
Abstract
Buntanetap is an orally available small RNA targeting molecule that inhibits the translation of multiple neurotoxic aggregating proteins, including amyloid precursor protein (APP) and Tau. It has been evaluated in 13 clinical trials involving over 1000 participants, including healthy volunteers, patients with Alzheimer’s disease (AD) and Parkinson’s disease (PD), and has shown a favorable safety and tolerability profile. In two small studies in early AD, buntanetap demonstrated a trend toward cognitive improvement, despite being underpowered for efficacy. In a Phase2/3 study in early PD, it also improved PD patients’ cognitive functions. We evaluated safety and efficacy of buntanetap in treating mild to moderate AD patients in this 3-month randomized double-blind dose-ranging study (NCT05686044). Total of 351 Patients were equally randomized to either 7.5 mg, 15 mg, 30 mg buntanetap or placebo. Buntanetap had a favorable safety profile. The study did not meet its primary endpoints (ADAS-Cog-11 and ADCS-CGIC), as 40% of participants lacked amyloid pathology. However, in amyloid biomarker-positive mild AD patients, buntanetap demonstrated nominally statistically significant dose-dependent cognitive benefits, supported by biomarker evidence of target and pathway engagement. Further evaluation of buntanetap in this patient population are warranted. A Phase 3 trial is currently underway to confirm these findings (NCT06709014).
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder, affecting over 7-million people in the United States alone and represents the most common cause of dementia in older adults1,2. AD, along with many other neurodegenerative diseases, is characterized by memory loss, cognitive decline, and changes in personality and behavior1,3. Currently available symptomatic treatments do not delay disease progression4, while recently approved monoclonal antibodies targeting amyloid, Leqembi and Kisunla, slow cognitive decline by 27–35% in 18 months5,6. However, despite recent progress in the field, AD continues to represent a significant unmet need.
In the past 10 years, studies have shown that multiple neurotoxic protein aggregates are found in the brains of people with AD7,8,9. Amyloid plaques and tau neurofibrillary tangles are clearly associated with disease10, but other misfolded proteins such as alpha-synuclein (αSYN) and TDP43 have also been found to be elevated in the brain of AD patients11,12,13,14. Although their exact role in AD is still not fully understood, current research supports that patients with multiple misfolded proteins beyond amyloid and tau may have faster disease progression14,15. Therefore, therapeutic strategies that target multiple neurotoxic proteins may potentially be more beneficial than those targeting a single neurotoxic protein.
The unique mechanism of action of buntanetap (also known as Posiphen) strongly positions it as a promising therapy for AD. Buntanetap is an orally bioavailable small molecule that inhibits the translation of several neurotoxic aggregating proteins, such as APP, Tau, αSYN, huntingtin, TDP43, and prion proteins16,17,18,19,20,21,22,23,24,25,26,27. In the Discover study run by the Alzheimer’s Disease Cooperative Study (ADCS), buntanetap demonstrated a dose-dependent statistically significant reduction in APP and Aβ40 synthesis as shown by Stable Isotope Labeling Kinetic (SILK) analysis that used 13C6-leucine to label all newly synthesized proteins22. This observation supports the mechanism of action that buntanetap is an inhibitor of APP synthesis in humans. Further, in multiple animal and human studies, buntanetap treatment effectively reduced APP and its downstream products, total Tau (t-Tau), phosphorylated Tau (pTau), and αSYN, as measured in mouse or rat brains and in patients’ CSF, further supporting its mechanism of action18,20,22,26,28,29,30,31.
Buntanetap has been tested in thirteen clinical trials with over 1000 dosed participants, including healthy volunteers and patients with AD and Parkinson’s disease (PD) and is generally safe and well-tolerated across studies. There have been no serious adverse events (SAEs) associated with buntanetap treatment in any of the clinical trials to date.
Buntanetap’s efficacy in treating both AD and PD patients has been confirmed in three studies. In a Phase 1b study in early AD patients and a Phase 2a study, which included participants with early AD and early PD, participants were treated with buntanetap or placebo once a day (QD) for 25 days28. While both studies were not powered for efficacy, buntanetap (60 and 80 mg) demonstrated improvements in cognition (Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog11)) and speed (Wechsler Adult Intelligence Scale Coding (WAIS coding)) in AD patients (80 mg vs placebo ADAS-Cog11 −4.4 ± 2.04 vs −1.1 ± 2.63; 60 + 80 mg vs placebo ADAS-Cog11-3.93 ± 1.58 vs −1.41 ± 2.04; in both groups, buntanetap treatment statistically improved ADAS-Cog11 compared with baseline). In the same Phase 2a study, PD patients were given from 5 to 80 mg of buntanetap QD. Amongst them, participants treated with 10 and 20 mg showed the strongest statistically significant improvements in mobility (Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS)) and speed (WAIS coding), outperforming the 80 mg treatment group. In the recently completed Phase 2/3 study, 20 mg buntanetap improved PD patients’ cognition (data in preparation). The comparable pharmacokinetics (PK) of buntanetap in AD and PD patients suggested the need to further explore its dose range for the treatment of AD.
The current study builds upon our earlier studies and was designed to examine the safety and efficacy of three different doses of buntanetap (7.5, 15, and 30 mg) compared to placebo in participants with mild and moderate AD in 3 months (NCT05686044).
Results
Participant disposition and demographics
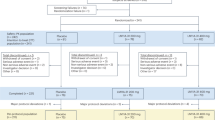
A total of 701 participants with mild to moderate AD were screened for study participation in 54 sites across the United States. Overall, 351 participants were enrolled in the study across the buntanetap and placebo cohorts. The study randomized 88, 87, and 87 participants across the three buntanetap dosage groups of 7.5 mg, 15 mg, and 30 mg, respectively, and 89 participants to placebo, where 95.5%, 92.0%, and 90.8% of participants from the respective buntanetap groups and 92.1% from the placebo group completed the 12-week study (Fig. 1). The primary reason for discontinuation across all cohorts was similar—withdraw by participant (n = 6 (2.3%) in all buntanetap treatment groups combined; n = 2 (2.2%) in placebo group). Study discontinuation due to adverse events (AEs) was 1.9% of buntanetap recipients (n = 5 (1.9%) total; n = 1 for 15 mg, n = 4 for 30 mg). All reasons for discontinuation and the participant flow from screening through study completion for each treatment group are illustrated in Fig. 1.

