
Abstract
This report concerns a benchtop prototype instrument containing a gas chromatographic microanalytical system (μGC) designed for the selective determination of multiple airborne volatile organic compounds (VOCs) at concentrations in the vicinity of recommended occupational exposure limits. The core microsystem consists of a set of discrete Si-microfabricated devices: a dual-cavity, adsorbent-packed micro-preconcentrator-focuser (μPCF) chip that quantitatively captures and thermally desorbs/injects VOCs with vapor pressures between ~0.03 and 13 kPa; tandem micro-column (μcolumn) chips with cross-linked PDMS wall-coated stationary phases capable of temperature-programmed separations; and an integrated array of five μchemiresistors (μCR) coated with different thiolate-monolayer protected gold nanoparticle (MPN) interface films that quantifies and further differentiates among the analytes by virtue of the response patterns generated. Other key components include a pre-trap for low-volatility interferences, a split-flow injection valve, and an onboard He carrier–gas canister. The assembled unit measures 19×30×14 cm, weighs ~3.5 kg, operates on AC power, and is laptop/LabVIEW controlled. Component- and system-level tests of performance demonstrated injection bandwidths <1 s, a μcolumn capacity of ≥8 μg injected mass, linear calibration curves, no humidity effects, excellent medium-term (that is, 1 week) reproducibility, autonomous operation for 8 h, detection limits below Threshold Limit Values (TLV) for 10 mL air samples collected in 1 min, and response patterns that enhanced vapor recognition. The determination of a 17-VOC mixture in the presence of seven interferences was performed in 4 min. Results augur well for adapting the microsystem to an all-MEMS wearable μGC currently under parallel development.
Similar content being viewed by others

Introduction
Gas chromatographic instrumentation constructed using Si-microfabricated analytical components (μGC) may afford the means to quantitatively measure individual worker exposures to multiple specific volatile organic compounds (VOC) in near-real time. Such multi-VOC measurements are currently only possible with portable GCs1–4 and transportable FTIR5 and GC–MS6,7 instruments, which are too large and expensive for routine evaluations of personal exposures. Although significant advances have been reported recently in the design and development of individual μGC components for preconcentration8–11, separation12–20,detection21–25, and systems that combine one or more such microdevices with conventional GC components26–29, surprisingly few reports have appeared on integrated and/or packaged μGC systems in which the core analytical components were microfabricated30–36.
Members of our group have reported on a number of μGC and μGC×μGC components and systems in recent years37–50. We have also developed automated prototype instruments containing μGC systems for measuring low- to sub-ppb concentrations of trichloroethylene (TCE) in vapor-intrusion impacted homes51,52, and markers of explosives for transportation security53,54. Features common to these prototype instruments include a partially selective high-volume sampler of conventional design, a micromachined preconcentrator-focuser (μPCF) for focusing and injection, one or more μcolumns for temperature-programmed separations, and arrays of μchemiresistors (μCR) with thiolate-monolayer protected gold nanoparticle (MPN) interface films for multichannel detection and recognition of eluting VOCs.
Our current efforts are directed toward creating a μGC that we have termed a Personal Exposure Monitoring Microsystem (PEMM), which is being designed as a belt-mountable instrument for measuring worker exposures to numerous target VOCs simultaneously. Towards that end, we created a first-generation (Gen-1) bench-top PEMM-1 prototype that is AC powered and laptop controlled for the purpose of gathering experience and data to inform the design, assembly and operating conditions of the wearable Gen-2 PEMM (that is, PEMM-2).
The core microanalytical system in the PEMM-1 comprises a redesigned dual-bed μPCF with a new heater design, a dual-μcolumn separation stage, and a redesigned μCR array. These are combined with ancillary non-microfabricated fluidic components, interface circuitry, and data acquisition and control software, as described below. The μPCF and μCR array devices of the PEMM-1 unit are the same as those incorporated into the PEMM-2 design, and neither has been presented previously. The separation module differs from the monolithic PEMM-2 μcolumn55, and a dual-μcolumn ensemble of similar length has been used in our recent study of μGC×μGC separations45, but it has not yet been incorporated into a complete microsystem. Other unique aspects of the PEMM-1 design are the use of a pre-trap, split-flow injection, and He carrier gas. Our focus on VOCs at relatively high (that is, ~parts per million, ppm) concentrations is also new and obviates the need for high-volume samplers.
The instrument is intended to perform generalized VOC measurements in industrial workplaces, with a capability for quantitatively analyzing up to ~20 VOCs per measurement at a rate of ~6–8 measurements per hour. For practical reasons, we focused on VOCs falling within a moderate volatility window defined by their vapor pressures, pv. For quantification, it was assumed that concentrations would fall in the high parts per billion (ppb) to high ppm range, which is the most relevant range for workplace exposure monitoring56. Following the presentation of component-level test results intended to confirm critical performance capabilities, we present a comprehensive series of system-level test results intended to demonstrate reliable mixture-component determinations and autonomous operation. The implications of the results for the design and operation of the PEMM-2 are then assessed.
