
Abstract
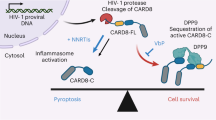
A strategy to functionally cure AIDS by eliminating latent HIV-1 reservoirs involves non-nucleoside reverse transcriptase inhibitors (NNRTIs) that promote pyroptosis of HIV-1 infected cells. These NNRTIs stimulate dimerization of the Gag-Pol polyprotein, resulting in premature HIV-1 protease (PR) dimerization and cleavage of intracellular CARD8. A unique cell-based high-throughput screen was developed to identify potent compounds activating the CARD8 inflammasome through Gag-Pol dimerization. Our in-house library of NNRTIs was evaluated, including a series of catechol diethers, which are potent, nontoxic antivirals. JLJ648 was identified as a promising dual-function antiviral and Gag-Pol dimerizer. Cryo-EM studies of HIV reverse transcriptase p66 bound to JLJ648 revealed populations of homodimers and, surprisingly, a homotetramer. This novel homotetramer structure resembling an ‘infinity knot’ revealed two JLJ648-bound homodimers forming an extensive interface and nucleated around a dimer of JLJ648 molecules. Structure-guided mutagenesis studies indicate that Gag-Pol homotetramerization may play a critical role in facilitating PR self-cleavage and triggering pyroptosis.
Similar content being viewed by others
Introduction
HIV-1 and AIDS remain major health crises around the globe, with ~41 million infections as of December 20241. While current treatments successfully suppress viral loads in over 70% of people living with HIV-12, these antiretroviral therapies (ART) are not curative. Shortly after patients are infected with HIV-1, the virus begins integrating its genome into infected cells, including CD4+ T cells, some of which can revert to a resting memory state and form a long-lasting latent viral reservoir which, upon later activation, is capable of producing infectious virus3,4. Infected macrophages, which are less susceptible to the cytotoxic effects of viral production, may also contribute to this viral reservoir5. This reservoir is nearly invisible to the immune system and cannot be targeted by conventional ART6. Patients relapse within 2–3 weeks of stopping therapy, even after years of successful suppression of the virus to undetectable levels7. To ensure continued viral suppression, even after a patient has eliminated active, circulating virus, ART must be continuously administered for the duration of the patient’s life. Thus, attempts to cure AIDS must target this latent HIV-1 reservoir.
HIV-1 reverse transcriptase (RT) was the first enzyme targeted by ART and remains an attractive target as it is responsible for converting the viral RNA into double-stranded DNA, a crucial step in retroviral replication. Nearly all previous drug development efforts have focused on the mature p66/p51 heterodimer, which is a traditional right-handed polymerase (Fig. 1a). The p66 subunit, comprised of fingers, palm, thumb, connection and RNaseH domains, performs the enzyme’s catalytic functions8. The p51 subunit, which provides structural support and has been implicated in primer-template loading, is formed by cleaving the C-terminal RNaseH domain from another p66 subunit9. Both subunits share a similar domain architecture10. In the p66 subunit, the thumb domain is close to the fingers and palm such that the thumb and fingers will close around the nucleic acid template and incoming dNTP to sequester the active site11,12, while the connection domain extends farther out toward the RNaseH domain. In the p51 subunit, the positions of the thumb and connection domains are switched such that the connection domain is close to the fingers and palm, while the thumb domain is extended to make contact with the connection and RNaseH domain of the p66 subunit.

a Crystal structure of JLJ636 bound to the RT heterodimer (PDB 5DTW) to show structural and domain organization of HIV-1 RT. Protein backbone is shown as a cartoon and colored according to the key below. The NNRTI JLJ636 is shown as magenta spheres. Figure prepared using PyMOL. b Schematic diagram of the predicted structure of the p66/p66’ homodimer. Subunits are colored according to the key used in panel a. Names are indicated for each domain and are abbreviated as follows: fingers (F), palm (P), thumb (T), connection (C), and RNaseH (RH). Dashed lines around the p66’ RNaseH domain indicate that this domain may be unfolded. Figure prepared using Adobe Illustrator. c General scaffold of the catechol diether compound series and the three groups replacing the phenyl ring. Figure prepared using ChemDraw. d Schematic diagram of the generation of the THP-1-based clone 14 cell line. Figure prepared using BioRender. e, f Comparison of single clones. Each clone was treated with either VbP (5 µM), RPV (5 µM), or RPV (5 µM) plus LPV (5 µM) for 3 h before cell death measurement by the LDH release assay. g, h Validation of the clone 14 cell line. In g, intracellular HIVp24 staining was performed and quantified by flow cytometry. Figure prepared using FlowJo v10. In h, induction of CARD8 expression was measured by immunoblotting with antibodies against the C-terminus of CARD8. Cell lysate was collected 24 h post-treatment with Dox (1 µg/mL). i Assay specificity. Clone 14 cells with or without Dox induction were treated with the indicated drugs at 5 µM for 3 h before cell death measurement by the LDH release assay. In f, p-values were calculated using two-way ANOVA and the Šídák test. In i, p-values were calculated with one-way ANOVA and the Dunnett test. ****p < 0.0001. j The Dox-treated or control clone 14 cells were treated with VbP (5 µM) alone, or in combination with LPV (5 µM) for 3 h before cell death measurement by the LDH release assay. Panels prepared using GraphPad Prism unless otherwise specified.
While this p66/p51 heterodimer is the most stable, RT can also form p66 homodimers with an affinity of ~4 µM13. These homodimers can form before the p66 subunit is cleaved from the Gag-Pol polyprotein, which also contains the viral protease (PR) and integrase (INT). NMR spectra suggest that the p66 homodimer adopts a conformation similar to that of the p66/p51 heterodimer, with the second p66 subunit (p66’) adopting the structural conformation typically observed in the p51 subunit14. In this case, the extra RNaseH domain of the p66’ subunit adopts a conformation like that of an RNaseH domain free in solution (Fig. 1b) and unfolds slowly over time to make the cleavage site available to the viral protease. This is supported by a cryo-EM structure of the entire Gag-Pol polyprotein, in which the usual RT heterodimer was the only portion of the map defined at high resolution15. Despite these similarities, it is unclear how RT inhibitors will interact with these immature RT homodimers.
Inhibitors of RT fall into two classes: nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs). NRTIs compete with natural dNTPs and are incorporated into the growing DNA chain, preventing further DNA synthesis. NNRTIs inhibit RT by binding in an allosteric pocket 10 Å away from the polymerase active site8 (Fig. 1a), locking the thumb domain in a more open conformation, which makes sequestering the active site difficult16. There are currently five FDA-approved NNRTIs, including first-generation nevirapine (NVP), second-generation efavirenz (EFV) and third-generation inhibitors etravirine (ETR), rilpivirine (RPV) and doravirine (DOR)17. RPV and DOR are structurally more flexible than NPV and EFV, allowing them to adopt active conformations in pockets shaped by common drug-resistant mutants18,19. However, dose-limiting toxicities and the emergence of new drug-resistant variants make long-term adherence to these therapies difficult19,20. Our previous virtual screening and computational and structure-guided design efforts have led to the development of a new family of NNRTIs built around a catechol diether scaffold (Fig. 1c). Initial efforts produced picomolar RT inhibitors by introducing a cyanovinyl group to the phenyl ring (Group I)21. As this cyanovinyl group had the potential to act as a Michael acceptor, further rounds of optimization produced two new families of highly potent inhibitors which replaced the phenyl with an indolizine (Group II)22 or naphthyl (Group III)23 group, respectively. Members of this catechol diether family have been extensively characterized and have proven effective at treating HIV-1 in humanized mice24.
Some NNRTIs, including RPV and EFV, are dual-function inhibitors that enhance the ability of RT to dimerize, driving the dimerization of uncleaved Gag-Pol, and therefore are termed Gag-Pol dimerizers25. This enhancement of Gag-Pol polyprotein dimerization leads to dimerization of PR and INT, promoting premature activation of the PR, which must be dimeric to be catalytically active26. PR, which is not typically dimeric until the polyproteins are concentrated at the cell membrane and the viral particle begins to bud, now has access to cellular proteins, including caspase recruitment domain-containing protein 8 (CARD8)27,28. Cleavage of CARD8 activates the inflammasome, leading to NNRTI-mediated pyroptosis of HIV-1 infected cells. This pyroptotic effect is extremely specific to HIV-1 infected cells. This newly discovered subset of NNRTIs has the potential to be a good component of “shock and kill” treatments, which activate the latent HIV-1 genome to eliminate the viral reservoir, as this mechanism does not rely on a patient’s compromised immune system to eliminate infected cells29. Moreover, this mechanism depends on the enzymatic activity of PR, which is highly conserved and essential for viral replication, making it effective against all major HIV-1 subtypes30. However, the mechanism by which these NNRTIs mediate RT dimerization is poorly understood, as a compound’s dimerization activity does not correlate with its ability to inhibit catalysis31,32. Additionally, pyroptotic activity is often observed at concentrations several orders of magnitude above those required for inhibition of DNA synthesis33. Strong dimerizers have been identified in several NNRTI families covering a broad range of chemical space, but small modifications to these molecules, which do not affect their antiviral potency or binding affinity for the mature RT heterodimer, have large effects on their abilities to enhance RT dimerization31.
