
Abstract
Aqueous zinc-ion batteries (AZIBs) offer a combination of safety and low cost, but they suffer from dynamic pH fluctuations that drive parasitic reactions and capacity fading. In this review, we report the current mechanistic understanding of the dynamic pH evolution across the electrolyte-electrode interface and its role in triggering interphase instability. We summarize advanced strategies to regulate pH, including electrolyte engineering, electrode surface modification, and interphase construction. We further discuss current methods for probing pH dynamics and outline potential future approaches for gaining deeper mechanistic and design insights.
Similar content being viewed by others


Introduction
With the increasing demand for electricity in modern society and the rapid growth of renewable energy sources such as solar and wind, energy production is becoming increasingly variable and decentralized1,2. However, the intermittent nature of these sources often leads to a mismatch between energy generation and consumption. This necessitates the development of large-scale energy storage systems enabling the rapid integration of renewable energy sources with the electric grid safely and reliably3. In this context, AZIBs have recently emerged as one of the most promising systems for next-generation energy storage, attracting increasing research attention4,5,6.
Compared with commercial lithium-ion batteries (LIBs), AZIBs offer multiple intrinsic advantages owing to their use of water as the electrolyte solvent. First, the aqueous medium is inherently non-flammable, eliminating the risk of thermal runaway and significantly enhancing system safety7,8. Second, both water and water-soluble zinc salts are inexpensive and widely available, and zinc itself is more abundant in the Earth’s crust and less expensive compared to lithium9,10. Additionally, the low reactivity of aqueous electrolytes toward atmospheric moisture and oxygen enables battery fabrication under ambient conditions, eliminating the need for stringent dry-room or inert-atmosphere environments11. These factors collectively reduce the overall cost of the battery system. Aqueous electrolytes also exhibit markedly higher ionic conductivity ( ~ 0.1 S cm−1) than typical organic systems ( ~ 1–10 mS cm−1), which facilitates rapid ion transport, leading to superior rate performance and power density12. The above-mentioned key advantages and the AZIB schematic are depicted in Fig. 1. These combined merits position AZIBs as a promising platform for developing safe, cost-effective, and scalable energy storage systems.

Schematic and key advantages of AZIBs, alongside the growing number of publications and citations highlighting rising research interest.
Nevertheless, the intrinsic nature of aqueous electrolytes also introduces critical challenges. Under an electrochemical field, water molecules may undergo decomposition or participate in electrode conversion reactions, leading to the local generation of protons (H⁺) or hydroxide anions (OH-) based on the operating potential. These species dynamically change the local chemical environment at the interface, often giving rise to undesirable side reactions such as the formation of zinc hydroxide sulfate and the corrosion of the zinc anode13,14. Such parasitic processes severely compromise the reversibility, efficiency, and longevity of AZIBs, thus hindering their practical deployment. According to data retrieved from the Web of Science using “zinc aqueous battery” as the primary keyword and filtering for publications with “pH” appearing in the abstract, research activity highlighting pH-related phenomena in AZIBs has grown steadily over the past decade (Fig. 1). This upward trend reflects the community’s increasing recognition that electrolyte pH plays a decisive role in dictating interfacial reactions and long-term battery stability15,16,17. This growing focus reflects a broader recognition that spatiotemporal pH fluctuations are not merely a process intrinsic to the electrochemical process, but also active contributors to failure mechanisms such as anode degradation, passivation, and loss of coulombic efficiency.
Here, we systematically examine the spatiotemporal origin and evolution of pH in AZIBs, highlighting its dynamic nature and far-reaching impact on interfacial chemistry and electrochemical performance. We summarize recent advances in understanding how local pH environments arise and evolve during cycling, and how these variations contribute to parasitic reactions and instability at the electrode–electrolyte interface. Furthermore, we discuss emerging advanced experimental techniques capable of probing pH evolution with increasingly high spatial and temporal resolution. By elucidating the fundamental mechanisms underlying pH fluctuations, we aim to provide insights that can guide the rational design of next-generation electrolyte systems with improved reversibility, interfacial stability, and cycle life.