aInclusion criterion # 3 required Mini Mental State Examination [MMSE] score 14–24 at Baseline; Exclusion criterion #17 excluded participants taking strong and moderate CYP3A4 inhibitors and/or inducer; participant was receiving Atorvastatin; Exclusion criterion #7 excluded participants with clinically significant renal (Chronic Kidney Disease-Epidemiology Collaboration [CKD-EPI] <60 mL/min/BSA [body surface area]) or hepatic impairment (alkaline phosphatase [ALP] > 2.0× upper limit of normal [ULN] and/or total bilirubin >2.0× ULN).
Population demographics were balanced across treatment groups and there were no significant differences in age, race, or BMI. Mean age was 73.6 years for all participants and ranged from 73.1 to 74.7 across treatment groups. Between 50.6 and 65.5% of participants were female. Racial and ethnic minority groups comprised more than 41% of study participants (for demographic data see Supplementary Table 1).
More than 99% of all participants received concomitant medications over the course of the study. The most reported concomitant medication classification was psychoanaleptics reported by 84.4% of participants across all treatment groups. Among those, the most reported medications were anticholinesterase inhibitors (51.7% of participants).
Buntanetap had a favorable safety profile in participants with mild and moderate AD
Safety was a primary study objective, and it was measured via AEs, concomitant medication monitoring, 12-lead ECGs, clinical laboratory testing, vital signs assessments, physical and neurological examinations, and the Columbia-Suicide Severity Rating Scale (C-SSRS). The safety population totaled 346 participants who received at least one dose of study drug.
Buntanetap had a favorable safety profile and was well-tolerated across all dosage groups. Treatment-emergent AEs (TEAEs) were experienced by 33.3% (86/262) of buntanetap recipients across all three doses combined and 26.1% (23/88) of those receiving placebo. TEAEs related to study drug were 8.9% (23/258) of buntanetap recipients across all three doses combined and 3.4% (3/88) for placebo. While the percentage of TEAEs related to study drug were numerically higher for the buntanetap doses, they were not statistically different than placebo, nor was there a dose-dependent relationship between the number of related TEAEs (10.5%, 9.4%, and 6.9% in 7.5 mg, 15 mg, and 30 mg, respectively). Three 30 mg buntanetap recipients (3/87, 3.4%) and three placebo recipients (3/88, 3.4%) experienced serious TEAE, but there were no serious TEAEs related to study drug in any treatment group. In assessing severity, all TEAEs across all study groups were mild and moderate (mild range: 19.5% to 24.4%; moderate range: 5.7% to 13.8%), and there were no severe TEAEs. The most frequent TEAEs (>5%) occurred in the system organ class of Infections and Infestations (e.g., urinary tract infection, COVID-19, etc.) with 10.1% (26/258) of buntanetap recipients across all three doses combined and 6.8% (6/88) in placebo, and in Nervous System Disorders (e.g., dizziness, headache, etc.) with 5.8% (17/258) of buntanetap recipients and 5.7% (5/88) in placebo (for AE summary data and TEAEs by system organ class see Supplementary Tables 2 and 3). For drug related TEAE reported in ≥ 2 participants in any individual group were dizziness (5/346, 1.4% in all buntanetap treatment group combined vs 1/88, 1.1% in placebo group), headache (3/346, 0.9% in all buntanetap treatment group combined vs 0/88, 0% in placebo group), and nausea (2/346, 0.6% in all buntanetap treatment group combined vs 0/88, 0% in placebo group).
One of the most significant genetic risk factors for developing AD is APOE432,33,34,35,36,37. Safety data was further stratified by APOE4 carrier status. There was no increased severity of AEs among APOE4 carriers. All TEAEs were categorized as either mild or moderate in nature, same was the case for non-carriers. Among APOE4 carriers, 42.1% of buntanetap recipients across all three doses combined had TEAEs compared to 34.2% placebo; participants with serious TEAEs were 0.8% on buntanetap across all three doses combined and 7.9% on placebo. No serious TEAEs were related to the study drug in APOE4 carriers or non-carriers (for AE summary data by APOE4 carrier status see Supplementary Tables 4 and 5).
Primary analysis results
The primary study hypothesis was that buntanetap treatment (pooling the 3 treatment arms together, the intent-to-treat (ITT) population) is superior to placebo in reducing the cognitive impairment and functional impairment in participants with mild to moderate AD, as assessed by the 2 co-primary endpoints (ADAS-Cog11 change from baseline to 12 weeks and Alzheimer’s Disease Study- Clinical Global Impression of Change (ADCS-CGIC) score at Week 12). For study power considerations, all three active treatment groups were combined and compared to the control for the primary and key secondary analyses.
In the ITT population, both buntanetap and placebo groups improved to the same level in ADAS-Cog11. Similar improvement was also observed in both groups in ADCS-CGIC and ADCS-ADL (Table 1). Because primary endpoints were not met, the remaining analysis were all considered exploratory.
Buntanetap improved cognition in biomarker positive (pTau217) mild AD participants (Post-hoc Analysis Results)
In this study, participants were enrolled based on the NIA-AA 2011 AD diagnosis criteria38,39, and neither CSF/plasma biomarker nor PET imaging was used for patient inclusion/exclusion criteria to confirm the AD diagnosis. Recent studies have shown that both CSF and plasma pTau217 are reliable indicators of tau and amyloid pathology40,41. During the conduct of this trial, diagnostic plasma biomarker testing for pTau217 became commercially available. Therefore, implementation of this biomarker to confirm AD pathology, as indicated by a pTau217/t-Tau ratio, was conducted prior to database lock and revealed that approximately 38% of participants did not have amyloid burden, thus not fulfilling the biological definition of AD. Only 62.1% (n = 216) of participants were identified to have AD pathology. These biomarker positive patients were evenly distributed in all dose groups with comparable baseline ADAS-Cog11 (Supplementary Table 6).