In this study, we present a new high-throughput assay for screening NNRTIs for pyroptotic cell killing. Through this assay, we identified JLJ648, a new member of our naphthyl catechol diethers as a Gag-Pol dimerizer, which has both highly potent antiviral and pyroptotic properties. To better understand why JLJ648 is so successful, we determined the structure of this compound bound to the p66/p66’ homodimer and a novel p66 homotetramer. Our investigation suggests that, while the affinity of the p66 homodimer in the presence of each NNRTI is the key determinant for the strength of its pyroptotic activity, this NNRTI-bound homotetramer has an important role in the pyroptotic pathway.
Results
A HTS cell-based assay to identify HIV-1 Gag-Pol dimerizers promoting HIV-1 protease activation of CARD8 inflammasome pathway and pyroptosis
We generated a unique cell-based system to search for more potent chemicals that activate the CARD8 inflammasome through HIV-1 Gag-Pol dimerization. We transduced the CARD8-KO THP-1 cells with two lentiviral vectors, which carry a codon-optimized HIV-1 Gag–Pol and a doxycycline-inducible CARD8 expression cassette, respectively (Fig. 1d). To evaluate the CARD8 function in selected single clones, we treated cells with either RPV or val-boroPro (VbP) and measured cell death. VbP is an inhibitor for dipeptidyl peptidase 9 (DPP9), which drives CARD8 activation in THP-1 cells34 independent of Gag-Pol dimerization. Clone #14 was one of the clones with the strongest response to both RPV and VbP (Fig. 1e), and as such the RPV response was blocked by an HIV-1 protease inhibitor lopinavir (LPV) (Fig. 1f). At steady state, this cell line expresses the Gag–Pol polyprotein with minimal protease activity and does not express CARD8 (Fig. 1g, h). Thus, at steady state, the cells did not exhibit a pyroptotic effect in response to RPV or VbP treatment. Cell death was only observed in VbP- or RPV-treated groups upon induction by doxycycline (Fig. 1i), suggesting that the cell killing was dependent on the HIV-1 protease and CARD8. Additionally, we confirmed that LPV does not block VbP-induced pyroptotic cell killing as previously observed35 (Fig. 1j). Therefore, we selected THP-1 clone 14 for our high-throughput screening (HTS) assay.
Identification of potent antiviral NNRTIs as Gag-Pol dimerizers
After optimization of this cell-based model and adaptation to the high-throughput platform, this screen was used to determine whether any of our novel NNRTIs can induce pyroptosis in HIV-1 infected cells. Our previous work over the years using a computational and structure-guided drug design approach led to the synthesis of a library (758 compounds) of novel NNRTIs. These compounds, optimized for favorable drug-like properties, represented a range of chemical scaffolds, including a series of catechol diethers with a terminal uracil. Each compound was evaluated in an MT-2 cellular assay using methylthiazolyldiphenyl-tetrazolium bromide (MTT) dye to measure cell viability in the presence and absence of HIV-1 to identify those compounds that were potent antiviral replication inhibitors with no general cytotoxicity. The previously introduced catechol diether series was among the most potent and least cytotoxic. As all known dimerizers are potent NNRTIs, we selected a subset of 113 NNRTI compounds with high antiretroviral potency (EC50 < 50 nM) and low cytotoxicity (CC50 > 20 µM) for screening in the HTS THP-1 assay to identify dual-function compounds that may also exhibit pyroptotic cell death in HIV-1 infected cells. Eight of these compounds, six of which are catechol diethers, induced pyroptosis more effectively than EFV at 5 µM (Fig. 2a). None of these compounds induced cell death in cells with autoprocessing-deficient (S297A) CARD8, suggesting that the cell killing effect observed in wildtype cells is due to NNRTI-mediated CARD8 cleavage rather than other cytotoxic effects (Fig. 2b). One of the most potent of these is JLJ648, a naphthyl catechol diether with a thiouracil ring. The pyroptotic cell killing effects of JLJ648 were abrogated by the addition of the HIV-1 protease inhibitor LPV (Fig. 2c), suggesting that, like RPV and EFV, JLJ648-induced pyroptosis is also HIV-1 protease dependent. JLJ600, the non-thiolated version of JLJ648, also induced pyroptosis in the THP-1 screen, but to a lesser degree than EFV. Interestingly, JLJ555, the indolizine version of JLJ600, appeared to have minimal cell killing activity in the HTS screen even though it is 4.2-fold more potent than JLJ648 in the MT-2-based antiviral assays assessing antiviral replication (Fig. 2d). These three compounds were further evaluated for their pyroptotic cell killing activity by determining dose response curves for JLJ648, JLJ600 and JLJ555 in primary CD4+ T cells, which have EC50 values of 86, 410 nM and 3 µM, respectively (Fig. 2d, e). The dose response in THP-1 cells for pyroptotic cell killing was also confirmed for JLJ648 in a similar mid-nanomolar range (Fig. S1c). The general cytotoxicity for each of the compounds in MT-2, THP-1, and CD4+ T cells is summarized in the table in Fig. 2d. The cytotoxicity data for THP-1 and CD4+ T cells are shown in Fig. S1a, b. These results suggest that both the naphthyl ring and thiolation of the uracil ring are important features for this cell-killing effect.

a, b Drug screening. Dox-treated cells were incubated with candidate drugs at 5 µM for 3 h before cell death measurement by the LDH release assay. In a, the clone 14 cells were used to determine drug potency. In b, a control CARD8-KO THP-1 cell line expressing HIV-1 Gag-Pol and auto-processing-deficient CARD8 (CARD8-S297A) was used to measure non-specific cell death. c The Dox-treated clone 14 cells were treated with the indicated compounds at 5 µM for 3 h before cell death measurement by the LDH release assay. Multiple comparisons were made using one-way ANOVA followed by Dunnett's test. ****p < 0.0001 d The table summarizes chemical structures of JLJ555, JLJ600, and JLJ648 and the activity values for antiviral replication, cytotoxicity, pyroptotic cell death, and p66 homodimerization using HTRF. The inhibition of antiviral replication (EC50) and cytotoxicity (CC50) were determined in MT-2 in the presence and absence, respectively, of HIV-1 infection using MTT to measure cell viability. In addition, general cytotoxicity was determined in THP-1 and CD4+ T cells. Pyroptotic cell killing in CD4+ T cells was determined as described in e. Table prepared using Microsoft PowerPoint. The cell killing dose responses. CD4+ T cells were isolated from PBMCs and co-stimulated with anti-CD3 and anti-CD28 antibodies for 3 days. Stimulated cells were infected with VSVG pseudotyped HIV-1 reporter virus NL4-3-ΔEnv-GFP. JLJ555, JLJ600, and JLJ648 in successive three-fold dilutions were used to treat infected cells for 2 days before GFP measurement by flow cytometry. f p66 homodimerization dose response curves. Serial dilutions of JLJ555, JLJ600, and JLJ648 were incubated with 12.5 nM Hp66 and 25 nM HTFp66 for 17 h prior to the addition of A-6HIS-Eu donor and anti-FLAG-M2-XL665 acceptor antibodies. Percent effect is calculated by normalizing the background-subtracted ratios of acceptor:donor fluorescence at each compound concentration to that with the highest acceptor:donor ratio. Data shown is the average of three separate assays performed in triplicate. Panels prepared using GraphPad Prism unless otherwise specified.
Next, we wanted to confirm that this cell-killing activity is due to enhanced Gag-Pol polyprotein dimerization. RT p66 homodimerization can be used as a proxy for the polyprotein in homogeneous time-resolved fluorescence (HTRF) dimerization assays; fluorescence transfer from donor to receptor conjugated antibodies, which recognize N-terminally His or C-terminally FLAG tagged RT p66, respectively, is used to quantify the number of His/FLAG dimers in the presence of NNRTIs. As expected, EC50 values for enhancement of p66 dimerization by JLJ648, JLJ600, and JLJ555 (Fig. 2f) correlate well with the compounds’ ability to induce pyroptosis in HIV-1 infected cells (Fig. 2d, e). The efficiency of JLJ648 in inducing p66 dimerization was ~10 times higher than JLJ555 and ~2.5 times higher than JLJ600.
Structural studies investigating JLJ648 as a Gag-Pol dimerizer
To understand why JLJ648 enhances dimerization more effectively than JLJ600 and JLJ555, we solved the structure of JLJ648 bound to the p66/p51 heterodimer by X-ray crystallography. This 2.4 Å structure had explicit density for the ligand in the canonical NNRTI site, between the palm and thumb domains of the p66 catalytic subunit (Fig. S2c). This structure was then compared to structures of the heterodimer bound to JLJ55522 or JLJ60023, which had been previously solved to 2.9 and 2.5 Å, respectively. Within the canonical binding site, there are a few differences in the binding of these three compounds (Fig. S3a, b). JLJ648 overlays almost exactly with JLJ600, as do all the protein residues within a 5 Å radius of the ligand, allowing for the maintenance of key binding interactions. Specifically, the naphthyl and catechol groups interact with Y181, Y188, and W229 through face-to-face and edge-to-face π interactions. The carbonyl oxygen in the thiouracil group interacts with the backbone nitrogen of K103 via a hydrogen bond. Notably, the well-ordered water often observed within hydrogen bonding distance of the opposite uracil carbonyl is not present near the thiouracil ring in the JLJ648 structure. As observed previously36, JLJ555 sits slightly higher in the NNRTI binding site to allow the indolizine ring to make similar π stacking interactions as those observed in the naphthyl compounds, resulting in slight rearrangements of the catechol and uracil rings. To accommodate these changes, Y181 and Y318 rotate slightly to maintain ideal π stacking interactions with the catechol and uracil rings, respectively. L234 also rotates out of the pocket in the JLJ555 structure as the indolizine ring would likely form a weaker π–CH interaction with the residue’s δ carbon. However, these slight changes do not seem likely to account for the nearly 10-fold decrease in dimerization enhancement observed for JLJ555 compared to JLJ648.