Spatiotemporal pH dynamics in AZIBs
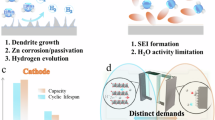
In simple terms, the working principle of AZIBs is straightforward: Zn is deposited and stripped at the anode, while at the cathode, Zn participates in intercalation or other conversion reactions. Zn ions migrate through the electrolyte, and electrons flow through the external circuit to deliver and store energy. However, under practical operating conditions, the electrochemical processes are far more complex and strongly influenced by local pH fluctuations, side reactions, and the dynamic between electrode and electrolyte interfacial chemistry. As illustrated in Fig. 2, using the widely studied Zn–Mn aqueous battery system as an example, Zn2+ ions in the electrolyte are typically coordinated with six H2O molecules. During Zn deposition at the anode, the standard electrode potential for the Zn2+/Zn couple is approximately –0.763 V versus the standard hydrogen electrode (SHE, at pH = 0), whereas the hydrogen evolution reaction (HER, 2H+ + 2e− → H2) has a standard potential of 0 V versus SHE18. The occurrence of HER generates OH⁻, raising the local pH near the anode surface19. At high pH, OH- can react with ZnSO4 (salt in the electrolyte) to form electrically insulating zinc hydroxide sulfate, which hinders Zn deposition and accelerates performance degradation. Over repeated cycling, these processes lead to morphological instability and non-uniform Zn plating, further compromising reversibility and charge efficiency20.

The schematic illustrates the time-dependent, spatially non-uniform pH distribution near the Mn-based cathode and Zn anode. Different ways of pH evolution at the MnO2 cathode (top image) are (1) proton intercalating into MnO2 and the free OH- increases the local pH, (2) Free H+ at the interface reduces the local pH, when Mn2+ reacts with H2O at higher potentials, and (3) At OER regimes, the protons are released, that decreases the local pH. In the case of anode (bottom image), free OH- are generated during the parasitic HER, thereby increasing the local pH.
On the cathode side, the reaction pathway is even more complex. At relatively low potentials, around 1.0 V versus SHE, proton intercalation occurs, with H⁺ inserting into the MnO2 lattice to form MnOOH and producing OH⁻, thereby increasing the local pH21,22. Under alkaline conditions, this may further trigger the reaction, leading to the formation of zinc hydroxide sulfate. At higher potentials, Mn2+ from the electrolyte reacts with H2O and deposits as MnO2, releasing protons and lowering the local pH23,24. Furthermore, under certain conditions, the potential can reach the oxygen evolution reaction (OER) regime, where water decomposition produces O2 and protons, further acidifying the electrolyte25,26. Such acidification can corrode the anode material and carbon current collectors, trigger dissolution of active Mn or Zn species, and exacerbate interfacial instability27,28. In recently developed electrode-free systems that require precise anode–cathode capacity balance, such as dual free configurations29,30. This corrosion-driven degradation can be particularly harmful, as it amplifies capacity mismatch and accelerates performance decay. These intertwined processes underscore the importance of electrolyte engineering, interface stabilization strategies, and optimized cycling protocols in mitigating local pH extremes and suppressing parasitic reactions.
Regulation of local pH evolution in AZIBs
pH plays a pivotal role in determining the electrochemical behavior of AZIBs, and extensive efforts have therefore been devoted to regulating it to enhance overall performance. These approaches include the use of pH buffers, rational design of solvation structures, engineering of the solid electrolyte interphase (SEI), and construction of artificial protective interfaces. Electrolyte optimization, as an integral component of electrolyte engineering, has attracted extensive attention as the most straightforward strategy to control pH, while offering conceptual simplicity, cost-effectiveness, and broad applicability.
The most direct strategy to mitigate pH evolution-induced degradation is the use of pH-buffering additives. Lyu et al.31 demonstrated that organic heterocycles such as pyridine and imidazole can effectively stabilize Zn anodes by moderating local pH fluctuations (Fig. 3a). The nitrogen lone pairs in these molecules are capable of forming hydrogen bonds with protons, which alleviates sharp variations in the local chemical environment and suppresses the generation of parasitic by-products such as Zn hydroxides. At the same time, preferential adsorption of these molecules on the Zn surface directs uniform nucleation. It guides dendrite-free growth along the Zn(002) facet, resulting in smoother and more compact deposits. This dual role of buffering and surface regulation effectively improves the reversibility of Zn plating/stripping and prolongs the lifetime of symmetric Zn cells. Similar effects have also been achieved with acetate–acetate salt pairs32, phosphate buffers such as KH₂PO₄33, and hydrolytic solid buffers like black phosphorus34, all of which stabilize the local pH environment and thereby suppress side reactions, improving Zn reversibility.

a Coulombic efficiency curves of Zn anodes with pyridine as a pH buffer and a schematic illustration of pyridine. b Relationship between solvation structure, pH, and Coulombic efficiency (left), and schematic of the Zn²⁺ solvation environment with corresponding battery performance at different temperatures (right). c Coulombic efficiency of Zn anodes with and without sodium glycerophosphate additive, and schematic illustration of in situ SEI formation on the Zn surface. d Coulombic efficiency of bare Zn and the CuIn@Zn, with interfacial pH mapping of bare Zn (insert, left) versus CuIn@Zn (insert, right) after 24 h soaking. a31, c43 adapted with permission from Wiley; panels b35 and d47 adapted with permission from Springer Nature.
