
Abstract
Patients with acute kidney injury (AKI) frequently suffer from extra-renal complications including hepatic dysfunction and systemic inflammation. We aimed to determine the mechanisms of AKI-induced hepatic dysfunction and systemic inflammation. Mice subjected to AKI (renal ischemia reperfusion (IR) or nephrectomy) rapidly developed acute hepatic dysfunction and suffered significantly worse hepatic IR injury. After AKI, rapid peri-portal hepatocyte necrosis, vacuolization, neutrophil infiltration and pro-inflammatory mRNA upregulation were observed suggesting an intestinal source of hepatic injury. Small intestine histology after AKI showed profound villous lacteal capillary endothelial apoptosis, disruption of vascular permeability and epithelial necrosis. After ischemic or non-ischemic AKI, plasma TNF-α, IL-17A and IL-6 increased significantly. Small intestine appears to be the source of IL-17A, as IL-17A levels were higher in the portal circulation and small intestine compared with the levels measured from the systemic circulation and liver. Wild-type mice treated with neutralizing antibodies against TNF-α, IL-17A or IL-6 or mice deficient in TNF-α, IL-17A, IL-17A receptor or IL-6 were protected against hepatic and small intestine injury because of ischemic or non-ischemic AKI. For the first time, we implicate the increased release of IL-17A from small intestine together with induction of TNF-α and IL-6 as a cause of small intestine and liver injury after ischemic or non-ischemic AKI. Modulation of the inflammatory response and cytokine release in the small intestine after AKI may have important therapeutic implications in reducing complications arising from AKI.
Similar content being viewed by others

Main
Acute kidney injury (AKI) is a major clinical problem.1 However, the morbidity and mortality from AKI is very high and remains virtually unchanged for the past 50 years in part because of a high incidence of extra-renal complications.1, 2 In particular, hepatic dysfunction is very frequent in patients suffering from AKI. Furthermore, development of liver injury in patients with AKI frequently leads to other extra-renal complications including intestinal barrier disruption, respiratory failure and the systemic inflammatory response syndrome with the eventual development of sepsis and a multi-organ failure.3, 4, 5 These extra-renal systemic complications secondary to AKI are the leading causes of mortality in the intensive care unit.6 Indeed, clinical studies show that patients with isolated AKI have significantly better prognosis than patients with AKI plus extra-renal organ dysfunction.7 In this study, we aimed to determine the mechanisms of ischemic or non-ischemic AKI-induced hepatic dysfunction.
Inflammation has a major function in the progression of AKI, and recent studies have shown that innate immunity contributes to the pathogenesis of renal injury.8, 9 Pro-inflammatory cytokines including TNF-α and IL-6 generated by injured renal tubule cells or from extra-renal cells (eg T-lymphocytes) have been implicated as the major contributors of renal ischemia reperfusion (IR) injury.10, 11, 12 Indeed, increased circulating pro-inflammatory cytokine levels were detected in mice and human beings with AKI.13 Taking these previous observations together, we tested the hypothesis that induction of AKI results in increased circulating pro-inflammatory cytokines (TNF-α and IL-6) and that these cytokines directly cause hepatic dysfunction. We used murine models of ischemic (renal IR injury) or non-ischemic (nephrectomy) AKI and examined (1) the effects of ischemic or non-ischemic AKI on liver structure and function, (2) whether circulating pro-inflammatory cytokines TNF-α and/or IL-6 increased after AKI and (3) whether the increased TNF-α and/or IL-6 directly contribute to hepatic injury after AKI using neutralizing antibodies and mice deficient for TNF-α or IL-6. We showed rapid hepatic injury, inflammation with increased systemic TNF-α as well as IL-6 after AKI. In particular, we observed striking hepatocyte vacuolization, necrosis and neutrophil infiltration concentrated near the portal venous drainage. Therefore, we also examined for small intestine injury, inflammatory changes and cytokine generation after ischemic or non-ischemic AKI. Our findings show that AKI rapidly results in intestinal generation of IL-17A leading to production of IL-6 and TNF-α to initiate a systemic-inflammatory response and acute hepatic dysfunction.
MATERIALS AND METHODS
Detailed methods (surgery and anesthesia protocols, immunohistochemistry, vascular permeability assays, DNA laddering assay, terminal deoxynucleotidyl transferase biotin-dUTP nick end-labeling (TUNEL) staining, RNA isolation and PCR) are available as Supplementary Methods.
Mice
All mice strains were bred or purchased on a C57BL/6 background. Male C57BL/6 mice (20–25 g) were obtained from Harlan, Indianapolis, IN, USA. IL-17A-deficient mice (IL-17A−/−) were obtained as a gift from Yoichiro Iwakura (University of Tokyo, Tokyo, Japan) and IL-17A receptor-deficient mice (IL-17R−/−) were generously provided by Amgen (Cambridge, MA, USA) through Taconic Farms (Hudson, NY, USA).14 In addition, C57BL/6J (TNF-α+/+ and IL-6+/+), TNF-α gene-deficient (TNF-α−/−) male mice (strain B6.129S-Tnftm1Gkl/J; stock no. 005540) and IL-6 gene-deficient (IL-6−/−) male mice (B6.129S2-Il6tm1Kopf/J; stock no. 002650) were purchased from the Jackson Laboratory (Bar Harbor, ME).