We also examined whether there were any noticeable differences along the dimerization interface between the p66/p51 heterodimer bound to JLJ648 or more weakly dimerizing compounds. Most residues in the JLJ648 structure had an RMSD value of 0.6 Å or less when compared to either JLJ555 or JLJ600 bound structures (Fig. S3c, d). Nearly all the residues with larger differences are located on the connection and RNaseH domains of the p66 subunit and the thumb domain of the p51 subunit, with very few differences in the fingers or palm domains near the NNRTI binding site. Counterintuitively, more of these differences were observed when comparing the JLJ648 structure to the JLJ600 structure than when comparing the JLJ648 structure to the JLJ555 structure, suggesting that the extra differences are more likely due to the inherent flexibility of the regions. Many of the residues with higher RMSD values have their side chains truncated due to poor density in one or both structures. The rearrangements highlighted in both comparisons, including the p66 C-terminus and the p51 flexible loop containing N318, P420, P421, and L422, are solvent-exposed regions and provide little insight into a mechanism for NNRTI-mediated dimerization.
Cryo-EM structures of RT homodimer bound to JLJ648
As structures of the p66/p51 heterodimer bound to NNRTIs appear almost identical, we pursued the structure of the p66 homodimer in complex with JLJ648 using cryo-electron microscopy (cryo-EM), as this is the RT species present in the Gag-Pol polyprotein and therefore most relevant for understanding the cell-killing effect. We resolved two oligomeric species of JLJ648 bound to p66, a homodimer and a homotetramer, at 3.1 and 3.4 Å, respectively. The p66 homodimer resembled the previously reported structures of the p66/p51 heterodimer, but the p66 homotetrameric species has never been reported before. The p66 homodimeric class was further classified and refined into two subclasses, both at 3.1 Å resolution. The two subclasses mainly differ by the presence of an additional region of cryo-EM density, only present in Subclass 2. This density is surrounded by the thumb, connection, and RNaseH domains of the p66 catalytic subunit (Fig. 3b, c). A JLJ648 molecule could be modeled into this additional density, but due to unfavorable ligand geometry and interactions with the protein, we decided to keep this density unmodeled. Similar to the RT heterodimer structure, the two p66 protomers fold into the catalytic (denoted by p66) and structural subunits (denoted by p66’), respectively, forming the asymmetric homodimer. The well-defined cryo-EM density of the p66’ subunit ends around residue 420, leaving the entire RNaseH domain in the p66’ unresolved. Presence of the RNaseH domain was confirmed by SDS-PAGE and protein intact mass spectrometry, suggesting that the missing RNaseH domain of p66’ in our reconstruction is due to it being either unstructured or highly flexible (Fig. S4).

a Cryo-EM data processing workflow for HIV-1 RT in complex with JLJ648. b–d Reconstructed maps for Subclass 1 homodimer (b), Subclass 2 homodimer with additional density indicated in circle (c), and Tetramer (d). Colors are used to distinguish p66 and p66’ subunits. Figure prepared using ChimeraX.
The cryo-EM structure of the Subclass 1 RT homodimer shows minimal differences from the RT heterodimer crystal structure (Fig. S5a), with RMSD > 2 Å primarily in the peripheral loops where the electron density is poorly defined. Similarly, minor structural differences were observed between the Subclass 2 RT homodimer cryo-EM structure and RT heterodimer crystal structure (Fig. S5b). Binding of NNRTIs to the canonical site is also very similar between these three structures. In both Subclass 1 and Subclass 2 structures, the NNRTI pocket is slightly expanded with β-sheet 9, containing V179, I180, and Y181, and β-sheet 13, containing L234, H235, and P236, shifted away from bound JLJ648 relative to the heterodimer crystal structure (Fig. S5c, d). The JLJ648 catechol ring shifts toward V179 to fill the extra space. All aromatic interactions are maintained in the homodimer structures as the slightly repositioned naphthyl group induces compensatory rotations of Y181, Y188, W229, and F227. The hydrogen bond between the carbonyl oxygen of the uracil ring and the backbone of K103 observed in the heterodimer structure is observed in both homodimer structures; in Subclass 1, the Nζ of K102 is close enough to the carbonyl oxygen to contribute an additional hydrogen bond. These similar binding positions correlate well with previous work, which suggests that the chemical environment around the NNRTI is similar when bound to either the heterodimer or p66 homodimer.
Discovery of JLJ648 bound to novel homotetramer
The surprising finding from our cryo-EM studies of JLJ648 with RT p66 was the presence of a homotetramer species that constituted approximately 30% of the final particle stack (Fig. 3a). The 2D class averages of this homotetrameric species resemble an “infinity knot” approximately double the size of the homodimer particles (Fig. 3a, d). The p66 homotetramer reconstruction was resolved at 3.4 Å resolution and revealed a dimer of homodimers. The homotetramer structure is assembled in a head-to-head manner with a central two-fold rotational symmetry, where the two p66 catalytic subunits from the homodimers closely contact each other, flanked by the p66’ structural subunits (Fig. 4a). The tetramer interface is exclusively formed between the p66 catalytic subunit in each homodimer, with a buried surface area of ~1100 Å (Figs. 4a and S6). The cryo-EM structure of the Subclass 1 RT homodimer is similar to that of dimer 1 (chains A/B) of the RT homotetramer, with small differences primarily in the loop regions (Fig. S7a). Notably, the helices in the p66 fingers and p66 RNase H of the homotetramer chain A/B (purple) were slightly shifted closer to the center compared to the RT homodimer structure (cyan), suggesting increased compactness during tetramerization. More significant structural differences were observed when comparing the homodimer structure to that of dimer 2 (chains C/D) of the RT homotetramer (Fig. S7b). The fingers and thumb domains move towards each other, exhibiting a more closed conformation. Additionally, the C-terminal half of the dimer becomes less structured and is averaged out during map reconstruction; most of the p66 RNaseH domain and the p66’ thumb and palm domains could not be modeled.

a Cryo-EM reconstruction of the HIV-1 RT tetramer in the presence of JLJ648. The map is colored by domains as in Fig. 1a. A 2-fold rotational symmetry is present between the two dimers. b Structural model of the HIV-1 RT tetramer colored by domains. JLJ648 molecules are shown in magenta. c Close-up view of the two copies of JLJ648 at the center of the tetramer, which is surrounded by lysine residues from the p66 catalytic subunits Chain A and C. Dashed lines indicate potential interactions. Figure prepared using ChimeraX.
In the center of the tetramer, between the two p66 catalytic subunits, there is an unmodeled cryo-EM density that could accommodate a JLJ648 dimer (Figs. 4c and S2d). This region is enclosed by the fingers and thumb domains of both homodimers and is ~10–15 Å away from their canonical sites (Fig. 4b). However, the resolution of our cryo-EM map prevented unambiguous modeling of the JLJ648 dimer into this density. We tentatively modeled several poses and refined a plausible pose that displays a rich network of face-to-face and edge-to-face π interactions among the naphthyl and catechol groups of the ligands, as well as hydrogen bond and electrostatic interactions between the thiouracil groups of the ligands and residues K65, K66 and K259 of p66 dimer 1 (Fig. 4c). The cyanonaphthyl group of one of the ligands also has potential interactions with K259 and K263 of dimer 2 p66. Notably, the lysine-rich loop containing K65 and K66 in p66 dimer 1 has clearer cryo-EM density in the tetramer structure than is observed for this region in the Subclass 1 homodimer, suggesting that binding of the JLJ648 dimer to the tetramer may stabilize this region.
In addition to binding at the dimer interface of the tetramer, JLJ648 is also bound to the canonical NNRTI binding site in each p66 catalytic subunit between the palm and thumb domains, exhibiting the same binding mode as in the homodimer (Fig. S7c,d). The pocket remains in the more open conformation as observed in the homodimer structures. However, the bound JLJ648 appears to be less rigidly positioned in the tetramer, as the A-chain and C-chain-bound inhibitors have strikingly different conformations. In the A chain, JLJ648 shifts up and away from V189, like its position in the crystal structure (Fig. S7c). F227 and W229 rotate slightly to accommodate the new position of the naphthyl ring. The uracil ring rotates slightly to maintain its hydrogen bond with the backbone of K103, and Y318 rotates slightly to maintain the stacking interaction. In the C chain, the naphthyl and catechol rings overlay well with their position in the homodimer structures (Fig. S7d). However, the thiouracil ring rotates away from K103, resulting in the loss of its hydrogen bond. Additionally, W229 rotates into the binding pocket, changing the angle of its edge-to-face π interactions with the naphthyl group. These variations have been observed in other structures of naphthyl compounds bound to RT mutants and appear to have little effect on the compound’s activity36.