In addition, tailoring the solvation structure to modulate interfacial reactions has emerged as an effective strategy for controlling pH evolution. As shown in Fig. 3b, Wang et al.35 reported a highly concentrated Zn-ion electrolyte (1 M Zn(TFSI)2 + 20 M LiTFSI), which remains neutral in pH and stable in ambient conditions, thereby enabling highly reversible Zn plating and stripping with nearly 100% Coulombic efficiency. Detailed structural analyses and molecular-scale modeling revealed that this performance originates from a carefully engineered solvation sheath: the high concentration of anions drives them into proximity with Zn2+ to form Zn–TFSI⁺ ion pairs, effectively replacing conventional [Zn(H2O)6]2+ species. By weakening the coordination of water and reducing its activity, the electrolyte design mitigates local pH fluctuations, suppresses parasitic reactions, and stabilizes the electrode–electrolyte interface. Similar strategies include the use of co-solvents36,37 to replace water in the Zn²⁺ coordination shell partially, or the application of high-entropy38 materials that reconfigure the solvation environment, both of which effectively lower water activity and mitigate interfacial pH fluctuations. Furthermore, Highly Concentrated Electrolytes (HCE) have been widely adopted for solvation structure design to suppress water-induced parasitic reactions. In HCE, the extreme salt-to-water ratio converts most [Zn(H2O)6]²⁺ into anion-coordinated Zn–anion complexes, significantly lowering water activity and stabilizing Zn plating and stripping39,40. However, their high viscosity and cost hinder practical use. To address these limitations, Localized Concentrated Hybrid Electrolytes (LCHE)41,42, retain the local anion-rich solvation environment of HCEs while introducing inert or weakly coordinating diluents to reduce viscosity and improve ionic transport. This localized concentration maintains low water activity and robust interfacial chemistry, enabling high reversibility and suppressed pH fluctuations in aqueous Zn systems.
In lithium-ion batteries, the SEI serves as a protective barrier to block direct contact between the electrode and the electrolyte. By analogy, similar concepts have been explored in aqueous Zn systems through the use of organic solvents or functional additives that can promote SEI formation29. For example, as shown in Fig. 3c, Hao et al.43 proposed sodium glycerophosphate (SG) as a multifunctional additive for AZIBs, in which the key stabilizing mechanism lies in the in situ generation of a protective SEI. Unlike conventional approaches, SG not only regulates electrolyte pH and lowers free-water activity before cycling, thereby suppressing HER and parasitic reactions, but also undergoes Zn-induced decomposition within the inner Helmholtz plane. This interfacial reaction produces a self-limiting SEI layer directly on the Zn surface, which halts further SG decomposition once the electrode is fully covered. Upon exposure of fresh Zn during cycling, the additive decomposes again, imparting the SEI with a dynamic self-repairing ability. Through this SEI-forming pathway, the Zn anode is effectively protected from dendrite growth and side reactions, resulting in highly reversible deposition and stripping, as well as extended cycling stability, even under lean-electrolyte conditions. Similar SEI-forming strategies have also been reported. Organic additives can decompose at the Zn surface to yield polymeric or organophosphate-rich layers that suppress hydrogen evolution and guide uniform Zn deposition44. Inorganic components such as fluorides or phosphates can generate ZnF2 or PO43-based interphases that block corrosion and by-product accumulation15,45. In addition, combined organic and inorganic systems create more robust and ion-conductive SEIs with enhanced mechanical integrity and even self-repairing capability46. Together, these approaches stabilize the interface, regulate ion transport, and enable long-term reversible cycling.
Beyond electrolyte engineering, regulating interfacial pH evolution can also be achieved through the design of electrode surfaces. Zhang et al.47 developed a readily accessible three-dimensional Cu and In alloy heterogeneous interface (CuIn@Zn) that combines an affinity for the zinc ions with significant mitigation of hydrogen evolution. The Cu component lowers the Zn nucleation barrier and guides uniform deposition with a Zn (002) orientation. At the same time, the incorporation of indium enhances the anti-HER activity, thereby mitigating uncontrolled Zn degradation and suppressing by-product formation. By simultaneously steering Zn nucleation and weakening HER activity, this alloyed interface effectively regulates local pH evolution at the Zn surface. During cycling, the gradual redox activity of indium enables the self-reconstruction of the CuIn alloy layer into a gradient hierarchical structure with both zincophilic and anti-HER activity, thereby maximizing the synergistic regulation of deposition and pH. Through this multifunctional interface design, dendrite-free Zn growth and improved reversibility were achieved, demonstrating the critical role of alloyed interfacial layers in stabilizing anodes. Besides the CuIn alloy interface, other strategies such as Zn–Al alloy anodes48 and crystallographically oriented substrates (e.g., Cu(111)49,50) have also been demonstrated to lower Zn nucleation barriers, suppress HER, and regulate local pH evolution for stable deposition.
