges in current cancer immunotherapies and then discuss how organoid technologies can be leveraged to address these challenges and inform the development of next-generation immunotherapeutic strategies (Fig. 1).

This platform integrates high-throughput screening, high-dimensional systems analyses to study tumor-immune interactions while mimicking the temporal dynamics. These unique features enable the evaluation of personalized therapies, addressing key challenges in cancer immunotherapy and accelerating the development of next-generation treatments. HLA human leukocyte antigen, TCR T cell receptor. Created in part with BioRender.com.
Challenges in current immunotherapies
Tumor type-specific heterogeneity
The clinical efficacy of Immunotherapy varies markedly across tumor types and among individual patients. This variation stems largely from distinct tumor-intrinsic features and immune escape mechanisms that modulate how tumor interact and evade the immune system.
One of the prominent challenges lies in the primary and secondary immune escape mechanisms. Primary immune escape occurs when the tumor evades immune detection before therapy, often resulting in a complete lack of responses in patients. Secondary immune escape arises when the tumor initially responds to treatment but later develops resistance, allowing disease progression despite continued therapy33. Although not yet fully understood, primary immune escape is often associated with deficient antigen presentation machinery or suboptimal T cell priming and activation, while second immune escape is thought to be driven by selective pressure imposed by immunotherapy, leading to tumor evolution, clonal selection, and immunoediting34,35,36,37. Across the different cancer types, tumor mutation burden (TMB) has emerged as a key biomarker for predicting response to immunotherapies. It is a measure of how many genetic mutations are present in a tumor, which influences the number of potential neoantigens, or mutant proteins that the immune system can recognize. Cancers with high TMB, such as melanoma and non-small cell lung cancer (NSCLC), generally respond better to checkpoint blockade therapies, while those with low TMB, like pediatric cancers, often show poor response38,39,40,41,42. However, TMB alone is insufficient to predict outcomes. Mutation patterns are also influenced by patient-specific factors, such as smoking or UV exposure, and additional mutations may arise during tumor evolution, including those driven by different enzymes or chemotherapy43. Tumor heterogeneity also impacts how the immune system recognizes neoantigens, further complicating the quantity and diversity of mutations when evaluating TMB as a biomarker44,45,46.
Beyond genetic heterogeneity, tumors exhibit substantial phenotypic heterogeneity driven by non-genetic mechanisms, such as epigenetic reprogramming and tumor microenvironment cues that dynamically reshape cellular states. Among these mechanisms, epithelial-mesenchymal transition (EMT) generates a continuum of cell states ranging from epithelial to mesenchymal, including hybrid epithelial/mesenchymal (E/M) phenotypes. These diverse phenotypes enhance intra-tumor diversity, and particularly, epithelial and mesenchymal phenotypes confer distinct advantages that facilitate tumor progression and metastasis. Increasing evidence indicates that the EMT spectrum closely aligns with variations in tumor immunogenicity and immune activity, highlighting EMT as a key mechanism linking phenotypic heterogeneity to tumor-immune interplay47,48. The interplay between TMB and immune microenvironments, including T cell infiltration, antigen-presenting cell (APC) function, and checkpoint molecule expression, also varies across cancer types. For example, in colorectal cancer (CRC), immune checkpoint inhibitors are effective in subtypes of the cancer such as CMS1 and CMS4, which have high CD8⁺ T cell and macrophage presence, while CMS2 tumors lack immune activation and show lower response hence reduced efficacy49,50. In pancreatic ductal adenocarcinoma (PDAC), characterized by dense stroma and severely immunosuppressive microenvironment paired with a high presence of transforming growth factor-β (TGFβ)51,52, limits immune cell activity and shows minimal response to checkpoint inhibitors alone. But combining chemotherapy with immunotherapy to modulate the microenvironment has shown improved efficacy, particularly in chemotherapy-naïve patients53.
Taken together, these findings highlight the heterogeneities in mutational landscape, immune phenotype, microenvironmental context across tumor types jointly shape the effectiveness of immunotherapies. Even when predictive markers like PD-L1, TMB, or CD8+ T cell infiltration suggest an effective response, clinical outcomes can be variable. To address these, more physiologically relevant models are needed to model diverse tumor types and immune environments in a patient-specific context.
Complexity of Tumor Microenvironment (TME) Across Different Organs
The distinct physical, structural, and immunological characteristics of tissue microenvironments across different organs contribute significantly to variations in tumor behavior and responses to immunotherapy. The tumor immune contexture54, encompassing immune cell density, spatial distribution, activation status, and cytokine milieu, varies considerably across organs due to variations in vascular structures, lymphatic networks, stromal composition, and resident immune cell populations55,56,57. Furthermore, the composition of the extracellular matrix (ECM), along with biomechanical properties such as tissue stiffness and viscoelasticity, plays a critical role in modulating tumor progression, immune infiltration and the efficacy of immunotherapy58.
Organ-specific immune environments profoundly influence therapeutic outcomes. For example, organs such as the liver exhibit a lower response rate to immunotherapies than other sites due to lower immune cell infiltration and a locally immunosuppressive environment59,60. Notably, a study of patients with advanced non-small cell lung cancer (NSCLC) treated with first-line anti-PD-1 or anti-PD-L1 therapy showed that while lung tumors showed the highest response rate at 67%, liver metastases showed the lowest at 30.6%. This highlights the heterogeneity of response depending on the anatomical site of metastasis and suggests that the liver’s dense and fibrotic ECM, combined with its immunosuppressive environment, may contribute to its lower responsiveness to ICB61.
Although T cells are central to the efficacy of ICB, B cells are emerging as important contributors, with their impact varying across different tissue and tumor microenvironments. In melanoma, some studies report that B cell depletion does not impact anti-PD-1 outcomes in melanoma62, but others link B cell presence, especially memory B cells and plasmablasts, to improved responses63,64. Higher levels of circulating plasmablasts have been associated with better outcomes in melanoma, lung, and renal cancers treated with checkpoint inhibitors. Memory B cell gene signatures also correlate with enhanced survival in melanoma and urothelial carcinoma patients receiving immune checkpoint blockade therapy65. This organ- and tumor-specific variability suggests B cell-mediated effects may be context-dependent. Moreover, B cell derived antibodies appear to support treatment responses, with increased levels of IgG and IgM observed in ICB responders. In renal cell carcinoma, antibody complexes may trigger the complement system and enhance immune activation. Additionally, B cells can express checkpoint molecules like PD-1 and CTLA-4, suggesting they may be directly modulated by ICB66,67,68.
Another critical dimension of TME complexity is vasculature heterogeneity. In most tumors, angiogenesis leads to blood vessels with abnormal structure and function, impairing perfusion that paradoxically supports malignancy. As tumors expand, their increasing metabolic demand triggers aberrant angiogenesis generating irregular, leaky, and poorly perfused vasculature. These abnormities impair the drug delivery and immune cell trafficking while fostering hypoxia that further activates fibroblasts69,70 and stimulates excessive ECM deposition to increase tissue stiffness and elevate solid stress71. The combination of leaky vessels and compression from cancer cell-induced solid stress further impairs perfusion, amplifying hypoxia and acidosis within TME. Under such conditions, malignant cells exploit the leaky vasculature to intravasate and disseminate to distant organs, promoting metastasis. Concurrently, immune surveillance becomes severely compromised as both tissue-resident and bone marrow-derived immune cells exhibit reduced activity and altered recruitment dynamicsv72,73. Collectively, these vasculature abnormalities promote fibrosis, hypoxia, and immune suppression that sustains tumor progression and limits therapeutic efficacy.
The anatomical location of a tumor and the microarchitectural complexity of the surrounding host tissue can impact immune cell infiltration74,75,76,77 and the efficiency of drug delivery78. Tumors situated in stiffer tissues or in regions with structural barriers, such as dense extracellular matrices, may hinder immune cell access and limit antigen presentation58,79. These physical and immunological constraints emphasize the need to account for both biological and mechanical features of the tumor microenvironment when designing effective immunotherapies.
Impact of aging
Aging has been reported to influence both cancer development and the effectiveness of immunotherapies. Elderly and aged individuals account for 60% of new cancer diagnoses and 70% of cancer-related deaths80, largely due to accumulation of genetic mutations, immune system decline, and a chronically pro-inflammatory systemic environment. Sexual dimorphism adds an additional layer of complexity, as aging-associated sex-specific metabolic, genetic, epigenetic, and immune differences are observed in both human and animal studies81,82. Recent studies have begun to dissect the multiple dimensions of aging to reveal the specific biological changes that shape immune responses and contribute to the heterogeneity in immunotherapy outcomes.
Cellular senescence is a physiological process in which proliferating cells undergo stable cell cycle arrest in response to stress or damage while remaining metabolically active. Senescent cells adopt a distinctive transcriptional and secretory state known as the senescence-associated secretory phenotype (SASP), characterized by the release of pro-inflammatory cytokines, growth factors, and ECM-remodeling proteins. Under general conditions, these secreted factors signal the immune system to clear senescent cells and promote tissue repair. However, with advancing age or consistent chronic tissue damage, immune surveillance becomes compromised, leading to accumulation of senescent cells and persistent SASP activity. The resulting chronic inflammation and microenvironment remodeling disrupt tissue homeostasis and promote cancer initiation and progression. Immune cells originate from bone marrow-resident hematopoietic stem cells (HSCs). These bone marrow–derived immune populations influence solid tumors through the hematopoiesis–cancer axis83,84,85. Aging generates profound alterations on hematopoiesis through systemic cytokine changes, somatic mutations, and epigenetic dysregulation. For older individuals, hematopoiesis often exhibits a myeloid bias, in which HSCs and progenitor cells preferentially differentiate into myeloid lineages (monocytes, macrophage, and neutrophils) at the expense of lymphoid lineages. This shift alters immune cell composition and function, impairing their anti-tumor responses. The elevated pro-inflammatory cytokines further perturb HSC homeostasis, reinforcing systemic inflammation and immune dysfunction. Together, these interactions remodel the TME, influencing tumor initiation, progression and therapeutic responses.
