
Abstract
A multistep model for pancreatic adenocarcinoma has been proposed recently. In this model, well-defined, noninvasive ductal lesions are recognized as precursors of invasive cancer and have been classified under the nomenclature of pancreatic intraepithelial neoplasia, or PanIN. Increasing evidence suggests that PanINs represent true neoplasms of the pancreatic ductal epithelium, accumulating histologic and genetic abnormalities in their progression toward invasive cancer. We have constructed a tissue microarray containing 55 PanIN lesions of all histologic grades in order to perform a multicomponent analysis of the pancreatic adenocarcinoma progression model. The protein products of 14 genes encompassing a variety of functional classes, such as tumor suppressor genes (p53, Smad4/Dpc4), oncogenes (β-catenin), cell cycle antigens (p16, cyclin D1), proliferation antigens (Ki-67, topoisomerase II alpha), and epithelial apomucins (MUC1, MUC2, MUC5), as well as “novel” genes described as differentially up-regulated in invasive pancreas cancer by global microarray expression analysis (mesothelin, prostate stem cell antigen, fascin, and 14-3-3σ), were analyzed by immunohistochemistry on the PanIN tissue microarray. Comparison of the results from the current study with previously published data performed on routine histologic sections of PanINs demonstrates that tissue microarrays are a valid platform for molecular analysis not only of invasive cancers but of precursor lesions as well. In addition, this study demonstrates that molecular abnormalities in PanINs are not random but can usually be stratified into “early” changes (e.g., expression of MUC5 and prostate stem antigen, or loss of p16), “intermediate” changes (e.g., expression of cyclin D1), and “late” changes (e.g., expression of p53, proliferation antigens, MUC1, mesothelin, and 14-3-3σ, or loss of Smad4/Dpc4). Understanding the molecular pathogenesis of precursor lesions of invasive pancreatic adenocarcinomas using a high-throughput tissue microarray–based approach is a valuable adjunct to designing rational strategies for early detection of this lethal neoplasm.
Similar content being viewed by others


INTRODUCTION
Adenocarcinoma of the pancreas affects approximately 29,000 individuals each year in the United States, and nearly all patients die within months of diagnosis (1). Only a minority of cases is amenable to surgery, and regrettably, surgical resection of localized pancreatic cancer is seldom curative. Thus, prevention and early detection of pancreatic adenocarcinoma remain the best hope for a cure, because once established, this disease is nearly always fatal (2).
A multistep model has recently been proposed for pancreatic adenocarcinomas, in which noninvasive precursor lesions in the pancreatic ducts undergo histologic and genetic progression toward invasive cancer (3, 4). These morphologically distinct, noninvasive lesions have been classified under a uniform nomenclature scheme termed pancreatic intraepithelial neoplasia, or PanIN. We and others have demonstrated that PanINs share many of the genetic aberrations associated with invasive adenocarcinomas, underscoring their classification as “neoplasms” rather than as a reactive/hyperplastic process (5, 6, 7, 8, 9). Some of the genetic alterations are nearly ubiquitous (e.g., telomere shortening) (10), suggesting that these are early events in the ductal epithelium, whereas others, such as loss of function of the tumor suppressor gene BRCA2, occur only in the most advanced PanIN lesions that precede invasive cancer (11). Understanding the molecular mechanisms that facilitate PanIN progression toward invasive adenocarcinomas is critical, because these noninvasive neoplasms represent one of the best targets available for early detection and chemoprevention strategies for pancreatic cancer (12).
We have recently explored the global expression profiles of invasive pancreatic adenocarcinomas with three different gene expression platforms (serial analysis of gene expression, oligonucleotide microarrays, and cDNA microarrays) (13, 14, 15). These studies have identified a multitude of genes that are overexpressed at the transcript level in pancreatic cancer compared with in normal ductal epithelium. We have subsequently validated the “cancer-specific” expression of a subset of these markers at the protein level by immunohistochemistry (16). However, the expression of these tumor markers in the noninvasive precursor ductal lesions remains largely unexplored. The advent of TMAs for high-throughput expression profiling in archival tissues has greatly facilitated the identification of novel diagnostic and prognostic markers in multiple cancer types (17, 18, 19, 20, 21, 22). We have constructed a TMA made up entirely of noninvasive ductal lesions (PanIN TMA) for multicomponent profiling of the pancreatic adenocarcinoma progression model. Using a TMA-based approach, we herein validate molecular abnormalities previously reported in routine histologic sections of PanINs, as well as examine the expression of novel cellular markers that have been discovered by global gene expression profiling of pancreatic adenocarcinomas.
MATERIALS AND METHODS
Tissue samples were obtained from the surgical pathology archives of the Department of Pathology at the Johns Hopkins University School of Medicine. Formalin-fixed, paraffin-embedded blocks were retrieved from 44 patients who underwent pancreaticoduodenectomy (Whipple resection) for pancreatic ductal adenocarcinoma. PanIN lesions were selected by two authors (AM and RHH) and classified into PanIN-1A, PanIN-1B, PanIN-2, and PanIN-3 using previously described criteria (3). For TMA construction, representative areas containing morphologically defined PanINs were circled on the glass slides and used as a template. The PanIN TMA was constructed using a manual Tissue Puncher/Arrayer (Beecher Instruments, Silver Spring, MD) as previously described (10). For each PanIN, a 1.4-mm core was punched from the donor block to ensure that the entire duct lesion and adequate surrounding tissue could be incorporated into the spot. A total of 99 cores (61 PanINs, 11 intra- and interlobular pancreatic ducts, and 27 control tissue cores from various extrapancreatic organs) were arrayed on the recipient block. Six PanIN lesions were not suitable for evaluation because of either loss of tissue cores or exhaustion of the duct lesion in deeper sections, and these were excluded from analysis. The remaining 55 PanIN lesions comprised 16 PanIN 1A, 18 PanIN 1B, 14 PanIN 2, and 7 PanIN 3 lesions).