Current strategies to probe pH dynamics in AZIBs
Local pH evolves spatiotemporally at both electrodes and strongly impacts performance, motivating operando measurements with high temporal and spatial sensitivity. A common approach is to insert a pH probe into an open working cell to sample the electrolyte36,51,52. For example, Boeun et al.16 employed this strategy in an α-MnO2 | |Zn cell system; the results show that the pH rose from ~4.6 to ~5.7 during discharge and decreased to ~5.2 on charge, indicating incomplete reversibility and the concomitant formation of zinc hydroxide sulfate. Methodologically, this configuration captures only the dynamics of the bulk electrolyte, whereas the operative chemistry at the interfaces is spatiotemporally complex, as outlined above. Accordingly, developing localized operando pH probes with high spatial resolution is crucial for resolving the dynamics at both electrodes. Christian et al.53 built an operando colorimetric pH cell using a standard ultraviolet-visible spectrophotometer cuvette filled with electrolyte containing bromocresol green, Zn, and MnO2 sheets, which were mounted on opposite cuvette walls and cycled at 40 mA g−1 between 0.9 and 1.9 V. Figure 4a, b schematize the mentioned setup and the observed results, respectively. Initially, the electrolyte is uniformly green. During discharge and through the first charge, the bulk electrolyte turns deep blue, indicating a net pH increase driven by OH⁻ generation at the Zn anode during Zn2+ reduction and concomitant hydrogen evolution, with the blue intensity tracking state of charge in the potential-mapped frames. At higher charge potentials around 1.7 V vs Zn/Zn2+, a localized yellow band appears at the MnO2 cathode–electrolyte interface, evidencing acidification via H3O+ formation or OH- consumption confined to the positive-electrode region. To quantify and verify the local pH evolution during cycling, they propose a microelectrode-tilted strategy, as shown in Fig. 4c. The results, as demonstrated in Fig. 4d, indicate a pH drop at higher potentials during charging, as indicated by the bromocresol green above. Notably, the local pH of the cathode suddenly drops from 5 to 3, as the cell was charged at a high potential ( > 1.5 V), and it eventually returns to its initial value of 5. This behavior indicates that the local pH drop occurs due to the combination of mechanisms that often happen at high potentials: free protons generated as an OER byproduct, and when Mn2+ dissolved from the cathode reacts with OH− ions from H2O, leaving the dissociated H+ ions at the interface. Similarly, Liu et al.54 constructed an operando pH cell that presses a flat membrane microelectrode against the carbon paper backing of a cathode, allowing for sensitive near-contact tracking of local pH during coupled H+ and Zn2+ intercalation. A concise comparison of these representative operando pH probing methods is summarized in Table 1. However, these measurements primarily rely on coarse physical repositioning of a pH electrode among the electrode, separator, and electrolyte, which limits spatial resolution and slows the response; the readout is also susceptible to liquid junction potentials and perturbations induced by the probe, making it challenging to resolve transient pH gradients within the diffusion boundary layer and the pore-scale microenvironment. Therefore, more precise operando probe tools are needed.

a UV–vis cuvette setup. b Time-lapse images aligned to potential: initial green (pH≈4.3) turns deep blue during discharge and early charge; a localized yellow zone appears at the MnO2 side near 1.7 V vs Zn/ Zn2⁺, indicating acidification. c Tilted strategy for local pH probe. d Operando pH at these locations with the cell potential. Adapted from ref. 53, published on behalf of The Electrochemical Society by IOP Publishing Limited.