Immune system aging, termed immunosenescence, is the age-related decline in immune function, marked by structural deterioration of immune organs and impaired innate and adaptive responses86. Research has identified quantifiable shifts in TCR repertoire that are associated with Immunosenescence. Using a framework called Repertoire Functional Units (RFUs), the study revealed specific RFUs that consistently decline with age in over 6500 blood-derived TCR sequencing samples, indicating large alterations in immune composition over time. Moreover, individuals with immunosuppressive conditions exhibited an accelerated loss of these RFUs, suggesting that immune suppression may add to the age-related immune decline of patients87. A recent study leveraging advanced single-cell technologies mapped age-associated changes in the human peripheral immune system across the lifespan. T cells undergo the most pronounced age-related alterations, with effector memory CD8+ T cells expanding in both young and elderly individuals, while CD8+ cells peak in adolescence and decrease with age, suggesting diminished immune defense in older individuals61. The study also identifies a novel cytotoxic B cell subset unique in children, highlighting the immune heterogeneity in the development.
Aging is also linked to chronic low-grade inflammation, known as inflammaging, which causes changes in gut microbiota composition and impacts the immune responses. Researchers have transferred microbiota from young and old conventional mice into young germ-free (GF) mice. Results showed that aged microbiota induced inflammatory responses in young GF mice, including increased gut permeability, elevated systemic T cell activation, and higher levels of pro-inflammatory bacterial taxa. The aged microbiota also promoted an increase in splenic Th1, Th2, and Treg cells, indicating that microbial changes associated with aging can drive immune alterations88.
Aging also reshapes the tumor tissue microenvironment by promoting the accumulation of senescent stromal cells that release factors altering the ECM, increasing tissue stiffness and promoting tumor progression, particularly in soft tissues83. While overall ECM degradation occurs with age, localized stiffening can create microenvironments favorable for cancer. In lungs and breast tissue, age-related increases in collagen and crosslinking have been linked to greater cancer risk89,90. Age-related vasculature abnormalities further perturb immune infiltration and nutrient exchange, while alterations in adipocytes and neural regulation impact immune modulation. Despite growing evidence linking aging-related ECM remodeling on immune-tumor interactions, the precise effects remain poorly defined and need further investigation.
Moreover, aging is closely correlated with therapy responses83,91,92. Interestingly, age does not uniformly impair immunotherapy efficacy. An analysis of 538 patients revealed that individuals over 60 demonstrated better responses to anti-PD-1 therapy, regardless of prior inhibitor treatment. In genetically identical murine melanoma tumors, older mice responded more favorably to anti-PD-1 therapy compared to younger mice, potentially owing to a more favorable CD8+ T cell to Treg ratio in aged tumor microenvironment93. However, age-related immune responses have yet to be studied thoroughly due to the extremely limited database on age-specific immunotherapy outcomes. Of the 30 clinical trials reported in ClinicalTrials.gov, only 11 include results for patients over 65. Additionally, most of these were early-phase trials, underscoring the lack of comprehensive data on how aging affects immunotherapy effectiveness. Notably, severe side effects (Grades 3 or 4) occurred in around 40% of elderly patients. These rates are higher than those seen in younger patients, suggesting a need for more detailed studies that assess both the safety and effectiveness of immunotherapies in older adults94. Currently, older adults remain underrepresented in clinical trials95. Among these trials, inconsistent definitions of “older” age and selective enrollment of healthier individuals introduce bias that limits generalizability. Incorporation of geriatric and frailty assessments to estimate biological age rather than chronological age may improve the interpretation of immunotherapy outcomes96,97.
Cross-species limitations
One major challenge in immunotherapy development is the disconnect between preclinical models and human immune systems. While murine models are widely used for mechanistic insights and therapeutic screening, significant differences in tumor biology and immune function often limit the translational relevance of preclinical findings98. In mice, tumors are typically implanted and progress rapidly over days to weeks, whereas in humans, tumor arise and evolve over years through extensive interaction with the immune system. These differences significantly influence how tumors evade immune surveillance and respond to therapy. Both innate and adaptive immune responses contribute to the disconnection between mouse and humans, understanding their species-specific differences is critical for improving the design and translation of immunotherapeutic strategies.
Innate immunity serves the body’s first line of defense, offering rapid and often nonspecific responses against pathogens and foreign invaders. In cancer, this includes the activity of macrophages, NK cells, and dendritic cells. However, these innate cells can functionally differ between mice and humans. For example, Toll-like receptors (TLRs), a type of pattern recognition receptor (PRR) in the innate immune system, detect pathogen-associated molecular patterns (PAMPs) from infectious agents and damage-associated molecular patterns (DAMPs)99,100. These receptors have played an instrumental role in cancer immunity, as they are expressed on various immune cells to activate signaling pathways that stimulate cytokines to attack invaders101. However, several TLRs are expressed differently in mice and humans, with differences in which cell types produce them and how their activity is controlled when cells are activated102,103. Certain molecular regulators of immunity, such as tumor suppressor p53 protein, also shows species-specific functional differences in innate immunity. P53 helps manage cellular stress responses by regulating genes involved in DNA repair, metabolism, and cell cycle control, which are essential for maintaining immune cell function. However, studies have shown that the specific DNA regions p53 binds to vary greatly between mice and humans, leading to different gene expression patterns104. This variation is due to evolutionary changes in DNA elements that control gene activity, making it challenging to directly apply findings from mouse models to human biology.
Adaptive immune responses, particularly T cell-mediated immunity, central to cancer immunotherapy by providing antigen-specific responses critical for long-term protection and tumor control. T cells mount antigen-specific responses through highly diverse TCRs recognizing peptide presented by major histocompatibility complex (MHC) molecules105. MHC class I molecules present intracellular antigens to CD8+ T cells, while MHC class II molecules present extracellular antigens to CD4+ T cells. The polymorphic nature of human MHC genes, coupled with structural differences in MHC molecules across species, pose significant challenges for linking murine T cell data to humans. Furthermore, the phenomenon of TCR cross-reactivity, where a single receptor can bind multiple pMHC complexes, adding complexity to repertoire analysis and antigen validation. The intricate nature of TCR-pMHC interactions helps explain why traditional models often struggle to accurately forecast immunotherapy results. The immense diversity of T cell receptors, the individualized structure of MHC molecules, and the tendency of TCRs to recognize multiple pMHC targets all underscore the need for testing platforms tailored to each patient.
Importantly, species-specific differences in both skin and spleen structure also influence immune system function. Human skin is approximately four times thicker than that of mice106 and lacks the murine unique population of dendritic epidermal T cells107. The increased thickness of human skin enables more direct access to the lymphatic system, which may influence immune cell trafficking and surveillance108,109,110. The structural organization of the spleen also varies notably between the two species. While both humans and mice have red pulp and white pulp regions, the spatial arrangement of immune cells within these compartments differs. In mice, T cells are typically localized in well-defined zones, whereas in humans, they are more diffusely distributed throughout germinal centers. Moreover, mice exhibit a clearly delineated marginal zone that is not present in human spleens. This murine marginal zone includes layered populations of macrophages, such as CD169+ cells, a feature not yet confirmed in humans111. These structural differences highlight the limitations of directly translating immunological findings from mice to humans.
In addition to anatomical and functional disparities, there is the genetic uniformity of laboratory mice. Inbred strains lack the genetic diversity of humans, who also have the added factor of genetic predisposition to disease and environmental factors, whereas mice are raised in highly controlled conditions with little genetic variation and exposure to different environments112,113,114. As a result, immune responses in murine models may fail to capture the full spectrum of human variation. Researchers have developed various strategies to address the limitations of inbred mouse models, including the use of humanized mice115, collaborative cross strains116, and even exposure to more environmentally diverse “pet shop” mice117,118. These alternative models offer valuable insights and certain advantages, yet they also face notable challenges that have limited their widespread implementation.
Leveraging organoids to develop next-generation immunotherapeutic strategies
As advanced cancer treatment increasingly moves toward precision immunotherapy, the need for sophisticated and physiologically relevant models has become critical. Addressing tumor type-specific heterogeneity, organ-dependent complexity of the tumor TME, aging-associated immune alterations (Table 1), and the translational gap between murine and human systems requires advanced modeling approaches. Organoids have emerged as a powerful tool to address these challenges. Unlike traditional 2D cell culture models, which are widely utilized for preliminary drug screening but lack the structural complexity and cellular heterogeneity of in vivo systems, organoids recapitulate the architectural, cellular diversity, and molecular features of the original tissues. This allows for more faithful modeling tumor-immune interactions and more accurate evaluation of therapeutic responses in a tissue-specific and patient-relevant manner119. Compared to murine models, organoids could overcome the limitation in cross-specifies limitations presented in translational research, providing a human-relevant system that better predicts clinical outcomes.
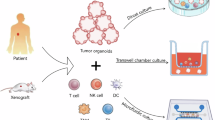
In this section, we explore how organoid-based systems contribute to the development of next-generation cancer immunotherapies. By enabling in-depth modeling the tumor-immune interactions within a controlled and tunable environment, organoids offer a scalable, human-relevant and physiologically accurate platform to advance cancer immunotherapy research and clinical translation (Fig. 2).