Immunohistochemistry was performed as previously described (16). Briefly, unstained 5-μm sections were cut from the paraffin block selected and deparaffinized by routine techniques. The TMAs were placed in 200-mL of Target Retrieval Solution, pH 6.0 (Envision Plus Detection Kit, DAKO) for 20 minutes at 100° C. After cooling for 20 minutes, slides were quenched with 3% H2O2 for 5 minutes before incubating with primary antibody using the Dako Autostainer. The list of primary antibodies used, with respective dilutions and source of antibodies, is given in Table 1. Labeling was detected with the Dako Envision system according to the manufacturer's protocol. Negative controls (primary antibody replaced by serum from appropriate species) were used for each antibody in each run. Internal positive controls were available from the plethora of pancreatic and extrapancreatic control cores on the PanIN TMA itself (for example, basal cells in the prostate express prostate stem cell antigen; normal pancreatic ductal epithelium expresses Dpc4, etc.); additionally, external controls were examined with each run (e.g., breast carcinoma for MUC1, colonic adenocarcinoma for MUC2, gastric tissue for MUC5, etc.). For all immunohistochemical stains, the pattern of staining in the normal pancreatic ductal epithelium was considered to be the baseline for comparison with progressive PanIN lesions.
The following scoring criteria were used for assessment of immunolabeling: for the 11 proteins that are not expressed or minimally expressed in the normal pancreatic ductal epithelium (p53, cyclin D1, Ki-67, topoisomerase II α [topo II], MUC1, MUC2, MUC5, mesothelin, fascin, prostate stem cell antigen [PSCA], and 14-3-3σ), immunolabeling in <5% of cells within a PanIN lesion was considered negative; immunolabeling in 5–25% was considered focal, and >25% labeling was considered diffuse. Only appropriate subcellular localization of detectable signal was considered for assessment of percentage staining (for example, only nuclear staining was counted for p53, cyclin D1, Ki-67, and topo II, and cytoplasmic or membrane labeling was counted for the rest). Complete absence of cytoplasmic and nuclear expression of Dpc4, or complete absence of nuclear expression of p16, was considered negative (abnormal) for the respective stain, as previously described (8, 23, 24). Finally, the expression of β-catenin, which is normally present only in a membranous distribution, was graded based on percentage of cells with strong nuclear and cytoplasmic labeling, as previously described (25).
Statistical analyses for significant differences in immunohistochemical abnormalities between high-grade (PanIN 2 and 3) and low-grade (PanIN 1A and 1B) lesions were performed using two-tailed Fisher's exact test, with a significant P value set as < .05. All statistical comparisons were performed using the Analyze-it add-in for Microsoft Excel.
RESULTS AND DISCUSSION
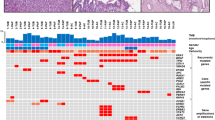
A summary of immunohistochemical abnormalities in the 55 PanIN lesions in this study is tabulated in Table 2 and is pictorially illustrated using Eisen's TREEVIEW software (http://rana.lbl.gov/EisenSoftware.htm) in Figure 1. The results for the 14 individual proteins are described in the ensuing discussion, in the context of each protein's functional class and relevant literature.

Pictorial representation of progressive molecular abnormalities in PanIN lesions depicted using Eisen's TREEVIEW software (http://rana.lbl.gov/EisenSoftware.htm). Individual PanIN lesions are in rows, and the 14 proteins examined are in columns. PanIN 1A lesions are designated as A1–A16; PanIN 1B lesions, as B1–B18; PanIN 2 lesions, as C1–C14, and PanIN 3 lesions, as D1–D7. The original TREVIEW color scheme for relative gene expression is retained for designating relative protein expression in this figure. Thus, green implies no expression, red designates diffuse expression, and brown designates focal expression. Depending on the protein of interest, therefore, red color could imply retained normal expression (e.g., Dpc4/Smad4), or aberrant overexpression (MUC5, PSCA, etc.).
Tumor Suppressor Genes (p53 and Dpc4)
P53 is commonly inactivated by mutations in invasive pancreatic adenocarcinomas, with a frequency of 50–75% of cases, depending on the series (26, 27, 28). Immunohistochemical detection of nuclear p53, as a surrogate marker for mutation, usually demonstrates a slightly lower frequency of abnormalities (∼30–50%) (29, 30, 31), consistent with the lower sensitivity of this technique. Nuclear overexpression of p53 has also been reported in PanIN lesions in the pancreas. For example, DiGiuseppe et al. (31) found abnormalities in p53 labeling in 2 of 17 (12%) histologically high-grade lesions (carcinomas in situ, equivalent to PanIN 3 in the current classification scheme). In contrast, all histologically lower-grade duct lesions labeled normally, suggesting that p53 gene inactivation is a late event in genetic progression in pancreatic ducts. Similarly, Apple et al. (32) reported p53 accumulation in 20% of “dysplastic” ductal lesions but in no “ductal hyperplasias.” In concert with these findings, we report diffuse nuclear overexpression of p53 (>25% nuclei) in 4 of 7 (57%) PanIN 3 lesions but in no normal ducts, PanIN 1A, PanIN 1B, or PanIN 2 foci. Thus, we confirm that p53 mutation, as assessed by nuclear overexpression of p53 protein, is a “late” event in the progression model of pancreatic adenocarcinoma, occurring only in the most advanced PanIN lesions (Fig. 2).

Representative photomicrographs illustrating immunohistochemical abnormalities in various histologic categories of PanIN lesions. The protein of interest is designated in rows, and the PanIN histologic category is designated in columns. Row 1, absence of nuclear p53 labeling in PanIN 1A, 1B, and 2, with diffuse nuclear labeling in PanIN 3. Row 2, absence of nuclear MIB-1 labeling in PanIN 1A and 1B, with focal nuclear labeling in PanIN 2 and 3. Row 3, absence of MUC1 labeling in PanIN 1A and 1B; note the apical labeling in normal ducts subjacent to PanIN lesions. In contrast, strong and diffuse apical labeling is seen in PanIN 2 and 3 lesions. Row 4, unlike the case with MUC1, MUC5 labeling is present in low-grade PanIN 1A and 1B lesions, with persistent expression in PanIN 2 and 3 lesions. Row 5, absence of fascin expression in PanIn1A, with focal or diffuse labeling in subsequent PanIN 1B, 2 and 3 lesions; note intense diffuse cytoplasmic labeling in PanIN 3 lesion. Row 6, absence of 14-3-3σ labeling in PanIN 1A and 1B lesions, with expression in PanIN 2 and 3 lesions.