Mature toolbox to probe pH dynamics in AZIBs
In fact, mapping local pH is already a widely studied topic across electrocatalysis, corrosion, electrochemical manufacturing, and the biological sciences, among other fields, which offers a mature toolbox of operando probes that can be adapted to AZIBs. Guo et al.,55 in their electrodeposited IrOx on the ring of a rotating ring disk electrode, and exploited the pH sensitivity of its open-circuit potential to read near-surface pH operando, then calibrated the voltage in natural seawater and used the collection efficiency to convert the ring readout to the disk interfacial pH, enabling quantitative tracking of catalyst-surface acidity during electrolysis. Similarly, Ryu et al.56 used a concurrent nonfaradaic probe reaction on nanostructured Pt/C, where hydrogenation of cis-2-butene-1,4-diol yields 1,4-butanediol and n-butanol with a product selectivity that varies linearly with bulk pH. Because both products branch from a common surface-bound intermediate, the product ratio reports the interfacial pH within molecular length scales of the electrode. These examples illustrate that local pH can be indirectly probed by converting the pH signal into other measurable readouts, such as electrode potentials via electrochemical self-reporters or product distributions via concurrent probe reactions. For AZIBs, adopting these methods can decouple anode alkalization from cathode acidification, thereby quantifying transient gradients across electrodes in specific regimes of Zn plating, Mn dissolution, proton intercalation, and basic zinc sulfate formation. We also summarize a broad set of potential operando methods for AZIBs (see Table 2). It should be noted, however, that because different indicators respond indirectly to pH, their achievable spatial and temporal resolutions may vary. Although these operando probes span a broad range of spatial and temporal resolutions, most have not yet been directly applied to AZIBs. Nevertheless, they offer a promising toolbox: ultrafast vibrational and plasmonic methods can resolve interfacial water structure, while fluorescence and ISFET-based sensors can capture mesoscale proton gradients. Looking forward, adapting these techniques to Zn-based environments will require careful consideration of cell geometry, optical or electronic accessibility, and minimal perturbation of the underlying electrochemistry. With proper integration, these probes could enable a more complete picture of pH evolution during Zn plating, Mn dissolution, proton intercalation, and zinc hydroxide sulfate formation.
Outlook
Progress in pH diagnostics for AZIBs will hinge on tools that reveal what truly matters in batteries, that is, when and where pH changes occur, whether a gradient develops across the interface, and how these variations couple to solvation structure, interfacial reactions, and the electrical double layer. Interfacial pH gradients can reorganize the Zn²⁺ solvation shell by changing the participation of water and anions, thereby shifting ion transport and deposition pathways. They also reshape the electrical double layer by modulating charge screening and ionic distributions, which in turn govern nucleation, corrosion, and parasitic side reactions. The goal is to capture these dynamic gradients across the different components of the cells, with spatial and temporal resolution matched to the electrical double layer and bulk diffusion. Beyond diagnosis, such knowledge lays the groundwork for rational regulation strategies, where electrolyte design, interfacial engineering, and materials selection can be tuned to control solvation and stabilize pH landscapes.
A pragmatic path forward is correlated to the other state-of-the-art characterization tools. For example, cryogenic electron microscopy (cryo-EM) can preserve interfacial morphologies associated with recent pH fluctuations, as reported in cryo-EM works on different battery material interfaces57,58. Three-dimensional ToF-SIMS can map ions and byproducts that serve as pH proxies, such as basic zinc sulfate or protonated manganese oxides. Synchrotron X-ray absorption spectroscopy can track oxidation states and local coordination that respond to pH, and pairing with Raman or infrared spectroscopy helps assign intermediates. When two independent probes are co-located and registered to the same site, the confidence in the inferred pH field increases substantially. Employing operando fluorescent imaging methods by adding ratiometric fluorescent pH indicators or nanoparticles in the electrolyte could also provide quantitative pH maps near the electrode interfaces20,59.
The increased use of artificial intelligence (AI) in battery research has undergone tremendous improvement in recent years, from its pattern-finding ability to complete automation, underscoring its undeniable importance and the significant potential for providing meaningful insights on the pH mapping in AZIBs60,61,62. As shown in Fig. 5, by integrating the above operando and correlative measurements with physics-based models and machine learning, quantitative pH fields can be reconstructed in space and time. With comprehensive datasets and advanced AI, the relationships between pH landscapes and efficiency, rate capability, and lifetime can be learned, enabling recommendations on operating windows and materials modifications. Beyond diagnosis, such models can suggest rational regulation strategies, for instance, tailoring electrolyte composition to reshape solvation shells, engineering electrode surfaces to buffer interfacial reactions, or designing separators that mitigate pH gradients. In this way, pH evolution becomes not only a diagnostic indicator but also a controllable design handle rather than a hidden failure mode.

The diagram highlights future direction for probing and regulating pH in AZIBs.
Data availability
No datasets were generated or analysed during the current study.
References
Alstone, P., Gershenson, D. & Kammen, D. M. Decentralized energy systems for clean electricity access. Nat. Clim. Change 5, 305–314 (2015).
Etacheri, V., Marom, R., Elazari, R., Salitra, G. & Aurbach, D. Challenges in the development of advanced Li-ion batteries: a review. Energy Environ. Sci. 4, 3243–3262 (2011).