Murine Model of Ischemic or Non-ischemic AKI and Hepatic IR
All protocols were approved by the Institutional Animal Care and Use Committee of Columbia University. Male mice under pentobarbital anesthesia were subjected to sham-kidney manipulations, ischemic AKI (20 or 30 min renal ischemia) or non-ischemic AKI (unilateral or bilateral nephrectomy) as described.15, 16 Separate cohorts of mice were subjected to sham operation or 45 min liver IR injury as described previously17 (Supplementary Methods). During hepatic ischemia, we performed sham-kidney manipulations, 20 min renal ischemia, unilateral nephrectomy or bilateral nephrectomy. Five hours after reperfusion, the liver and small intestine tissues were collected to examine tissue injury, pro-inflammatory cytokine mRNA expression (with RT–PCR), leukocyte infiltration (with immunohistochemistry for neutrophils, T-lymphocytes or macrophages), vascular permeability (Evans blue dye (EBD) extravasation) and apoptosis (with terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling staining and DNA laddering). We also collected portal venous and systemic plasma as well as the liver and small intestine tissues for the measurement of alanine aminotransferase (ALT), bilirubin, creatinine and cytokine levels (TNF-α, IL-6 and IL-17A) at 5 and 24 h after reperfusion. Our previous studies showed that plasma levels of cytokines, creatinine, neutrophil infiltration and ALT peaked between 5 and 24 h after reperfusion.16, 17, 18
Plasma ALT Activity, Bilirubin and Creatinine Level
The plasma ALT activities were measured using the InfinityTM ALT assay kit according to the manufacturer's instructions (Thermo Fisher Scientific, Waltham, MA, USA). The plasma bilirubin levels were measured by the QuantiChromTM Bilirubin Assay kit according to the manufacturer's instructions (BioAssay Systems, Hayward, CA, USA). Plasma creatinine was measured by an enzymatic creatinine reagent kit according to the manufacturer's instructions (Thermo Fisher Scientific). This method of creatinine measurement largely eliminates the interferences from mouse plasma chromagens well known to the Jaffe method.19
Histological Analysis of Hepatic and Intestine Injury
For histological preparations, tissues from liver and small intestine (jejunum and ileum) were washed in ice-cold PBS and fixed overnight in 10% formalin. After automated dehydration through a graded alcohol series, tissues were embedded in paraffin, sectioned at 4 μM and stained with hematoxylin–eosin (H&E). Liver H&E sections were evaluated blindly by a pathologist (VDD’A). Intestine H&E sections were also blindly evaluated (by VDD’A) for intestinal epithelial cell necrosis, development of a necrotic pannus over the mucosal surface, villous endothelial cell apoptosis and swelling and blunting of villi because of villous mononuclear cell mucosal inflammation and edema.
Intestinal Permeability
Intestinal permeability was assessed by enteral administration of FITC-dextran 4000 (200 mg/kg) 5 min before induction of AKI as described.20
Enzyme-Linked Immunosorbent Assay for Plasma TNF-α, IL-6 or IL-17A
After 5 and 24 h induction of AKI, plasma, liver and small intestine TNF-α, IL-6 and IL-17A levels were measured with mouse-specific ELISA kits according to the manufacturer's instructions (eBiosciences, San Diego, CA, USA). Liver and intestine tissues were homogenized in ice-cold RIPA buffer (150 mM NaCl, 50 mM Tris–HCl, 1 mM EDTA and 1% Triton-X [pH 7.4]) and samples processed for mouse-specific ELISA kits.
Critical Functions for TNF-α, IL-6 and IL-17A in Generating Liver and Intestine Injury After Ischemic or Non-ischemic AKI
To test the hypothesis that TNF-α, IL-6 and/or IL-17A have important functions in inducing liver and intestine injury after ischemic or non-ischemic AKI, we used complementary approaches using neutralizing antibodies specific for TNF-α, IL-6 and IL-17A (eBiosciences) and TNF-α, IL-6, IL-17A and IL-17A receptor (IL-17R)-deficient mice (sources described above). For the neutralizing antibody studies, C57BL/6 mice were injected with 0.1 mg of TNF-α, IL-6 or IL-17A antibody i.v. immediately after induction of ischemic or non-ischemic AKI. TNF-α−/−, IL-6−/−, IL-17A−/− or IL-17R−/− mice were subjected to ischemic or non-ischemic AKI as described above. A separate cohort of mice were treated with a combination of recombinant mouse TNF-α (2 μg/mouse), IL-6 (1 μg/mouse) plus IL-17A (1 μg/mouse) in lieu of AKI induction to determine whether these cytokine injections alone can result in hepatic injury.
Assessment of Liver and Intestine Inflammation
Liver and intestine inflammation was determined by detection of neutrophil, (7/4), T-lymphocyte (CD-3) and macrophage (F4/80) infiltration by immunohistochemistry 5 or 24 h after ischemic and non-ischemia AKI as described previously17 and by measuring mRNA encoding markers of inflammation, including intercellular adhesion molecule-1 (ICAM-1), monocyte chemoattractive protein-1 (MCP-1), macrophage-inflammatory protein-2 (MIP-2) and TNF-α, IL-6, IL-17A and keratinocyte-derived cytokine (KC), 5 h after ischemic or non-ischemic AKI as described17 (Table 1) (Supplementary Methods).
Vascular Permeability of Liver and Intestine Tissues
Changes in liver and kidney vascular permeability were assessed by quantitating extravasation of EBD into the tissue as described21 (Supplementary Methods).
Detection of Liver and Intestine Apoptosis
We used two independent assays to assess the degree of liver and intestine apoptosis 5 h after sham surgery or AKI: in situ TUNEL assay and the detection of DNA laddering18, 22 (Supplementary Methods). For DNA laddering, liver and small intestine tissues were removed, apoptotic DNA fragments were extracted according to the methods of Herrmann et al.23 This method of DNA extraction selectively isolates apoptotic, fragmented DNA and leaves behind the intact chromatin.
Statistical Analysis
The data were analyzed with t-test when means between two groups were compared or with one-way (eg plasma creatinine or ALT) ANOVA plus Tukey post hoc multiple comparison test to compare mean values across multiple treatment groups. In all cases, P<0.05 was taken to indicate significance. All data are expressed as mean±s.e.m.
Reagents and Protein Determination
Recombinant mice TNF-α, IL-6 and IL-17A were obtained from eBiosciences. Unless otherwise specified, all chemicals were obtained from Sigma (St Louis, MO, USA). Protein contents were determined with a bicinchoninic acid protein assay kit (Pierce Chemical, Rockford, IL, USA), using bovine serum albumin as a standard.
RESULTS
Acute Renal and Hepatic Dysfunction After Ischemic or Non-ischemic AKI
At 20 or 30 min of renal IR injury led to significant and graded rise in serum creatinine relative to sham-operated animals at 5 or 24 h (Figure 1a). Unilateral nephrectomy resulted in a small and transient rise in plasma creatinine after 5 h, which returned to sham-operated levels at 24 h after surgery. In contrast, bilateral nephrectomy resulted in significant elevations in serum creatinine levels at 5 and 24 h after injury (Figure 1a).

Acute renal and hepatic dysfunction after ischemic or non-ischemic AKI. Plasma creatinine (a), alanine aminotransferase (b) and bilirubin (c) levels after ischemic (renal IR) or non-ischemic (nephrectomy) AKI induction in mice. Mice were subjected to sham operation (Sham, N=4), unilateral nephrectomy (UNx, N=6–8), bilateral nephrectomy (BNx, N=6–8), 20 min renal IR (RIR, N=8) or 30 min renal IR (N=8). *P<0.05 vs sham-operated mice. Data presented as mean±s.e.m.