Cryo-EM of RT homodimer bound to JLJ555
We also tested the weaker Gag-Pol dimerizer, JLJ555, for its ability to induce homotetramerization. Interestingly, in the presence of JLJ555, p66 existed in the homodimeric as well as the homotetrameric conformation (Fig. S8a). No major conformational changes were observed between the homodimeric species of p66 in the presence of JLJ555 compared to JLJ648. The homotetrameric species of p66 in the presence of JLJ555 showed significantly higher disorder and resolved to a much lower resolution compared to the p66 homotetramer in the presence of JLJ648 (Fig. S8b). This suggests that the JLJ555-bound p66 homotetramers are more dynamic compared to their JLJ648-bound counterparts. The presence of homotetramers in the presence of JLJ555, a weaker Gag-Pol dimerizer with modest pyroptotic cell killing, is not surprising, as the experimental conditions for cryo-EM necessitate the use of high concentrations of p66 and NNRTIs (JLJ555 and JLJ648). Experimental conditions used for cryo-EM led to JLJ555 concentrations approximately 50-fold greater than its EC50 for RT dimerization, and at these concentrations, both the homodimeric and homotetrameric populations of JLJ555 bound p66 are expected. Since the NNRTI-bound homodimer is an essential building block for the homotetrameric form, the ~10-fold higher efficiency of JLJ648 over JLJ555 in inducing Gag-Pol dimerization places it as a superior NNRTI for inducing p66 homotetramerization.
Examining homotetramer formation in solution
The presence of the tetramer in solution was confirmed using size exclusion chromatography coupled with multi-angle light scattering (SEC-MALS) (Fig. S9). In the absence of JLJ648, RT exists in an equilibrium between monomer and dimer. However, in the presence of JLJ648, the monomer population completely disappeared, and the peak shifted to a higher molecular weight of 264 kDa, corresponding to a tetramer population. The broad peak indicates there may exist a transient dimer–tetramer equilibrium unresolvable in the chromatography. These results are consistent with our results from cryo-EM studies of p66 with JLJ648, wherein we resolved homodimeric and homotetrameric species of p66.
As an orthologous approach, we also used mass photometry to study the conformational dynamics of p66 in response to the various NNRTIs. Initial experiments in which p66 was incubated with either JLJ648 or an equivalent amount of DMSO under conditions similar to those used for cryo-EM and diluted directly onto the mass photometer resulted in the expected distribution of monomers and dimers, but no tetramers were observed. As the dimer is highly stable (t1/2 = 2 days13), we expect that the 330-fold dilution required for the mass photometry data acquisition will have little impact on the monomer/dimer equilibrium, but the tetramer is more transient and readily dissociates into dimers at nanomolar concentrations. To capture the equilibrium at micromolar protein concentrations, the samples were cross-linked with ethylene glycol bis(succinimidyl succinate) (EGS) prior to dilution. Successful crosslinking was verified with SDS–PAGE, and the resulting populations were quantified using mass photometry (Fig. S10a, b). In the absence of JLJ648, p66 is distributed into two populations with molecular weights of 66 kDa (monomer), and 133 kDa (dimer), which make up 61% and 26% of the population, respectively. In the presence of saturating amounts of JLJ648, JLJ600, and JLJ555, similar to the amount used for cryo-EM studies, the monomer population largely disappears, the dimer population increases to ~80% and a tetramer population appears, representing 6–7% of the total counts. These results confirm that the addition of saturating amounts of NNRTI results in a nearly identical enhancement of dimer and tetramer populations.
In order to more comprehensively understand the propensity of JLJ555 and JLJ648 to induce homodimerization, mass photometry experiments were carried out at lower protein concentrations in the absence of crosslinking. As illustrated in Fig. S11, the EC50 value for homodimerization for JLJ555 was approximately 10-fold weaker than JLJ648, in accord with the HTRF and pyroptotic cell killing assays.
Cryo-EM of HIV-1 p66/p66’ in absence of ligand
To confirm structurally that the tetramer is only formed in the presence of a dimerization-enhancing NNRTI, an RT homodimer sample was prepared using the same process but with DMSO instead of NNRTIs. After following the standard cryo-EM data processing pipeline, only monomers and homodimers were resolved from this dataset (Fig. 5a). This suggests that tetramer formation is specifically induced by NNRTI binding. The overall resolution for the apo RT homodimer was 3.85 Å. The refined cryo-EM structure of the apo RT homodimer (Fig. 5b) was overlaid and compared with the single JLJ648-bound homodimer structure (Subclass 1 dimer) (Fig. 5c). The most prominent structural change was observed in the thumb domain (residues 237–318). In the apo RT homodimer, the thumb domain (light green) adopts a closed conformation, which is located closer to the fingers domain (light blue). Upon JLJ648 binding, the thumb domain (dark green) shifts to an open conformation, moving away from the fingers domain (dark slate blue). This NNRTI-induced repositioning of the thumb domain correlates well with similar movement often observed in crystal structures of the p66/p51 heterodimer obtained in the presence or absence of ligand16. In the absence of NNRTI (apo RT), we did not resolve any species of the p66 homodimer in the thumb “open” conformation. Other domains in p66, including the fingers, palm (purple), connection (pink), and RNase H (orange), showed slight movements, possibly to accommodate the thumb movement (Fig. 5c). In contrast, p66’ exhibited minimal structural changes. The cryo-EM density for both p66’ thumb domains (light gray for the RT apo homodimer versus dark gray for the Subclass 1 JLJ648-bound homodimer) was slightly weak, limiting confident modeling and providing less evidence for domain movement. The RNase H domain of p66’ was not modeled due to incomplete density in that region. Disorder in this region is consistent with a prior cryo-EM structure of RT as part of the Gag-Pol polyprotein15. To further understand the absence of p66 homotetramers in the apo RT cryo-EM dataset, two copies of the apo RT homodimer were aligned to the JLJ648-bound homotetramer structure (Fig. 5d). Steric clashes between the thumb domains of the two apo RT homodimers are observed in the central region, as shown in the zoomed-in view (right panel). As the NNRTI-bound conformation of p66 homodimer is locked in the thumb “open” conformation, there are no steric clashes in the central region, and instead the p66 thumb domains coordinate a dimer of NNRTI molecules in this central region, further stabilizing the extensive inter-homodimeric interface. This further emphasizes that RT homotetramer formation is specifically induced by NNRTI binding.

a Representative 2D classes of the RT homodimer apo and the density volume obtained after ab initio modeling, heterogeneous refinement, and 3D reconstruction. b The final refined cryo-EM structure of the RT homodimer apo is shown in yellow cartoon representation, with its cryo-EM density displayed as a gray surface. c Structural comparison between the RT homodimer apo (in light colors) and the homodimer with JLJ648 bound (in dark colors). The p66 domains—fingers, palm, thumb, connection, and RNase H—are colored blue, purple, green, pink, and orange, respectively, while the corresponding domains in p66’ are shown in gray. The distinct movement in the thumb domain from the open conformation to the closed conformation was indicated by a blue arrow. d Two copies of the apo homodimer were aligned to the JLJ648-bound homotetramer structure (left panel). Steric clashes between the thumb domains from two homodimers are observed in the central region, as shown in the zoomed-in view (right panel), suggesting that the apo homodimer may preclude tetramer formation in the absence of JLJ648. Figures prepared using ChimeraX.
Investigating the importance of homotetramer for Gag-Pol dimerizer-induced pyroptotic cell killing
Our cryo-EM structure of JLJ648 bound p66 homotetramer revealed that JLJ648 is central to the nucleation of this oligomeric species, and structural analyses revealed several lysines in the p66 catalytic domain that mediate the coordination of JLJ648 at the heart of the homotetramer. One of the key residues important in this interaction is K259 (Fig. 4c). Guided by the JLJ648-bound homotetramer structure, we mutated K259 where the well-defined cryo-EM density indicated its crucial contribution to the homotetramer but minimal involvement in the homodimeric interface (Fig. 4c). The effects of this mutation were examined at both the cellular and biochemical levels by preparing both mutant virus and recombinant protein. The resultant K259A pseudotyped HIV-1 reporter virus NL4-3-ΔEnv-GFP was then used to infect the anti-CD3 and anti-CD28 co-stimulated CD4+ T cells. An ~20–30% weaker pyroptotic cell killing was observed for the mutant than for the WT across all tested compounds (EFV, JLJ648, JLJ600) at both 500 nM and 1.6 µM (Fig. 6a). Using the recombinantly expressed K259A mutant, the effect of JLJ648 to promote p66 homodimerization was compared with that of WT RT using mass photometry. This mutant does not have a significant effect on the ability of p66 to dimerize in response to JLJ648; 500 nM JLJ648 produces 58 ± 1% dimer and 52 ± 4% dimer in WT and K259A populations, respectively (Fig. 6b). This effect is consistent across a wide range of drug concentrations as the drug has a similar EC50 in both WT and mutant p66 (Fig. 6c). Similar dose response curves were obtained using an HTRF assay (Fig. S12). Therefore, the impaired cell killing effect in the K259A mutant does not appear to be attributable to p66 dimerization as indicated by these findings. These data imply that pyroptotic cell killing effects of Gag-Pol dimerizers such as JLJ648 are enhanced by the formation of the p66 homotetramer.