Grey, C. P. & Tarascon, J. M. Sustainability and in situ monitoring in battery development. Nat. Mater. 16, 45–56 (2017).
Ma, L. et al. Realizing high zinc reversibility in rechargeable batteries. Nat. Energy 5, 743–749 (2020).
Heo, J., Dong, D., Wang, Z., Chen, F. & Wang, C. Electrolyte design for aqueous Zn batteries. Joule 9, https://doi.org/10.1016/j.joule.2025.101844 (2025).
Gourley, S. W. D., Brown, R., Adams, B. D. & Higgins, D. Zinc-ion batteries for stationary energy storage. Joule 7, 1415–1436 (2023).
Li, W., Dahn, J. R. & Wainwright, D. S. Rechargeable lithium batteries with aqueous electrolytes. Science 264, 1115–1118 (1994).
Wu, X. & Ji, X. Aqueous batteries get energetic. Nat. Chem. 11, 680–681 (2019).
Li, C. et al. Roadmap on the protective strategies of zinc anodes in aqueous electrolyte. Energy Storage Mater. 44, 104–135 (2022).
Zampardi, G. & La Mantia, F. Open challenges and good experimental practices in the research field of aqueous Zn-ion batteries. Nat. Commun. 13, 687 (2022).
Li, H., Chen, J. & Fang, J. Recent advances in wearable aqueous metal-air batteries: from configuration design to materials fabrication. Adv. Mater. Technol. 8, 2201762 (2023).
Lv, Y., Xiao, Y., Ma, L., Zhi, C. & Chen, S. Recent advances in electrolytes for “beyond aqueous” zinc-ion batteries. Adv. Mater. 34, 2106409 (2022).
Ma, L. et al. Toward practical high-areal-capacity aqueous zinc-metal batteries: quantifying hydrogen evolution and a solid-ion conductor for stable zinc anodes. Adv. Mater. 33, 2007406 (2021).
Cai, Z. et al. Chemically resistant Cu–Zn/Zn composite anode for long cycling aqueous batteries. Energy Storage Mater. 27, 205–211 (2020).
Wang, W. et al. Regulating interfacial reaction through electrolyte chemistry enables gradient interphase for low-temperature zinc metal batteries. Nat. Commun. 14, 5443 (2023).
Lee, B. et al. Critical role of pH evolution of electrolyte in the reaction mechanism for rechargeable zinc batteries. ChemSusChem 9, 2948–2956 (2016).
Liu, M. et al. Strategies for pH regulation in aqueous zinc ion batteries. Energy Storage Mater. 67, 103248 (2024).
Krężel, A. & Maret, W. The biological inorganic chemistry of zinc ions. Arch. Biochem. Biophys. 611, 3–19 (2016).
Fu, J. et al. Electrically rechargeable zinc–air batteries: progress, challenges, and perspectives. Adv. Mater. 29, 1604685 (2017).
Lim, W.-G., Li, X. & Reed, D. Understanding the role of zinc hydroxide sulfate and its analogues in mildly acidic aqueous zinc batteries: a review. Small Methods 8, 2300965 (2024).
Ke, X. et al. Mn-oxide cathode material for aqueous Zn-ion battery: structure, mechanism, and performance. Energy 2, 100095 (2024).
Seo, J. K. et al. Intercalation and conversion reactions of nanosized β-MnO2 cathode in the secondary Zn/MnO2 alkaline battery. J. Phys. Chem. C. 122, 11177–11185 (2018).
Paudel, N., Ale Magar, B., Acharya, K., Lambert, T. N. & Vasiliev, I. Influence of defects and surfaces on the electrochemical performance of MnO2 cathodes in rechargeable alkaline Zn/MnO2 batteries: a first-principles study. ACS Appl. Energy Mater. 7, 2767–2778 (2024).
Yang, H. et al. Protocol in evaluating capacity of Zn–Mn aqueous batteries: a clue of pH. Adv. Mater. 35, 2300053 (2023).
Feng, Y. et al. A rechargeable neutral hybrid Zn-Air/MnO2 battery with high energy efficiency. Adv. Energy Mater. n/a, 2501294, .
Wang, N. et al. A review of zinc-based battery from alkaline to acid. Mater. Today Adv. 11, 100149 (2021).
Yuan, Y. et al. Identifying the role of Zn self-dissolution in the anode corrosion process in Zn-ion batteries. Energy Environ. Sci. 18, 5610–5621 (2025).
Ravi, A. et al. Revealing and quantifying carbon corrosion in aqueous manganese-based batteries. Nano Lett. 25, 10834–10839 (2025).