The tumor suppressor gene SMAD4 (Small Mothers Against Decapentaplegic 4) or DPC4 (Deleted in Pancreatic Carcinoma 4) located on chromosome 18q, is a member of the transforming growth factor β (TGFβ) family (33). Inactivation of SMAD4/DPC4 by homozygous deletions, or a combination of intragenic mutation and allelic loss of heterozygosity, is present in approximately 55% of invasive pancreatic adenocarcinomas (33). Immunohistochemical labeling for the SMAD4/DPC4 gene product has recently been shown to mirror SMAD4/DPC4 gene status (34). Wilentz et al. (8) studied a large series of routine histologic sections from pancreata using the B-8 anti-Dpc4 antibody. Dpc4 expression was intact in all of the low-grade duct lesions that they examined, whereas 30% of the histologically high-grade lesions had a complete loss of Dpc4 expression. By using the PanIN classification scheme on our PanIN TMA, we detected complete loss of Dpc4 expression in 2 of 7 (28%) PanIN 3 lesions. Dpc4 expression was intact in all of the intra- or interlobular ducts, PanIN 1A, PanIN 1B, and PanIN 2 foci examined. Our findings are virtually identical to those findings of Wilentz et al. (8) and confirm that, similar to the case of the p53 gene, inactivation of the DPC4 gene appears to be a “late” event in the genetic progression of pancreatic cancer.
Cell Cycle Regulatory Genes (p16 and Cyclin D1)
The p16 gene on chromosome 9p encodes for a regulator of the cell cycle, and it is inactivated by homozygous deletions, mutations, or methylation of the p16 promoter in approximately 95% of invasive pancreatic adenocarcinomas (35, 36). Recent evidence suggests that similar mechanisms for p16 gene inactivation are also active in the noninvasive pancreatic ductal lesions, and that there is a progressive accumulation of these genetic abnormalities with histologic progression in the ducts (9, 37). Loss of nuclear p16 protein expression is considered to be a reliable surrogate for complete inactivation of p16 gene function (23, 24). Wilentz et al. (7), using immunohistochemical labeling for the p16 gene product on routine histologic sections, showed that 30% of “flat duct lesions without significant atypia” (equivalent to PanIN 1A), 27% of “papillary duct lesions without significant atypia” (equivalent to PanIN 1B), 55% of “papillary duct lesions with atypia” (equivalent to PanIN 2), and 71% of carcinoma in situ (PanIN 3) lesions had loss of expression of nuclear p16. In our TMA-based study, we found that 5 of 16 (31%) PanIN 1A, 8 of 18 (44%) PanIN 1B, 7 of 14 (50%) PanIN 2, and 6 of 7 (85%) PanIN 3 lesions had loss of nuclear p16. Thus, we were able to confirm that p16 inactivation is a common and “early” event in the multistep model of pancreatic adenocarcinoma, that the frequency of this loss increases with increasing grades of dysplasia, and that it precedes both p53 and Dpc4 inactivation. Nevertheless, loss of one cell cycle checkpoint protein appears to be inadequate to initiate uncontrolled cell division in PanIN lesions, as demonstrated by the concomitantly examined proliferation antigens (see below). It is likely that because of the redundancy of cell cycle checkpoint controls, more than one “hit” is required for initiation of deregulated proliferation.
The cyclin D1 or PRAD1 gene, located on chromosome 11q13, is a positive regulator of the cell cycle. Overexpression of cyclin D1 protein has been documented in several human cancers, such as breast, lung, and colon carcinoma, and in mantle cell lymphoma (38, 39, 40, 41). Between 60% and 85% of invasive pancreatic adenocarcinomas demonstrate nuclear overexpression of cyclin D1 protein by immunohistochemistry (42, 43, 44). Cyclin D1 overexpression in pancreatic cancer is associated with a poor prognosis and decreased survival (42, 45), whereas inhibition of cyclin D1 expression results in increased chemosensitivity and decreased expression of multiple chemoresistance genes (46). In the current study, focal or diffuse nuclear overexpression of cyclin D1 was seen in 0 of 11 normal pancreatic ducts, 0 of 16 PanIN 1A, 0 of 18 PanIN B, 4 of 14 (29%) PanIN 2, and 4 of 7 (57%) PanIN 3 lesions. Based on nuclear cyclin D1 expression in a third of PanIN 2 lesions, cyclin D1 abnormalities would best be classified as an “intermediate” event in the multistage progression of pancreatic adenocarcinoma, preceding p53 mutations and inactivation of Dpc4 in a subset of cases.
Oncogene (β-Catenin)
Beta-catenin is a target of the canonical wnt signaling pathway, implicated in both development and tumorigenesis. Oncogenic mutations of the β-catenin gene, which result in stabilization and nuclear translocation of β-catenin protein, have been reported in a variety of human cancers. β-catenin mutations are uncommon in pancreatic adenocarcinomas (47) but are frequent in nonductal neoplasms of the pancreas (solid pseudopapillary tumors, pancreatoblastomas, and acinar cell carcinomas) (25, 48, 49). All 11 normal and 55 PanIN foci examined in this study demonstrated normal membranous expression of β-catenin, with no convincing cytoplasmic or nuclear expression in any lesion. The complete absence of abnormal β-catenin expression in PanIN lesions reiterates the existence of two distinct, genetically divergent pathways of neoplasia in the pancreas, one resulting in the more common, conventional ductal adenocarcinomas and the other resulting in the less common nonductal neoplasms.