Subgroup analyses based on biomarker positive participants (pTau217/t-Tau ≥4.2%) were further stratified by MMSE score; mild AD was represented by MMSE scores of 21–24 (n = 95) and moderate AD was represented by MMSE scores of 14–20 (n = 115) with 6 participants whose baseline MMSE score fell out of 14–24 range and hence were not included. In moderate AD participants, ADAS-Cog11 did not show a treatment-related change from baseline (Fig. 2a). However, in mild AD participants, a dose-dependent response was observed, and buntanetap demonstrated nominally statistically significant improvement compared to baseline (Fig. 2b). The improvement was already observed after 6 weeks’ treatment although not statistically significant at that time. By 12 weeks, the placebo effect appeared to diminish in the placebo group, while the treatment group continued to show improvement. (Supplementary Fig. 1). Subgroup analysis of ADCS-CGIC did not demonstrate treatment-related improvement compared to placebo (Fig. 2c, d).

a ADAS-Cog11 results for moderate AD participants; b ADAS-Cog11 results for mild AD participants; c ADCS-CGIC results for moderate AD participants; d ADCS-CGIC results for mild AD participants. The ADAS-Cog11 and ADCS-CGIC endpoints were analyzed using mixed models for repeated measures. Error bars represent standard error.
Buntanetap had similar treatment effects across different demographics in mild AD, including in both APOE4 carriers and non-carriers (Post-hoc analysis results)
Further subgroup analyses comparing 30 mg buntanetap to placebo are presented in the forest plot (Fig. 3). These analyses evaluate buntanetap’s effects across various participant subgroups, including age, BMI, ethnicity, gender, race, and concomitant medications. Encouragingly, buntanetap demonstrated a consistent favorable effect over placebo across all groups.

Forest plot of change from baseline to week 12 for ADAS-Cog11 in Biomarker Positive, Mild AD patients (MMSE 21-24).
Buntanetap demonstrated a trend of reducing neurotoxic proteins and inflammation and increasing axonal integrity (Post-hoc analysis results)
Several plasma biomarkers (Tau, TDP43, IL5, IL6, S100A12, IFN-γ, IGF1R, GFAP, and NFL) were examined in biomarker-positive mild AD patients (MMSE 21-24) and in all biomarker-positive AD patients (MMSE 14-24) to assess buntanetap’s target and pathway engagement (Fig. 4).

a–i Biomarker-positive mild AD patients (MMSE 21-24); j–r all biomarker-positive AD patients (MMSE 14-24). The error bar represents standard error.
To test buntanetap’s effects on neurotoxic aggregating proteins, we tested plasma t-Tau and TDP43 (Fig. 4a, b). In mild AD patients, plasma t-Tau levels increased in placebo after 12 weeks compared to baseline, while the 30 mg buntanetap group remained relatively unchanged, indicating a potential reduction of Tau.
Although TDP43 is mostly involved in amyotrophic lateral sclerosis, frontotemporal lobar degeneration, and limbic-predominant age-related TDP-43 encephalopathy, TDP43 inclusions have been found in up to 57% of AD cases14. Currently, there is no established method to detect brain-derived TDP43. We used Quanterix assay to detect plasma TDP43, which recognizes both full-length and pathologically truncated forms of the protein. Interestingly, plasma TDP43 levels had a slight increase in the placebo group while it was profoundly reduced in the 30 mg buntanetap group.
Since there is no AD specific inflammatory marker, plasma IL5, IL6, S100A12, IFG-γ, IGF1R, and GFAP were tested (Fig. 4c–h).
Pro-inflammatory pathways, including the interleukin 6 (IL6) pathway, are well documented to play a role in AD42. Genetic predisposition to higher circulating levels of IL6 was reported to be associated with an increased risk for AD43. In animal models, reducing IL6 led to alleviated amyloid burden, attenuated inflammation, and improved cognition44. Although less studied, research has also shown IL5 involvement in neurodegenerative diseases45. In mild AD patients, interestingly placebo group showed a slight decrease in IL6, while the 30 mg group demonstrated approximately three times greater reduction. For IL5, both groups exhibited elevated levels, but the increase in the placebo group was almost twice that observed in the 30 mg group.
S100A12, also known as calgranulin C, is a protein that belongs to the S100 family of calcium-binding proteins46. It plays a role in several cellular processes, including inflammation, and is reported as a potential inflammatory biomarker for AD. A sizable increase was detected in placebo group after 3 months, suggesting elevated inflammation. In contrast, there was almost no change in the 30 mg buntanetap group compared to baseline.
Interferon gamma (IFN- γ) is a soluble cytokine that plays an important role in inducing and modulating an array of immune responses. In the AD mouse model, IFN- γ was shown to enhance amyloid deposition and suppress microglial degradation of amyloid47. In this study, IFN- γ levels increased in the placebo group while reduced in the 30 mg treatment group.
Insulin-like growth factor 1 receptor (IGF1R) is a receptor tyrosine kinase. In AD animal models, inhibiting this pathway was shown to attenuate insoluble amyloid48. Reduction in IGF1R protein also improved cognitive performance in AD mice. Similar to IFN- γ, its level increased in the placebo group and reduced in the 30 mg treatment group.
Plasma levels of GFAP have been suggested to show a robust association with AD pathophysiology, especially in the early stage49,50. In this study, plasma GFAP levels increased in both the treatment and placebo groups, and no treatment effect was detected.