Li, Y. et al. In situ formation of liquid crystal interphase in electrolytes with soft templating effects for aqueous dual-electrode-free batteries. Nat. Energy 9, 1350–1359 (2024).
Li, Y. et al. A high-energy-density aqueous dual-ion anode-free Zn battery under cryogenic conditions. Energy Storage Mat. 76, 104159 (2025).
Lyu, Y. et al. Organic pH buffer for dendrite-free and shuttle-free Zn-I2 batteries. Angew. Chem. Int. Ed. 62, e202303011 (2023).
Li, L. et al. Dual-additive-based electrolyte design for aqueous zinc ion batteries with high plating/stripping efficiency. Chem. Commun. 60, 6809–6812 (2024).
Liu, W. et al. pH buffer KH2PO4 boosts zinc ion battery performance via facilitating proton reaction of MnO2 cathode. J. Colloid Interface Sci. 657, 931–941 (2024).
Cheng, H. et al. Hydrolysis of solid buffer enables high-performance aqueous zinc ion battery. Adv. Sci. 11, 2307052 (2024).
Wang, F. et al. Highly reversible zinc metal anode for aqueous batteries. Nat. Mater. 17, 543–549 (2018).
Fitz, O. et al. Electrolyte study with in operando pH tracking providing insight into the reaction mechanism of aqueous acidic Zn//MnO2 batteries. ChemElectroChem 8, 3553–3566 (2021).
Cao, L. et al. Solvation structure design for aqueous Zn metal batteries. J. Am. Chem. Soc. 142, 21404–21409 (2020).
Chang, L. et al. High-entropy solvation chemistry towards affordable and practical Ah-level zinc metal battery. Nat. Commun. 16, 6134 (2025).
Zhu, Y. et al. Concentrated dual-cation electrolyte strategy for aqueous zinc-ion batteries. Energy Environ. Sci. 14, 4463–4473 (2021).
Dong, D., Wang, T., Sun, Y., Fan, J. & Lu, Y.-C. Hydrotropic solubilization of zinc acetates for sustainable aqueous battery electrolytes. Nat. Sustain. 6, 1474–1484 (2023).
Chen, S. et al. Highly reversible aqueous zinc metal batteries enabled by fluorinated interphases in localized high concentration electrolytes. J. Mater. Chem. A 9, 22347–22352 (2021).
Lyu, H. et al. Localized Eutectic Electrolytes for Stable Aqueous Zinc-Ion Batteries. ACS Energy Lett. 10, 2924–2933 (2025).
Hao, J., Yuan, L., Zhu, Y., Jaroniec, M. & Qiao, S.-Z. Triple-function electrolyte regulation toward advanced aqueous Zn-Ion batteries. Adv. Mater. 34, 2206963 (2022).
Xie, K. et al. In situ construction of zinc-rich polymeric solid–electrolyte interface for high-performance zinc anode. eScience 3, 100153 (2023).
Huang, Y. et al. Ultra-stable aqueous zinc anodes: enabling high-performance zinc-ion batteries via a ZnSiF6-derived protective interphase. Adv. Sci. 11, 2407201 (2024).
Di, S. et al. Zinc anode stabilized by an organic-inorganic hybrid solid electrolyte interphase. Energy Storage Mater. 43, 375–382 (2021).
Zhang, M. et al. Synergetic bifunctional Cu-In alloy interface enables Ah-level Zn metal pouch cells. Nat. Commun. 15, 9455 (2024).
Sun, J. et al. Scalable production of hydrogen evolution corrosion resistant Zn-Al alloy anode for electrolytic MnO2/Zn batteries. Energy Storage Mater. 54, 570–578 (2023).
Wang, M. et al. Crystal facet correlated Zn growth on Cu for aqueous Zn metal batteries. Energy Storage Mater. 56, 424–431 (2023).
Xiao, X. et al. Epitaxial Electrodeposition of zinc on different single crystal copper substrates for high-performance aqueous batteries. Nano Lett. 25, 1305–1313 (2025).
Chamoun, M., Brant, W. R., Tai, C.-W., Karlsson, G. & Noréus, D. Rechargeability of aqueous sulfate Zn/MnO2 batteries enhanced by accessible Mn2+ ions. Energy Storage Mater. 15, 351–360 (2018).
Li, L. et al. Functioning mechanism of the secondary aqueous Zn-β-MnO2 battery. ACS Appl. Mater. Interfaces 12, 12834–12846 (2020).
Bischoff, C. F. et al. Revealing the local pH value changes of acidic aqueous zinc ion batteries with a manganese dioxide electrode during cycling. J. Electrochem. Soc. 167, 020545 (2020).