Materials and methods
Materials
Most test compounds and reagents were purchased from Sigma-Aldrich/Fluka (Milwaukee, WI, USA) or Acros/Fisher (Pittsburgh, PA, USA) in >95% (most >99%) purity and were used as received. MPNs used as μCR interface films had core diameters in the range of 3.5–5 nm and were derived from the following thiols: n-octanethiol (C8), 6-phenoxyhexane-1-thiol (OPH), isooctyl-3-mercaptopropionate (EOE), methyl-6-mercaptohexanoate (HME) and 1-mercapto-(triethylene glycol) methyl ether (TEG). TEG was purchased from Nanoprobes, Inc. (Yaphank, NY, USA). The remaining MPNs were synthesized in house by known methods57,58. The graphitized carbon adsorbents Carbopack X (C-X, 240 m2 g−1), Carbopack B (C-B, 100 m2 g−1), Carbopack C (C-C, 10 m2 g−1), and Carbopack F (C-F, 5 m2 g−1) were all 60/80 mesh and were obtained from Supelco (Bellefonte, PA, USA). The C-X and C-B were sieved such that only the fractions with nominal diameters between 212 and 250 μm were used. Polydimethylsiloxane (PDMS) was purchased from Ohio Valley Specialty Chemicals (OV-1, Marietta, OH, USA). Pressurized gas canisters (95 mL, 4.0 cm outer diameter (o.d.) ×13 cm) of He (purity >99.5%; 2500 PSI) and a custom regulator were obtained from Leland (South Plainfield, NJ, USA).
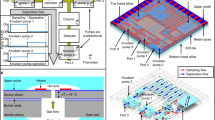
A block diagram of the analytical subsystem of the PEMM-1 prototype is presented in Figure 1.

Layout diagram of fluidic and analytical components of the PEMM-1 prototype μGC.
Pre-trap
Two configurations of pre-traps for precluding low-volatility interferences from entering the system were explored. The first, referred to as pre-trap A, consisted of a thin-walled stainless-steel tube (1.58 mm, inner diameter (i.d.)) packed with up to 5.4 mg of either C-F or C-C and retained with glass wool. The second consisted of short sections cut from commercial capillary columns, 250 μm or 530 μm i.d., with stationary phases of PDMS (Rtx-1; pre-trap B1) or polymethylphenylsiloxane (Rtx-20; 80:20 methyl:phenyl; pre-trap B2), respectively (Restek, Bellefonte, PA, USA). The effects of several relevant variables on the breakthrough volumes of representative test compounds were evaluated.
Micro-machined preconcentrator-focuser (μPCF)
The μPCF chip, shown in Figure 2a, has dimensions of 13.6×4.1 mm and was fabricated from Si using deep-reactive-ion etching (DRIE). Each cavity is 380 μm deep, has a volume of ~4.7 μL, and is separated from the adjacent cavity by a row of 150-μm-diameter pillars spaced 150 μm from one another. Filling ports were etched into the sidewalls of each cavity for adsorbent loading. Fluidic ports were etched to snugly accommodate a 250 μm i.d. (380 μm, o.d.) fused silica capillary affixed with a flexible, high-temperature silicone adhesive (Duraseal 1531, Cotronics, Brooklyn, NY, USA). The fluidic inlet channel features a tee junction to permit sample loading through one branch and back-flushed injection through the other. A 200 μm thick Pyrex plate was anodically bonded to the top surface at wafer level to seal the cavities and channels. A Ti/Pt resistive heater and resistive temperature device (RTD) were patterned onto the backside of the Si after growing a thin oxide layer for electrical isolation (Figure 2a). SolidWorks (R2014, Dassault Systems, Waltham, MA, USA) and COMSOL Multiphysics (R4.2, Burlington, MA, USA) packages were used for heater design and simulations, respectively, to minimize expected temperature gradients across the cavities.

Photographs of (a) the front and back side of an unloaded μPCF fitted with capillaries (U.S. quarter for scale); (b) the front and back side of a μcolumn (U.S. quarter for scale); (c) the μCR array with backside heater and RTD (U.S. quarter for scale); (d) assembled PCBs (sans fluidic components) and valve manifold; and (e) the fully assembled PEMM-1 prototype with lid removed μPCF, micro-preconcentrator-focuser; μCR, micro-chemiresistor; resistive temperature device; RTD, resistive temperature device; PCBs, printed circuit boards; PEMM-1, first-generation Personal Exposure Monitoring Microsystem’s.
Using mild suction, the front cavity (that is, during sampling) was loaded with 2.0 mg of C-B and the rear cavity was loaded with 2.3 mg of C-X, as determined gravimetrically to ±0.1 mg. Filling ports were subsequently sealed with Duraseal. The device was inverted, mounted on a custom printed circuit board (PCB) using epoxy (Hysol 1C, Rocky Hill, CT, USA), and wire-bonded to the PCB bonding pads for electrical connections.