a, b RT bearing K259A, a putative tetramer-deficient mutation predicted from the tetramer structure, treated by EFV, JLJ648 and JLJ600 at 500 nM or 1.6 μM. Multiple comparisons were made using the Welch t-tests followed by the Holm–Šídák method to calculate adjusted p values: 0.1234 (ns), 0.0332 (*), 0.0021 (**), 0.0002 (***), <0.0001 (****). c Representative mass photometry measurement of 75 nM p66 homodimerization of WT and K259 variants in the presence of 500 nM JLJ648. Quantification of all three replicates for this concentration is shown in the inset bar graph. The difference between populations is not significant. Figure prepared using Refeyn Discover MP. d p66 homodimerization dose response curves. Serial dilutions of JLJ648 were incubated with 75 nM HTp66 WT or K259A for 18 h prior to performing mass photometry. Data shown is the average of triplicate measurements. Panels prepared using GraphPad Prism unless otherwise specified.
Discussion
The concept of a “shock and kill” approach to attack latent HIV-1 reservoirs offers a strategy for a functional cure for AIDS. Earlier studies21,22,23 demonstrate that some NNRTIs, such as EFV and RPV, act as Gag-Pol dimerizers and exhibit pyroptotic cell killing. Current FDA-approved NNRTIs dimerize intracellular HIV-1 Gag-Pol suboptimally at physiologically relevant concentrations33 and do not fully account for NNRTI-associated resistant viral variants present in the reservoir37,38. Building upon this work, we described the development of a new class of dual-function inhibitors that are potent antivirals and also potentiate CARD8 activation and clearance of HIV-1 infected cells. This effort to discover more potent Gag-Pol dimerizers was enabled by the development of a novel HTS assay with inducible lentiviral vectors for HIV-1 Gag-Pol and CARD8 expression, which specifically identifies compounds that exhibit pyroptotic cell killing dependent on PR activation of CARD8. The HTS focused on evaluating our in-house synthesized NNRTI library for potent antiviral activity combined with a lack of general cytotoxicity. Follow-up screening in primary CD4+ T cells was used to establish dose–response relationships. The majority of potent compounds identified contained a catechol diether scaffold with a cyano naphthyl ring as one arm of the diether and an ethoxy linker with a terminal uracil on the other, as noted in Fig. 1c, with JLJ648 being the most potent at 80 nM. A surprising structure–activity relationship was noted for three catechol diether compounds with similar chemical scaffolds: JLJ555, JLJ600, and JLJ648 with aryl ring substitution: JLJ555 (cyano indolizinyl), JLJ600 (cyano napthyl), and JLJ648 (JLJ600 with 4-thiouracil). While JLJ555 with the indolizine ring had a modest pyroptotic cell killing effect (EC50 = ~3 µM), the napthyl derivatives, JLJ600 and JLJ648, were over 9 and 37 times, respectively, more potent, even though all have antiviral activity in the low nanomolar or high picomolar range. Moreover, the high potency of JLJ648 emphasizes the importance of the 4-thiouracil versus the uracil in JLJ600. Further cellular and biochemical assays confirmed that the propensity of each NNRTI towards enhancing p66 homodimerization tracked very well with their pyroptotic cell killing ability.
It is possible that there might be distinct structural differences in how each compound binds to HIV-1 RT that would, in turn, be reflected in the propensity to enhance p66 homodimerization. The structural characterization of the interactions between these three compounds and the RT heterodimer confirms that minor changes in binding to the canonical NNRTI binding site are not responsible for their very different phenotypes. This suggested that dimerization enhancement and pyroptotic activity may be determined by drug binding to an altered canonical site in immature RT or to a new nascent site remote from the allosteric site. As the most relevant structural species for understanding the physiological consequences of Gag-Pol dimerization brought about by JLJ648 binding, the cryo-EM structure of the p66 homodimer-inhibitor complex was determined, yielding several surprising findings. One Subclass of JLJ648-bound RT p66 dimer particles has a region of density between the thumb, connection and RNase H domains, which could potentially fit an additional drug molecule. Additionally, we present a novel RT structure in which two JLJ648-bound p66 homodimers further dimerize around a region of strong electron density. While this density is not sufficiently well defined to allow for confident ligand fitting, modeling of various poses suggests that this density is likely fit by a JLJ648 dimer held together by face-to-face and edge-to-face π interactions between the naphthyl and catechol rings. This interaction is also facilitated by hydrogen bonding and electrostatic interactions of the 4-thiouracil to serve as a nucleation site for tetramer formation.
To determine whether the tetramer was specifically formed in the presence of strong dimerizers like JLJ648, the cryo-EM structures of the homodimer were determined under identical experimental conditions in either the presence of JLJ555 or the absence of any NNRTI. Similar volumes were obtained for both the dimer and tetramer in the presence of JLJ555 (Fig. S8), though there was no evidence for tetramerization in the absence of NNRTIs (Fig. 5). As saturating amounts of both JLJ648 and JLJ555 produce similar tetramer populations, the key determinant of whether a compound will have strong pyroptotic activity is likely its efficiency in promoting p66 homodimerization. Given that all NNRTI-bound p66 homodimers are locked in the thumb open conformation, every NNRTI-bound homodimer is capable of forming a homotetrameric species.
Our studies with K259A RT suggest that the homotetramerization may play an important role in the pyroptotic cell killing effect. This mutation, designed to disrupt interactions between the tetramer and the two JLJ648 molecules at its center, results in a significant disruption of pyroptotic activity that cannot be attributed to a decrease in its ability to dimerize (Fig. 6).
A working model based on our structural and biochemical studies for describing how JLJ648 might enhance p66 homodimer and homotetramer formation is illustrated in Fig. 7. In the model, the p66 and p66’ represent the catalytic and structural subunits that can form an asymmetric homodimer. Two of these monomers (left) can bind to form a p66 homodimer in either the presence (bottom middle) or absence (top middle) of dimerizing NNRTIs. If there is a sufficient amount of ligand-bound homodimer available, they can further dimerize around additional NNRTI molecules to form a homotetramer (bottom right). Formation of an apo homotetramer is unfavorable, as the two thumb domains, positioned much closer to their respective finger domains, would clash in a tetramer. The affinity of the dimer in the presence of a specific NNRTI determines the amount of dimer available to form an NNRTI-bound homotetramer and likely determines the strength of its pyroptotic activity.

RT is shown divided into domains. p66 subunits are colored according to the keys below. p66’ subunits are shown in gray, or the colors used for the p66 subunit are overlaid with gray. Regions bordered by dashed lines are not visible in previously published monomer or polyprotein structures or our homodimer/tetramer structures. Protease and integrase subunits are shown as translucent, as they were not present in the constructs used for this work. Two p66 monomers (left) can form either apo (top middle) or NNRTI-bound (bottom middle) homodimers. Only NNRTI-bound dimers may form stable tetramers (bottom right) as the positioning of the thumb in the apo structure (shown circled in orange) is not compatible with tetramer formation. These NNRTI-bound tetramers may, in turn, seed a Gag-Pol tetramer (black box). Integrase subunits are shown as a functional tetramer, which may stabilize that of the polyprotein. Protease dimer 1 is illustrated in a position to cleave protease dimer 2 from the polyprotein as tetramerization may facilitate this reaction. Figure prepared using Adobe Illustrator.
Our studies with the K259A mutant virus suggest that the homotetramer is required for the full pyroptotic effect. One possible mechanism is that the p66 homotetramer might facilitate premature HIV-1 protease activation at a cellular level. The immature uncleaved Gag-Pol polyprotein in HIV-1 infected cells contains p6, PR, RT and INT. PR and RT are active as dimers, while INT is active as a tetramer. In a stepwise fashion, activation occurs when PR forms a catalytically active dimer and cleaves p6 from its N-terminus, RT from its C-terminus and INT from RT. While the p66 dimer and tetramer appear to exchange rapidly, an uncleaved PR-RT construct forms a stable population of tetramers in the presence of RPV39. As noted in the model, the formation of the NNRTI-bound Gag-Pol tetramer, further stabilized by the RT and PR dimers and integrase tetramer in the Gag-Pol polyprotein, could bring one PR dimer close enough to the other to facilitate cleavage from polyprotein, producing the fully activated HIV-1 protease.
In summary, the discovery of JLJ648 as a dual-function Gag-Pol dimerizer that inhibits viral replication and induces pyroptosis in HIV-1 infected cells offers a promising preclinical candidate and a strategy for the clearance of HIV-1 reservoirs in an approach for a functional cure for AIDS.
Methods
Preparation of HIV-1 reporter viruses and lentiviruses
HIV-1 reporter viruses and lentiviruses were packaged by co-transfecting HEK293T cells with viral vectors, a packaging vector, and pVSV-G using Lipofectamine 2000 (Thermo Fisher Scientific # 11668019). Lenti-X Concentrator (TaKaRa #631232) was used to concentrate the supernatant-containing virus.
Chemicals and reagents
The chemicals and reagents used in this study were of reagent-grade purity. The syntheses of JLJ555 and JLLJ600 have previously been reported22,23. The synthesis of JlJ648 was prepared in a similar manner, and a complete description of preparation and characterization is provided in Supplementary Note 1.
Compound screening
The CARD8-KO THP-1 cell line was generated previously. This cell line was transduced with two lentiviral vectors expressing a codon-optimized HIV-1 Gag-Pol and a doxycycline-inducible CARD8 expression cassette, respectively. Transduced cells were maintained in the RPMI media with the presence of 500 µg/mL G418 and 200 µg/mL Hygromycin B. Cells were pre-treated with 1 µg/mL Doxycycline for 24 h to induce CARD8 expression. Dox-induced cells were seeded in a 96-well plate at a density of 75,000 cells per well and then treated with NNRTIs or other compounds for 4 h before cell death measurement by the invitrogen™ CyQUANT™ LDH Cytotoxicity Assay (#C20301).