Liu, X. et al. Operando pH measurements decipher H+/Zn2+ intercalation chemistry in high-performance aqueous Zn/δ-V2O5 batteries. ACS Energy Lett. 5, 2979–2986 (2020).
Guo, J. et al. Direct seawater electrolysis by adjusting the local reaction environment of a catalyst. Nat. Energy 8, 264–272 (2023).
Kim, Y., Seo, M. & Baek, S. Ion-selective electrode-based sensors from the macro- to the nanoscale. Sens. Actuators Rep. 9, 100258 (2025).
Zhang, Z. et al. Cathode-electrolyte interphase in lithium batteries revealed by cryogenic electron microscopy. Matter 4, 302–312 (2021).
Weng, S., Li, Y. & Wang, X. Cryo-EM for battery materials and interfaces: Workflow, achievements, and perspectives. iScience 24, 103402 (2021).
Chen, Y. et al. In situ visualization of ion transport processes in aqueous batteries. ACS Appl. Mater. Interfaces 16, 42321–42331 (2024).
Li, H., Hao, J. & Qiao, S.-Z. AI-driven electrolyte additive selection to boost aqueous Zn-ion batteries stability. Adv. Mater. 36, 2411991 (2024).
Xu, G. et al. Toward stable zinc anode: an AI-assisted high-throughput screening of electrolyte additives for aqueous zinc-ion battery. Angew. Chem. Int. Ed. 64, e202511389 (2025).
Wu, Y. et al. Machine learning-guided discovery of high-efficiency electrolyte additives for aqueous magnesium-air batteries. Energy Storage Mater. 76, 104120 (2025).
Shen, Y. R. & Ostroverkhov, V. Sum-frequency vibrational spectroscopy on water interfaces: polar orientation of water molecules at interfaces. Chem. Rev. 106, 1140–1154 (2006).
Richmond, G. L. Molecular bonding and interactions at aqueous surfaces as probed by vibrational sum frequency spectroscopy. Chem. Rev. 102, 2693–2724 (2002).
Kubota, J. & Domen, K. Study of the dynamics of surface molecules by time-resolved sum-frequency generation spectroscopy. Anal. Bioanal. Chem. 388, 17–27 (2007).
Niemann, R. et al. Long-wave infrared super-resolution wide-field microscopy using sum-frequency generation. Appl. Phys. Lett. 120, https://doi.org/10.1063/5.0081817 (2022).
Ataka, K. & Heberle, J. Biochemical applications of surface-enhanced infrared absorption spectroscopy. Anal. Bioanal. Chem. 388, 47–54 (2007).
Huo, S.-J. et al. Extending in situ attenuated-total-reflection surface-enhanced infrared absorption spectroscopy to Ni electrodes. J. Phys. Chem. B 110, 4162–4169 (2006).
Hicks, M. H. et al. Electrochemical CO2 reduction in acidic electrolytes: spectroscopic evidence for local pH gradients. J. Am. Chem. Soc. 146, 25282–25289 (2024).
Homola, J. Surface plasmon resonance sensors for detection of chemical and biological species. Chem. Rev. 108, 462–493 (2008).
Mayer, K. M. & Hafner, J. H. Localized surface plasmon resonance sensors. Chem. Rev. 111, 3828–3857 (2011).
Joshi, G. K., Johnson, M. A. & Sardar, R. Novel pH-responsive nanoplasmonic sensor: controlling polymer structural change to modulate localized surface plasmon resonance response. RSC Adv. 4, 15807–15815 (2014).
Axelrod, D. Total internal reflection fluorescence microscopy in cell biology. Traffic 2, 764–774 (2001).
Nakata, E. et al. A ratiometric fluorescent probe for pH measurement over a wide range composed of three types of fluorophores assembled on a DNA scaffold. Chemistry 5, 1832–1842 (2023).
Zhang, Y. et al. High-resolution label-free 3D mapping of extracellular pH of single living cells. Nat. Commun. 10, 5610 (2019).
Acknowledgements
The authors thank the members of the Aqueous Battery Consortium for insightful discussion. This work was inspired by the Aqueous Battery Consortium, an energy innovation hub under the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering.
Author information
Authors and Affiliations
Contributions
Z.X., S.J., X.Z., and J.N. wrote the main manuscript text. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xue, Z., Jagadeesan, S.N., Zheng, X. et al. Probing and tuning spatiotemporal pH evolution in aqueous zinc ion batteries. npj Energy Mater. 1, 3 (2026). https://doi.org/10.1038/s44456-026-00003-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44456-026-00003-7