The injection bandwidth was characterized using a bench-scale GC-FID (7890 Agilent, Santa Clara, CA, USA) with the μPCF connected across two ports of a 6-port valve. Test atmospheres containing selected VOCs in N2 were generated in Flex-foil inert gas sample bags (Supelco). A suction pump (model UMP015, KNF Neuberger, Trenton, NJ, USA) was used to draw a sample from the bag through the μPCF to load 50–100 ng of analyte, after which the valve was switched, and He from the GC injection port was backflushed through the μPCF while it was heated. An initial fast (uncontrolled) ramp of 400 °C s−1 was used to heat from 30 to 100 °C, followed by a pulse-width modulated (PWM) ramp of 150 °C s−1 to 225 °C, which was maintained for 40 s, as subsequently applied for the injection step with the prototype. Passive cooling to 30 °C required ~90 s. Injected samples were passed to an FID via a section of capillary maintained at 100 °C.
μColumns
Each of the two μcolumn chips (Figure 2b; 3.1×3.1 cm) contains a single DRIE channel (240 μm (h)×150 μm (w); 3.1 m long) that spirals in to the center and back out to the edge of the chip in a square pattern with chamfered corners45. A 500 μm thick Pyrex cap was anodically bonded to the top face to seal the fluidic channel at wafer scale. A pair of heaters and an RTD were patterned from Ti/Pt onto the backside. Deactivated capillaries (250 μm i.d.) inserted into the inlet and outlet ports of each μcolumn were sealed with Duraseal.
A previously described static method was used to deposit and crosslink the PDMS stationary phase on the inner walls of the channels to a nominal thickness of 0.20 μm (Refs. 14, 40). After coating, the inlet and outlet capillaries were replaced with fresh uncoated (deactivated) capillaries sealed to the chip with Hysol. The capillaries emanating from the outlet of the first μcolumn and the inlet of the second μcolumn were cut to ~5 mm, inserted into opposite ends of a short (that is, ~1.2 cm) Pyrex/Si conduit interconnect with a Ti/Pt meander-line heater patterned on the Pyrex, and sealed (Hysol). The assembly was inverted, affixed with Hysol to a custom PCB with rectangular cutouts below the μcolumns, and wire-bonded.
μCR array
The μCR array chip (33×20×0.5 mm; Figure 2c) has a set of 5 primary and 5 back-up Au (300 nm)/Cr (30 nm) interdigital electrodes (IDE) in a single row, with a Au/Cr RTD at the center that was deposited and patterned via a standard lift-off process onto a Pyrex substrate. Each μCR contains 27 pairs of IDEs 5 μm wide, spaced 4 μm apart, and overlapping by 210 μm. A meander-line Ti/Pt heater was patterned on the backside. Header pins were bent to 90° and low-temperature soldered to each of the 22 IDE bonding pads. Cables were soldered to the heater bonding pads. A Si lid (33×10×0.5 mm) was also fabricated with a linear, recessed DRIE channel, 150 μm deep ×350 μm wide, running lengthwise down the center to align directly above the sensors in the array. The linearity of the interconnection with external capillaries was an improvement over previous μCR array configurations47,51. The array was plugged into a socket on a carrier PCB, and the entire assembly was covered with a grounded Cu Faraday cage.
MPNs were dissolved in suitable solvents (toluene for C8, OPH, and TEG; chloroform for EOE, dichloromethane for HME) at concentrations of ~5 mg mL−1. Each sensor was coated with a film of one type of MPN by drop casting from a 1 μL syringe to create multilayer films with baseline resistances between 100 kΩ and 10 MΩ, depending on the MPN and the film thickness. The film thicknesses were not uniform and were not measured but were roughly estimated to be on the order of 200–500 nm on average on the basis of previous work59.
After coating, 0.3×2.5 cm strips of 50-μm thick double-sided adhesive tape (VHB tape, 3 M, St Paul, MN, USA) were mounted along both sides of the array, the Si lid was pressed down onto the tape, and a narrow bead of Hysol was applied along the outer seam. Short segments of deactivated inlet/outlet capillary (250 μm i.d.) were sealed to the fluidic ports (Hysol) for connection to other devices via press-tight unions. Although the integrated heater on the μCR array could be used to control temperature via a constant voltage bias, it was eventually found to be unnecessary (vide infra).
System integration and prototype assembly
Two custom PCBs were designed, fabricated (Advanced Circuits, Aurora, CO, USA), populated, and checked for proper performance. One PCB was dedicated to the microsystem components and the other to the mini-pump, mini-valves, and fans. Individual carrier PCBs for each microsystem device were mounted on stand-offs to the baseplate board. For more details, please refer to the Supplementary Section S1.
Figures 2d and e show the arrangement of the PCBs and fluidic components prior to final assembly and the fully assembled PEMM-1 prototype, respectively. The PEMM-1 measures 15.2 (h)×30.5 (w)×14 (d) cm and weighs 3.18 kg. The rear-panel-mounted He canister and regulator increase the net depth by 3.8 cm and the weight by 0.34 kg. In Figure 2d, the three carrier PCBs are shown (sans microcomponents) mounted on stand-offs to a single base-plate PCB. Beneath this PCB, arranged in order, are the microsystem control PCB, DAQ board, and fluidic-control PCB. The custom stainless-steel valve manifold accepts zero-dead-volume fittings for capillary interconnects and five face-mounted, 3-way, latching solenoid valves (Model LHLA1221111H, Lee Co., Westbrook, CT, USA). A wall-mounted diaphragm mini-pump (NMP-09M, KNF Micro AG, Reiden, Switzerland) collected the air samples. Two miniature needle valves (Beswick, Greenland, NH, USA) were mounted to the front panel (Figure 2e) for manual adjustment of the sampling flow rate and injection split-flow ratio.