Current organoid models have been established across cancer types and organs, enabling comprehensive investigation of tumor heterogeneity, tissue-specific tumor-immune interactions, cross-species differences, and age-related effects on immune responses. These systems have been leveraged for high-throughput drug screening, toxicity assessment, and the development of precision immunotherapy. Future advancements aim to integrate multi-system crosstalk, scalable and controllable production, AI-assisted prediction and real-time monitoring to improve physiological relevance, reproducibility and prediction accuracy. These innovations are expected to accelerate the development of next-generation, more effective immunotherapies. Created in part with BioRender.com.
Organoid Models Across Cancer Types and Organs
Given the extensive tumor heterogeneity and the distinct biological characteristics of cancers originating in different organs, it is a critical need to develop robust in vitro models can reliably assess therapeutic efficacy, predict clinical outcomes, and inform precision medicine strategies including the treatment consistency and adverse effects. To meet this need, organoid models have been developed for a wide range of cancers including pancreatic ductal adenocarcinoma (PDAC)120,121, brain cancers such as glioblastoma (GBM)122,123, breast cancer124,125, colorectal cancer (CRC)126,127, gastric cancer128, lymphoma129, bladder cancer130, and oral cancer131. These platforms offer physiologically relevant platforms for studying tumor biology, with many incorporating features such as vasculature, immune cell infiltration and tissue-specific microenvironments. For example, GBM organoids have been developed to rebuild the GBM microenvironment, with the introduction of vasculature, blood-brain barrier (BBB), and immune cells, facilitating more predictive drug screening and therapeutic evaluation132.
Large-scale tumor organoid libraries further expand the applications of these platforms. Fujii et al. established a library of 55 colorectal tumor organoid lines that represent diverse histological subtypes and clinical stages, faithfully reproducing the histopathological grades and differentiation capacities of parental tumors. These lines have been genetically characterized and optimized based on niche factor dependencies, enabling comprehensive genotype-phenotype analyses and advancing personalized oncology research126. Similarly, Yan et al. established an organoid biobank for human gastric cancer, comprising 17 normal and 46 tumor-derived organoid lines that preserve key genomic and transcriptomic features, offering a powerful platform for high-throughput drug screening and therapeutic development133.
The human immune system functions as a defense network that orchestrates protective responses against pathogens and cancer. A comprehensive understanding of immune regulation is essential not only for maintaining health but also for diagnosis, treatment, and prevention of cancer. Recent advances in organoid technology have led to the development of immune organoids, derived from key lymphoid tissues such as tonsil134, lymph nodes135, bone marrow136,137, spleen138, thymus139, as well as intestine with immune compartments140. These models are opening new doors for studying human immune systems in physiologically relevant contexts and hold great potential for advancing cancer immunotherapy. For example, Zhong et al. engineered immune organoids by integrating PBMCs or tonsil cells into a synthetic hydrogel matrix to facilitate B cell maturation and antibody production135. Wagar et al. developed human tonsil organoids starting from a single-cell suspension to accurately model key germinal center (GC) responses, including antigen-specific antibody production, somatic hypermutation, affinity maturation, class-switch recombination and interactions between T follicular helper (Tfh) and B cells upon stimulation with different forms of influenza vaccines134,141,142. Loskill and colleague further advanced the tonsil organoid system into lymphoid-tissue-on-chip model that enable active transmigration of monocyte-derived dendritic cells (moDCs) into tonsillar microenvironment using centrifugal microfluidics under continuous dynamic flows143. Mebius et al. integrate lymphatic vasculature into human lymph node-on-chip model to recapitulate lymph node architecture and fluid dynamics. This system maintains continuous perfusion, homeostatic chemokine gradients and B cell organization, supporting dynamic immune cell trafficking and cytokine signaling144. Zhou and Clevers et al. established nasal organoids as reliable in vitro surrogates for airway tissues in viral neutralization assays. These organoids accurately recapitulate native viral entry mechanisms, including ACE2/TMPRSS2 expression, mucus layer formation, and functional ciliated cells, which are absent in conventional cell lines145,146. Another notable example is that human intestinal immune-organoids (IIOs) developed by Recaldin et al. through self-organization of epithelial organoids and autologous tissue-resident memory T (TRM) cells, a T cell subset that integrates within the epithelium and continuously surveil the mucosal barrier. This organoid model can respond to inflammatory stimuli including T cell-engaging bispecific molecules and allow for studying immune responses related to tumorigenesis, infectious and autoimmune disease140.
Organoids for Modeling Tumor Immune Microenvironment (TIME) Complexity
Traditional tumor organoid models often neglecting the critical role played by immune components in shaping tumor development and thus significantly limiting their applications for predicting immunotherapeutic outcomes. Growing efforts have been made to incorporate physiological relevant immune components into organoids systems to reflect the complexity and heterogeneity of TIME. A recent review on lymphatic and immune micro-system also highlights the necessity of integrating lymphatic components into in vitro models to preserve immune functionality147. Incorporation of immune cells, including tumor-infiltrating lymphocytes (TILs), natural killer (NK) cells, macrophages, dendritic cells (DCs), eosinophils, mast cells, and myeloid-derived suppressor cells (MDSCs), into the organoids enables to study the dynamic immune-tumor interactions in vitro. These immune cells can be sourced from autologous tumor tissue, peripheral blood, or secondary lymphoid organs. There phenotypic and functional states (e.g. activation, differentiation, exhaustion, proliferation, migration, and metabolism) can be dynamically influenced by the TME148. Several strategies have been employed to integrate immune components into organoids models149,150,151,152,153,154,155. One example is co-culturing immune cells from that derived from a different donor. This allogeneic co-culture approach is technically simple and scalable but carries the risk of non-specific immune response due to human leukocyte antigen (HLA) mismatch, which may overshadow tumor antigen-specific responses. Alternatively, autologous co-culture approach uses patient-matched tumor and immune cells. For example, Dijkstra et al. co-cultured autologous tumor organoids with peripheral blood lymphocytes (PBLs) to induce and expand tumor-reactive CD8⁺ T cells. The resulting cytotoxic T lymphocytes exhibited tumor-specific reactivity, demonstrated by selective killing of matched tumor organoids but not unrelated controls55. As an advancement of earlier work, Cattaneo et al. reported a detailed protocol for personalized tumor-T cell co-culture system, which enables generation and functional assessment of tumor-reactive T cells from autologous PBLs in patient-specific tumor organoids156. Liu et al. developed a gel–liquid interface (GLI) co-culture system combining lung cancer organoids with autologous PBMCs to investigate systemic anti-tumor immune responses to immune checkpoint inhibitors (ICIs)157. Compared to allogeneic co-culture approach, these approaches can better mimic in vivo condition and avoid artificial immune activation. However, it is often limited by the availability of both tumor tissues and immune cells from the same patient. Another notable approach is the holistic approach, which preserves endogenous immune cells directly within tumor organoids. For example, Neal et al. developed an air-liquid interface (ALI) approach that preservers multiple immune cells including T cells, B cells, macrophages and NK cells within patient-derived tumor tissues from multiple cancer types including colon, pancreas and lung cancers, as well as uncommon forms such as ampullary carcinoma, schwannomas, and pleomorphic adenomas of the salivary gland. This system retained TCR diversity and successfully predicted immune checkpoint blockade responses ex vivo158. Beyond immune-cell incorporation, inclusion of vasculature further enhances the physiological relevance and complexity of the model. Huh et al. developed a tumor-on-a-chip featuring vascularized human tumor explants that enable controlled immune-cell perfusion for real-time monitoring of CAR-T cell dynamics159.
Collectively, these immune-integrated organoid models provide robust platforms for analyzing specific immune signaling pathways within TME and generate important insights into immune evasion mechanisms for identifying therapeutic targets for effective cancer immunotherapy. Cell motility and cell-cell interaction can be observed in real-time160,161. Moreover, it is also powerful to model therapy resistance by tracking reversible phenotypic changes and irreversible mutational changes during treatment exposure helping to uncover mechanisms of escape from ICB or other immunotherapies.
Organoids for studying aging-associated changes
Aging is a critical yet often underrecognized factor influencing tumor progression, immune response, and therapeutic outcome. As aforementioned, tumor in aged patients often exhibit more aggressive phenotypes, which is strongly linked to immunosenescence—the progressive decline of immune function with age. Immunosenescence is characterized by shifts in immune cell populations, reduced innate immune activity, skewed adaptive immune cell repertoire, and increased chronic inflammation162, which compromises immune surveillance and facilitate tumor initiation and development.
Organoids present a promising solution to study age-associated immune alterations. As three-dimensional systems that retain the architecture and functional characteristics from their tissue of origin, organoids enable long-term tracking of cellular behavior and molecular changes under physiologically relevant conditions. While most existing organoid models are still based on young or non-aged tissues, aging-related organoid platforms are emerging as powerful tools for studying how aging influences cancer biology and immune responses. Particularly, patient-derived organoids provide a unique opportunity to model human-specific aging trajectories, including mechanical stress responses and epigenetic memory, offering novel insights into cancer risk and therapeutic sensitivity across the lifespan. One example is using brain organoids for modeling immune-driven neurodegeneration of aging such as Alzheimer’s disease (AD) features including age-related hallmark accumulation and immune activation163. Ao et al. developed a 3D printed human brain organoid microphysiological analysis platform (MAP) that revealed the increased infiltration of aged monocytes and overexpression of aging-related markers (e.g., p16) involved in the dynamic process of immune-driven brain aging in human cortical organoids164.