Lastly, neurofilament light (NFL) was tested as a marker of neuronal damage. It has been reported in multiple studies to be elevated in various neurodegenerative diseases, including AD51,52. As expected, plasma NFL levels increased in the placebo group over time. Notably, in the 30 mg buntanetap group, plasma NFL levels showed a decrease at 12 weeks compared to baseline, indicating a potential reduction of neuronal injury (Fig. 4i).
Similar trends across all the aforementioned biomarkers were observed in biomarker-positive AD patients (MMSE 14–24) (Fig. 4j–r).
Overall, biomarker results showed that buntanetap treatment (30 mg) decreased plasma t-Tau, TDP43, IL5, IL6, S100A12, IFN- γ, IGF1R, and NFL.
Discussion
Results of this dose ranging study showed that buntanetap had a favorable safety profile in participants with mild and moderate AD. In the ITT population, buntanetap did not meet the primary nor co-primary endpoints in AD patients over placebo. However, post-hoc analyses showed nominally statistically significant, dose-dependent improvement in cognition as measured by ADAS-Cog11 compared to baseline in biomarker-positive mild AD patients.
Identifying the proper patient population is imperative for clinical trials, and that was evidenced in this study, where ~40% of participants, despite meeting inclusion/exclusion criteria, did not fulfill the biological definition of AD. This data is consistent with reports that showed that 30–40% clinically diagnosed AD patients do not have amyloid pathology53. Moreover, each group has rather balanced numbers of biomarker-positive patients, which further supports the discrepancy was mostly due to the misdiagnosis. This further emphasizes the importance of biomarkers in patient identification and stratification.
Recent discovery of plasma biomarker (pTau217) as a diagnostic tool for AD54 allowed for post-hoc stratification of the population to focus solely on patients with confirmed disease. While acknowledging its limitations, we conducted post-hoc analysis in this mild AD group. Encouragingly, buntanetap showed a treatment effect across all subgroups (age, BMI, gender, ethnicity, APOE4 carrier status, concomitant medication) regardless of baseline characteristics in mild AD patients. This is consistent with the results from our previous studies. In our Phase 1b Discover study and Phase 2a AD/PD study, despite both studies not powered for efficacy, buntanetap treatment showed improvement in early AD patients’ ADAS-Cog28. We are validating these findings in our ongoing 760-patient 18-month pivotal study in early AD.
A treatment effect in ADCS-CGIC was not detected in mild AD patients, which most likely was due to the limited number of patients. Because of the subjective nature of the scale, a larger number of patients would be needed to detect any meaningful difference. In our original power analysis, we specified that we would pool all three arms of buntanetap treatment group (N = 240) in order to detect a 0.22-point difference with a standard deviation of 0.5. Proper power was ensured in the ongoing early AD study.
Consistent with what we have observed before, buntanetap has a favorable safety profile and is well-tolerated in mild to moderate AD patients. One of the leading genetic risk factors for developing AD is APOE434,35. APOE4 carriers are prone to AD with over 90% probability in homozygous population36. Further, APOE4 homozygotes are at much higher risk for developing amyloid-related imaging abnormalities (ARIA), such as swelling (ARIA-E) and microhemorrhages (ARIA-H), from currently available monoclonal antibodies6,55,56. In our study, buntanetap treatment showed a remarkably similar safety profile and efficacy in all AD patients despite their APOE4 status, positioning it as a promising treatment for this highly vulnerable population.
Due to buntanetap’s unique mechanism of actions, standard of care symptomatic AD drugs were allowed in this study, and more than 84% of all participants took psychoanaleptics (Ache inhibitors or other anti-dementia drugs). Buntanetap’s excellent safety profile supports its use both as a standalone treatment and in combination with other symptomatic therapies. This is especially important so patients can continue their existing medications while adding buntanetap, without the need to discontinue either. In our future studies, it will be of interest to test buntanetap treatment effects together with the recently approved amyloid monoclonal antibodies.
Several plasma biomarkers were tested to demonstrate target and pathway engagement. First, we tested the levels of neurotoxic aggregating proteins. Current standard for AD diagnosis and staging use of amyloid and Tau as the core AD biomarkers defining AD, while the TDP-43 and synuclein reflect common concurrent pathologies or co-morbidities.
Due to the limitations of plasma biomarkers, we were not able to measure plasma APP, although our previous Phase 2a study showed that buntanetap reduced APP levels in CSF28. Because of the short duration of the study, plasma Aβ42/40 ratio did not change in either treatment or placebo group. Plasma Aβ42/40 ratio has been used as a surrogate biomarker of cortical amyloid deposition but not as a prognostic biomarker. However, we observed a reduction of t-Tau in plasma after treatment with buntanetap. Higher plasma t-Tau was reported to be associated with AD dementia, and higher CSF Tau, but the correlations were weak57. A recent study highlighted a novel biomarker, brain-derived Tau (BD Tau), that can be useful in evaluating the AD-dependent neurodegenerative processes58, and we plan to evaluate BD Tau in the future study. TDP43 is another marker that has been implicated in AD pathology and is linked with more severe cognitive impairment and is particularly prevalent in APOE4 carriers14. Although the relationship between plasma TDP43 and brain TDP43 aggregates is not yet clear, our data indicate that buntanetap reduces plasma TDP43. Further studies will be needed to fully understand the correlation of plasma TDP43 with AD progression.
Furthermore, we evaluated six inflammatory biomarkers associated with Alzheimer’s disease (IL-5, IL-6, S100A12, IFN-γ, IGF1R, and GFAP). Encouragingly, the 30 mg treatment group exhibited a consistent reduction across all markers except GFAP, indicating a potential anti-inflammatory effect. These data suggest a reduction inflammation, a hallmark of AD.
Lastly, we tested NFL, a well-established biomarker for neuronal health51,52. Buntanetap treatment showed a trend of decrease in NFL, suggesting a reduction of neuronal damage.