Proliferation Antigens (Ki-67 and Topo II)
Unchecked proliferation is an intrinsic feature of neoplasia. The proliferation antigen Ki-67, as detected by the monoclonal antibody MIB-1, is expressed in the majority of invasive adenocarcinomas of the pancreas (50, 51), whereas we have demonstrated up-regulation of topo II in invasive pancreatic cancers by using both oligonucleotide and cDNA microarrays (13, 15). The expression of one of these proliferation antigens—Ki-67—has been systematically examined by Klein et al. (52) in routine histologic sections of PanINs. In their study, Klein et al. (52) found increasing Ki-67–labeling indices with increasing grades of dysplasia in PanINs; thus, PanIN 1A had a labeling index of 0.69%, PanIN 1B, of 2.33%, PanIN 2, of 14.08%, and PanIN 3, of 22.01%. There are no studies to date examining topo II expression in the precursor lesions of pancreatic adenocarcinoma. In the current study, no normal pancreatic ducts, PanIN 1A, or PanIN 1B expressed Ki-67 or topo II, whereas only focal nuclear Ki-67 and topo II expression was seen in 1 of 14 (7%) PanIN 2 and 5 of 7 (71%) PanIN 3 lesions; there was complete concordance between Ki-67 and topo II expression in the PanIN lesions. Our percentages for Ki-67 labeling are discordant from those in the study of Klein et al. (52) because we did not use a nuclear labeling index but rather calculated expression based on a two-tier focal (5–25%) versus diffuse (>25%) labeling scheme. In their study, Klein et al. (52) found a mean Ki-67–labeling index of 22% for PanIN 3, which is consistent with the absence of diffuse labeling in the PanIN 3 lesions in our series. Cyclin D1 expression demonstrated a marginally better concordance with proliferation than p53; 5 of 6 (83%) of Ki-67– and topo II–positive lesions also overexpressed nuclear cyclin D1, whereas 4 of 6 (67%) concomitantly overexpressed p53. There was no correlation between p16 loss and Ki-67/topo II overexpression, given the discordance in the timing of their occurrence (early versus late) in the multistep model of pancreatic cancer progression.
Epithelial Mucins (MUC1, MUC2, and MUC5)
The apomucins MUC1, MUC2 and MUC5 are frequently overexpressed in epithelial cancers, particularly those arising in the gastrointestinal tract and pancreas (53, 54, 55, 56). MUC1 is the principal pancreatic mucin, being expressed in normal pancreatic ducts and acini (57). MUC1 expression is commonly observed in invasive pancreatic adenocarcinomas at both the transcript and protein levels (55, 56, 57, 58), approaching 100% of cases in some series. In our study, MUC1 expression was present in 11 of 11 (100%) normal intra- and interlobular pancreatic ducts, 6 of 14 (43%) PanIN 2, and 6 of 7 (85%) PanIN 3 lesions but in only 1 of 16 (6%) PanIN 1A and 1 of 18 (5%) PanIN 1B lesions. Thus, in the multistep progression of pancreatic adenocarcinomas, MUC1 expression within normal intra- and interlobular ducts appears to be decreased in the low-grade PanINs (PanIN 1A and 1B). MUC1 appears to be subsequently re-expressed in the advanced PanIN lesions, and this expression persists into invasive adenocarcinoma. The expression of MUC1 is detected primarily in the apical membranes of normal ducts and PanINs, but only in the latter is occasional cytoplasmic expression also present. The transfection of MUC1-expressing construct into MUC1-negative tumor cell lines results in increased tumorigenicity and invasiveness in vivo (59), whereas functional studies have demonstrated MUC1 being upstream of the oncogenic Grb2-SOS-ras signaling pathway (60, 61). Thus, irrespective of the mechanism, re-expression of MUC1 in advanced PanIN lesions would appear to favor cell growth and invasion.
Unlike MUC1, the expression of the apomucin MUC2 is uncommon in both normal pancreas and in invasive ductal adenocarcinomas (57, 58). In contrast, MUC2 expression is commonly seen in intraductal papillary mucinous neoplasms (IPMNs) and their associated invasive colloid carcinomas (56). Adsay et al. (55) have proposed a dichotomy in the pathways to tumorigenesis within the pancreas, with MUC1-expressing invasive adenocarcinomas arising from PanINs and MUC2-expressing invasive adenocarcinomas arising in the backdrop of IPMNs. Consistent with these previous observations, none of the 66 normal or PanIN lesions in our study expressed MUC2. Thus, MUC2 expression is unlikely to play a significant role in the progression model of ductal adenocarcinoma of the pancreas. The basis for absence of MUC2 expression remains largely unknown, although it may be attributed to epigenetic inactivation by methylation of the MUC2 gene promoter (62).
The apomucin MUC5 is a gastric foveolar mucin that is normally expressed in the gastric epithelium but is down-regulated in the course of intestinal metaplasia (54). In our study, MUC5 was focally or diffusely expressed in 0 of 11 normal pancreatic ducts, in 12 of 16 (75%) PanIN 1A, in 13 of 18 (72%) PanIN 1B, in 13 of 14 (93%) PanIN 2, and in 7 of 7 (100%) PanIN 3 lesions. These findings are comparable to the findings of Kim et al. (63), who detected MUC5 expression in 71–90% of PanIN lesions in regular histologic sections. MUC5 is similar to MUC1 in that it is also expressed in the majority of invasive ductal adenocarcinomas (56, 57, 63). In contrast to MUC1, however, MUC5 is not expressed in normal ducts, but its expression is up-regulated even in the earliest PanIN lesions and persists thereafter in the majority of lesions of all histologic grades.
Novel Tumor Markers (PSCA, Mesothelin, Fascin, and 14-3-3σ)
Our recent global gene expression analyses of pancreatic adenocarcinomas have identified a large number of transcripts that are differentially up-regulated in primary pancreatic cancers and in pancreatic cancer cell lines (13, 14, 15, 64). We have subsequently validated a subset of these genes at the protein level in routine tissue sections of invasive pancreatic adenocarcinomas. For example, prostate stem cell antigen (PSCA) is a glycosylphosphatidyl inositol (GPI)–anchored protein with a limited tissue distribution, being most strongly expressed in the basal cells of the prostate (65). Argani et al. (66) have demonstrated up-regulation of PSCA transcripts in pancreatic adenocarcinomas by SAGE analysis (http://www.ncbi.nlm.nih.gov/SAGE) and have demonstrated overexpression of PSCA gene product in 60% of infiltrating adenocarcinomas by immunohistochemistry. In the current study, focal or diffuse PSCA expression was present in 0 of 11 normal ducts, in 7 of 16 (43%) PanIN 1A, in 5 of 18 (28%) PanIN 1B, in 6 of 14 (43%) PanIN 2, and in 4 of 7 (57%) PanIN 3 lesions. The pattern of immunolabeling was similar to that seen in invasive adenocarcinomas, being primarily cytoplasmic with focal luminal accentuation. Thus, up-regulation of PSCA can be classified as an “early” event in the progression model of pancreas cancer.