Sections of stainless-steel tubing (750 μm, i.d.) and fused silica capillary (250 μm, i.d.) were used for fluidic interconnections. For the latter, press-tight unions (Supelco) were used, with a subset wrapped in polyimide-embedded resistive film heaters (Omega, Stamford, CT, USA) held at ~80 °C. Voltage regulators for the pump and solenoid valves were mounted to the inside of the front panel. Small axial fans were used for cooling. The 24-V DC power was supplied through an AC-to-DC converter connected to wall power. For most initial testing, a separate compressed gas cylinder of He was connected through the bench-scale GC for pressure regulation and subsequently to the fitting on the prototype used later to connect the on-board He canister regulator. The head pressures necessary to achieve a flow rate of 3 mL min−1 through the analytical system (both splitless and with a 2:1 split) were 18 and 23 psi, respectively.
System control, data acquisition and processing
The instrument was operated from a laptop computer running a custom LabVIEW (Ver. 14.0, National Instr., Austin, TX, USA) program. Operating parameter settings and control functions were entered through a graphical user interface for either manual operation of each run or multiple continuous runs. Independent proportional-integral-derivative feedback loops were designed to control heating rates and temperatures via solid-state relays and PWM of the generated signals. Although not critical for this prototype, power efficiency was considered in the design of the components. The power and energy per analysis are provided in the Supplementary Section S1.
Raw chromatogram traces were stored as text files and analyzed using OriginPro (Ver. 9.1, OriginLab, Northampton, MA, USA). Calibration curve regression models and response patterns were generated in Excel. Monte Carlo simulations coupled with extended disjoint principal components regression (MC-EDPCR) analyses were implemented in Visual Basic via custom programs43.
Results and discussion
System design and operation specifications
By design, air samples are drawn by the mini-pump through the inlet, and low volatility interfering VOCs are retained by the wall-coated-capillary pre-trap (Figure 1). The air sample then passes through the μPCF, where target VOCs are quantitatively captured in one of the two adsorbent beds. After turning off the pump and switching the valves to start the flow of He through the microsystem, the μPCF is rapidly heated to thermally desorb the captured VOCs for passage to the μcolumns in a narrow band. Temperature-programmed separation of the VOC mixture components is performed, with detection provided by the transient changes in the resistance of the MPN-coated μCRs in the array as VOCs reversibly sorb into the MPN films to different extents, giving rise to a response pattern. The flow is then redirected to backflush the pre-trap and the fore-line to purge any residual VOCs, and the μPCF and μcolumns are allowed to cool in preparation for the next cycle.
Several performance criteria were used to rationalize the design, integration, and operation of the instrument components39. The primary constraint placed on the target VOCs was that they fall within a pv range of ~0.03 to 13 kPa. Less volatile compounds would tend to adhere to surfaces in the (unheated) fluidic pathways, and more volatile compounds would be difficult to trap, separate, and detect because these functions rely on partitioning phenomena. Where possible, target VOCs were chosen that also had assigned Threshold Limit Values (TLV®)56, which serve as reference values for specifying the ranges of concentrations to be encountered. To assess the selectivity of the pre-trap and μPCF, we included several potentially interfering compounds outside of the designated pv range. For the target VOCs, a set of 17 common workplace VOCs was selected with the understanding that actual workplace exposures might involve greater or fewer VOCs in practice. Table 1 lists the set of 24 target and interfering VOCs selected, together with pv values and assigned TLVs.
Results from previous studies were considered in the selection of the μPCF adsorbents, the μcolumn stationary phase and total length, the number of sensors and nature of MPN coatings for the μCR array, and the 5- and 10-ml air sample volumes used at the outset39,40,47,50,58,61. For the latter, we assumed a working limit of detection (LOD) of ~5 ng for the μCR sensors47,50,58,61. A 40-fold concentration range limit was imposed, indexed to the TLV Time-Weighted Average (TLV-TWA, 8 h average) for each VOC. That is, it was assumed that 0.1×TLV was a suitable LOD-level concentration to measure and that 4×TLV was a suitable maximum quantification limit for any given measurement. Several target compounds also have an assigned Short-Term Exposure Limit TLV (that is, TLV-STEL), which is a 15 min average limit set to protect against acute health effects, and is usually within 4× the TLV-TWA (Table 1). The challenge of specifying a fixed sample volume for mixtures of VOCs with widely different TLV values is discussed in the Supplementary Section S2.
Pre-trap characterization
Our working goal was to pre-trap compounds with pv values similar to or lower than that of n-tridecane (C13, pv=0.0075 kPa) while allowing compounds with pv values similar to or greater than that of C12 (pv=0.027 kPa) to pass through with negligible retention. Compounds of intermediate volatility would be partially retained, as a practical concession. Additionally, the pre-trap had to be regenerable via backflushing without heating.