These results are consistent with what we have observed before. In the Discover study, we reported a real-time decline in CSF of newly synthesized APP and Aβ4022. In our Phase 2a study, we further saw a reduction in CSF of multiple neurotoxic proteins, a lowering of multiple inflammatory factors, and preservation of neuronal synaptic functions28. These biomarker data supported buntanetap’s mechanism of action and pathway engagement as a translational inhibitor of neurotoxic proteins.
Importantly, we also observed the same trend in the plasma of all biomarker-positive AD patients, not just mild AD patients, further validating the drug’s mechanism of action. While buntanetap treatment showed a reduction in inflammatory factors in moderate AD patients, it did not show clinical efficacy in this patient population in this study, which could be due to the short duration of the study. We plan to further investigate this in future studies.
As previously noted, this study was not powered to detect the treatment effect of individual doses compared to placebo on the functional outcome measure ADCS-CGIC. Furthermore, the absence of biomarker confirmation for Alzheimer’s disease pathology resulted in approximately 40% of enrolled participants likely not having AD. Our analysis focused on a post hoc subgroup of patients with confirmed or probable AD; however, these analyses were not prespecified and were not adjusted for multiplicity, despite the fact that baseline characteristics within this subgroup were generally well balanced. In our current Phase 3 study, we have addressed these limitations by incorporating biomarker-based patient selection, conducting appropriate power calculations, and applying rigorous, prespecified statistical analyses to ensure robust and interpretable results.
Overall, in the biomarker positive mild AD patients, buntanetap showed promising results in improving patients’ cognition. Because of its unique mechanisms of action, it has the potential to improve cognitive function while also modifying the underlying disease pathology. Therefore, we are validating the symptomatic findings from this study as well as testing its potential disease modifying effects in 760 early AD patients in a large Phase 3 study, which is currently ongoing. It is a dual randomized, placebo-controlled, multi-center study in biomarker-confirmed patients with early AD with 6-month and 18-month data read-outs (NCT06709014).
Methods
Investigational drug
Buntanetap, (3aR)-1, 3a, 8-trimethyl-1, 2, 3, 3a, 8, 8a-hexahydropyrrolo (2, 3-b) indol-5-yl phenylcarbamate tartrate (investigational new drug #72,654), was manufactured according to Good Manufacturing Practice (GMP) regulations (Wilmington PharmaTech, Newark, DE). The investigational drug product and matching placebo, containing a standard pharmaceutical excipient, were provided as an immediate release solid oral dosage form. They were prepared in hard capsule shells and manufactured in accordance with the GMP regulations by Frontida BioPharm (Philadelphia, PA).
Participants
The following inclusion criteria were applied: (1) diagnosis of AD according to NIA and NIA-AA criteria for probable AD; (2) individuals between 55 and 85 years old; (3) MMSE 14-24; (4) have a study partner who will provide written informed consent to participate, is in frequent contact with the participant (at least 10 h per week) and will accompany the participant to study visits; (5) female participants of childbearing potential must have a negative urine pregnancy test, must be non-lactating, and must agree to use a highly effective method of contraception during the trial and for 4 weeks after the last dose of trial treatment; non-childbearing potential includes surgically sterilized or postmenopausal with no menstrual bleeding for at least one year; (6) male participants must be sterile, sexually inactive, or agree not to father a child during the study and one month after the last dose of study medication, and must agree to use a barrier method for contraception; female partners of male participants must adopt a highly effective method of contraception; (7) participants can provide written informed consent; (8) no evidence of current suicidal ideation or previous suicide attempt in the past two months as evaluated by the Columbia Suicide Severity Rating Scale nor suicidal behavior in the past six months; (9) stability of permitted medications for at least 4 weeks prior to screening; (10) adequate visual and hearing ability (physical ability to perform all study assessments); (11) good general health with no disease expected to interfere with the study. Please see Supplementary Method 1 for exclusion criteria. Written informed consent was obtained from all participants and their study partners, and the study protocol was approved by the central Institutional Review Boards Advarra (original approval on January 23, 2023).
Randomization
Participants who signed an informed consent and met screening eligibility requirements were randomly assigned to the active (3 buntanetap dose levels: 7.5 mg, 15 mg, and 30 mg) and placebo treatment groups in a 1:1:1:1 ratio using the Rave RTSM system via permuted block randomization. This was a quadruple (participants, care providers, investigators, and outcomes assessors) blinded study.
Trial design
A sample of 320 patients with mild and moderate AD was targeted for 12 weeks of treatment. All patients consented to voluntarily participate in the clinical trial. The sample size was determined to assess the efficacy and safety of buntanetap when pooling all three active treatment arms together. Adequately characterizing PK and supporting potential dose proportionality analyses, along with plasma biomarkers focused on buntanetap’s mechanism of action and pathway engagement, were also explored in this study.
Participants were treated at home with 7.5 mg, 15 mg, or 30 mg buntanetap QD or placebo for 12 weeks. The co-primary (ADAS-Cog 11 & ADCS-CGIC) and the key secondary (ADCS-ADL) efficacy endpoints were assessed at baseline before treatment, at 6 weeks, and after 12 weeks treatment. Blood samples for PK and biomarkers were collected at the baseline and end-of-trial visits before treatment and for 4 h after treatment.
Biomarker assays
The pTau217 and t-Tau peptide concentrations were measured in plasma samples using the immunoprecipitation-mass spectrometry (IP/MS) workflow by C2N. Two separate measurements were performed on each sample, one that measured a pTau217-specific peptide and one for a t-Tau-specific peptide. Both peptides were measured from the same sample and analyzed in a 96-well plate format on Waters Acquity M-Class liquid chromatography (LC) units interfaced to Thermo Fusion Lumos Tribrid mass spectrometers (MS/MS)59. The pTau217 protein was enriched through immunoprecipitation with a tau-specific antibody. LC-MS/MS was used to separate, identify, and quantify the concentrations of tau peptides (tau amino acids 212–221) phosphorylated at Thr-217 (pTau217) or not phosphorylated at Thr-217 in human plasma. Plasma aliquots were combined where necessary, and the final sample volumes were prepared using a Hamilton STAR® liquid handler and a Kingfisher Flex device.