Along the same lines, mesothelin is a membrane-bound GPI-anchored protein that plays a role in cell adhesion and is normally most abundantly overexpressed by mesothelium (67). Mesothelin is also overexpressed in ovarian carcinomas and in squamous carcinomas of the lung, cervix, and esophagus (68, 69). Argani et al. (70) reported up-regulation of mesothelin transcripts in SAGE libraries of pancreatic adenocarcinomas and confirmed the overexpression of mesothelin protein in 100% (83% diffuse, 17% focal) of primary pancreatic adenocarcinomas (70). In the current study, focal or diffuse mesothelin labeling was seen in 0 of 11 normal ducts, 1 of 16 (6%) PanIN 1A, 0 of 18 PanIN 1B, 2 of 14 (14%) PanIN 2, and 1 of 7 (14%) PanIN 3 lesions. The labeling pattern of mesothelin resembles that of PSCA, being primarily cytoplasmic with luminal accentuation; in addition, luminal contents are frequently positive. In contrast to PSCA, however, mesothelin expression is uncommon in PanINs but is strongly up-regulated in the process of, or after tissue invasion by neoplastic ductal cells. Thus, mesothelin up-regulation would be classified as a “late” event in the progression model of pancreas cancer.
The human homolog of the sea urchin fascin is an actin-binding cytoskeletal protein that has been implicated in cell motility (71, 72). Iacobuzio-Donahue et al. (13) have reported the up-regulation of fascin transcripts in pancreatic adenocarcinoma tissues and cell lines using oligonucleotide microarrays, whereas Maitra et al. (16) have confirmed the overexpression of fascin protein in 95% of primary pancreatic adenocarcinomas. In the current study, focal or diffuse cytoplasmic fascin expression was seen in 0 of 11 normal ducts, 4 of 16 (25%) PanIN 1A, 5 of 18 (28%) PanIN 1B, 8 of 14 (57%) PanIN 2, and 4 of 7 (57%) PanIN 3 lesions. Therefore, fascin up-regulation would be considered an “intermediate” event in pancreatic adenocarcinoma progression, not uncommon in early PanIN lesions, but substantially up-regulated in advanced PanINs and nearly universal in invasive cancers.
14-3-3σ is a p53-regulated gene that belongs to the family of 14-3-3 proteins, which plays a role in a variety of cellular processes like signal transduction, cell cycle regulation, apoptosis, stress response, and cytoskeletal organization (73, 74). Several lines of evidence reiterate the role of 14-3-3σ as a classic tumor suppressor gene in that loss of expression of 14-3-3σ mRNA or protein has been reported in breast carcinomas (75), squamous cell carcinomas of the head and neck (76), primary bladder cancers (77), and hepatocellular carcinomas (78). Iacobuzio-Donahue et al. (15) demonstrated paradoxical up-regulation of 14-3-3σ transcripts in pancreatic adenocarcinomas by cDNA microarray analysis and confirmed the overexpression of the 14-3-3σ protein in 90% of invasive pancreatic adenocarcinomas. Additionally, they were able to demonstrate cancer-specific hypomethylation of the 14-3-3σ promoter compared with normal pancreatic ductal epithelium, which lacks expression of 14-3-3σ. We have subsequently demonstrated the overexpression of 14-3-3σ in other gastrointestinal adenocarcinomas by immunohistochemistry (unpublished data). In the current study, 0 of 11 normal pancreatic ducts, 2 of 16 (13%) PanIN 1A, 2 of 18 (11%) PanIN 1B, 3 of 14 (21%) PanIN 2, and 6 of 7 (85%) PanIN 3 demonstrated focal or diffuse expression of 14-3-3σ. Thus, up-regulation of 14-3-3σ is another example of a “late” molecular event in the multistep progression of pancreas cancers, possibly occurring in concert with p53 abrogation and/or significant chromosomal instability within the ductal epithelium.
In summary, we have examined 14 cellular proteins encompassing a variety of cellular pathways and functions in the precursor lesions of pancreatic ductal adenocarcinomas using a TMA-based approach. Although the numbers of PanIN lesions in individual histologic categories, particularly PanIN 3, are small, we have demonstrated that frequency of abnormalities for a given marker using TMAs is quite comparable, if not remarkably similar, to the frequency detected using routine histologic sections. This study underscores the overall reliability of a TMA-based approach demonstrated in cancers and extends this concept to the study of precancerous lesions as well. Needless to say, the logistical benefits and cost-effectiveness of studying >50 ductal lesions on a single slide are enormous. In addition, the use of a TMA approach allows one to compare multiple parameters in the same precancerous lesions, a prerequisite for dissecting the molecular pathways to carcinogenesis. Figure 3 demonstrates our current understanding of molecular abnormalities in the multistep progression model of pancreas cancer (“PanINgram”) and underscores the tremendous advances made in our understanding of these precursor lesions since the earliest version of this illustration appeared, merely 3 years ago (79). The separation of early molecular events in PanIN lesions (up-regulation of PSCA) from late changes (up-regulation of mesothelin and 14-3-3σ) demonstrated in this study will hopefully lead to rational early-detection strategies in patients at risk for developing pancreatic cancer (for example, patients belonging to high-risk pancreatic cancer families or patients with one of the known pancreas cancer syndromes). Thus, one could envision that in such patients undergoing screening for pancreas cancer, the presence of elevated mesothelin levels in the pancreatic juice is likely to suggest a more advanced ductal lesion, if not occult carcinoma, as compared with the presence of elevated PSCA alone. Invasive pancreatic adenocarcinoma, once established, is almost always fatal, and therefore, the systematic identification and targeting of molecular abnormalities in the precursor lesions of invasive cancer remains one of our strongest avenues for combating this lethal disease.