Details of the tests performed and results obtained are given in the Supplementary Section S3. Initial tests using packed tubes containing ~mg quantities of either C-F or C-C (that is, pre-trap A) challenged with n-alkanes C11 to C13 at ~200 ppm failed to yield acceptable results. In subsequent tests with capillary-column pre-traps, B1 and B2 gave similar results, the best of which were obtained with segments 6.5 cm in length. Representative results from pre-trap B2 are shown in Supplementary Figure S2, which produced a 10% breakthrough volume of 5 ml for C13 but a 90% breakthrough volume of only 1.2 mL for C11. The breakthrough volume of C12 was ~2.6 mL. For a sample of 5–10 mL, this pre-trap was considered to offer an acceptable trade off in performance. Backflushing/cleaning of the pre-trap required ~2× the sample volume.
μPCF characterization
The dynamic adsorption capacities and desorption efficiencies for representative VOCs were determined previously using the predecessor to the current μPCF device, which had the same fluidic layout39. A sampling rate of 5 or 10 mL min−1 made no difference in capacity. Because the same mass of C-B and a greater mass of C-X were used in the new μPCF, the capacities were not re-confirmed.
Since a new heater design was used in the current μPCF, a limited set of tests of the injection band width was conducted at an analytical-path flow rate of 3 mL min−1 for both splitless injections and injections with a 2:1 split ratio (vent:analysis). Benzene, toluene, and C12 (~5 ppm each) were tested to span the range of target-VOC pv values. The values of full width at half maximum (fwhm) of the FID peaks measured with splitless injection were 0.94, 1.0, and 1.4 s, respectively, and the asymmetry factors were 1.1, 1.7 and 2.3, respectively. Desorption efficiencies were >99%. With a 2:1 split (that is, desorption, 9 mL min−1; analysis, 3 mL min−1), the fwhm values decreased to 0.59, 0.79, and 0.85 s, respectively, and the asymmetry factors for toluene and C12 decreased by approximately 10% (Supplementary Figure S3 and Supplementary Section S4). These fwhm values are somewhat smaller than those observed with the previous μPCF39, attesting to the improved heater of the new device.
The sharp injections for benzene and toluene are more important for achieving good chromatographic resolution because C12 and compounds of similar volatility benefit from on-column focusing. Thus, in those cases where separation of earlier eluting compounds is more important than sensitivity, split injection is advantageous39,50.
μColumn characterization
Helium was chosen as the carrier gas because of its superior chromatographic performance relative to scrubbed ambient air at typical operating flow rates62. The 2.4 g of He in each canister is projected to allow 228 analyses, corresponding to 30 h of continuous operation at 8 min (and 63 ml of He) per cycle. Tests of the separation efficiency of the dual μcolumn ensemble with n-octane yielded optimal average volumetric flow rates of 0.17 and 0.56 mL min−1 for N2 and He, respectively (Supplementary Figure S4 and Supplementary Section S5) and a maximum plate count N of ~4300 plates/m. At our operating flow rate of 3 mL min−1 (that is, much greater than the optimal flow rate), the values of N were 150 and 570 plates/m for N2 and He, respectively, which justifies the use of He as the carrier gas.
Tests of the μcolumn capacity indicated that the fwhm values of the peaks from several test compounds started to increase significantly and resolution started to degrade significantly at injected masses >8 μg (Supplementary Figure S5 and Supplementary Section S5). For an air sample of 10 mL, 8 μg corresponds to an air concentration of 800 mg m−3, which is >4×TLV for most, but not all, compounds with assigned TLVs (Table 1 and Ref. 56). For reference, toluene has a TLV of 20 ppm, or 75 mg m−3, whereas heptane has a TLV of 400 ppm, or 1600 mg m−3. Thus, the capacity of the μcolumns, while sufficient for the vast majority of VOCs of interest, might be exceeded for VOCs with high TLVs at concentrations >TLV, with a resulting slight decrease in chromatographic resolution. In such cases, a modest injection split could be used.
PEMM-1 temperature control, sample throughput, and humidity effects
The assembled PEMM-1 prototype was first run through a series of 22 discrete sampling and analytical cycles over a 3-h period using blanks of N2: 1 min sample (10 mL min−1), splitless injection and 4 min separation (3 mL min−1) using a typical μcolumn temperature program, with a 3-min cool-down period. See Supplementary Figure S6 and Supplementary Section S6 for typical temperature profiles. The internal (that is, baseplate) temperature rose by only ~3 °C during μcolumn heating and returned to baseline during cool down. The run-to-run variability in the temperature ramps of the μcolumns was <3% (relative s.d. (RSD)). The μCR array temperature varied randomly by <1.7% (range <0.4 °C) around its average of 30 °C within a run, without active heating, and showed no net drift. Tests of the two press-tight union heaters showed no effect on eluting peak widths with the upstream heater and significant effects with the downstream heater only for C11 and C12, where the fwhm values were 12% and 21% larger, respectively, without heating. Therefore, the upstream press-tight heater was not used.
A limited test of humidity effects was run on a mixture of benzene and toluene, assuming that the only effect might be overlap of the water vapor peak with peaks from early-eluting compounds41. Comparison of a dry test atmosphere with one at 60% relative humidity showed that water vapor eluted as small broad peaks ~20 s before benzene with no significant changes in the magnitudes of the benzene (or toluene) peaks from any sensor. Consistent with the MPN polarities, the ratios of water peak magnitudes were 1:1:2.3:4.6:3.7 for C8, EOE, OPH, TEG, and HME. Rejection of most atmospheric water vapor is attributable to the use of hydrophobic μPCF adsorbents.