A number of studies have leveraged intestinal organoids to explore how aging alert gut immunity, epithelial integrity and tumorigenesis. Age-related immune decline in gut has been highlighted by impaired regeneration, increased inflammation and shifts in microbiome composition165. The IFNgamma-STAT1 axis has been identified as the driving force of epithelial deterioration and homeostasis loss in aged intestinal organoids166. On the molecular level, aging-like spontaneous epigenetic silencing within intestinal organoids promotes Wnt activation and facilitatce BrafV600E-driven tumor genesis167, while Lgr5 silencing reduces stemness and induces senescence168. Canonical Wnt signaling has been shown to reverse some aging phenotypes by restoring intestinal stem cell function169. In a word, these organoid-based evaluations dissect the cellular and molecular consequences of gut aging, as well as identifying potential targets for age-related decline. In addition, aging organoids have been used to study epigenetic reprogramming and metabolic dysfunction. For example, hepatic organoids from aged donors show functional heterogeneity among senescent hepatocytes, likely linked to diverse epigenetic lanscapes170. Studies have shown that PDO can retain tissue-specific aging signatures, such as epigenetic and transcriptomic changes171. Cellular reprogramming approaches now enable modeling of age-related methylation and chromatin remodeling patterns, providing a platform to evaluate aging clocks and rejuvenation strategies in vitro172. Moreover, Immune-metabolic aging has been linked to elevated CD38 expression in aged immune cells, which depletes NAD+ and perturbs metabolic hemostasis173.
In addition to biological alterations, aged-associated mechanical and physical changes can be recapitulated using engineered biomaterials within organoid systems. Organoids with tunable stiffness and viscoelasticity have been used to model aged tissue microenvironments. These models can mimic the structural properties of aged tissues to dissect how physical stressors modulate cellular senescence, differentiation and tissue remodeling174. Furthermore, integration of microfluidics and spatial omics technologies has allowed the decoding of senescence program at single-cell resolution within engineered contexts, highlighting the spatial heterogeneity of aging responses175. Given the profound impact of aging on tumor-immune interactions, organoid platforms provide a promising strategy to screen and evaluate anti-aging drugs, such as senolytics176, mTOR inhibitors and metformin, for potential use in combination immunotherapies.
Organoids for Evaluating Immunotherapy Efficacy and Safety
Organoids are able to model various therapeutic outcomes across cancer types by recapitulating various tumor stages in different types and capturing inter- and intra- patient heterogeneity. For example, Papargyrious et al. developed branched organoid models of PDAC capable of reflecting PDAC subtypes and intratumoral phenotypic diversity. These model revealed that the branched structure formation relies on classical TGF-β-associated mechano-transduction pathway120. Organoid libraries of colorectal tumors have been established to monitor the progression and dependency on stem cell niche factors, such as epithelial growth factor (EGF), Wnt, and TGF-β. With these organoids, diverse genetic heterogeneity was captured and variation of niche factor dependency was analyzed based on a phenotype-genotype paired study126.
Multiple studies demonstrate that organoids enable biobank profiling and evaluation of anti-tumor T cell activity. Kong et al. developed an organoid-based assay to evaluate TIL cytotoxicity and functional restoration after ICB treatment. Six of the 17 tested patients achieved an objective pathological complete response (pCR) after treatment177; PDOs from 21 mismatch repair-deficient (dMMR) and 20 mismatch repair-proficient (pMMR) early-stage colon cancers demonstrated that neoadjuvant ICB could induce deep or complete pathologic responses, particular in dMMR tumors. Notably, peripheral blood lymphocytes collected post-treatment from responders is capable of recognizing autologous organoids, even in some pMMR tumors, highlighting the potential of organoids in characterizing patient-specific anti-tumor T cell activity178.
Mechanistic studies using organoids have play a critical role in investigating signaling pathways of immunotherapies for therapeutic prediction179. ICB therapy aims to restore cytotoxic T cell function by inhibiting checkpoint pathways such as PD-1, PD-L1 or CTLA-4 signaling. In chordoma, organoids derived from 24 surgical specimens treated with nivolumab showed dose-dependent size reductions and increased apoptosis from both PD-L1-positive and PD-L1-negative organoids, suggesting organoids can provide a more sensitive functional prediction of ICB response than PD-L1 IHC alone180. Voabil et al. developed a patient-derived tumor fragment (PDTF) platform from five cancer types, preserving native TME and architecture. Within 24–48 h of PD-1 blockade treatment, early intratumoral responses, characterized by resident T cell reactivation and increased chemokine (CXCL9, CXCL10, CCL5, CXCL13) release, were observed and strongly correlated with clinical outcomes181. Organoids have also contributed to combination therapy optimization. Mouse-derived gastric cancer organoids showed that the inhibition of Hedgehog signaling with GANT61 reduced PD-L1 expression and enhanced the effectiveness of chemotherapy and anti-PD-L1 by promoting CTL-mediated tumor apoptosis182. PDOs derived from NSCLC patients showed that combined inhibition of CDK4/6 enhances ICB response by promoting T cell activation183. PDOs from patients with metastatic cutaneous melanoma and microsatellite instability (MSI)-high CRC revealed inhibition of immune-evasion gene TBK1 sensitized tumors to PD-1 blockade by lowering resistance to TNF and IFN-gamma associated cell death184. Zhou et al. developed a high-throughput platform incorporating tumor-specific T cells into pancreatic tumor organoids, enabling stromal barriers, hypoxia, and immune exclusion. Using this system, the HDAC inhibitor ITF2357 and BET inhibitor I-BET151 were identified as potent sensitizer of anti-PD-1 therapy185. Zhou et al. utilized ALI-based organoid models to evaluate the efficacy of pan-cancer immunotherapies by velcro-like, density-dependent targeting of tumor-associated carbohydrate antigens186.
Adoptive cell transfer (ACT) therapies, including CAR-T cell therapy, benefit significantly from organoid systems. These systems enable precise in vitro evaluation of engineered immune cells to assess therapeutic effectiveness across diverse cancer types including lung adenocarcinoma, glioblastoma and leukemia159,187 as well as T cell-mediated immune toxicity188,189. Logun et al. developed a patient-matched glioblastoma organoid (GBO) model derived from six patients enrolled in a phase I CAR-T cell therapy trial, enabling real-time ex vivo assessment alongside clinical treatment. CAR-T cell treatment led to target antigen reduction, tumor cytolysis, and cytokine responses in GBOs that closely reflected those observed in patient cerebrospinal fluid profiles, supporting the use of GBOs to predict and interpret individual immunotherapy responses190.
Organoids serve as a powerful platform to evaluate antigen immunogenicity, adjuvant potency, and B and T cell activation and function, facilitating the rational design of next-generation tumor vaccines. For example, Kastenschmidt et al. utilized human tonsil organoids to assess how antigen format influences vaccine efficacy regarding the magnitude, diversity, phenotype, function, and breadth of vaccine-induced B and T cell responses191. This same organoid platform has also been used to evaluate novel adjuvants. Yin et al. demonstrated that a nanoparticle-based adjuvant efficiently promotes human B cell differentiation and enhances antigen-specific antibody generation192. Demmers et al. uncovered heterogeneity in HLA class I peptide presentation across tumor clones from a single patient using an organoid system. Despite identical genomic background and culture conditions, clonal organoids displayed distinct HLA ligand repertoires not predictable from proteomic data alone. This clonal organoid system enabled high-resolution profiling of the tumor immunopeptidome, highlighting its value for antigen discovery and vaccine design193.
Beyond efficacy assessment, organoids offer a robust platform for identifying biomarkers and defining molecular predictors of treatment response, opening new avenues for therapeutic strategies. Dost et al. developed murine and human alveolar epithelial organoids that faithfully model early-stage KRAS-driven lung adenocarcinoma (LUAD). These organoids recapitulated key transcriptional changes including the loss of AT2 lineage identity and upregulation of developmental markers such as SOX9 and HMGA2, which provides insight into early molecular events in KRAS-mutant LUAD and potential targets for early intervention194. A murine endometrial organoid was developed to reveal that oncogenic KRASG12D expression induces cellular transformation and epithelial-mesenchymal transition (EMT). Malignant phenotypes occurs more strongly when KRAS activation combined with Cdkn2a loss or p53 mutation195. A HER2-overpressing human iPSC-derived lung organoid exhibited HER2-driven transformation activates downstream oncogenic pathway including MAPK and PIK3, also contributes to pre-neoplastic changes of early LUAD196. Toth et al. used murine alveolar organoids to investigate how Nkx2-1 maintains the epigenomic identity of alveolar epithelial progenitor cells. Loss of Nkx2-1 in organoids disrupted progenitor maintenance and promote the emergence of aberrant cell states197.
Advancing Organoid Technology: Multi-Zonal and Multi-Organ Systems
Recent advances in organoid engineering have expanded potential for modeling complex tissue organization, cross-tissue interaction, systemic physiology, and disease progression. For example, Takebe’s team recently developed multi-zonal human liver organoids that successfully reconstruct multi-zonal hepatic architecture, enabling the functional gradient from the portal vein to the central vein and demonstrating zone-specific metabolic activities. This system provides a promising platform for studying liver physiology and modeling liver diseases198. Frenkel et al. explored lymphatic-tumor interactions by integrating lymphatic endothelial cells with colorectal cancer organoids using a microfluidic co-culture system. This model allows dynamic analysis of cancer-driven lymphangiogenesis and cancer cell motility, offering valuable insights into mechanism of tumor metastasis199. Multi-organoid systems, when combined with engineered strategies, provide a more robust and accurate platform to study inter-tissue communication and systemic process200,201. Functional matrices and scaffolds202,203,204 support the structural and functional recapitulation of native tissues, such as the reconstruction of the lung crystal ribcage205. Manufacturing innovations including bioprinting206,207, microfluidics208,209,210, genetic perturbation211 and micro-patterning212,213 enable the generation of spatially organized tissue architectures and facilitate large-scale, high-throughput fabrication. Advanced spatial profiling techniques214 and integrative spatial multi-omics analyses215, provide quantitative insights into how faithfully organoids recapitulate native tissue architecture and cellular organization. Advanced imaging technologies160,161,216,217 allow real-time monitoring and quantitative assessment, while AI-assisted tools enhance predictive modeling and data analysis218. Downstream molecular analyses, such as genomic profiling and spatial transcriptomics, further enable detailed characterization of cellular heterogeneity, lineage trajectories, and microenvironmental dynamics219,220.