A “PanINgram” illustrating our current understanding of the molecular changes in the multistep progression model of pancreas adenocarcinomas.
References
Greenlee RT, Hill-Harmon MB, Murray T, Thun M . Cancer statistics, 2001. CA Cancer J Clin 2001; 51: 15–36.
Wilentz RE, Hruban RH . Pathology of cancer of the pancreas. Surg Oncol Clin N Am 1998; 7: 43–65.
Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol 2001; 25: 579–86.
Hruban RH, Wilentz RE, Goggins M, Offerhaus GJ, Yeo CJ, Kern SE . Pathology of incipient pancreatic cancer. Ann Oncol 1999; 10: 9–11.
Hruban RH, Wilentz RE, Kern SE . Genetic progression in the pancreatic ducts. Am J Pathol 2000; 156: 1821–5.
Luttges J, Galehdari H, Brocker V, Schwarte-Waldhoff I, Henne-Bruns D, Kloppel G, et al. Allelic loss is often the first hit in the biallelic inactivation of the p53 and DPC4 genes during pancreatic carcinogenesis. Am J Pathol 2001; 158: 1677–83.
Wilentz RE, Geradts J, Maynard R, Offerhaus GJ, Kang M, Goggins M, et al. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res 1998; 58: 4740–4.
Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res 2000; 60: 2002–6.
Yamano M, Fujii H, Takagaki T, Kadowaki N, Watanabe H, Shirai T . Genetic progression and divergence in pancreatic carcinoma. Am J Pathol 2000; 156: 2123–33.
van Heek NT, Meeker AK, Kern SE, Yeo CJ, Lillemoe KD, Cameron JL, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol 2002; 161: 1541–7.
Goggins M, Hruban RH, Kern SE . BRCA2 is inactivated late in the development of pancreatic intraepithelial neoplasia: evidence and implications. Am J Pathol 2000; 156: 1767–71.
Hruban RH, Canto MI, Yeo CJ . Prevention of pancreatic cancer and strategies for management of familial pancreatic cancer. Dig Dis 2001; 19: 76–84.
Iacobuzio-Donahue CA, Maitra A, Shen-Ong GL, van Heek T, Ashfaq R, Meyer R, et al. Discovery of novel tumor markers of pancreatic cancer using global gene expression technology. Am J Pathol 2002; 160: 1239–49.
Ryu B, Jones J, Blades NJ, Parmigiani G, Hollingsworth MA, Hruban RH, et al. Relationships and differentially expressed genes among pancreatic cancers examined by large-scale serial analysis of gene expression. Cancer Res 2002; 62: 819–26.
Iacobuzio-Donahue C, Maitra A, Olsen M, Lowe A, van Heek NT, Rosty C, et al. Exploration of global expression patterns in pancreatic adenocarcinomas using cDNA microarrays. Am J Pathol 2003; 162: 151–62.
Maitra A, Iacobuzio-Donahue C, Rahman A, Sohn TA, Argani P, Meyer R, et al. Immunohistochemical validation of a novel epithelial and a novel stromal marker of pancreatic ductal adenocarcinoma identified by global expression microarrays: sea urchin fascin homolog and heat shock protein 47. Am J Clin Pathol 2002; 118: 52–9.
Rubin MA, Zhou M, Dhanasekaran SM, Varambally S, Barrette TR, Sanda MG, et al. alpha-Methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. JAMA 2002; 287: 1662–70.
Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, et al. Delineation of prognostic biomarkers in prostate cancer. Nature 2001; 412: 822–6.
Bouras T, Southey MC, Chang AC, Reddel RR, Willhite D, Glynne R, et al. Stanniocalcin 2 is an estrogen-responsive gene coexpressed with the estrogen receptor in human breast cancer. Cancer Res 2002; 62: 1289–95.
Torhorst J, Bucher C, Kononen J, Haas P, Zuber M, Kochli OR, et al. Tissue microarrays for rapid linking of molecular changes to clinical endpoints. Am J Pathol 2001; 159: 2249–56.
Schraml P, Kononen J, Bubendorf L, Moch H, Bissig H, Nocito A, et al. Tissue microarrays for gene amplification surveys in many different tumor types. Clin Cancer Res 1999; 5: 1966–75.
Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med 1998; 4: 844–7.
Maitra A, Roberts H, Weinberg AG, Geradts J . Loss of p16(INK4a) expression correlates with decreased survival in pediatric osteosarcomas. Int J Cancer 2001; 95: 34–8.
Maitra A, Roberts H, Weinberg AG, Geradts J . Aberrant expression of tumor suppressor proteins in the Ewing family of tumors. Arch Pathol Lab Med 2001; 125: 1207–12.
Abraham SC, Klimstra DS, Wilentz RE, Yeo CJ, Conlon K, Brennan M, et al. Solid-pseudopapillary tumors of the pancreas are genetically distinct from pancreatic ductal adenocarcinomas and almost always harbor beta-catenin mutations. Am J Pathol 2002; 160: 1361–9.
Scarpa A, Capelli P, Mukai K, Zamboni G, Oda T, Iacono C, et al. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol 1993; 142: 1534–43.
Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994; 54: 3025–33.
Barton CM, Staddon SL, Hughes CM, Hall PA, O'Sullivan C, Kloppel G, et al. Abnormalities of the p53 tumour suppressor gene in human pancreatic cancer. Br J Cancer 1991; 64: 1076–82.
Ruggeri B, Zhang SY, Caamano J, DiRado M, Flynn SD, Klein-Szanto AJ . Human pancreatic carcinomas and cell lines reveal frequent and multiple alterations in the p53 and Rb-1 tumor-suppressor genes. Oncogene 1992; 7: 1503–11.