PEMM-1 calibrations and LODs
Prior to generating the calibration curves, the throughput of a 10-VOC mixture was determined by bypassing the μCR array and connecting the output of the second μcolumn to an FID via a heated transfer line. A 10-ml sample of the mixture (100 ppm each in a Flex-foil bag) was sampled and analyzed in triplicate with and without the pre-trap installed. The results were also compared with those obtained from the bench scale GC-FID for samples drawn from the same test atmosphere but with sample-loop injection.
For benzene through C12, a <3% reduction in average peak area was observed with the pre-trap installed. For C13, a 72% reduction in peak area was noted with the pre-trap, indicating substantial retention of this low-volatility interference per the design of the pre-trap. However, the results from the reference GC-FID indicated that some loss of C13 to surface adsorption in the flow path is also involved (see Supplementary Figure S7 and Supplementary Section S7 for details).
Calibrations were performed using a 2:1 split-flow injection setting over a 40-fold range of sample mass (that is, from 0.1 to 4× TLV-TWA) on two subsets of compounds, that is, those with TLVs of 0.5 to 20 ppm and those with TLVs of 25 to 400 ppm. Samples of 10 and 20 ml were drawn from each of three test atmospheres in bags to span the injection masses corresponding to these concentration ranges. The analysis proceeded as described above using the same temperature program as in all subsequent testing, and all compounds within a subset were fully resolved chromatographically.
Peak shapes differed significantly among VOCs and sensors, and tailing (that is, peak asymmetry) generally increased with elution time. The peaks for 2-hexanone (MBK, compound 13 in Table 1), which had a mid-range retention time tR of 67 s, are shown in Figure 3 in order of increasing asymmetry. Sensors coated with HME, EOE, and C8 showed much less tailing than did TEG and OPH, and asymmetry factors ranged from 1.4 to 4.3. Data collected from one of the back-up sensors ruled out location along the flow path as a contributing factor to peak broadening. The trends with VOC elution time indicate that vapor sorption and desorption rates in/out of the MPN films contribute to both peak broadening and tailing, which is consistent with previous reports47,51,53.

Range of peak shapes among the μCR sensors in the array for the representative compound 2-hexanone (MBK, compound 13 in Table 1, tR=67 s) from calibration test series. From left to right, the sensor and corresponding peak asymmetry factor (at 10% of peak max.) are listed as follows: HME, 1.4; EOE, 1.7; C8, 1.8; TEG, 2.3; and OPH, 4.3. Each small tick mark on the x axis is 1 s. Peaks have been scaled vertically to have similar heights such that the y axis units are arbitrary.
Plotting of peak height versus sample mass yielded straight lines with linear regression R2 values ⩾0.99 (forced-zero y-intercept) for most VOC-sensor pairs and >0.98 for all pairs. Calibration curves for each compound on all four μCR sensors are presented in Supplementary Figure S8, and the slope sensitivities and R2 values are compiled in Supplementary Table S2 (Supplementary Section S8). As shown, the EOE sensor exhibited the highest sensitivity for most VOCs due apparently to its amphiphilicity and the accessibility and flexibility of the thiolate monolayers, which could enhance the rates of sorption and desorption of VOCs in the MPN film63. The non-polar C8-coated sensor showed high sensitivity for aromatic and aliphatic hydrocarbons, as expected. Among the more polar VOCs, the TEG-coated sensor was more sensitive than the OPH- and HME-coated sensors, particularly for ketones and butyl acetate. The generally low sensitivity exhibited by the OPH sensor can be attributed to the greater thickness of the MPN film compared with the others, since previous work with this material as a sensor coating resulted in peak widths similar to those with C8 and HME coatings47. This would reduce VOC sorption-desorption rates and thus promote shorter, tailing peaks (Figures 3 and 4).

(a) Reference GC-FID chromatogram of the 24-VOC mixture. Conditions: 6-m capillary column (PDMS); He carrier gas; 50 μL loop injection; and each vapor at ~100 ppm except for acetone (500 ppm) in a Flex-foil bag in N2. (b) Corresponding PEMM-1 chromatograms from the 5 μCR sensors. Conditions: 1 min sample at 5 mL min−1; 60 s desorption at 225 °C; 2:1 split injection (3 mL min−1 for analysis); He carrier gas; μcolumns at 28 °C for 0.5 min, followed by 10 °C min−1 to 33 °C then 50 °C min−1 to 125 °C, then hold; and μCR array at 30 °C. See Table 1 for peak assignments and text for sensor acronym definitions. Normalized response patterns (pk. ht.) shown for 7 vapors (order, l-to-r: HME/EOE/C8/TEG/OPH).