Together, these technologies advance the organoid models to model distinct stages of carcinogenesis across tumor types, uncover therapeutic vulnerabilities and novel targets, and identify predictive biomarkers and molecular signatures.
Discussion and outlook
Organoid models represent a transformative advancement in immunotherapy research by enabling in-depth exploration of complex immune mechanisms and providing clinically relevant insights into therapeutic efficacy. As highly tunable systems, organoids facilitate the dissection of cancer-immune interactions, evaluation of treatment responses, and informed clinical decision-making.
The application of organoids has expanded beyond fundamental tumor biology and immunology to drug discovery and development. Their capacity for high-throughput screening and clinical relevance makes them especially valuable for evaluating drug formulations, combination therapies and immune toxicity. They also offer critical insights into synergistic or additive effects of immunotherapy with other cancer treatments, such as chemotherapy, radiotherapy, or targeted therapy.
Regulatory shifts have further accelerated the employment of organoid models. Following the FDA Modernization Act 2.0 in 2022, which allows new drug approvals without mandatory animal testing, organoids emerged as pivotal tools in preclinical testing for drug discovery, sensitivity, and immunotoxicity evaluation. More recently (April 2025), FDA announced a strategic shift to phase out mandatory animal testing requirements for monoclonal antibodies and other biologics, promoting the adoption of New Approach Methodologies (NAMs) including organoid systems, artificial intelligence (AI) based computational models, and other human cell-based assays. A new pilot program has also been launched to assess the performance of these alternative methods, signaling a transformative evolution in preclinical evaluation frameworks. Currently, the rapid progress of AI is also fueling the translation of organoid technologies, which can process the high-throughput organoid data in a more sufficient way221,222,223.
Among organoid platforms, patient-derived tumor organoid (PDO) shows great potential in personalized precision medicine. This model preserves the genetic, phenotypic and microenvironmental features of a patient’s tumor, allowing for ex vivo testing of patient-specific responses to therapies. The rapid increase in studies utilizing PDOs, especially in tumor immunology and individualized drug sensitivity assays, highlights their potential clinical value. In the past five years, over 50 ongoing clinical trials worldwide have incorporated PDOs, either to compare clinical treatment responses, or to guide clinical decision-making, which covers the applications across multiple cancer types and treatment modalities, including immunotherapy, radiotherapy, chemotherapy, targeted therapy and combination therapy.
Despite these promising advances, significant challenges remain. Current organoid models face issues such as limited longevity, instability and batch-to-batch variation. The incorporation of immune cells, while critical, is still challenged by poor viability, limited lifespan and functional decline. Maintaining robust and reproducible immune-competent organoids with accurately reflecting the complexity of human immune responses is clinical unmet needs. Lack of protocol standardization, particularly for co-cultured organoids, remains a limitation in the field, highlighting the future need for standardized methodologies to improve reproducibility and cross-study comparison.
Strategies for addressing these challenges are emphasized in the future research. Examples like advanced biomaterials and engineered scaffolds that support sustained immune responses and innovative device designs, including microfluidic systems, bioprinting and multi-system assemblies, further prolonging the organoids’ longevity and functional integrity.
Furthermore, establishing standardized evaluation criteria is essential to rigorously assess the clinical relevance and predictive accuracy of organoids models. While most tumor-centric studies have primarily focused on local responses, there is growing recognition that systemic immune interactions must also be considered. Thus, expanding organoid systems to model multi-tissue dynamics and systemic immune response is a key direction for future development.
Data availability
No datasets were generated or analyzed during the current study.
References
Liu, Y. et al. Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Sci. Transl. Med. 13, eabb5191 (2021).
Magré, L. et al. Emerging organoid-immune co-culture models for cancer research: from oncoimmunology to personalized immunotherapies. J. Immunother. Cancer 11, e006290 (2023).
Duan, Z. et al. CAR-T cells based on a TCR mimic nanobody targeting HPV16 E6 exhibit antitumor activity against cervical cancer. Mol. Ther.: Oncol. 32, 200892 (2024).
Liu, L. et al. Engineering sonogenetic EchoBack-CAR T cells. Cell 188, 2621–2636.e20 (2025).
Lu, Y.-C. et al. Treatment of patients with metastatic cancer using a major histocompatibility complex class II–restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. JCO 35, 3322–3329 (2017).
Hong, D. S. et al. Phase I dose escalation and expansion trial to assess the safety and efficacy of ADP-A2M4 SPEAR T cells in advanced solid tumors. JCO 38, 102–102 (2020).
Liu, M. et al. MAGE-A4 pMHC-targeted CAR-T cells exploiting TCR machinery exhibit significantly improved in vivo function while retaining antigen specificity. J. Immunother. Cancer 12, e010248 (2024).
Van Norman, G. A. Limitations of animal studies for predicting toxicity in clinical trials. JACC: Basic Transl. Sci. 4, 845–854 (2019).
Atkins, J. T. et al. Pre-clinical animal models are poor predictors of human toxicities in phase 1 oncology clinical trials. Br. J. Cancer 123, 1496–1501 (2020).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017).
Galon, J. & Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218 (2019).
Gu, S. S. et al. Clonal tracing reveals diverse patterns of response to immune checkpoint blockade. Genome Biol. 21, 263 (2020).
Morad, G., Helmink, B. A., Sharma, P. & Wargo, J. A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 184, 5309–5337 (2021).
Fennema, E., Rivron, N., Rouwkema, J., Van Blitterswijk, C. & De Boer, J. Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol. 31, 108–115 (2013).
Fang, G., Chen, Y., Lu, H. & Jin, D. Advances in spheroids and organoids on a chip. Adv. Funct. Mater. 33, 2215043 (2023).
Zhang, B. & Radisic, M. Organ-on-a-chip devices advance to market. Lab Chip 17, 2395–2420 (2017).
Leung, C. M. A guide to the organ-on-a-chip. Nat. Rev. Methods Primers 2 (2022).
Bar-Ephraim, Y. E., Kretzschmar, K. & Clevers, H. Organoids in immunological research. Nat. Rev. Immunol. 20, 279–293 (2020).
Hofer, M. & Lutolf, M. P. Engineering organoids. Nat. Rev. Mater. 6, 402–420 (2021).
Zhao, Z. et al. Organoids. Nat. Rev. Methods Prim. 2, 94 (2022).
Wagar, L. E. Human immune organoids: a tool to study vaccine responses. Nat. Rev. Immunol. 23, 699–699 (2023).
Polak, R., Zhang, E. T. & Kuo, C. J. Cancer organoids 2.0: modelling the complexity of the tumour immune microenvironment. Nat. Rev. Cancer 24, 523–539 (2024).
Tang, X.-Y. et al. Human organoids in basic research and clinical applications. Sig Transduct. Target Ther. 7, 168 (2022).
Ingber, D. E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 23, 467–491 (2022).
Nakamura, T. & Sato, T. Advancing intestinal organoid technology toward regenerative. Med. Cell. Mol. Gastroenterol. Hepatol. 5, 51–60 (2018).
Huang, R., Gao, F., Yu, L., Chen, H. & Zhu, R. Generation of neural organoids and their application in disease modeling and regenerative medicine. Adv. Sci. 12, e01198 (2025).
Zheng, M., Erice, E., Wang, H., Zhang, L. & Lawrie, C. H. Organoid-on-a-chip (OrgOC): advancing cystic fibrosis research. Mater. Today Bio 34, 102148 (2025).
Zhao, J. et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat. Commun. 11, 5540 (2020).
Liu, X. et al. Analogous comparison unravels heightened antiviral defense and boosted viral infection upon immunosuppression in bat organoids. Sig Transduct. Target Ther. 7, 392 (2022).
Li, P. et al. Mpox virus infection and drug treatment modelled in human skin organoids. Nat. Microbiol. 8, 2067–2079 (2023).
Takebe, T. & Wells, J. M. Organoids by design. Science 364, 956–959 (2019).
Zhao, Y. et al. Integrating organoids and organ-on-a-chip devices. Nat. Rev. Bioeng. 2, 588–608 (2024).
Kim, J. M. & Chen, D. S. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann. Oncol. 27, 1492–1504 (2016).
Kim, R., Emi, M. & Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 121, 1–14 (2007).
Angelova, M. et al. Evolution of metastases in space and time under immune selection. Cell 175, 751–765.e16 (2018).
TRACERx consortium, T. he et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485 (2019).
Philip, M. & Schietinger, A. CD8+ T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 22, 209–223 (2022).
Wolf, Y. et al. UVB-induced tumor heterogeneity diminishes immune response in melanoma. Cell 179, 219–235.e21 (2019).
Klempner, S. J. et al. Tumor mutational burden as a predictive biomarker for response to immune checkpoint inhibitors: a review of current evidence. Oncologist 25, e147–e159 (2020).
Ricciuti, B. et al. Association of high tumor mutation burden in non–small cell lung cancers with increased immune infiltration and improved clinical outcomes of PD-L1 blockade across PD-L1 expression levels. JAMA Oncol. 8, 1160 (2022).
Wolf, Y. & Samuels, Y. Intratumor heterogeneity and antitumor immunity shape one another bidirectionally. Clin. Cancer Res. 28, 2994–3001 (2022).
Aggarwal, C. et al. Assessment of tumor mutational burden and outcomes in patients with diverse advanced cancers treated with immunotherapy. JAMA Netw. Open 6, e2311181 (2023).