Sham-operated C57BL/6 mice had normal plasma ALT at 5 h (ALT=53±6 mg/dl, N=5 and Cr=0.48±0.02 mg/dl, N=5) and 24 h after surgery (ALT=61±8 U/L, N=4 and Cr=0.50±0.03 mg/dl, N=4; Figures 1a and b). However, mice developed acute hepatic dysfunction at 5 h after ischemic or non-ischemic AKI injury with significantly higher plasma ALT levels (Figure 1b; P<0.05 compared with sham-operated mice). In mice subjected to 30 min renal IR, the plasma ALT continued to increase, whereas in other groups, the ALT levels peaked at 5 h and then declined (Figure 1b). We also measured plasma bilirubin levels as a marker of hepatic dysfunction. As shown in Figure 1c, we detected significant rises in plasma bilirubin 5 and 24 h after bilateral nephrectomy or 30 min renal IR.
Acute Hepatic Injury with Increased Hepatic Necrosis and Vacuolization After Ischemic or Non-ischemic AKI
Liver histology was also assessed by H&E staining of liver sections. As shown in Figure 2a, sham-operated mice had normal liver histology. Five hours after 30 min renal IR, nuclear and cytoplasmic degenerative changes, cellular vacuolization, leukocyte infiltration and congestion were observed (Figures 2b and c). Similar hepatic injury was observed 5 h after 20 min renal IR, unilateral or bilateral nephrectomy (data not shown). The severity of tissue injury was increased at 24 h after 30 min renal IR (as reflected in plasma ALT) as manifested by the extent of cellular degenerative changes including individual hepatocyte necrosis and focal apoptotic, pyknotic nuclei. Some degree of centrilobular necrosis was observed in this group. At 24 h after bilateral nephrectomy, the severity of injury was milder relative to 5 h, although areas of congestion could be detected. Vascular congestion and leukocyte (mainly neutrophil) infiltration were also detected after bilateral nephrectomy or 30 min renal IR.

Acute hepatic injury with increased hepatic necrosis and vacuolization after ischemic AKI. Representative photomicrographs of liver from six experiments (hematoxylin and eosin staining, magnification, × 600) of mice subjected to sham operation (Sham) or to 30 min renal ischemia and 5 h of reperfusion (30 min renal IR). Sham-operated animals show normal-appearing hepatocyte parenchyma (a). Five hours after 30 min renal IR (b, c), nuclear and cytoplasmic degenerative changes, centrilobular necrosis (b, arrows), marked hepatocyte vacuolization (c) and congestion were observed.
Increased Hepatic Inflammation, Apoptosis and Vascular Permeability After Ischemic or Non-ischemic AKI
Hepatic as well as intestinal inflammation, apoptosis and vascular permeability were assessed at 5 h after ischemic or non-ischemic AKI, as plasma ALT and bilirubin levels were significantly elevated at this time point. With RT–PCR, we measured the expression of pro-inflammatory cytokine mRNAs in the liver 5 h after 30 min renal IR or bilateral nephrectomy (Figure 3a). We show significant upregulation of all of the pro-inflammatory mRNAs examined (ICAM-1, TNF-α, IL-6, IL-17A, KC, MCP-1 and MIP-2) in the liver 5 h after 30 min renal IR or bilateral nephrectomy. We also show significantly increased PMN infiltration (dark brown stain, especially near the portal venous drainage) in mice subjected to 30 min renal IR or bilateral nephrectomy (Figure 3b). In sham-operated mice, we were unable to detect any neutrophils in liver. Five hours after unilateral (13±3 neutrophils/field, × 200 magnification, N=5) or bilateral (24±4 neutrophils/field, × 200 magnification, N=5) nephrectomy, increased neutrophil infiltration occurred. Similarly, we observed increased neutrophil infiltration into the liver 5 h after 20 min (23±6 neutrophils/field, × 200 magnification, N=5) or 30 min (28±6 neutrophils/field, × 200 magnification, N=5) renal IR. In contrast, we failed to observe increased T-lymphocyte (CD3) or macrophage (F4/80) infiltration after ischemic or non-ischemic AKI compared with sham-operated animals (data not shown).

Increased hepatic inflammation after ischemic or non-ischemic AKI. (a) Representative gel images and band intensity quantifications of semi-quantitative RT–PCR of the pro-inflammatory markers ICAM-1, TNF-α, IL-6, IL-17A, KC, MCP-1 and MIP-2 from liver tissues of mice subjected to sham operation (Sham), bilateral nephrectomy (BNx) or 30 min renal IR (RIR). Liver tissues were harvested 5 h after sham operation or AKI induction. *P<0.05 vs sham-operated mice. Error bars represent 1 s.e.m. (b) Representative photomicrographs ( × 200 and × 400) of four experiments of immunohistochemistry for neutrophil infiltration (dark brown stain) in the liver tissues harvested from mice subjected to sham operation (Sham) or to bilateral nephrectomy (BNx) 5 h prior.
We also observed increased number of apoptotic nuclei in the liver of mice subjected to 30 min renal IR (TUNEL-positive cells; Supplementary Figure 1) or bilateral nephrectomy (data not shown). TUNEL-positive cells were heavily localized to the peri-portal area. DNA laddering experiments confirmed increased apoptosis in the liver after 30 min renal IR or bilateral nephrectomy (Supplementary Figure 2). We also measured liver vascular permeability after 30 min renal IR or bilateral nephrectomy with EBD injection. EBD binds to plasma proteins and its appearance in extravascular tissues reflects an increase in vascular permeability. Analysis showing increased EBD extravasations in livers of mice subjected to ischemic (30 min renal IR) or non-ischemic (bilateral nephrectomy) AKI is shown in Figure 4.

Increased hepatic vascular permeability after ischemic or non-ischemic AKI. (Left) Representative photographs of liver isolated from mice subjected to sham operation or to bilateral nephrectomy (BNx) 5 h prior and injected with Evans blue dye (EBD). (Right) Quantification of liver EBD extravasations in mice subjected to sham operation (N=6), bilateral nephrectomy (BNx) or 30 min renal IR (RIR). Five hours after surgery, EBD was extracted in formamide and the amount of extravasated EBD in the liver was calculated against a standard curve. *P<0.05 vs sham-operated mice. Error bars represent 1 s.e.m.
Ischemic or Non-ischemic AKI Exacerbates Hepatic IR Injury
We also induced liver IR injury in mice subjected to sham operation, unilateral or bilateral nephrectomy or 20 min renal IR injury. We determined that mice subjected to hepatic IR together with ischemic (20 min renal IR) or non-ischemic (unilateral or bilateral nephrectomy) AKI developed significantly exacerbated hepatic injury with significant higher plasma ALT at 24 h after liver IR (Figure 5a). Exacerbation of hepatic injury (20 min renal IR) or non-ischemic (bilateral nephrectomy) AKI was associated with the increased hepatic necrosis (Figure 5b), apoptosis (Figure 5c) and inflammation (Figure 5d) compared with mice subjected to hepatic IR alone.