Zhang SY, Ruggeri B, Agarwal P, Sorling AF, Obara T, Ura H, et al. Immunohistochemical analysis of p53 expression in human pancreatic carcinomas. Arch Pathol Lab Med 1994; 118: 150–4.
DiGiuseppe JA, Hruban RH, Goodman SN, Polak M, van den Berg FM, Allison DC, et al. Overexpression of p53 protein in adenocarcinoma of the pancreas. Am J Clin Pathol 1994; 101: 684–8.
Apple SK, Hecht JR, Lewin DN, Jahromi SA, Grody WW, Nieberg RK . Immunohistochemical evaluation of K-ras, p53, and HER-2/neu expression in hyperplastic, dysplastic, and carcinomatous lesions of the pancreas: evidence for multistep carcinogenesis. Hum Pathol 1999; 30: 123–9.
Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996; 271: 350–3.
Wilentz RE, Su GH, Dai JL, Sparks AB, Argani P, Sohn TA, et al. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol 2000; 156: 37–43.
Hilgers W, Kern SE . Molecular genetic basis of pancreatic adenocarcinoma. Genes Chromosomes Cancer 1999; 26: 1–12.
Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997; 57: 3126–30.
Fukushima N, Sato N, Ueki T, Rosty C, Walter KM, Wilentz RE, et al. Aberrant methylation of preproenkephalin and p16 genes in pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am J Pathol 2002; 160: 1573–81.
Yu Q, Geng Y, Sicinski P . Specific protection against breast cancers by cyclin D1 ablation. Nature 2001; 411: 1017–21.
Anton RC, Coffey DM, Gondo MM, Stephenson MA, Brown RW, Cagle PT . The expression of cyclins D1 and E in predicting short-term survival in squamous cell carcinoma of the lung. Mod Pathol 2000; 13: 1167–72.
Palmqvist R, Stenling R, Oberg A, Landberg G . Expression of cyclin D1 and retinoblastoma protein in colorectal cancer. Eur J Cancer 1998; 34: 1575–81.
Raffeld M, Jaffe ES . bcl-1, t(11;14), and mantle cell-derived lymphomas. Blood 1991; 78: 259–63.
Gansauge S, Gansauge F, Ramadani M, Stobbe H, Rau B, Harada N, et al. Overexpression of cyclin D1 in human pancreatic carcinoma is associated with poor prognosis. Cancer Res 1997; 57: 1634–7.
Poch B, Gansauge F, Schwarz A, Seufferlein T, Schnelldorfer T, Ramadani M, et al. Epidermal growth factor induces cyclin D1 in human pancreatic carcinoma: evidence for a cyclin D1-dependent cell cycle progression. Pancreas 2001; 23: 280–7.
Biankin AV, Kench JG, Morey AL, Lee CS, Biankin SA, Head DR, et al. Overexpression of p21(WAF1/CIP1) is an early event in the development of pancreatic intraepithelial neoplasia. Cancer Res 2001; 61: 8830–7.
Kornmann M, Ishiwata T, Itakura J, Tangvoranuntakul P, Beger HG, Korc M . Increased cyclin D1 in human pancreatic cancer is associated with decreased postoperative survival. Oncology 1998; 55: 363–9.
Kornmann M, Danenberg KD, Arber N, Beger HG, Danenberg PV, Korc M . Inhibition of cyclin D1 expression in human pancreatic cancer cells is associated with increased chemosensitivity and decreased expression of multiple chemoresistance genes. Cancer Res 1999; 59: 3505–11.
Gerdes B, Ramaswamy A, Simon B, Pietsch T, Bastian D, Kersting M, et al. Analysis of beta-catenin gene mutations in pancreatic tumors. Digestion 1999; 60: 544–8.
Abraham SC, Wu TT, Klimstra DS, Finn LS, Lee JH, Yeo CJ, et al. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am J Pathol 2001; 159: 1619–27.
Abraham SC, Wu TT, Hruban RH, Lee JH, Yeo CJ, Conlon K, et al. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am J Pathol 2002; 160: 953–62.
Makinen K, Hakala T, Lipponen P, Alhava E, Eskelinen M . Clinical contribution of bcl-2, p53 and Ki-67 proteins in pancreatic ductal adenocarcinoma. Anticancer Res 1998; 18: 615–8.
Linder S, Parrado C, Falkmer UG, Blasjo M, Sundelin P, von Rosen A . Prognostic significance of Ki-67 antigen and p53 protein expression in pancreatic duct carcinoma: a study of the monoclonal antibodies MIB-1 and DO-7 in formalin-fixed paraffin-embedded tumour material. Br J Cancer 1997; 76: 54–9.
Klein WM, Hruban RH, Klein-Szanto AJ, Wilentz RE . Direct correlation between proliferative activity and dysplasia in pancreatic intraepithelial neoplasia (PanIN): additional evidence for a recently proposed model of progression. Mod Pathol 2002; 15: 441–7.
Yonezawa S, Sato E . Expression of mucin antigens in human cancers and its relationship with malignancy potential. Pathol Int 1997; 47: 813–30.
de Bolos C, Real FX, Lopez-Ferrer A . Regulation of mucin and glycoconjugate expression: from normal epithelium to gastric tumors. Front Biosci 2001; 6: D1256–63.
Adsay NV, Merati K, Andea A, Sarkar F, Hruban RH, Wilentz RE, et al. The dichotomy in the preinvasive neoplasia to invasive carcinoma sequence in the pancreas: differential expression of MUC1 and MUC2 supports the existence of two separate pathways of carcinogenesis. Mod Pathol 2002; 15: 1087–95.
Luttges J, Zamboni G, Longnecker D, Kloppel G . The immunohistochemical mucin expression pattern distinguishes different types of intraductal papillary mucinous neoplasms of the pancreas and determines their relationship to mucinous noncystic carcinoma and ductal adenocarcinoma. Am J Surg Pathol 2001; 25: 942–8.
Terada T, Ohta T, Sasaki M, Nakanuma Y, Kim YS . Expression of MUC apomucins in normal pancreas and pancreatic tumours. J Pathol 1996; 180: 160–5.