McGrail, D. J. et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 32, 661–672 (2021).
Hellmann, M. D. et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 33, 843–852.e4 (2018).
Samstein, R. M. et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 51, 202–206 (2019).
Chan, T. A. et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol. 30, 44–56 (2019).
Terry, S. et al. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 11, 824–846 (2017).
Malagoli Tagliazucchi, G., Wiecek, A. J., Withnell, E. & Secrier, M. Genomic and microenvironmental heterogeneity shaping epithelial-to-mesenchymal trajectories in cancer. Nat. Commun. 14, 789 (2023).
He, R. et al. Progress in the application of immune checkpoint inhibitor-based immunotherapy for targeting different types of colorectal cancer. Front. Oncol. 11, 764618 (2021).
Chowdhury, S. et al. Consensus molecular subtyping of metastatic colorectal cancer expands biomarker-directed therapeutic benefit for patients with CMS1 and CMS2 tumors. Br. J. Cancer 131, 1328–1339 (2024).
Pickup, M., Novitskiy, S. & Moses, H. L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 13, 788–799 (2013).
Karamitopoulou, E. Tumour microenvironment of pancreatic cancer: immune landscape is dictated by molecular and histopathological features. Br. J. Cancer 121, 5–14 (2019).
Bear, A. S. et al. Biochemical and functional characterization of mutant KRAS epitopes validates this oncoprotein for immunological targeting. Nat. Commun. 12, 4365 (2021).
Fridman, W. H., Pagès, F., Sautès-Fridman, C. & Galon, J. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer 12, 298–306 (2012).
Dijkstra, K. K. et al. Generation of tumor-reactive t cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 174, 1586–1598.e12 (2018).
Andel, D. et al. Pre-existing subclones determine radioresistance in rectal cancer organoids. Cell Rep. 43, 113735 (2024).
Kratz, J. D. et al. Subclonal response heterogeneity to define cancer organoid therapeutic sensitivity. Sci. Rep. 15, 12072 (2025).
Mai, Z., Lin, Y., Lin, P., Zhao, X. & Cui, L. Modulating extracellular matrix stiffness: a strategic approach to boost cancer immunotherapy. Cell Death Dis. 15, 307 (2024).
Tumeh, P. C. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014).
Lee, J. C. et al. The liver-immunity nexus and cancer immunotherapy. Clin. Cancer Res 28, 5–12 (2022).
Wang, Q. et al. Differential organ-specific tumor response to first-line immune checkpoint inhibitor therapy in non-small cell lung cancer—a retrospective cohort study. Transl. Lung Cancer Res. 12, 312–321 (2023).
Damsky, W. et al. B cell depletion or absence does not impede anti-tumor activity of PD-1 inhibitors. J. Immunother. Cancer 7, 153 (2019).
Griss, J. et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 10, 4186 (2019).
Helmink, B. A. et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020).
Thorsson, V. et al. The Immune landscape of cancer. Immunity 48, 812–830.e14 (2018).
Boussiotis, V. A. et al. Activated human B lymphocytes express three CTLA-4 counterreceptors that costimulate T-cell activation. Proc. Natl Acad. Sci. USA 90, 11059–11063 (1993).
Keir, M. E., Butte, M. J., Freeman, G. J. & Sharpe, A. H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704 (2008).
Yang, Y. et al. CTLA-4 expression by B-1a B cells is essential for immune tolerance. Nat. Commun. 12, 525 (2021).
Jain, R. K. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26, 605–622 (2014).
Kabir, A. U., Subramanian, M., Kwon, Y. & Choi, K. Linking tumour angiogenesis and tumour immunity. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-025-01211-z (2025).
Levental, K. R. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906 (2009).
Martin, J. D., Seano, G. & Jain, R. K. Normalizing function of tumor vessels: progress, opportunities, and challenges. Annu. Rev. Physiol. 81, 505–534 (2019).
D’Andrea, M. R. et al. Propensity for early metastatic spread in breast cancer: role of tumor vascularization features and tumor immune infiltrate. Cancers 13, 5917 (2021).
Ley, K., Laudanna, C., Cybulsky, M. I. & Nourshargh, S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689 (2007).
Hamzah, J. et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 453, 410–414 (2008).
Ene–Obong, A. et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 145, 1121–1132 (2013).
Woods, A. N. et al. Differential expression of homing receptor ligands on tumor-associated vasculature that control CD8 effector t-cell entry. Cancer Immunol. Res. 5, 1062–1073 (2017).
Heldin, C.-H., Rubin, K., Pietras, K. & Östman, A. High interstitial fluid pressure—an obstacle in cancer therapy. Nat. Rev. Cancer 4, 806–813 (2004).
Gabrilovich, D. I., Ostrand-Rosenberg, S. & Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 12, 253–268 (2012).
Anczuków, O. et al. Challenges and opportunities for modeling aging and cancer. Cancer Cell 41, 641–645 (2023).
Gal-Oz, S. T. et al. ImmGen report: sexual dimorphism in the immune system transcriptome. Nat. Commun. 10, 4295 (2019).
Márquez, E. J. et al. Sexual-dimorphism in human immune system aging. Nat. Commun. 11, 751 (2020).
Fane, M. & Weeraratna, A. T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 20, 89–106 (2020).
Chen, A. C. Y. et al. The aged tumor microenvironment limits T cell control of cancer. Nat. Immunol. 25, 1033–1045 (2024).
Dolan, M., Libby, K. A., Ringel, A. E., Van Galen, P. & McAllister, S. S. Ageing, immune fitness and cancer. Nat. Rev. Cancer 25, 848–872 (2025).
Liu, Z. et al. Immunosenescence: molecular mechanisms and diseases. Sig Transduct. Target Ther. 8, 200 (2023).
Hu, J., Pan, M., Reid, B., Tworoger, S. & Li, B. Quantifiable blood TCR repertoire components associate with immune aging. Nat. Commun. 15, 8171 (2024).
Fransen, F. et al. Aged gut microbiota contributes to systemical inflammaging after transfer to germ-free mice. Front. Immunol. 8, 1385 (2017).
Acerbi, I. et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 7, 1120–1134 (2015).
Calhoun, C. et al. Senescent cells contribute to the physiological remodeling of aged lungs. GERONA 71, 153–160 (2016).
Tsukita, Y. et al. Immunotherapy or chemoimmunotherapy in older adults with advanced non–small cell lung cancer. JAMA Oncol. 10, 439 (2024).
Kao, C. et al. Age-related divergence of circulating immune responses in patients with solid tumors treated with immune checkpoint inhibitors. Nat. Commun. 16, 3531 (2025).
Kugel, C. H. et al. Age correlates with response to anti-PD1, reflecting age-related differences in intratumoral effector and regulatory T-cell populations. Clin. Cancer Res. 24, 5347–5356 (2018).
Hamilton, J. A. G. & Henry, C. J. Aging and immunotherapies: new horizons for the golden ages. Aging Cancer 1, 30–44 (2020).
Johnson, D. B., Sullivan, R. J. & Menzies, A. M. Immune checkpoint inhibitors in challenging populations. Cancer 123, 1904–1911 (2017).
Loizides, S. & Papamichael, D. Considerations and challenges in the management of the older patients with gastric cancer. Cancers 14, 1587 (2022).
Tran Van Hoi, E. et al. Blood based immune biomarkers associated with clinical frailty scale in older patients with melanoma receiving checkpoint inhibitor immunotherapy. Immun. Ageing 21, 83 (2024).
Day, C.-P., Merlino, G. & Van Dyke, T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 163, 39–53 (2015).
Rehli, M. Of mice and men: species variations of Toll-like receptor expression. Trends Immunol. 23, 375–378 (2002).
Mestas, J. & Hughes, C. C. W. Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731–2738 (2004).
Huang, L., Xu, H. & Peng, G. TLR-mediated metabolic reprogramming in the tumor microenvironment: potential novel strategies for cancer immunotherapy. Cell Mol. Immunol. 15, 428–437 (2018).
Huang, B., Zhao, J., Unkeless, J. C., Feng, Z. H. & Xiong, H. TLR signaling by tumor and immune cells: a double-edged sword. Oncogene 27, 218–224 (2008).
Song, I. J. et al. The contribution of toll-like receptor signaling to the development of liver fibrosis and cancer in hepatocyte-specific TAK1-deleted mice. Int. J. Cancer 142, 81–91 (2018).
Fischer, M. Mice are not humans: the case of p53. Trends Cancer 7, 12–14 (2021).
Frank, M. L. et al. T-cell receptor repertoire sequencing in the era of cancer immunotherapy. Clin. Cancer Res. 29, 994–1008 (2023).
Zomer, H. D. & Trentin, A. G. Skin wound healing in humans and mice: challenges in translational research. J. Dermatol. Sci. 90, 3–12 (2018).
Allison, J. P. & Havran, W. L. The immunobiology of T cells with invariant gammadelta antigen receptors. Annu. Rev. Immunol. 9, 679–705 (1991).
Kupper, T. S. & Fuhlbrigge, R. C. Immune surveillance in the skin: mechanisms and clinical consequences. Nat. Rev. Immunol. 4, 211–222 (2004).
Nagao, K. et al. Stress-induced production of chemokines by hair follicles regulates the trafficking of dendritic cells in skin. Nat. Immunol. 13, 744–752 (2012).
Pasparakis, M., Haase, I. & Nestle, F. O. Mechanisms regulating skin immunity and inflammation. Nat. Rev. Immunol. 14, 289–301 (2014).
Lewis, S. M., Williams, A. & Eisenbarth, S. C. Structure and function of the immune system in the spleen. Sci. Immunol. 4, eaau6085 (2019).
Sittig, L. J. et al. Genetic background limits generalizability of genotype-phenotype relationships. Neuron 91, 1253–1259 (2016).