Exacerbated hepatic IR injury (necrosis, apoptosis and inflammation) after ischemic or non-ischemic AKI. (a) Plasma alanine aminotransferase (ALT) levels in mice subjected to sham operation (Sham, N=4), 45 min hepatic ischemia and reperfusion (HIR, N=6), HIR coupled with unilateral nephrectomy (UNx+HIR, N=6), HIR coupled with bilateral nephrectomy (BNx+HIR, N=6) or HIR coupled with 20 min renal ischemia and reperfusion (RIR+HIR, N=8) 24 h prior. *P<0.05 vs HIR mice. Error bars represent 1 s.e.m. (b) Representative hematoxylin and eosin staining photomicrographs in liver sections (magnification, × 40) in mice subjected to sham operation, 45 min hepatic ischemia and reperfusion (HIR), HIR coupled with bilateral nephrectomy or HIR coupled with 20 min renal ischemia and reperfusion 24 h prior. Necrotic hepatic tissue appears as light pink. Arrows indicate vascular congestion and inflammation. (c) Representative fluorescence photomicrographs (of four experiments) of liver sections illustrating apoptotic nuclei (terminal deoxynucleotidyl transferase biotin-dUTP nick end-labeling (TUNEL) fluorescence staining, × 100) from mice subjected to 45 min hepatic ischemia and reperfusion (HIR) or HIR coupled with 20 min renal ischemia and reperfusion 24 h prior. (d) Representative gel images (top) and band intensity quantifications (bottom) of semi-quantitative RT–PCR of the pro-inflammatory markers ICAM-1, IL-6 and MCP-1 from liver tissues of mice subjected to sham operation (Sham), 20 min renal ischemia and reperfusion (RIR), 45 min hepatic ischemia and reperfusion (HIR) or 20 min RIR plus HIR. Liver tissues were harvested 5 h after sham operation or AKI induction. *P<0.05 vs sham-operated mice. Error bars represent 1 s.e.m. #P<0.01 vs HIR mice.
Increased Small Intestine Villous Capillary Endothelial Apoptosis, Necrosis and Inflammation After Ischemic or Non-ischemic AKI
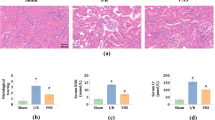
Small intestine histology was also assessed 5 h after bilateral nephrectomy in H&E stained sections, which showed profound villous endothelial cell apoptosis (right panel, Figure 6a and magnified insert), individual epithelial cell necrosis of villous lining cells and the development of a necrotic epithelial pannus over the mucosal surface (middle panel of Figure 6a). We also observed congestion of villous capillaries and swelling and blunting of villi because of villous mononuclear cell mucosal inflammation and edema. Histological changes were qualitatively similar, but inflammatory changes were more severe in the ileum than the jejunum (Supplementary Figure 3). Similar intestine histopathology was observed after 30 min renal IR (Figure 6a, middle panel) or unilateral nephrectomy (Supplementary Figure 4). In addition, we show increased pro-inflammatory mRNA (ICAM-1, TNF-α, IL-6, IL-17A, KC, MCP-1 and MIP-2) expression in the small intestine after ischemic (30 min renal IR) or non-ischemic (bilateral nephrectomy) AKI with RT–PCR (Figure 6b). We also show significant influx of neutrophils (Figure 6c), macrophages (Figure 6d) and T-lymphocytes (Figure 6e) into the small intestine compared with the sham-operated mice 5 h after 30 min renal IR or bilateral nephrectomy in mice. Neutrophil infiltration was scant in the intestines from sham-operated mice; however, infiltration was easily visible in the small intestines from mice subjected to ischemic (30 min renal IR) or non-ischemic (bilateral nephrectomy) AKI.





Increased small intestine necrosis, apoptosis and inflammation after ischemic or non-ischemic AKI. (a) Representative photomicrographs of ileum from five experiments (hematoxylin and eosin staining, magnification, × 200 and × 400) of mice subjected to sham operation (Sham), to 30 min renal ischemia and 5 h of reperfusion (RIR) or to bilateral nephrectomy (BNx). Sham-operated animals show normal-appearing intestine histology (left panel). As shown in middle upper panel, lower power images ( × 200) show full thickness of the ileal wall and villi that appear thickened, blunted and inflamed compared with sham. In addition, 5 h after 30 min renal IR or bilateral nephrectomy, severe intestine epithelial cell necrosis of villous lining cells and the development of a necrotic epithelial pannus (arrows) over the mucosal surface were observed (middle panel). Finally, we observed prominent capillary endothelial apoptosis within the central villi of ileum (right panel). Enlarged insert shows several apoptotic endothelial cells (red arrow heads) within a villus. (b) Representative gel images and band intensity quantifications of semi-quantitative RT–PCR of the pro-inflammatory markers ICAM-1, TNF-α, IL-6, IL-17A, KC, MCP-1 and MIP-2 from ileum of mice subjected to sham operation (Sham), bilateral nephrectomy (BNx) or 30 min renal IR (RIR). Tissues were harvested 5 h after sham operation or AKI induction. *P<0.05 vs sham-operated mice. Error bars represent 1 s.e.m. (c–e) Representative photomicrographs ( × 400, dark brown stain indicated by arrows) of five experiments of immunohistochemistry for neutrophils (c), macrophages (d) and T-lymphocytes (e) in the small intestine tissues harvested from mice subjected to sham operation (Sham), bilateral nephrectomy (BNx) or 30 min renal IR (RIR) 5 h prior. (f) Representative fluorescence photomicrographs (of five experiments) of ileum sections illustrating apoptotic nuclei (terminal deoxynucleotidyl transferase biotin-dUTP nick end-labeling (TUNEL) fluorescence staining, × 100). Mice were subjected to sham operation, bilateral nephrectomy (BNx) or 30 min renal ischemia reperfusion (RIR) 5 h prior. Enlarged insert (of ileum from mice subjected to 30 min RIR) shows prominent capillary endothelial apoptosis. (g) (Left) Representative photographs of small intestine tissues (duodenum, jejunum and ileum) isolated from mice subjected to sham operation or to bilateral nephrectomy (BNx) 5 h prior and injected with Evans blue dye (EBD). (Right) Quantification of small intestine EBD extravasations in mice subjected to sham operation (N=6), bilateral nephrectomy (BNx) or 30 min renal IR (RIR). Five hours after surgery, EBD was extracted in formamide and the amount of extravasated EBD in the intestine was calculated against a standard curve. *P<0.05 vs sham-operated mice. Error bars represent 1 s.e.m.