Plasma biomarkers Neurofilament -Light (NFL) and Glial Fibrillary Acidic Protein (GFAP), and TDP-43 were tested at Quanterix, (Billerica, MA, USA). NFL and GFAP were tested using their Simoa® Neurology 2-Plex B kit (N2PB). Commercially available Simoa® Neurology 2-Plex B kits were used according to manufacturer’s instructions. The assays were performed on the Simoa HD-X analyzer using Single Molecule Array (Simoa) technology60. Plasma samples were diluted at 4× and ran in duplicate. The mean of replicates for each sample was calculated, and results were included in the analysis if the coefficient of variation across replicates was <25%. All measurements were conducted on the automated HD-X analyzers (Quanterix, Billerica, MA, USA). For plasma TDP-43 levels, samples (n = 501) from human patients were diluted 4× and assayed in duplicate. The mean of the replicates for each sample was calculated, and results were included in the analysis if the coefficient of variation (%CV) across replicates was <25%.
Plasma IL5 and IL6 were tested by DiamiR Biosciences. The assays were performed using the Ella automated immunoassay platform (Bio-Techne), which operated without manual intervention once the run was initiated. The system interfaced pneumatically with microfluidic Simple Plex™ cartridges, which were pre-loaded with validated reagents, including matched antibody pairs, thereby streamlining assay setup and eliminating the need for standard curve preparation. For each assay, 50 μL of diluted sample and wash buffer were added to the designated wells of the cartridge, following the manufacturer’s instructions. The cartridge was then inserted into the Ella instrument for fully automated processing, including reagent handling, washing, and incubation under tightly controlled conditions. Each cartridge contained multiple fluidic channels housing three glass nanoreactors (GNRs) per analyte, each pre-coated with capture antibodies, enabling triplicate measurements for IL5 and IL6. During analysis, the sample flowed through the microfluidic channels, where target analytes were captured and detected using a pre-loaded detection reagent. Unbound components were washed away, and signal generation occurred within the GNRs. Quantification was achieved using pre-calibrated standard curves embedded on each cartridge, providing fully analyzed results in under 90 min.
Plasma IFN- γ, IGF-1R, S100A12 were tested by Dr. Laurie Sander’s lab in Duke University. Levels of cytokines and/or receptors were measured in plasma samples by a sandwich enzyme-linked immunosorbent assay (ELISA). To quantify IFN-γ levels, the Human High Sensitivity ELISA kit was used (Abcam, ab46048). The Human ELISA Kit was used for IGF-1 R (RayBiotech, ELH-IGF1R), and the Human ELISA kit was used for S100A12 (Abcam, ab282299). A standard curve was performed and plotted for each analyte and plate. All determinations were performed according to the manufacturer’s instructions. Assay sensitivity was 69 pg/mL, 6 pg/mL, 1.89 pg/mL for IFN-γ, IGF-1 R and S100A12, respectively.
Pharmacokinetics (PK)
To determine the concentration of buntanetap in human plasma, a high-performance liquid chromatographic mass spectrometric detection method was validated at Charles River. The method was later transferred to Sannova Analytical LLC, where partial validation and sample analysis were performed. Deuterated buntanetap was used as the internal standard to ensure accurate concentration assessments. The bioanalytical method was performed in accordance with current FDA, Industry Guidelines, and OECD Principles. The determination of buntanetap in human plasma was performed using an assay range of 9.989–24972.908 pg/mL. The sample analysis was conducted in accordance with current GCP and GLP principles, and the results were presented for PK profiling.
Sample size
Sample size was determined to provide adequate statistical power for the primary efficacy endpoint, ADAS Cog11. Assuming a two-sidedαof0.05, a mean difference of2.7points between pooled buntanetap and placebo groups, and a commonSDof6.0, a total of 280participants (210active,70placebo) were required to achieve 90% power. For the co-primary endpoint ADCS CGIC, this sample size provided 89% power to detect a 0.23-point difference. Allowing for an anticipated 12.5% dropout, approximately 320 participants (80 per arm) were planned for enrollment using a 1:1:1:1 randomization design.
Statistical analysis
Statistical analyses were performed using SAS version 9.4. The co-primary endpoints are the change from baseline in ADAS-Cog11 scores and ADCS-CGIC scores at 12 weeks. ADAS-Cog11 was analyzed via mixed models for repeated measures (MMRM). The ADAS-Cog11 model included treatment, timepoint, and treatment-by-timepoint interaction as the fixed effects, baseline ADAS-Cog11 as the covariate, and participant as a random effect. The ADCS-CGIC model included treatment, timepoint, treatment-by-timepoint interaction as the fixed effects, and participant as a random effect. An unstructured covariance matrix was used, and the Kenward-Roger approximation was used to adjust the denominator degrees of freedom. Missing data were assumed to be missing at random for the primary analyses. To assess the robustness of this MAR assumption, a conservative control-based multiple imputation method based on missing not at random (MNAR) assumption was applied to impute missing data arising from intercurrent events for the two co-primary endpoints analyses. Similar models were used for subgroup analyses. The change from baseline and the differences in the change from baseline between treatments were based on the Least Square Means from this model. The two-sided 95% confidence intervals of the treatment differences were reported.PK endpoints were based on the assessments of buntanetap levels in plasma. Samples were collected at pre-dosing (0 h) and at 1, 2, and 4 h for baseline and end-of-trial visits.
Data availability
Study results and study protocol have been published on CT.gov (NCT05686044), including overall study design, baseline characteristics, outcome measures, and adverse events. Raw data cannot be shared until the company receives a New Drug Application for buntanetap in treating Alzheimer’s disease.