Beura, L. K. et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516 (2016).
Rosshart, S. P. et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, eaaw4361 (2019).
Ito, R., Takahashi, T., Katano, I. & Ito, M. Current advances in humanized mouse models. Cell Mol. Immunol. 9, 208–214 (2012).
The Complex Trait Consortium The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet 36, 1133–1137 (2004).
Kitching, A. R. & Ooi, J. D. From bench to pet shop to bedside? The environment and immune function in mice. Kidney Int. 90, 1142–1143 (2016).
Medetgul-Ernar, K. & Davis, M. M. Standing on the shoulders of mice. Immunity 55, 1343–1353 (2022).
Daly, A. C., Prendergast, M. E., Hughes, A. J. & Burdick, J. A. Bioprinting for the Biologist. Cell 184, 18–32 (2021).
Papargyriou, A. et al. Heterogeneity-driven phenotypic plasticity and treatment response in branched-organoid models of pancreatic ductal adenocarcinoma. Nat. Biomed. Eng. https://doi.org/10.1038/s41551-024-01273-9 (2024).
Cakir, B. et al. Expression of the transcription factor PU.1 induces the generation of microglia-like cells in human cortical organoids. Nat. Commun. 13, 430 (2022).
Ogawa, J., Pao, G. M., Shokhirev, M. N. & Verma, I. M. Glioblastoma model using human cerebral organoids. Cell Rep. 23, 1220–1229 (2018).
Jacob, F. et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 180, 188–204.e22 (2020).
Sachs, N. et al. A Living Biobank of breast cancer organoids captures disease heterogeneity. Cell 172, 373–386.e10 (2018).
Dekkers, J. F. et al. Long-term culture, genetic manipulation and xenotransplantation of human normal and breast cancer organoids. Nat. Protoc. 16, 1936–1965 (2021).
Fujii, M. et al. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 18, 827–838 (2016).
Drost, J. et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 521, 43–47 (2015).
Steele, N. G. et al. An organoid-based preclinical model of human gastric cancer. Cell. Mol. Gastroenterol. Hepatol. 7, 161–184 (2019).
Shah, S. B. et al. Combinatorial treatment rescues tumour-microenvironment-mediated attenuation of MALT1 inhibitors in B-cell lymphomas. Nat. Mater. 22, 511–523 (2023).
Lee, S. H. et al. Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell 173, 515–528.e17 (2018).
Lee, M. R. et al. Organoid morphology-guided classification for oral cancer reveals prognosis. Cell Rep. Medi. 6, 102129 (2025).
Pawlowski, K. D., Duffy, J. T., Babak, M. V. & Balyasnikova, I. V. Modeling glioblastoma complexity with organoids for personalized treatments. Trends Mol. Med. 29, 282–296 (2023).
Yan, H. H. N. et al. A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell 23, 882–897.e11 (2018).
Wagar, L. E. et al. Modeling human adaptive immune responses with tonsil organoids. Nat. Med. 27, 125–135 (2021).
Zhong, Z. et al. Human immune organoids to decode B cell response in healthy donors and patients with lymphoma. Nat. Mater. 24, 297–311 (2025).
Griffin, K. H. et al. Engineered bone marrow as a clinically relevant ex vivo model for primary bone cancer research and drug screening. Proc. Natl Acad. Sci. USA 120, e2302101120 (2023).
Olijnik, A.-A. et al. Generating human bone marrow organoids for disease modeling and drug discovery. Nat. Protoc. 19, 2117–2146 (2024).
Gee, K. et al. Spleen organoid units generate functional human and mouse tissue-engineered spleen in a murine model. Tissue Eng. Part A 26, 411–418 (2020).
Seet, C. S. et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat. Methods 14, 521–530 (2017).
Recaldin, T. et al. Human organoids with an autologous tissue-resident immune compartment. Nature 633, 165–173 (2024).
Mallajosyula, V. et al. Coupling antigens from multiple subtypes of influenza can broaden antibody and T cell responses. Science 386, 1389–1395 (2024).
Wagoner, Z. W. et al. Systems immunology analysis of human immune organoids identifies host-specific correlates of protection to different influenza vaccines. Cell Stem Cell 32, 529–546.e6 (2025).
Teufel, C. et al. Lymphoid-tissue-on-chip recapitulates human antibody responses in vitro. https://doi.org/10.1101/2025.01.14.632762 (2025).
Morrison, A. I. et al. Integration of lymphatic vasculature to a human lymph node-on-chip enhances physiological immune properties. Mater. Today Bio 35, 102326 (2025).
Li, C. et al. Human airway and nasal organoids reveal escalating replicative fitness of SARS-CoV-2 emerging variants. Proc. Natl Acad. Sci. USA. 120, e2300376120 (2023).
Wan, Z. et al. Organoid-based neutralization assays reveal a distinctive profile of SARS-CoV-2 antibodies and recapitulate the real-world efficacy. Proc. Natl Acad. Sci. USA. 122, e2509616122 (2025).
Jain, I., Singh, A. & García, A. J. Microphysiological Systems of Lymphatics and Immune Organs. Adv. Healthcare Mater. e03201 (2025).
Yuki, K., Cheng, N., Nakano, M. & Kuo, C. J. Organoid models of tumor immunology. Trends Immunol. 41, 652–664 (2020).
Rogoz, A., Reis, B. S., Karssemeijer, R. A. & Mucida, D. A 3-D enteroid-based model to study T-cell and epithelial cell interaction. J. Immunol. Methods 421, 89–95 (2015).
Nozaki, K. et al. Co-culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. J. Gastroenterol. 51, 206–213 (2016).
Schreurs, R. R. C. E. et al. Human fetal TNF-α-cytokine-producing CD4+ effector memory T cells promote intestinal development and mediate inflammation early in life. Immunity 50, 462–476.e8 (2019).
Collin De l’Hortet, A. et al. Generation of human fatty livers using custom-engineered induced pluripotent stem cells with modifiable SIRT1 metabolism. Cell Metab. 30, 385–401.e9 (2019).
Skardal, A. et al. Drug compound screening in single and integrated multi-organoid body-on-a-chip systems. Biofabrication 12, 025017 (2020).
Vazquez-Armendariz, A. I. et al. Multilineage murine stem cells generate complex organoids to model distal lung development and disease. EMBO J. 39, e103476 (2020).
Schreurs, R. R. C. E., Baumdick, M. E., Drewniak, A. & Bunders, M. J. In vitro co-culture of human intestinal organoids and lamina propria-derived CD4+ T cells. STAR Protoc. 2, 100519 (2021).
Cattaneo, C. M. et al. Tumor organoid–T-cell coculture systems. Nat. Protoc. 15, 15–39 (2020).
Li, K. et al. An organoid co-culture model for probing systemic anti-tumor immunity in lung cancer. Cell Stem Cell 32, 1218–1234.e7 (2025).
Neal, J. T. et al. Organoid modeling of the tumor immune microenvironment. Cell 175, 1972–1988.e16 (2018).
Liu, H. et al. A tumor-on-a-chip for in vitro study of CAR-T cell immunotherapy in solid tumors. Nat. Biotechnol. https://doi.org/10.1038/s41587-025-02845-z (2025).
Dekkers, J. F. et al. Uncovering the mode of action of engineered T cells in patient cancer organoids. Nat. Biotechnol. 41, 60–69 (2023).
Alieva, M. et al. BEHAV3D: a 3D live imaging platform for comprehensive analysis of engineered T cell behavior and tumor response. Nat. Protoc. 19, 2052–2084 (2024).
Aw, D., Silva, A. B. & Palmer, D. B. Immunosenescence: emerging challenges for an ageing population. Immunology 120, 435–446 (2007).
Hossain, M. K., Kim, H.-R. & Chae, H. J. Aging phenotype in AD brain organoids: Track to success and challenges. Ageing Res. Rev. 96, 102256 (2024).
Ao, Z. et al. Understanding immune-driven brain aging by human brain organoid microphysiological analysis platform. Adv. Sci. 9, 2200475 (2022).
Walrath, T. et al. Age-related changes in intestinal immunity and the microbiome. J. Leukoc. Biol. 109, 1045–1061 (2021).
Omrani, O. et al. IFNγ-Stat1 axis drives aging-associated loss of intestinal tissue homeostasis and regeneration. Nat. Commun. 14, 6109 (2023).
Tao, Y. et al. Aging-like spontaneous epigenetic silencing facilitates wnt activation, stemness, and BrafV600E-Induced tumorigenesis. Cancer Cell 35, 315–328.e6 (2019).
Uchida, R. et al. Epigenetic silencing of Lgr5 induces senescence of intestinal epithelial organoids during the process of aging. npj Aging Mech. Dis. 4, 12 (2018).
Nalapareddy, K. et al. Canonical Wnt signaling ameliorates aging of intestinal stem cells. Cell Rep. 18, 2608–2621 (2017).
Kumar, P., Hassan, M., Tacke, F. & Engelmann, C. Delineating the heterogeneity of senescence-induced-functional alterations in hepatocytes. Cell. Mol. Life Sci. 81, 200 (2024).
Torrens-Mas, M. et al. Organoids: an emerging tool to study aging signature across human tissues. modeling aging with patient-derived organoids. IJMS 22, 10547 (2021).
Pitrez, P. R. et al. Cellular reprogramming as a tool to model human aging in a dish. Nat. Commun. 15, 1816 (2024).
Chini, C. C. S. et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD+ and NMN levels. Nat. Metab. 2, 1284–1304 (2020).
Madl, C. M. Accelerating aging with dynamic biomaterials: recapitulating aged tissue phenotypes in engineered platforms. iScience 26, 106825 (2023).