When the small intestines isolated from mice subjected to 30 min renal IR or bilateral nephrectomy were examined for apoptosis, we observed significantly increased number of apoptotic nuclei (TUNEL-positive cells; Figure 6f). TUNEL-positive cells were heavily localized to the central villi consistent with perivascular capillary endothelial cells rather than to the intestinal epithelial lining cells (magnified insert image in Figure 6f). We further confirmed that the TUNEL-positive cells are endothelial cells by staining parallel jejunum sections with TUNEL and CD34 (an endothelial cell marker, Abcam Inc., Cambridge, MA, USA) and confirmed that TUNEL-positive cells also stained for CD34 (Supplementary Figure 5). DNA laddering experiments confirmed increased apoptosis in the small intestine after ischemic (30 min renal IR) or non-ischemic (bilateral nephrectomy) AKI (Supplementary Figure 6). We also show increased small intestine vascular permeability after ischemic and non-ischemic AKI with EBD injection. Analysis of EBD extravasations in sham-operated mice and mice subjected to 30 min renal IR or bilateral nephrectomy is shown in Figure 6g. Finally, mice subjected to bilateral nephrectomy (serum FITC dextran=7538±778 ng/ml, N=4, P<0.0001) or 30 min renal IR (serum FITC dextran=6213±353 ng/ml, N=4, P<0.0001) showed severe disruption of intestinal permeability 5 h after AKI induction compared with sham-operated mice (serum FITC dextran=8.3±6.0 ng/ml, N=4).
Increased TNF-α, IL-6 and IL-17A Levels After Ischemic or Non-ischemic AKI
Using mouse-specific ELISA kits, we show that IL-17A, but not TNF-α and IL-6, is derived from the small intestine. We first measured systemic and portal venous plasma TNF-α, IL-17A and IL-6 levels after ischemic or non-ischemic AKI in mice. Table 1 shows that in mice subjected to ischemic (20 or 30 min renal IR) or non-ischemic (unilateral or bilateral nephrectomy) AKI, both systemic and portal venous cytokine levels increased over time compared with the sham-operated mice. However, the rise in portal venous levels of IL-17A was significantly greater than the levels detected in the systemic circulation at 5 h after bilateral nephrectomy or 30 min renal IR. In contrast, systemic levels of IL-6 were higher than the levels observed in the portal vein. In addition, we treated mice with specific neutralizing antibody for TNF-α, IL-17A or IL-6. Neutralizing antibody treatment not only selectively and specifically reduced the plasma levels of cytokine targeted, but also reduced other cytokine levels 24 h after bilateral nephrectomy or 20 min renal IR (Table 1). We also measured hepatic and small intestine tissue TNF-α, IL-6 and IL-17A levels after ischemic or non-ischemic AKI in mice. Table 2 shows that the levels of all three cytokines increased after ischemic (30 min renal IR) or non-ischemic (bilateral nephrectomy) AKI induction. However, hepatic IL-6 and TNF-α levels were higher than the levels observed in the small intestine. Small intestine IL-17A levels on the other hand were greater than the levels determined from the liver (Table 2).
Critical Functions for TNF-α, IL-6 and IL-17A in Generating Hepatic and Intestine Injury After Ischemic or Non-ischemic AKI
We determined whether the increased pro-inflammatory cytokines TNF-α, IL-17A and/or IL-6 have a critical function in hepatic and intestine injury after ischemic or non-ischemic AKI. Mice genetically deficient for TNF-α, IL-17A, IL-17R or IL-6 were significantly protected against hepatic (Table 3) and small intestine (Figure 7a) injury after ischemic (30 min renal IR; data not shown) or non-ischemic AKI (bilateral nephrectomy; Figure 7b). We complemented our knockout mice studies by treating the wild-type mice with specific neutralizing antibody for TNF-α, IL-17A or IL-6. Each neutralizing antibody treatment was very effective in protecting against hepatic (Table 3) and small intestine injury after ischemic (30 min renal IR; Figure 7c) or non-ischemic (bilateral nephrectomy; data not shown) AKI. Furthermore, TNF-α, IL-17A or IL-6 antibody treatment also significantly attenuated the exacerbation of hepatic injury because of ischemic (30 min renal IR) or non-ischemic (bilateral nephrectomy) AKI (Table 3). Finally, mice injected with a combination of recombinant mouse TNF-α, IL-6 plus IL-17A in lieu of AKI induction developed acute hepatic (ALT=368±67 U/L, N=5, P<0.001 vs sham) and intestine dysfunction (Figure 7d).




(a) Representative gel images (top) and band intensity quantifications (bottom) of semi-quantitative RT–PCR of the pro-inflammatory markers ICAM-1, TNF-α, IL-6, KC, MCP-1 and MIP-2 from ileum of C57BL/6 (WT) mice subjected to sham operation (Sham) or bilateral nephrectomy (BNx) compared with pro-inflammatory gene expression from the ileum of TNF-α, IL-6 or IL-17A-deficient (KO) mice. Tissues were harvested 5 h after sham operation or AKI induction. *P<0.05 vs sham-operated mice. #P<0.05 vs WT mice subjected to BNx. Error bars represent 1 s.e.m. (b–d) Representative photomicrographs of ileum of hematoxylin and eosin staining (magnification, × 200 and × 400) of WT, TNF-α, IL-6 or IL-17A KO mice subjected to BNx 5 h prior (b), WT mice treated with isotype control IgG, TNF-α, IL-6 or IL-17A neutralizing antibody (Ab) and subjected to RIR 24 h prior (c) and WT mice treated with a combination of recombinant mouse TNF-α, IL-6 plus IL-17A in lieu of AKI 5 h prior (d). Cytokine-deficient mice or WT mice treated with cytokine neutralizing antibodies (single antibody) were protected against small intestine injury, whereas WT mice treated with cytokine cocktails show severe injury (*). Representative of four experiments.