References
2025 Alzheimer’s disease facts and figures. Alzheimers Dement. 21, 1474043 (2025).
Plassman, B. L. et al. Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 29, 125–132 (2007).
Safiri, S. et al. Alzheimer’s disease: a comprehensive review of epidemiology, risk factors, symptoms diagnosis, management, caregiving, advanced treatments and associated challenges. Front. Med. 11, 1474043 (2024).
Doody, R. S., Geldmacher, D. S., Gordon, B., Perdomo, C. A. & Pratt, R. D. Open-label, multicenter, phase 3 extension study of the safety and efficacy of donepezil in patients with Alzheimer disease. Arch. Neurol. 58, 427 (2001).
Sims, J. R. et al. Donanemab in early symptomatic Alzheimer disease. JAMA 330, 512 (2023).
van Dyck, C. H. et al. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 388, 9–21 (2023).
Petersen, R. C. How early can we diagnose Alzheimer disease (and is it sufficient)? Neurology 91, 395–402 (2018).
Wu, J., Wu, J., Chen, T., Cai, J. & Ren, R. Protein aggregation and its affecting mechanisms in neurodegenerative diseases. Neurochem. Int. 180, 105880 (2024).
Moda, F. Secondary protein aggregates in neurodegenerative diseases: almost the rule rather than the exception. Front. Biosci. Landmark 28, 255 (2023).
Chen, X.-Q. & Mobley, W. C. Alzheimeras disease pathogenesis: insights from molecular and cellular biology studies of oligomeric Aá and Tau species. Front. Neurosci 13, 659 (2019).
Twohig, D. & Nielsen, H. M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 14, 23 (2019).
Tosun, D. et al. Identifying individuals with non-Alzheimer’s disease co-pathologies: A precision medicine approach to clinical trials in sporadic Alzheimer’s disease. Alzheimers Dement. 20, 421–436 (2024).
Higashi, S. et al. Concurrence of TDP-43, tau and α-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 1184, 284–294 (2007).
Meneses, A. et al. TDP-43 pathology in Alzheimer’s disease. Mol. Neurodegener. 16, 84 (2021).
Thomas, D. X. et al. Association of TDP-43 proteinopathy, cerebral amyloid angiopathy, and Lewy bodies with cognitive impairment in individuals with or without Alzheimer’s disease neuropathology. Sci. Rep. 10, 14579 (2020).
Chen, X. Q. et al. Posiphen reduces the levels of huntingtin protein through translation suppression. Pharmaceutics 13, 2109 (2021).
Cho, H. H. et al. Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J. Biol. Chem. 285, 31217–31232 (2010).
Kuo, Y.-M., Nussbaum, R. L., Rogers, J. & Maccecchini, M. L. Translational inhibition of α-synuclein by Posiphen normalizes distal colon motility in transgenic Parkinson mice. Am. J. Neurodegener. Dis. 8, 1–15 (2019).
Rogers, J. T., Xia, N., Wong, A., Bakshi, R. & Cahill, C. M. Targeting the iron-response elements of the mRNAs for the Alzheimer's amyloid precursor protein and ferritin to treat acute lead and manganese neurotoxicity. Int. J. Mol. Sci 20, 994 (2019).
Teich, A. F. et al. Translational inhibition of APP by posiphen: efficacy, pharmacodynamics, and pharmacokinetics in the APP/PS1 mouse. Alzheimers Dement. Transl. Res. Clin. Interv. 4, 37–45 (2018).
Yu, Q.-S. et al. Synthesis of the Alzheimer drug Posiphen into its primary metabolic products (+)-N1-norPosiphen, (+)-N8-norPosiphen and (+)-N1, N8-bisnorPosiphen, their inhibition of amyloid precursor protein, α-Synuclein synthesis, interleukin-1β release, and cholinergic action. Antiinflamm. Antiallergy Agents Med. Chem. 12, 117–128 (2013).
Galasko, D. et al. A multicenter, randomized, double-blind, placebo-controlled ascending dose study to evaluate the safety, tolerability, pharmacokinetics (PK) and pharmacodynamic (PD) effects of Posiphen in subjects with early Alzheimer’s Disease. Alzheimers Res. Ther. 16, (2024).
Mikkilineni, S. et al. The anticholinesterase phenserine and its enantiomer posiphen as 5′untranslated-region-directed translation blockers of the Parkinson’s alpha synuclein expression. Parkinsons Dis. https://doi.org/10.1155/2012/142372 (2012).
Rogers, J. T. et al. The alpha-synuclein 5’untranslated region targeted translation blockers: anti-alpha synuclein efficacy of cardiac glycosides and Posiphen. J. Neural Transm. 118, 493–507 (2011).
Cahill, C. M., Lahiri, D. K., Huang, X. & Rogers, J. T. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim. Biophys. Acta Gen. Subj. 1790, 615–628 (2009).
Bandyopadhyay, S. et al. Novel 5′ untranslated region directed blockers of iron-regulatory protein-1 dependent amyloid precursor protein translation: implications for Down syndrome and Alzheimer’s disease. PLoS ONE 8, e65978 (2013).
Lahiri, D. K. et al. The experimental Alzheimer’s Disease drug posiphen [(+)-phenserine] lowers amyloid-β peptide levels in cell culture and mice. J. Pharmacol. Exp. Ther. 320, 386–396 (2007).
Fang, C. et al. Buntanetap, a novel translational inhibitor of multiple neurotoxic proteins, proves to be safe and promising in both Alzheimer’s and Parkinson’s patients. J. Prev. Alzheimers. Dis. 10, 25–33 (2022).
Maccecchini, M. L. et al. Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-β peptide and τ levels: target engagement, tolerability and pharmacokinetics in humans. J. Neurol. Neurosurg. Psychiatry 83, 894–902 (2012).
Chen, X. Q. et al. Targeting increased levels of APP in Down syndrome: Posiphen-mediated reductions in APP and its products reverse endosomal phenotypes in the Ts65Dn mouse model. Alzheimers Dement. 17, 271–292 (2021).