Hollingsworth MA, Strawhecker JM, Caffrey TC, Mack DR . Expression of MUC1, MUC2, MUC3 and MUC4 mucin mRNAs in human pancreatic and intestinal tumor cell lines. Int J Cancer 1994; 57: 198–203.
Suwa T, Hinoda Y, Makiguchi Y, Takahashi T, Itoh F, Adachi M, et al. Increased invasiveness of MUC1 and cDNA-transfected human gastric cancer MKN74 cells. Int J Cancer 1998; 76: 377–82.
Pandey P, Kharbanda S, Kufe D . Association of the DF3/MUC1 breast cancer antigen with Grb2 and the Sos/Ras exchange protein. Cancer Res 1995; 55: 4000–3.
Meerzaman D, Shapiro PS, Kim KC . Involvement of the MAP kinase ERK2 in MUC1 mucin signaling. Am J Physiol Lung Cell Mol Physiol 2001; 281: L86–91.
Siedow A, Szyf M, Gratchev A, Kobalz U, Hanski ML, Bumke-Vogt C, et al. De novo expression of the Muc2 gene in pancreas carcinoma cells is triggered by promoter demethylation. Tumour Biol 2002; 23: 54–60.
Kim GE, Bae HI, Park HU, Kuan SF, Crawley SC, Ho JJ, et al. Aberrant expression of MUC5AC and MUC6 gastric mucins and sialyl Tn antigen in intraepithelial neoplasms of the pancreas. Gastroenterology 2002; 123: 1052–60.
Ryu B, Jones J, Hollingsworth MA, Hruban RH, Kern SE . Invasion-specific genes in malignancy: serial analysis of gene expression comparisons of primary and passaged cancers. Cancer Res 2001; 61: 1833–8.
Reiter RE, Gu Z, Watabe T, Thomas G, Szigeti K, Davis E, et al. Prostate stem cell antigen: a cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci U S A 1998; 95: 1735–40.
Argani P, Rosty C, Reiter RE, Wilentz RE, Murugesan SR, Leach SD, et al. Discovery of new markers of cancer through serial analysis of gene expression: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res 2001; 61: 4320–4.
Chang K, Pastan I . Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci U S A 1996; 93: 136–40.
Fan D, Yano S, Shinohara H, Solorzano C, Van Arsdall M, Bucana CD, et al. Targeted therapy against human lung cancer in nude mice by high-affinity recombinant antimesothelin single-chain Fv immunotoxin. Mol Cancer Ther 2002; 1: 595–600.
Hassan R, Lerner MR, Benbrook D, Lightfoot SA, Brackett DJ, Wang QC, et al. Antitumor activity of SS(dsFv)PE38 and SS1(dsFv)PE38, recombinant antimesothelin immunotoxins against human gynecologic cancers grown in organotypic culture in vitro. Clin Cancer Res 2002; 8: 3520–6.
Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res 2001; 7: 3862–8.
Grothey A, Hashizume R, Sahin AA, McCrea PD . Fascin, an actin-bundling protein associated with cell motility, is upregulated in hormone receptor negative breast cancer. Br J Cancer 2000; 83: 870–3.
Hu W, McCrea PD, Deavers M, Kavanagh JJ, Kudelka AP, Verschraegen CF . Increased expression of fascin, motility associated protein, in cell cultures derived from ovarian cancer and in borderline and carcinomatous ovarian tumors. Clin Exp Metastasis 2000; 18: 83–8.
van Hemert MJ, Steensma HY, van Heusden GP . 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. Bioessays 2001; 23: 936–46.
Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, et al. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell 1997; 1: 3–11.
Ferguson AT, Evron E, Umbricht CB, Pandita TK, Chan TA, Hermeking H, et al. High frequency of hypermethylation at the 14-3-3 sigma locus leads to gene silencing in breast cancer. Proc Natl Acad Sci U S A 2000; 97: 6049–54.
Gasco M, Bell AK, Heath V, Sullivan A, Smith P, Hiller L, et al. Epigenetic inactivation of 14-3-3 sigma in oral carcinoma: association with p16(INK4a) silencing and human papillomavirus negativity. Cancer Res 2002; 62: 2072–6.
Ostergaard M, Rasmussen HH, Nielsen HV, Vorum H, Orntoft TF, Wolf H, et al. Proteome profiling of bladder squamous cell carcinomas: identification of markers that define their degree of differentiation. Cancer Res 1997; 57: 4111–7.
Iwata N, Yamamoto H, Sasaki S, Itoh F, Suzuki H, Kikuchi T, et al. Frequent hypermethylation of CpG islands and loss of expression of the 14-3-3 sigma gene in human hepatocellular carcinoma. Oncogene 2000; 19: 5298–302.
Hruban RH, Goggins M, Parsons J, Kern SE . Progression model for pancreatic cancer. Clin Cancer Res 2000; 6: 2969–72.
Acknowledgements
Supported in part by the Specialized Program of Research Excellence (SPORE) in Gastrointestinal Cancer, Grant P50-CA62924, and the Michael Rolfe Foundation for Pancreatic Cancer Research.
The authors gratefully acknowledge Helen Fedor and Marcela Southerland for assistance with TMA construction.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Maitra, A., Adsay, N., Argani, P. et al. Multicomponent Analysis of the Pancreatic Adenocarcinoma Progression Model Using a Pancreatic Intraepithelial Neoplasia Tissue Microarray. Mod Pathol 16, 902–912 (2003). https://doi.org/10.1097/01.MP.0000086072.56290.FB
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1097/01.MP.0000086072.56290.FB
Keywords
This article is cited by
-
Calligraphy tool offers clues to the origin of pancreatic cancer
Nature (2023)
-
S100PBP is regulated by mutated KRAS and plays a tumour suppressor role in pancreatic cancer
Oncogene (2023)
-
p53 inhibitor iASPP is an unexpected suppressor of KRAS and inflammation-driven pancreatic cancer
Cell Death & Differentiation (2023)
-
Mouse models of Kras activation in gastric cancer
Experimental & Molecular Medicine (2022)
-
Cancer Immunoprevention: Current Status and Future Directions
Archivum Immunologiae et Therapiae Experimentalis (2021)