DISCUSSION
The major new findings of this study are that mice subjected to moderate (20 min) or severe (30 min) renal IR injury or nephrectomy (unilateral or bilateral) not only suffered from acute liver dysfunction but also developed severe small intestinal injury. Liver injury after ischemic or non-ischemic AKI was characterized by focused peri-portal hepatocyte vacuolization, necrosis and apoptosis with inflammatory changes. Small intestinal injury after ischemic or non-ischemic AKI was characterized by villous lacteal capillary endothelial apoptosis, epithelial necrosis and increased leukocyte (neutrophils, macrophages and lymphocytes) infiltration. Vascular permeability was severely impaired in both liver and small intestine. After ischemic or non-ischemic AKI, TNF-α, IL-17A and IL-6 levels in plasma, liver and small intestine increased significantly. Furthermore, mice deficient in TNF-α, IL-17A or IL-6 and wild-type mice treated with TNF-α, IL-17A or IL-6 neutralizing antibodies were protected against liver and intestinal injury after ischemic or non-ischemic AKI, suggesting a critical function for these cytokines in causing liver and intestinal damage. Our data suggest that increased IL-17A after AKI originates in the small intestine with subsequent induction of IL-6 and TNF-α.
AKI is a strong, independent predictor of mortality regardless of the degree or etiology of renal impairment.1 Unfortunately, interventions to enhance kidney function or recovery in patients with AKI have been largely unsuccessful limiting treatment to supportive measures (eg hemodialysis). Initiation or exacerbation of remote organ injury in patients suffering from AKI contributes significantly to mortality and morbidity.24 Previous studies have shown that significant pulmonary, cardiac and cerebral injury can occur after AKI.6, 24, 25 Indeed, the prognosis of AKI is closely related to the severity of extra-renal complications that arise.26, 27 Therefore, a better understanding of the mechanisms of remote organ injury because of AKI would lead to improved therapeutic approaches for patients suffering from AKI.
Our study shows that acute hepatic injury and dysfunction occur early after AKI (∼5 h) with significant increases in plasma ALT levels in mice (Figure 1). Although hepatic injury in animals subjected to AKI has been previously shown by others,28, 29, 30, 31 the mechanisms that initiate hepatic injury to AKI have not been clearly elucidated. Previous study in rats showed that after bilateral nephrectomy or renal IR, significant hepatic injury occurred early that was associated with increased TNF-α, oxidative stress with decreased anti-oxidant levels and hepatic apoptosis detected by caspase-3 activation.28 Interestingly, unlike this study in mice in which hepatic apoptosis occurred after bilateral nephrectomy and renal IR, caspase-3 activation was not detected after renal IR in rats. We also show that significant inflammatory changes occur in the liver after ischemic or non-ischemic AKI with induction of all pro-inflammatory genes studied including TNF-α, IL-6 and IL-17A. Furthermore, we observed striking hepatic injury near the portal venous drainage areas with profound hepatocyte vacuolization, necrosis and apoptosis. However, the most novel finding of this study is the significant small intestinal injury after both ischemic and non-ischemic AKI. The specific peri-portal localization of hepatic injury led us to hypothesize that the injury mediators originated in the small intestine. This is the first report to show small intestinal injury after AKI and to implicate the small intestine as the source of cytokines generated and released after ischemic or non-ischemic AKI. In addition, we propose that cytokines generated in the small intestine and draining into the liver cause hepatic and perhaps extra-renal organ injury.
We also determined that in addition to the increased vascular permeability in the liver, small intestine vascular permeability was also severely disrupted after AKI (Figure 6g). We examined the histology of the small intestine (jejunum and ileum) and identified villous endothelial capillary apoptosis, intestinal barrier disruption (leukocyte infiltration) and mucosal epithelial necrosis in the ileum and jejunum. Therefore, after ischemic and non-ischemic AKI, it appears that intestinal endothelial and epithelial apoptosis and necrosis occur rapidly with subsequent intestine barrier and vascular disruption. As the small intestinal dysfunction with barrier interruption has been implicated as the source of multi-organ dysfunction, cytokine generation and sepsis,32 increased incidence of systemic inflammation and sepsis after AKI may be due to the injury to the small intestine. Interestingly, we observed greater increases in pro-inflammatory mRNA expression in the ileum compared with jejunum after bilateral nephrectomy. Perhaps, abundant concentrations of Peyer's patches containing a large numbers of pro-inflammatory lymphocytes in the ileum compared with jejunum may explain this difference.33
Closer examination of cytokine generation revealed several intriguing findings. We show in this study that in mice subjected to ischemic or non-ischemic AKI, levels of IL-17A were higher in the portal vein than in the systemic circulation. Furthermore, tissue cytokines measured by ELISA in mice subjected to AKI showed greater increases in IL-17A levels in the small intestine, whereas TNF-α and IL-6 levels were greater in the liver. Taken together, we propose that the small intestinal generation of IL-17A leads to hepatic-inflammatory changes including hepatic generation of TNF-α and IL-6 further potentiating hepatic injury and systemic-inflammatory responses. However, we acknowledge that our studies cannot rule out other potential sources of increased portal venous IL-17A including pancreas, spleen and visceral fat.
We show in this study that mice deficient in TNF-α, IL-17A or IL-6 or wild-type mice treated with TNF-α, IL-17A or IL-6 neutralizing antibodies were significantly protected against ischemic or non-ischemic AKI-induced hepatic and intestinal injury (Table 3). These findings suggest that the generation of cytokines after injury is not a redundant process, but rather individual cytokine (eg IL-17A) appears to propagate the generation of another cytokine (eg IL-6). Blockade of one cytokine was sufficient to attenuate hepatic and small intestinal injury and circulating levels of other cytokines after AKI. Furthermore, these findings have significant clinical implications as patients suffering from AKI may benefit from drugs already used to counteract chronic-inflammatory diseases (eg monoclonal antibody for TNF-α to treat Crohn's disease or rheumatoid arthritis).34 Of interest, after hepatic IR without AKI, cytokine neutralizing antibodies did not provide protection against liver injury at 5 h (Table 3), suggesting that these cytokines do not have a major function in early hepatocyte injury after ischemia and reperfusion. At 24 h after IR, however, these antibodies protected against liver injury. We propose two possible explanations: (1) these cytokines promote hepatic injury at a later time (after 5 h) after hepatic IR (eg 24 h) or (2) these cytokines have a function in extra-hepatic injury after hepatic IR (eg intestinal injury) that occurs later and further promote hepatocyte necrosis.