Venkataraman, A. et al. Decoding senescence of aging single cells at the nexus of biomaterials, microfluidics, and spatial omics. npj Aging 10, 57 (2024).
Aguado, J. et al. Senolytic therapy alleviates physiological human brain aging and COVID-19 neuropathology. Nat. Aging 3, 1561–1575 (2023).
Kong, J. C. H. et al. Tumor-infiltrating lymphocyte function predicts response to neoadjuvant chemoradiotherapy in locally advanced rectal cancer. JCO Precision Oncol. 2, 1–15 (2018).
Chalabi, M. et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 26, 566–576 (2020).
Jenkins, R. W. et al. Ex vivo profiling of PD-1 Blockade using organotypic tumor spheroids. Cancer Discov. 8, 196–215 (2018).
Scognamiglio, G. et al. Patient-derived organoids as a potential model to predict response to PD-1/PD-L1 checkpoint inhibitors. Br. J. Cancer 121, 979–982 (2019).
Voabil, P. et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat. Med. 27, 1250–1261 (2021).
Chakrabarti, J. et al. Hedgehog signaling induces PD-L1 expression and tumor cell proliferation in gastric cancer. Oncotarget 9, 37439–37457 (2018).
Deng, J. et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 8, 216–233 (2018).
Sun, Y. et al. Targeting TBK1 to overcome resistance to cancer immunotherapy. Nature 615, 158–167 (2023).
Zhou, Z. et al. A T cell-engaging tumor organoid platform for pancreatic cancer immunotherapy. Adv. Sci. 10, 2300548 (2023).
Zhou, R. W. et al. Safe immunosuppression-resistant pan-cancer immunotherapeutics by velcro-like density-dependent targeting of tumor-associated carbohydrate antigens. Cell https://doi.org/10.1016/j.cell.2025.09.001 (2025).
Ma, C. et al. Bioengineered immunocompetent preclinical trial-on-chip tool enables screening of CAR T cell therapy for leukaemia. Nat. Biomed. Eng. https://doi.org/10.1038/s41551-025-01428-2 (2025).
Zhang, C. J. et al. A human liver organoid screening platform for DILI risk prediction. J. Hepatol. 78, 998–1006 (2023).
Soussi, F. E. A. et al. Autologous organoid-T cell co-culture platform for modeling of immune-mediated drug-induced liver injury. Adv. Sci. https://doi.org/10.1002/advs.202508584e08584 (2025).
Logun, M. et al. Patient-derived glioblastoma organoids as real-time avatars for assessing responses to clinical CAR-T cell therapy. Cell Stem Cell 32, 181–190.e4 (2025).
Kastenschmidt, J. M. et al. Influenza vaccine format mediates distinct cellular and antibody responses in human immune organoids. Immunity 56, 1910–1926.e7 (2023).
Yin, Q. et al. A TLR7-nanoparticle adjuvant promotes a broad immune response against heterologous strains of influenza and SARS-CoV-2. Nat. Mater. 22, 380–390 (2023).
Demmers, L. C. et al. Single-cell derived tumor organoids display diversity in HLA class I peptide presentation. Nat. Commun. 11, 5338 (2020).
Dost, A. F. M. et al. Organoids model transcriptional hallmarks of oncogenic KRAS activation in lung epithelial progenitor cells. Cell Stem Cell 27, 663–678.e8 (2020).
Maru, Y. et al. Kras activation in endometrial organoids drives cellular transformation and epithelial-mesenchymal transition. Oncogenesis 10, 46 (2021).
Miura, A. et al. Oncogenic potential of human pluripotent stem cell-derived lung organoids with HER2 overexpression. Int. J. Cancer 149, 1593–1604 (2021).
Toth, A. et al. Alveolar epithelial progenitor cells require Nkx2-1 to maintain progenitor-specific epigenomic state during lung homeostasis and regeneration. Nat. Commun. 14, 8452 (2023).
Reza, H. A. et al. Multi-zonal liver organoids from human pluripotent stem cells. Nature https://doi.org/10.1038/s41586-025-08850-1 (2025).
Frenkel, N. et al. Long-lived human lymphatic endothelial cells to study lymphatic biology and lymphatic vessel/tumor coculture in a 3D microfluidic model. ACS Biomater. Sci. Eng. 7, 3030–3042 (2021).
Votanopoulos, K. I. et al. Model of patient-specific immune-enhanced organoids for immunotherapy screening: feasibility study. Ann. Surg. Oncol. 27, 1956–1967 (2020).
Ronaldson-Bouchard, K. et al. A multi-organ chip with matured tissue niches linked by vascular flow. Nat. Biomed. Eng. 6, 351–371 (2022).
Adu-Berchie, K. et al. Generation of functionally distinct T-cell populations by altering the viscoelasticity of their extracellular matrix. Nat. Biomed. Eng. 7, 1374–1391 (2023).
LeSavage, B. L. et al. Engineered matrices reveal stiffness-mediated chemoresistance in patient-derived pancreatic cancer organoids. Nat. Mater. 23, 1138–1149 (2024).
Ballerini, M. et al. A gut-on-a-chip incorporating human faecal samples and peristalsis predicts responses to immune checkpoint inhibitors for melanoma. Nat. Biomed. Eng. https://doi.org/10.1038/s41551-024-01318-z (2025).
Etesami, N. S. et al. B cells in the pneumococcus-infected lung are heterogeneous and require CD4+ T cell help including CD40L to become resident memory B cells. Front. Immunol. 15, 1382638 (2024).
Lawlor, K. T. et al. Cellular extrusion bioprinting improves kidney organoid reproducibility and conformation. Nat. Mater. 20, 260–271 (2021).
Bliley, J. M., Shiwarski, D. J. & Feinberg, A. W. 3D-bioprinted human tissue and the path toward clinical translation. Sci. Transl. Med. 14, eabo7047 (2022).
Schuster, B. et al. Automated microfluidic platform for dynamic and combinatorial drug screening of tumor organoids. Nat. Commun. 11, 5271 (2020).
Choi, D. et al. Microfluidic organoid cultures derived from pancreatic cancer biopsies for personalized testing of chemotherapy and immunotherapy. Adv. Sci. 11, 2303088 (2024).
Xue, X. et al. Generation of spatially patterned human neural tube-like structures using microfluidic gradient devices. Nat. Protoc. https://doi.org/10.1038/s41596-025-01266-1 (2025).
Lehrich, B. M. et al. Precision targeting of β-catenin induces tumor reprogramming and immunity in hepatocellular cancers. Nat. Commun. 16, 5009 (2025).
Deglincerti, A. et al. Self-organization of human embryonic stem cells on micropatterns. Nat. Protoc. 11, 2223–2232 (2016).
Velasco, V., Shariati, S. A. & Esfandyarpour, R. Microtechnology-based methods for organoid models. Microsyst. Nanoeng. 6, 76 (2020).
Betjes, M. A., Kok, R. N. U., Tans, S. J. & Van Zon, J. S. Cell tracking with accurate error prediction. Nat. Methods https://doi.org/10.1038/s41592-025-02845-6 (2025).
Johnson, J. A. I. et al. Human interpretable grammar encodes multicellular systems biology models to democratize virtual cell laboratories. Cell 188, 4711–4733.e37 (2025).
Kok, R. N. U., Spoelstra, W. K., Betjes, M. A., Van Zon, J. S. & Tans, S. J. Label-free cell imaging and tracking in 3D organoids. Cell Rep. Phys. Sci. 6, 102522 (2025).
Hu, M. et al. SPACE: spatially resolved multiomic analysis for high-throughput CRISPR screening in 3D models. https://doi.org/10.1101/2025.09.14.675819 (2025).
Diosdi, A. et al. HCS-3DX, a next-generation AI-driven automated 3D-oid high-content screening system. Nat. Commun. 16, 8897 (2025).
He, Z. et al. An integrated transcriptomic cell atlas of human neural organoids. Nature 635, 690–698 (2024).
Xu, Q. et al. An integrated transcriptomic cell atlas of human endoderm-derived organoids. Nat. Genet. https://doi.org/10.1038/s41588-025-02182-6 (2025).
Kong, J. et al. Network-based machine learning in colorectal and bladder organoid models predicts anti-cancer drug efficacy in patients. Nat. Commun. 11, 5485 (2020).
Kong, J. et al. Network-based machine learning approach to predict immunotherapy response in cancer patients. Nat. Commun. 13, 3703 (2022).
Cai, H. et al. Brain organoid reservoir computing for artificial intelligence. Nat. Electron 6, 1032–1039 (2023).
Bjornson-Hooper, Z. B. et al. A comprehensive atlas of immunological differences between humans, mice, and non-human primates. Front. Immunol. 13, 867015 (2022).
Huminiecki, L. & Wolfe, K. H. Divergence of spatial gene expression profiles following species-specific gene duplications in human and mouse. Genome Res. 14, 1870–1879 (2004).
Liu, T. et al. Cross-species single-cell transcriptomic analysis reveals pre-gastrulation developmental differences among pigs, monkeys, and humans. Cell Discov. 7, 8 (2021).
Acknowledgements
This study is supported by the University of Texas at Austin Startup Fund, Texas Health Catalyst and the Cancer Prevention Research Institute of Texas (CPRIT RR230066) to Q.Y.
Author information
Authors and Affiliations
Contributions
X.K. conceived the idea, wrote the initial draft, prepared figures, and edited the draft. S.K.C. wrote part of the initial draft. Q.Y. conceived the idea and edited the draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kang, X., Cheemalamarri, S.K. & Yin, Q. Organoid: a promising solution to current challenges in cancer immunotherapy. npj Biomed. Innov. 2, 49 (2025). https://doi.org/10.1038/s44385-025-00051-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44385-025-00051-9