The LODs were estimated from the slopes of the regression models (peak height vs. injected mass) of each sensor from the equation 3σ/slope, where σ is the RMS baseline noise level from each sensor, and these values are compiled in Supplementary Table S3 (Supplementary Section S8). Because a 2:1 split injection was used, for the purpose of deriving LODs in terms of air concentrations for a given sample volume, it was necessary to divide the slope by a factor of 3 to account for the loss of 2/3 of the captured mass. Assuming a 10-mL air sample, the LODs range from 0.1 ppm for TCB with the EOE sensor to 36 ppm for C12 with the OPH sensor (Table 1). The LODs for a given VOC generally differ by <5-fold among the sensors in the array, with the notable exceptions of the alkanes, for which the range is higher due to low sensitivity from the polar sensor films.
Importantly, our decision to use 2:1 split injections gave priority to chromatographic resolution over sensitivity and resulted in a loss in sensitivity of approximately threefold (somewhat less for early-eluting compounds that benefit from peak compression at the higher desorption flow rate39). Thus, for splitless injections, the sensitivity should increase ~2.5–3-fold, and the LODs should decrease proportionally from the values listed in Table 1. Assuming that splitless injections are used and further assuming that detectable signals from only 4 of 5 sensors are required for vapor recognition (OPH sensor omitted, vide infra)42,43, all compounds could be detected and recognized well below their TLV-STEL values, all but benzene could be detected and recognized well below their TLV-TWA values, and all but benzene, MIBK, and MBK could be detected and recognized at or below 0.1×TLV.
PEMM-1 response stability and autonomous operation
Table 2 presents the RSDs around the average values of peak area obtained for 9 selected compounds from the C8- and OPH-coated MPN sensors each day for 7 consecutive days on the basis of four analyses per day (note: RSD values for peak heights were very similar to those for peak area with the exceptions of those of the last three compounds for the OPH-coated sensor, where the peak height variability was consistently a few percentage higher, undoubtedly due to the broadness of the peaks). The RSD values for the other sensors fell within the ranges spanned by these two sensors, with a few exceptions for the TEG-coated sensor. For the C8 sensor, the intra-day RSDs ranged from 1.0 to 8.0% among the VOCs, and the OPH sensor RSDs were generally higher and ranged from 0.8 to 9.9%. Later-eluting compounds generally gave higher values, but no trend over time was noted in any case. The greater variability observed for the OPH (and TEG) sensors is consistent with responses that are more highly dependent on sorption/desorption rates and are therefore more sensitive to small flow rate fluctuations. The corresponding RSDs for tR values, which are presented in Supplementary Table S4 (Supplementary Section S9), were <1.2% for all compounds except for the earliest eluting C7 (RSD <3.6%).
Also shown in Table 2 are the inter-day RSD values calculated from the 7 daily average peak area values. For reference, the RSD values are given for the peak areas from separate analyses of the same test atmosphere performed with a bench-scale GC-FID (loop injection, 1 sample per day). As shown, the PEMM-1 peak area measurements showed excellent stability/reproducibility, with RSD values consistently higher than but comparable to those from the GC-FID for all compounds (that is, 2.7–9.6% and 1.3–7.5%, respectively). The values of tR and fwhm were similarly stable (see Supplementary Table S5 and Supplementary Section S9).
The PEMM-1 prototype was then operated continuously and autonomously for 8 h on each of two consecutive days (that is, 8 min per cycle, 60 cycles per day). For this series, the 6-VOC mixture was analyzed in triplicate at the outset, 54 blank analyses were performed, and the same mixture was re-analyzed in triplicate at the end of the day. As shown in Supplementary Table S6 (Supplementary Section S9), the tR values and sensitivities were notably stable, decreasing only slightly over the course of each day, which we attribute to small increases in flow rate and/or baseline temperature. More specifically, tR values decreased by <1.6%, and peak heights and peak areas decreased by ⩽9.8 % (typically <6%) over the course of 8 h. Although the average values of these variables were lower on the second day than on the first, differences were again small and sufficiently stable for any practical application.
PEMM-1 24 VOC analysis with vapor recognition
Figure 4 shows a set of 24-VOC chromatograms generated from the reference GC-FID (Figure 4a) and the five μCR sensors of the PEMM-1 prototype (Figure 4b) for a test atmosphere containing a mixture of these compounds at ~100 ppm each (500 ppm for acetone). The separation required only 3 min with PEMM-1. Although the retention order was the same, the specific retention times differed between the reference GC-FID and PEMM-1 traces due to differences in linear velocities. The lower overall resolution for the PEMM-1 traces relative to the GC-FID can be attributed to a combination of lower μcolumn separation efficiency and longer sensor response times. Consistent with Figure 3, the C8, EOE, and HME sensors gave relatively sharp peaks. Values of fwhm were <2.7 s in all cases and ≤1.5 s for the early-eluting target compounds 8–13. With the TEG and OPH sensors, the peaks were broader and more asymmetric, and fwhm values were 4.0 s for C12 and <2.5 for compounds 8–13.