Cell killing in primary CD4+ T cells
CD4+ T cells were isolated from healthy donor peripheral blood mononuclear cells (PBMCs) using the BioLegend human CD4+ T cell isolation kit (BioLegend #480010). Purified CD4+ T cells were co-stimulated with plate-bound CD3 antibody (Biolegend #300333) and soluble CD28 antibody (Biolegend #302943) in the presence of 20 ng/mL IL-2 (Biolegend #589106) for 3 days. Stimulated cells were infected by an HIV-1 reporter virus NL4-3-ΔEnv-GFP at a multiplicity of infection (MOI) of 10. Infection was performed by spin inoculation (1200×g) for 2 h at 25 °C. NNRTIs were added to infected cells 3 days post-infection. Cell death was measured 2 days post NNRTI treatment based on the elimination of GFP+ cells as described previously33.
Expression and purification of RT52a p66/p51 heterodimer
Heterodimeric RT52a was expressed and purified as previously described40.
Cloning, expression and purification of HIV-1 RT p66 for CryoEM (Xp66)
The HIV-1 RT p66 sequence (UniPROT P03366 residues 600–1154) was amplified from a clone generously provided by Dr. Stephen Hughes and Dr. Paul Boyer (Frederick Cancer Research and Development Center, MD). The primers used for amplification (Table S1) introduced Nde1 and Xho1 restriction sites to the 5’ and 3’ end of the amplicon, respectively. These restriction sites were used to clone the amplicon into a PET29a vector. Positive colonies were screened using Sanger sequencing at the Keck DNA Sequencing Facility at Yale. Sequencing results for the final plasmid were confirmed with the full-plasmid sequence (Plasmidsaurus).
This construct was transformed into BL21(DE3)-pRIL cells and grown in LB broth containing kanamycin (50 mg/L) at 37 °C to an optical density of 0.6–0.9 at 600 nm. The cultures were induced with 3 mM IPTG, harvested after an additional 3 h at 37 °C and frozen at −80 °C until needed for purification.
RT p66 was purified as previously described for the mature HIV-1 RT heterodimer (p66/p51) with the following modifications40. Frozen pellets were thawed and resuspended in Lysis Buffer. 1 mM PMSF was added to the resuspended cells to minimize proteolytic processing of the protein. The cells were lysed by passing through the cell disruptor three times, and the lysed cells were centrifuged at 16,000 rpm for 1 h. The supernatant was loaded onto two tandem 5 mL HisTrap FF columns (Cytiva), preequilibrated in Buffer A. The column was washed with 6 column volumes of Buffer A before eluting RT with 6 column volumes of Buffer B on the FPLC. Fractions containing RT were pooled and concentrated for dialysis into Dialysis Buffer, and the C-terminal 6X His Tag was cleaved with TEV Protease overnight at 4 °C. TEV L56V/S135G/S219V was expressed and purified in-house as previously described41. Uncleaved protein was removed by running the dialyzed protein over the tandem 5 mL HisTrap FF columns, preequilibrated in Buffer C. Cleaved RT p66 was collected in the flow-through and Buffer C wash fractions. TEV protease and residual uncleaved RT p66 were eluted with Buffer B. Cleaved RT p66 was loaded directly onto a 5 mL HiTrap Heparin column (Cytiva), preequilibrated in Buffer D. The column was washed with 15% Buffer E, followed by step elution of RT p66 at 18%, 20%, 22%, 24%, 26%, 28%, 30%, and 40% Buffer E on the FPLC. This elution method was optimized to separate several p51-like 55–60 kDa bacterial cleavage products from the true RT p66. Fractions containing only RT p66 with no visible contaminant bands on an SDS–PAGE gel were pooled and buffer exchanged into Storage Buffer. The resulting protein was aliquoted and stored at −80 °C until needed for freezing grids. Buffer recipes are available in Table S2.
Cloning, expression and purification of HIV-1 RT p66 for all other assays (Hp66, HTp66 and HTFp66)
pET30a_HIV-RT was a gift from Bryan Dickinson & Drew Endy (Addgene plasmid # 153311). Mutations at positions 172, 361, 368, 471 and 511 were corrected to match the UniPROT P03366 sequence. This construct produces RTp66 with an N-terminal His tag (Hp66). The 5-residue linker between the N-terminal His tag and the start of the RT p66 sequence was replaced with a TEV protease cleavage site (HTp66). A FLAG tag was then added to the construct’s C-terminus (HTFp66). All modifications were made with Q5 mutagenesis (NEB). Sequences of mutagenic primers are available in Table S1.
All constructs were transformed into BL21(DE3) cells and grown in LB broth containing kanamycin (50 mg/L) at 37 °C to an optical density of 0.6–0.8 at 600 nm. The cultures were induced with 1 mM IPTG, harvested after an additional 16–18 h at 17 °C and frozen at −80 °C until needed for purification.
For Hp66 purification, frozen pellets were thawed and resuspended in Lysis Buffer. 1 mM PMSF was added to the resuspended cells to minimize proteolytic processing of the protein. The cells were lysed by passing through the cell disruptor three times, and the lysed cells were centrifuged at 16,000 rpm for 1 h. The supernatant was loaded onto two tandem 5 mL HisTrap FF columns (Cytiva), preequilibrated in Buffer A. The column was washed with 6 column volumes of Buffer A before eluting RT with 6 column volumes Buffer B on the FPLC. Fractions containing RT were pooled and concentrated for dialysis into Buffer D. Dialyzed p66 was loaded directly onto a 5 mL HiTrap Heparin column (Cytiva), preequilibrated in Buffer D. RT p66 was eluted using a gradient from 0% to 55% Buffer E over 31 column volumes on the FPLC. Fractions containing only RT p66 with no visible contaminant bands on an SDS–PAGE gel were pooled and loaded onto a Superdex 200 Increase 10/300 GL column to remove aggregated protein and buffer exchange into Storage Buffer. The resulting protein was aliquoted and stored at −80 °C until needed for HTRF assays.
HTp66 was purified like Xp66 with the following modifications. Fractions from the heparin column containing only RT p66 and no visible contaminant band were loaded onto a Superdex 200 Increase 10/300 GL column to remove aggregated protein and buffer exchange into SEC-MALS Buffer for use in SEC-MALS and mass photometry experiments.
HTFp66 was purified like Xp66 with the following modifications. RT p66 was eluted using a gradient from 0% to 70% Buffer E over 40 column volumes on the FPLC. Fractions containing only RT p66 and no visible contaminant band were loaded onto a Superdex 200 Increase 10/300 GL column to remove aggregated protein and buffer exchange into Storage Buffer. Remaining uncleaved protein was collected from the second nickel column elution, buffer exchanged into Buffer D and purified using the same protocol as the cleaved protein for use as a positive control for the HTRF assay. Buffer recipes are available in Table S2.
The K259A mutation was introduced into all three constructs using Q5 mutagenesis. Sequences of mutagenic primers are available in Table S1. Each mutant construct was purified using the same method as its wild-type counterpart.
Protein crystallization, data collection, and refinement
Heterodimeric RT52A was cocrystallized with JLJ648 as previously described42. Briefly, 10–15 mg/mL RT52A was incubated with 500 µM JLJ648 for 1 h on ice, after which the solution was centrifuged at 15,500 rcf for 10 min to pellet precipitated NNRTI. This supernatant was combined 1:1 with the well solution (50 mM citric acid, MES, or HEPES, pH 5.0–7.5, 12.5–20% (w/v) PEG 8000, 100 mM ammonium sulfate, 15 mM magnesium sulfate, and 5 mM spermine) in 24-well hanging drop plates (Hampton Research). For freezing, crystals were transferred to a cryosolution containing 27% (vol/vol) ethylene glycol, an additional 2% PEG 8000 and 0.6 mM JLJ648 and flash frozen with liquid nitrogen.
X-ray diffraction data sets on frozen crystals were collected at the Brookhaven National Laboratory's National Synchrotron Light Source II on the FMX beamline. X-ray diffraction data were processed with AutoProc43. Phases were determined by molecular replacement with the program PHASER44 using the previously solved structure PDB 5TW345 as the initial search model. The model was built into electron density using COOT46, and the structure was refined using Phenix Refine47. For each refinement, 5% of all reflections were omitted and used for the calculation of Rfree. Successive rounds of XYZ coordinate and individual B-factor refinement were performed until acceptable geometry and refinement statistics were achieved. Atomic coordinates and structure factors are deposited in the PDB with code 9MXB. Crystallography data collection and refinement statistics are available in Supplementary Table S3.