The function of IL-17A in host immune defense and inflammation has been increasingly recognized recently. Six members of IL-17A ligands and receptors have been described (IL-17A to IL-17F). IL-17A was originally detected in a subset of T cells designated as Th17 cells distinct from Th1 or Th2 cells.35, 36 However, other cell types are now known to produce IL-17A, including CD8 T cells, natural killer cells and neutrophils.37 IL-17A has been found to be elevated in a variety of inflammatory conditions, such as rheumatoid arthritis, pneumonia, systemic lupus erythematosus, sepsis, allograft rejection and Crohn's disease.35, 37 Therefore, it is not surprising that IL-17A acts on various cell types, including neutrophils, fibroblasts, epithelial cells and endothelial cells, inducing the expression of pro-inflammatory mediators such as IL-6, CXC chemokines and matrix metalloproteinases.37 Furthermore, IL-17A induces expression of inflammatory genes, such as IL-8, and synergizes with TNF-α.37 Interestingly, the intestinal lamina propria was shown to be a unique site for the presence of Th17 cells in healthy animals.14 Atarashi et al38 confirmed these findings and showed high amounts of Th17 cells in the intestinal lamina propria, but not in the spleen, mesenteric lymph nodes or Peyer's patches of a healthy mouse. Recently, Takahashi et al39 showed that IL-17A produced by small intestinal Paneth cells drive TNF-α-induced inflammation and shock. The unique position of IL-17A as a regulator of both innate and acquired immunity makes this cytokine a crucial signal for the reinforcement and crosstalk of host defense systems.
In this study, we show that IL-6 and TNF-α are involved in further promoting hepatic and intestinal injury after AKI. TNF-α has been regarded as a ‘master regulator’ of the cytokine cascade that provides a rapid form of host defense against infection, but is fatal in excess. IL-17A and TNF-α appear to have a synergistic function in each other's production and function, and it has been shown that IL-17A can potentiate and synergize with TNF-α to upregulate the expression of target genes.40 In addition, it has been proposed that functional cooperation between IL-17A and TNF-α occurs at the level of IL-6 transcriptional activity.41, 42 IL-17A augments TNF-α-induced IL-6 and G-CSF and GM-CSF secretion in human colonic subepithelial myofibroblasts.41, 43 IL-6 is another multi-functional cytokine known to be involved in the regulation of immunity and pro-inflammatory responses.12, 42, 44 IL-6 is not only generated by T- and B-lymphocytes, but also by other cell types, including endothelial cells, renal tubule cells and hepatocytes.12, 44 IL-6 regulates the expression of adhesion molecules and other cytokines in endothelial cells, including IL-1β and TNF-α, which in turn potently enhance the inflammatory response, and also forms part of a positive feedback cycle in which TNF-α stimulates IL-6 production.41, 45
This study illustrates the important effects of renal failure independent of renal ischemia with the use of the bilateral nephrectomy model, as the kidney itself may be a source of IL-17A, TNF-α or IL-6 after ischemic AKI.46 Improved liver and intestinal protection in mice treated with neutralizing antibodies or mice deficient in TNF-α, IL-17A or IL-6 are independent of an effect on the kidney, as evidenced by bilateral nephrectomy. Moreover, the elevation in plasma TNF-α, IL-17A or IL-6 levels after bilateral nephrectomy indicates that the kidney is not the only source of these cytokines after AKI. As the kidney contributes to cytokine clearance, reduced renal cytokine elimination may contribute to increased plasma and tissue levels of TNF-α, IL-17A and IL-6. We observed continuous increases in plasma ALT and hepatic damage (histology) 24 h after renal IR in contrast to mice subjected to unilateral or bilateral nephrectomy (Figure 1). It is possible that kidney subjected to IR may be a continuous source of pro-inflammatory cytokines that can maintain hepatic injury. In contrast, mice subjected to bilateral nephrectomy were able to recover from hepatic damage earlier.
In summary, we show in this study that both ischemic and non-ischemic renal injury initiate IL-17A generation in the small intestine resulting in small intestinal and liver inflammation, apoptosis and necrosis (Figure 8). We show crucial functions for TNF-α, IL-17A and IL-6 in generating these injuries. We provide evidence that small intestine-derived IL-17A causes further cytokine generation to induce hepatic injury and systemic inflammation. Further studies are required to determine the specific cell type(s) responsible for intestinal IL-17A generation and to elucidate the initial stimulus for producing intestine injury after AKI.

Proposed mechanisms of acute kidney injury-induced liver dysfunction and systemic inflammation. Acute kidney injury causes small intestinal generation of IL-17A and subsequent intestinal injury (villous endothelial apoptosis, epithelial necrosis, increased pro-inflammatory cell translocation and cytokine flux to the liver). These events cause hepatic injury (inflammation, apoptosis and necrosis) with increased generation and release of TNF-α and IL-6 systemically causing further multi-organ injury and systemic inflammation.
References
Jones DR, Lee HT . Perioperative renal protection. Best Pract Res Clin Anaesthesiol 2008;22:193–208.
Bove T, Calabro MG, Landoni G, et al. The incidence and risk of acute renal failure after cardiac surgery. J Cardiothorac Vasc Anesth 2004;18:442–445.
Elapavaluru S, Kellum JA . Why do patients die of acute kidney injury? Acta Clin Belg Suppl 2007;2:326–331.
Paladino JD, Hotchkiss JR, Rabb H . Acute kidney injury and lung dysfunction: a paradigm for remote organ effects of kidney disease? Microvasc Res 2009;77:8–12.
Grigoryev DN, Liu M, Hassoun HT, et al. The local and systemic inflammatory transcriptome after acute kidney injury. J Am Soc Nephrol 2008;19:547–558.
Faubel S . Pulmonary complications after acute kidney injury. Adv Chronic Kidney Dis 2008;15:284–296.
Bagshaw SM, Laupland KB, Doig CJ, et al. Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: a population-based study. Crit Care 2005;9:R700–R709.
Burne-Taney MJ, Rabb H . The role of adhesion molecules and T cells in ischemic renal injury. Curr Opin Nephrol Hypertens 2003;12:85–90.
Rabb H . Immune modulation of acute kidney injury. J Am Soc Nephrol 2006;17:604–606.
Donnahoo KK, Meldrum DR, Shenkar R, et al. Early renal ischemia, with or without reperfusion, activates NFkappaB and increases TNF-alpha bioactivity in the kidney. J Urol 2000;163:1328–1332.
Donnahoo KK, Meng X, Ayala A, et al. Early kidney TNF-alpha expression mediates neutrophil infiltration and injury after renal ischemia-reperfusion. Am J Physiol 1999;277:R922–R929.
Kielar ML, John R, Bennett M, et al. Maladaptive role of IL-6 in ischemic acute renal failure. J Am Soc Nephrol 2005;16:3315–3325.
Liu KD, Altmann C, Smits G, et al. Serum interleukin-6 and interleukin-8 are early biomarkers of acute kidney injury and predict prolonged mechanical ventilation in children undergoing cardiac surgery: a case-control study. Crit Care 2009;13:R104.
Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006;126:1121–1133.
Lee HT, Gallos G, Nasr SH, et al. A1 adenosine receptor activation inhibits inflammation, necrosis, and apoptosis after renal ischemia-reperfusion injury in mice. J Am Soc Nephrol 2004;15:102–111.