Chen, X.-Q. A pilot exploration with Posiphen to normalize amyloid precursor protein in Down syndrome. Neural Regen. Res. 16, 2420 (2021).
Farrer, L. A. & APOE and Alzheimer Disease Meta Analysis Consortium Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. JAMA 278, 1349–1356 (1997).
Corder, E. H. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science (1979) 261, 921–923 (1993).
Kim, J., Basak, J. M. & Holtzman, D. M. The role of apolipoprotein E in Alzheimer’s Disease. Neuron 63, 287–303 (2009).
Bertram, L., Lill, C. M. & Tanzi, R. E. The genetics of Alzheimer disease: back to the future. Neuron 68, 270–281 (2010).
Fortea, J. et al. APOE4 homozygosity represents a distinct genetic form of Alzheimer’s disease. Nat. Med. 30, 1284–1291 (2024).
Raulin, A.-C. et al. ApoE in Alzheimer’s disease: pathophysiology and therapeutic strategies. Mol. Neurodegener. 17, 72 (2022).
McKhann, G. M. et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269 (2011).
Jack, C. R. et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 257–262 (2011).
Ashton, N. J. et al. Diagnostic accuracy of a plasma phosphorylated Tau 217 immunoassay for Alzheimer disease pathology. JAMA Neurol. 81, 255 (2024).
Khalafi, M. et al. Diagnostic accuracy of phosphorylated tau217 in detecting Alzheimer’s disease pathology among cognitively impaired and unimpaired: a systematic review and meta-analysis. Alzheimers Dement. 21, e14458 (2025).
Lyra e Silva, N. M. et al. Pro-inflammatory interleukin-6 signaling links cognitive impairments and peripheral metabolic alterations in Alzheimer’s disease. Transl. Psychiatry 11, 251 (2021).
Charisis, S. et al. Genetic predisposition to high circulating levels of interleukin 6 and risk for Alzheimer’s disease. Discovery and replication. J. Prev. Alzheimers Dis. 12, 100018 (2025).
Liu, M. et al. Interleukin-6 deficiency reduces neuroinflammation by inhibiting the STAT3-cGAS-STING pathway in Alzheimer’s disease mice. J. Neuroinflammation 21, 282 (2024).
Wood, L. B. et al. Identification of neurotoxic cytokines by profiling Alzheimer’s disease tissues and neuron culture viability screening. Sci. Rep. 5, 16622 (2015).
Di Molfetta, G. et al. Inflammation biomarkers and Alzheimer’s disease: a pilot study using NULISAseq. Alzheimers Dement. 17, e70079 (2025).
Browne, T. C. et al. IFN-γ production by amyloid β–specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J. Immunol. 190, 2241–2251 (2013).
Westwood, A. J. et al. Insulin-like growth factor-1 and risk of Alzheimer dementia and brain atrophy. Neurology 82, 1613–1619 (2014).
Kim, K. Y., Shin, K. Y. & Chang, K.-A. GFAP as a potential biomarker for Alzheimer’s disease: a systematic review and meta-analysis. Cells 12, 1309 (2023).
Peretti, D. E. et al. Association of glial fibrillary acid protein, Alzheimer’s disease pathology and cognitive decline. Brain 147, 4094–4104 (2024).
Gaetani, L. et al. Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 90, 870–881 (2019).
Jung, Y. & Damoiseaux, J. S. The potential of blood neurofilament light as a marker of neurodegeneration for Alzheimer’s disease. Brain 147, 12–25 (2024).
Jack, C. R. et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14, 535–562 (2018).
Jack, C. R. et al. Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s Association Workgroup. Alzheimers Dement. 20, 5143–5169 (2024).
Villain, N. et al. Lecanemab for early Alzheimer’s disease: appropriate use recommendations from the French federation of memory clinics. J. Prev. Alzheimers Dis. 12, 100094 (2025).
Rabinovici, G. D. et al. Donanemab: appropriate use recommendations. J. Prev. Alzheimers Dis. 12, 100150 (2025).
Mattsson, N. et al. Plasma tau in Alzheimer disease. Neurology 87, 1827–1835 (2016).
Gonzalez-Ortiz, F. et al. Plasma brain-derived tau is an amyloid-associated neurodegeneration biomarker in Alzheimer’s disease. Nat. Commun. 15, 2908 (2024).
Eastwood, S. M. et al. PrecivityAD2TM blood test: analytical validation of an LC-MS/MS assay for quantifying plasma phospho-tau217 and non-phospho-tau217 peptide concentrations that are used with plasma amyloid-β42/40 in a multianalyte assay with algorithmic analysis for detecting brain amyloid pathology. Diagnostics 14, 1739 (2024).
Rissin, D. M. et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 28, 595–599 (2010).
Acknowledgements
We extend our heartfelt gratitude to all the patients and their families for their participation and trust in this study. We are also deeply appreciative of the clinical and research staff, whose commitment and hard work were essential to the successful execution of this project. We gratefully acknowledge the support provided by our vendors, and finally, we sincerely thank the whole Annovis team for their valuable collaboration and contributions.
Author information
Authors and Affiliations
Contributions
C.F. and M.M. designed the study. C.F. wrote the paper. D.F. and M.C. led the data analysis. MG led the study operation. D.L. was the medical monitor. L.H.S. and K.A.W.B. were PIs from DCRI. K.L., E.P., A.R., R.K., R.G., R.L., and K.J. were the site PIs participated in the study. Alidad M and L.H.S. provided biomarker analysis. Alex M assisted with editing. M.F. was the chair of the data monitoring committee.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fang, C., Feng, D., Gaines, M. et al. Buntanetap treatment in mild to moderate Alzheimer’s disease: phase 2/3 study. npj Dement. 2, 26 (2026). https://doi.org/10.1038/s44400-026-00073-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44400-026-00073-z