HTRF dimerization assay
The HTRF dimerization assay protocol was performed as previously described with modifications31. Briefly, Hp66 and HTFp66 were diluted to 12.5 and 25 nM, respectively, in HTRF buffer and incubated at 22 °C for 6 h. Serial dilutions of each tested compound (final concentrations 60 µM–5 nM) were prepared in DMSO and diluted to 4% DMSO in HTRF buffer. A DMSO-only control was also included. 5 µL each of protein and diluted compound were mixed in a 384-well ProxiPlate (Revvity), sealed with TempPlate sealing film (USA Scientific) and incubated at 22 °C for 17 h. Anti-6HIS-Eu donor antibody (Revvity) and anti-FLAG-M2-XL665 acceptor antibody (Revvity) stocks were prepared according to the manufacturer’s instructions and diluted 200-fold in HTRF buffer. 10 µL of this mixture of diluted antibodies was added to each well and allowed to incubate for 1 h at 22 °C. Fluorescence of the donor and acceptor was measured at 620 and 665 nm, respectively, on a PHERAstar FSX plate reader (BMG Labtech). The two wavelengths were normalized (665 nm/620 nm*105) to correct for well-to-well variability and potential quenching by the tested compounds. Background was subtracted from each measurement, and each well was normalized to the highest value in that dataset. These normalized values were plotted against the compound concentration; EC50 values were determined by [Agonist] vs. normalized response–variable slope fitting in Prism version 10.4.0 for Mac. Each concentration was tested in triplicate, and the reported values are the mean of three separate assays, which were fit separately. Buffer recipes are available in Table S2.
CryoEM grid preparation and data collection
Purified Xp66 was buffer exchanged into CryoEM buffer and diluted to 10 µM with additional CryoEM buffer. Protein was incubated with 50 µM JLJ648, 50 µM JLJ555, or an equivalent amount of DMSO at 18 °C for 12 h prior to grid freezing. The assembled complex (4 µL) was applied to C-Flat R2/1 copper cryoEM grids (300 Grid Mesh, 20 nm Carbon Thickness) (EMS) after glow discharging the grids at 11 mA for 20 s (Pelco). Grids were blotted at blot force 1 for 7 s and plunge frozen in liquid ethane using the Vitrobot Mark IV system (Thermo Fisher Scientific) maintained at 10 °C. The detergent CHAPSO (0.15% final concentration) was added just before grid freezing to ensure uniform ice thickness and particle distribution. Buffer recipes are available in Table S2.
Grids were screened for optimal ice thickness and particle concentration. Data collection was carried out as previously described48. Briefly, the JLJ648 and apo RT dataset was collected from a single screened grid using a 300 kV Titan Krios cryo-transmission electron microscope (Thermo Fisher Scientific) equipped with a K3 camera (Gatan) and an imaging energy filter (Gatan) operated at a slit width of 15 eV. The dataset was collected in counting super-resolution mode with a nominal magnification of 81,000×, leading to a physical pixel size of 1.07 Å (super-resolution pixel size is 0.535 Å). The JLJ555 dataset was collected using a 200 kV Glacios cryo-transmission electron microscope (Thermo Fisher Scientific) equipped with a K3 camera (Gatan) at a nominal magnification of ×45,000 and a physical pixel size of 0.868 Å (super-resolution pixel size is 0.434 Å). All datasets were collected at a dose rate of 15 e−/pixel/s with a total electron dose of 50 e−/Å2 applied over 40 frames and a defocus range of −1.0 to −2.0 µm.
Cryo-EM data processing and model building
The cryo-EM data processing workflow was carried out using cryoSPARC v449. Movie frames were motion corrected using Patch motion correction and binned two-fold to yield a stack of motion-corrected micrographs with a pixel size of 1.07 Å. These micrographs were then manually curated, and an initial round of particle picking was performed using Blob picker on a subset of 500 micrographs. A 2D classification job on this initial particle stack yielded ideal 2D templates that were used to perform template-based particle picking on the entire dataset. The particle stack from the template picker was further curated to remove damaged/denatured particles using 2D classification. After 2D classification, selected particles were carried forward to Ab initio reconstruction for generating three different volumes and were used as 3D references for heterogeneous refinement of the particle stack. From the grid prepared with JLJ648, particles corresponding to two JLJ648-bound homodimers and a JLJ648-bound homotetramer were segregated and further refined using Non-Uniform (NU) refinement. From the grid prepared in the absence of JLJ648, particles corresponding to several monomer conformations and an apo p66 homodimer were segregated and further refined using Non-Uniform (NU) refinement. These particle stacks were further curated using 3D classification to improve compositional and conformational homogeneity. The final particle stacks were then polished using Reference-based motion correction and used for NU-refinement to create the final reconstructions. All datasets were processed as above, and the details are included in Supplementary Tables 4, 5. The final reconstructions were uploaded to the Electron Microscopy Data Bank (EMDB 48718, 48719, 48720, 48721).
Model building was done in Coot46 with the crystal structure of JLJ648 bound to the RT p66/p51 heterodimer as the starting template for all JLJ648-bound structures and the crystal structure of apo RT p66/p51 heterodimer (PDB: 1DLO) as the starting template for the apo RT p66 homodimer. After initial rigid-body docking, adjustments were made to manually fit into the high-resolution map using real-space refinement modules. Two rounds of real-space refinement in PHENIX47 were carried out, and the outliers were manually corrected in Coot before deposition in the Protein Data Bank (PDB 9MXQ, 9MXR, 9MXS, 9MXT).
Size exclusion chromatography linked to multi-angle light scattering (SEC-MALS)
Multiangle laser light scattering experiments were performed as previously described with modifications50. Briefly, HTp66 was diluted to 4.5 mg/mL in SEC-MALS buffer and incubated with either 50 µM JLJ648 or an equivalent volume of DMSO for 16 h at 18 °C. Immediately before injection, a Costar Spin-X 0.22 µm centrifuge tube filter was used to remove any protein aggregate or precipitated inhibitor. Samples (25 µL) were injected and run over a 4.6 mm TSKgel SuperSW3000 column (Tosoh Biosciences) at a 0.2 mL/min flow rate. SEC-MALS data were measured using a Dawn Heleos-II spectrometer (Wyatt Technology) coupled to an Opti-lab T-rEX interferometric refractometer (Wyatt Technology). Data on light scattering (690 nm laser), 280 nm absorbance and refractive index were measured. Prior to sample injection, the system was calibrated and normalized using a bovine serum albumin standard. These measurements were also used to determine the appropriate dn/dc value needed for molecular weight determination from light scattering, which was set to 0.1780 for sample analysis. Data was processed in ASTRA as previously described51 and the resulting graphs were replotted using Prism version 10.4.0 for Mac. Buffer recipes are available in Table S2. Additional notes about SEC-MALS data collection and analysis are provided in Supplementary Note 2.
Crosslinking and mass photometry
HTp66 was diluted to 7.5 µM in SEC-MALS buffer and incubated with either 50 µM JLJ648 or the equivalent volume of DMSO for 18 h at 18 °C. Ethylene glycol bis(succinimidyl succinate) (EGS) was added to a final concentration of 1 mM and allowed to crosslink the sample for 2 h on ice. At the end of the incubation, Tris was added to a final concentration of 70 mM to quench the reaction. Crosslinked samples were diluted to a final concentration of 22.5 nM in the well, which allowed for the measurement of approximately 3000 binding events within a 60 s recording.
Alternatively, WT or K259A HTp66 was diluted to 75 nM in Mass Photometry Buffer B and incubated with varying concentrations of JLJ648 or JLJ555 for 18 h at 18 °C. Samples for each drug concentration were prepared in triplicate. Each sample was loaded directly into the well without further dilution, which allowed for the measurement of 3000–6000 binding events within a 60 s recording, depending on the ratio of monomers and dimers in the sample. The percent dimer of each sample was determined by converting the number of counts for each species into its respective concentrations and dividing the dimer concentration by the total.
Mass photometry data were collected on a TwoMP mass photometer (Refeyn Ltd.). Well, cassettes were assembled onto carrier slides according to the manufacturer’s instructions. The instrument was calibrated using the thyroglobulin dimer (660 kDa) and the bovine serum albumin monomer (66.5 kDa) and dimer (133 kDa) diluted in the same buffer used for samples (Mass Photometry Buffer A for crosslinked samples and Mass Photometry Buffer B for non-crosslinked samples). 60 s recordings were processed using DiscoverMP software (Refeyn Ltd.). The populations of monomers, dimers and tetramers in each sample were calculated with the individual number of counts obtained for each population upon Gaussian fitting. Buffer recipes are available in Table S2.
Data availability
Crystallographic data for the structures reported in this article have been deposited in the Protein Data Bank under accession number 9MXB. CryoEM data for the structures reported in this article have been deposited in the Protein Data Bank under accession numbers 9MXQ, 9MXR, 9MXS, and 9MXT and the Electron Microscopy Data Bank with codes EMD-48718, 48719, 48720, and 48721. All other data supporting the findings of this study are available in the paper and its Supplementary Information.
References
UNAIDS. Fact Sheet—Latest Global and Regional Statistics on the Status of the AIDS Epidemic https://www.unaids.org/en/resources/documents/2024/UNAIDS_FactSheet (2025).
World Health Organization. HIV Data and Statistics https://www.who.int/teams/global-hiv-hepatitis-and-stis-programmes/hiv/strategic-information/hiv-data-and-statistics#:~:text=In%202023%2C%2072%25%20%5B65,and%20have%20viral%20load%20suppression (2025).
Finzi, D. et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300 (1997).
Zerbato, J. M., McMahon, D. K., Sobolewski, M. D., Mellors, J. W. & Sluis-Cremer, N. Naive CD4+ T cells harbor a large inducible reservoir of latent, replication-competent human immunodeficiency virus type 1. Clin. Infect. Dis. 69, 1919–1925 (2019).