Park SW, Chen SW, Kim M, et al. Human heat shock protein 27 overexpressing mice are protected against acute kidney injury after hepatic ischemia and reperfusion. Am J Physiol Renal Physiol 2009;297:F885–F894.
Lee HT, Park SW, Kim M, et al. Acute kidney injury after hepatic ischemia and reperfusion injury in mice. Lab Invest 2009;89:196–208.
Park SW, Chen SW, Kim M, et al. Human activated protein C attenuates both hepatic and renal injury caused by hepatic ischemia and reperfusion injury in mice. Kidney Int 2009;76:739–750.
SLOT C . Plasma creatinine determination. A new and specific Jaffe reaction method. Scand J Clin Lab Invest 1965;17:381–387.
Hart ML, Henn M, Kohler D, et al. Role of extracellular nucleotide phosphohydrolysis in intestinal ischemia-reperfusion injury. FASEB J 2008;22:2784–2797.
Chen SW, Park SW, Kim M, et al. Human heat shock protein 27 overexpressing mice are protected against hepatic ischemia and reperfusion injury. Transplantation 2009;87:1478–1487.
Park SW, Kim M, Chen SWC, et al. Sphinganine-1-phosphate attenuates both hepatic and renal injury induced by hepatic ischemia and reperfusion in mice. Shock 2009;33:31–42.
Herrmann M, Lorenz HM, Voll R, et al. A rapid and simple method for the isolation of apoptotic DNA fragments. Nucleic Acids Res 1994;22:5506–5507.
Faubel S . Acute kidney injury and multiple organ dysfunction sindrome. Minerva Urol Nefrol 2009;61:171–188.
Hassoun H, Grigoryev DN, Lie M, et al. Ischemic acute kidney injury induces a distant organ functional and genomic response distinguishable from bilateral nephrectomy. Am J Physiol Renal Physiol 2007;293:F30–F40.
Knaus WA, Draper EA, Wagner DP, et al. Prognosis in acute organ-system failure. Ann Surg 1985;202:685–693.
Seely AJ, Christou NV . Multiple organ dysfunction syndrome: exploring the paradigm of complex nonlinear systems. Crit Care Med 2000;28:2193–2200.
Golab F, Kadkhodaee M, Zahmatkesh M, et al. Ischemic and non-ischemic acute kidney injury cause hepatic damage. Kidney Int 2009;75:783–792.
Fadillioglu E, Kurcer Z, Parlakpinar H, et al. Melatonin treatment against remote organ injury induced by renal ischemia reperfusion injury in diabetes mellitus. Arch Pharm Res 2008;31:705–712.
Wang BY, Li QX, Li J, et al. Hepatotoxicity and gene expression down-regulation of CYP isozymes caused by renal ischemia/reperfusion in the rat. Exp Toxicol Pathol 2009;61:169–176.
Serteser M, Koken T, Kahraman A, et al. Changes in hepatic TNF-alpha levels, antioxidant status, and oxidation products after renal ischemia/reperfusion injury in mice. J Surg Res 2002;107:234–240.
Yang S, Koo DJ, Chaudry IH, et al. The important role of the gut in initiating the hyperdynamic response during early sepsis. J Surg Res 2000;89:31–37.
Pabst R . The anatomical basis for the immune function of the gut. Anat Embryol Berl 1987;176:135–144.
Shealy DJ, Visvanathan S . Anti-TNF antibodies: lessons from the past, roadmap for the future. Handb Exp Pharmacol 2008;181:101–129.
Kramer JM, Hanel W, Shen F, et al. Cutting edge: identification of a pre-ligand assembly domain PLAD; and ligand binding site in the IL-17 receptor. J Immunol 2007;179:6379–6383.
Nakae S, Komiyama Y, Nambu A, et al. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 2002;17:375–387.
Weaver CT, Hatton RD, Mangan PR, et al. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol 2007;25:821–852.
Atarashi K, Nishimura J, Shima T, et al. ATP drives lamina propria TH;17 cell differentiation. Nature 2008;455:808–812.
Takahashi N, Vanlaere I, de RR, et al. IL-17 produced by Paneth cells drives TNF-induced shock. J Exp Med 2008;205:1755–1761.
Gaffen SL, Kramer JM, Yu JJ, et al. The IL-17 cytokine family. Vitam Horm 2006;74:255–282.
Andoh A, Hata K, Araki Y, et al. Interleukin IL;-4 and IL-17 synergistically stimulate IL-6 secretion in human colonic myofibroblasts. Int J Mol Med 2002;10:631–634.
Shimada M, Andoh A, Hata K, et al. IL-6 secretion by human pancreatic periacinar myofibroblasts in response to inflammatory mediators. J Immunol 2002;168:861–868.
Hata K, Andoh A, Shimada M, et al. IL-17 stimulates inflammatory responses via NF-kappaB and MAP kinase pathways in human colonic myofibroblasts. Am J Physiol Gastrointest Liver Physiol 2002;282:G1035–G1044.
Wang W, Smail N, Wang P, et al. Increased gut permeability after hemorrhage is associated with upregulation of local and systemic IL-6. J Surg Res 1998;79:39–46.
Ding C, Cicuttini F, Li J, et al. Targeting IL-6 in the treatment of inflammatory and autoimmune diseases. Expert Opin Investig Drugs 2009;18:1457–1466.
Ramesh G, Reeves WB . TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 2002;110:835–842.
Acknowledgements
This work was supported by National Institute of Health grants RO1 DK-58547 and RO1 GM-067081.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Rights and permissions
About this article
Cite this article
Park, S., Chen, S., Kim, M. et al. Cytokines induce small intestine and liver injury after renal ischemia or nephrectomy. Lab Invest 91, 63–84 (2011). https://doi.org/10.1038/labinvest.2010.151
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/labinvest.2010.151
Keywords
This article is cited by
-
Acute kidney injury-associated delirium: a review of clinical and pathophysiological mechanisms
Critical Care (2022)
-
Immunopathophysiology of trauma-related acute kidney injury
Nature Reviews Nephrology (2021)
-
Effects of low versus standard pressure pneumoperitoneum on renal syndecan-1 shedding and VEGF receptor-2 expression in living-donor nephrectomy: a randomized controlled study
BMC Anesthesiology (2020)
-
Systemic and sustained thioredoxin analogue prevents acute kidney injury and its-associated distant organ damage in renal ischemia reperfusion injury mice
Scientific Reports (2020)
-
Recombinant thrombomodulin prevents acute lung injury induced by renal ischemia-reperfusion injury
Scientific Reports (2020)