Ganor, Y. et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 4, 633–644 (2019).
Shan, L. & Siliciano, R. F. From reactivation of latent HIV-1 to elimination of the latent reservoir: the presence of multiple barriers to viral eradication. Bioessays 35, 544–552 (2013).
Davey, R. T. et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 96, 15109–15114 (1999).
Sarafianos, S. G. et al. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 385, 693–713 (2009).
Harris, D., Lee, R., Misra, H. S., Pandey, P. K. & Pandey, V. N. The p51 subunit of human immunodeficiency virus type 1 reverse transcriptase is essential in loading the p66 subunit on the template primer. Biochemistry 37, 5903–5908 (1998).
Kohlstaedt, L. A., Wang, J., Friedman, J. M., Rice, P. A. & Steitz, T. A. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 256, 1783–1790 (1992).
Jacobo-Molina, A. et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å resolution shows bent DNA. Proc. Natl. Acad. Sci. USA 90, 6320–6324 (1993).
Huang, H., Chopra, R., Verdine, G. L. & Harrison, S. C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282, 1669–1675 (1998).
Venezia, C. F., Howard, K. J., Ignatov, M. E., Holladay, L. A. & Barkley, M. D. Effects of Efavirenz binding on the subunit equilibria of HIV-1 reverse transcriptase. Biochemistry 45, 2779–2789 (2006).
Zheng, X. et al. Selective unfolding of one Ribonuclease H domain of HIV reverse transcriptase is linked to homodimer formation. Nucleic Acids Res. 42, 5361–5377 (2014).
Harrison, J. J. E. K. et al. Cryo-EM structure of the HIV-1 Pol polyprotein provides insights into virion maturation. Sci. Adv. 8, eabn9874 (2022).
Hsiou, Y. et al. Structure of unliganded HIV-1 reverse transcriptase at 2.7 å resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure 4, 853–860 (1996).
Phanuphak, N. & Gulick, R. M. HIV treatment and prevention 2019: current standards of care. Curr. Opin. HIV AIDS 15, 4–12 (2020).
Poongavanam, V. et al. Integrative approaches in HIV -1 non-nucleoside reverse transcriptase inhibitor design. WIREs Comput. Mol. Sci. 8, https://doi.org/10.1002/wcms.1328 (2018).
Stanford University. Major Non-Nucleoside RT Inhibitor (NNRTI)Resistance Mutations https://hivdb.stanford.edu/dr-summary/resistance-notes/nnrti/ (2025).
Lai, M.-T. et al. In vitro characterization of MK-1439, a novel HIV-1 nonnucleoside reverse transcriptase inhibitor. Antimicrob. Agents Chemother. 58, 1652–1663 (2014).
Bollini, M. et al. Computationally-guided optimization of a docking hit to yield catechol diethers as potent anti-HIV agents. J. Med. Chem. 54, 8582–8591 (2011).
Lee, W.-G. et al. Picomolar inhibitors of HIV reverse transcriptase featuring bicyclic replacement of a cyanovinylphenyl group. J. Am. Chem. Soc. 135, 16705–16713 (2013).
Lee, W.-G. et al. Picomolar inhibitors of HIV-1 reverse transcriptase: design and crystallography of naphthyl phenyl ethers. ACS Med. Chem. Lett. 5, 1259–1262 (2014).
Kudalkar, S. N. et al. From in silico hit to long-acting late-stage preclinical candidate to combat HIV-1 infection. Proc. Natl. Acad. Sci. USA 115, E802–E811 (2018).
Tachedjian, G., Orlova, M., Sarafianos, S. G., Arnold, E. & Goff, S. P. Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 98, 7188–7193 (2001).
Figueiredo, A. et al. Potent Nonnucleoside Reverse Transcriptase Inhibitors Target HIV-1 Gag-Pol. PLoS Pathog. 2, e119 (2006).
Sudo, S. et al. Efavirenz enhances HIV-1 gag processing at the plasma membrane through Gag-Pol dimerization. J. Virol. 87, 3348–3360 (2013).
Wang, Q. et al. CARD8 is an inflammasome sensor for HIV-1 protease activity. Science 371, eabe1707 (2021).
Kim, J. G. & Shan, L. Beyond inhibition: a novel strategy of targeting HIV-1 protease to eliminate viral reservoirs. Viruses 14, https://doi.org/10.3390/v14061179 (2022).
Clark, K. M., Pal, P., Kim, J. G., Wang, Q. & Shan, L. The CARD8 inflammasome in HIV infection. Adv. Immunol. 157, 59–100 (2023).
Balibar, C. J. et al. Potent targeted activator of cell kill molecules eliminate cells expressing HIV-1. Sci. Transl. Med. 15, eabn2038 (2023).
Figueiredo, A., Zelina, S., Sluis-Cremer, N. & Tachedjian, G. Impact of residues in the nonnucleoside reverse transcriptase inhibitor binding pocket on HIV-1 reverse transcriptase heterodimer stability. Curr. HIV Res. 6, 130–137 (2008).
Clark, K. M., et al. Chemical inhibition of DPP9 sensitizes the CARD8 inflammasome in HIV-1-infected cells. Nat. Chem. Biol. 19, 431–439 (2023).
Johnson, D. C. et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med. 24, 1151–1156 (2018).
Sharif, H. et al. Dipeptidyl peptidase 9 sets a threshold for CARD8 inflammasome formation by sequestering its active C-terminal fragment. Immunity 54, 1392–1404 e1310 (2021).
Hollander, K. et al. Exploring novel HIV-1 reverse transcriptase inhibitors with drug-resistant mutants: a double mutant surprise. Protein Sci. 32, e4814 (2023).
Persaud, D. et al. Early archiving and predominance of nonnucleoside reverse transcriptase inhibitor-resistant HIV-1 among recently infected infants born in the United States. J. Infect. Dis. 195, 1402–1410 (2007).
Zaccarelli, M. et al. Genotypic resistance test in proviral DNA can identify resistance mutations never detected in historical genotypic test in patients with low level or undetectable HIV-RNA. J. Clin. Virol. 82, 94–100 (2016).
Chagas, B. C. A., Zhou, X., Guerrero, M., Ilina, T. V. & Ishima, R. Interplay between protease and reverse transcriptase dimerization in a model HIV-1 polyprotein. Protein Sci. 33, e5080 (2024).
Carter, Z. J., Hollander, K., Spasov, K. A., Anderson, K. S. & Jorgensen, W. L. Design, synthesis, and biological testing of biphenylmethyloxazole inhibitors targeting HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett. 84, https://doi.org/10.1016/j.bmcl.2023.129216 (2023).
Raran-Kurussi, S., Cherry, S., Zhang, D. & Waugh, D. S. Removal of Affinity Tags with TEV Protease. In Methods in Molecular Biology 221–230 (Springer, New York, 2017).
Prucha, G. R. et al. Covalent and noncovalent strategies for targeting Lys102 in HIV-1 reverse transcriptase. Eur. J. Med. Chem. 262, 115894 (2023).
Vonrhein, C. et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. Sect. D67, 293–302 (2011).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Kudalkar, S. N. et al. Structural and preclinical studies of computationally designed non-nucleoside reverse transcriptase inhibitors for treating HIV infection. Mol. Pharmacol. 91, 383–391 (2017).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. D75, 861–877 (2019).
Devarkar, S. C. et al. Structural basis for translation inhibition by MERS-CoV Nsp1 reveals a conserved mechanism for betacoronaviruses. Cell Rep. 42, 113156 (2023).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Shen, Q. et al. The capsid lattice engages a bipartite NUP153 motif to mediate nuclear entry of HIV-1 cores. Proc. Natl. Acad. Sci. USA 120, e2202815120 (2023).
Wyatt, P. J. Light scattering and the absolute characterization of macromolecules. Anal. Chim. Acta 272, 1–40 (1993).
Acknowledgements
Funding of this work was provided by NIH GM032136 to W.L.J., NIH/NIAID R37AI155072 to K.S.A., NIH grants R01AI162203 and R01AI176594 to L.S., and T32 GM148333 and F31 AI186613-01 to K.H. This research used facilities resources AMX (highly automated macromolecular crystallography) and FMX (frontier microfocusing macromolecular crystallography) of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The Center for BioMolecular Structure is primarily supported by the NIH, National Institute of General Medical Sciences (NIGMS) through a Center Core P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1605010). The Laboratory for BioMolecular Structure (LBMS) is supported by the DOE Office of Biological and Environmental Research (KP1607011).
Author information
Authors and Affiliations
Contributions
K.H.: Designing research studies, conducting experiments, analyzing data, writing the manuscript. S.C.D. and S.T.: Conducting experiments, analyzing data, writing the manuscript. S.M.: Analyzing data and writing the manuscript. R.T., E.D., Q.W., K.M.F.: Conducting experiments and analyzing data. W.L., K.A.S., J.A.R.: Conducting experiments. W.L.J., Y.X.: Designing research studies. K.S.A., L.S.: Designing research studies and writing the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hollander, K., Devarkar, S.C., Tang, S. et al. Mechanistic basis for a novel dual-function Gag-Pol dimerizer potentiating CARD8 inflammasome activation and clearance of HIV-infected cells. npj Drug Discov. 2, 22 (2025). https://doi.org/10.1038/s44386-025-00025-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44386-025-00025-2