The first (broad) peak in each PEMM-1 trace was water vapor presumably present at trace levels in the liquid samples from which the test atmosphere was generated. Peaks 1–7, most of which co-elute, were from the interferences, which were well resolved from the first target compound of benzene (peak 8). The separation of the 17 target mixture components with the PEMM-1 was excellent with the EOE, C8, and HME sensors (Rs>1.5), somewhat less good with TEG, and rather poor with the OPH sensor. The observed increases in peak width with increasing elution time were expected and were also observed in the GC-FID trace. Notwithstanding the OPH sensor, the excessive tailing of which renders it of less value as a detector, the speed and resolution obtained were quite good. Taken together with the peak capacities, which ranged from 80 to 103 among the sensors for a 4 min separation based on MBK, and the peak production rates, which ranged from 20–25 per min (also based on MBK), the chromatographic performance of the PEMM-1 exceeds that of other reported GC prototypes employing microfabricated separation components28,29,31–36,50–53.
The normalized response pattern for each compound was obtained by dividing the calibrated slope (that is, sensitivity) from each sensor by the slope from the sensor that gave the highest sensitivity in the μCR array for that compound. The patterns for all compounds are presented in Supplementary Figure S9 (Supplementary Section S10), and those for the four partially co-eluting target VOC pairs in Figure 4b are shown above the set of chromatograms. The ability to differentiate individual VOCs was assessed via Monte Carlo (MC) simulations coupled with extended disjoint principal components regression (EDPCR) classification models, as summarized in the Supplementary Section S10. This technique yields statistical estimates of single-vapor recognition rates (RR, %) based on the actual response variabilities and calibrated sensitivities41–43.
The resulting confusion matrix for all 17 target compounds obtained using the responses from all 5 sensors in the array (Supplementary Table S7 and Supplementary Section S10) shows that 8 of the 17 targets could be recognized with <10% error based on their response patterns alone if they were chromatographically resolved from other compounds. Even for the other 9 compounds with lower RR values, the use of tR values together with the response patterns could lead to unequivocal confirmation of their identities by reference to a calibration library. Removing the OPH sensor and re-running the MC-EDPCR analysis with the remaining four sensors resulted in relatively little change in RR values for most VOCs (with certain exceptions; see Supplementary Tables S7 and S8 and Supplementary Section S10) and no net loss in the effective vapor recognition capability. Regardless of the number of sensors used, it was not possible to differentiate m-xylene (XYL) from ethylbenzene (ETB) at a high rate based on their response patterns. The n-alkanes were also difficult to differentiate, as were 3-heptanone (EBK) and butyl acetate (BAC). Fortunately, homologous n-alkanes are always well resolved chromatographically, as were EBK and BAC (due to large differences in pv values).
For those pairs of peaks that were not fully resolved with all sensors (that is, those for which patterns were included in Figure 4b), MC-EDPCR analyses were run on each binary mixture to assess the capability for local discrimination. To simplify the analysis, the peaks from each pair were assumed to completely overlap. The RR values were ⩾95% for TCE+C7, TMB+C10, and XYL+EBK. Only the XYL+ETB pair (RR=77%) could not be effectively differentiated from its individual component compounds. This exceptional case notwithstanding, it is clear that this feature of the PEMM-1 prototype significantly enhances its analytical power.
Conclusions
We have demonstrated that the PEMM-1 prototype μGC described in this work is capable of direct, autonomous, multi-VOC determinations at concentrations relevant to workplace applications. The speed, reliability, selectivity, limits of detection, dynamic ranges, low operating power, and types of VOCs amenable to accurate detection and recognition render the PEMM-1 an effective new addition to the repertoire of quantitative exposure assessment tools available to occupational health scientists. Reconciling the trade offs among VOC mixture pre-selection, pre-concentration, separation, and recognition/detection functions was central to realizing effective system-level performance. Collectively, the operational features and performance characteristics of the PEMM-1 prototype demonstrated in this study exceed those demonstrated with other prototype μGCs reported to date28,29,31–36,50–53. Future work on optimizing sensor coating strategies should yield improvements in peak shapes and reductions in LODs. The results obtained from this study have been used to inform the design of a battery-powered, wearable prototype (PEMM-2), from which promising preliminary data have recently been generated. The results from that effort will be the subject of a forthcoming article.
References
SEER Technology. Accusense Chemical Recognition System. Salt Lake City: SEER Technology. 2017. Available at http://www.seertechnology.com/accusense (accessed June 2017).
INFICON. Micro GC Fusion Gas Analyzer. Basel: INFICON. 2017. Available at http://products.inficon.com/en-us/nav-products/Product/Detail/Micro-GC-Fusion-Gas-Analyzer?path=Products%2Fpg-ChemicalDetection (accessed July 2017).
Defiant Technologies. Hand Held Gas Chromatography. Albuquerque: Defiant Technologies. 2017. Available at www.defiant-tech.com (accessed July 2017).
Electronic Sensor Technology. zNose® Portfolio. Newbury Park: Electronic Sensor Technology. 2009. Available at http://www.estcal.com/products (accessed July, 2017).
Gasmet Technologies. Portable DX4040 FTIR Gas Analyzer. Helsinki: Gasmet. 2017 Available at http://www.gasmet.com/products/portable-gas-analyzers/dx4040 (accessed April 2017).
INFICON. HAPSITE® ER Chemical Identification System. East Syracuse: INFICON. 2017. Available at http://products.inficon.com/en-us/Product/Detail/HAPSITE-ER-Identification-System?path=Products%2Fpg-ChemicalDetection (accessed April 2017).
FLIR. Griffin G510. Wilsonville: FLIR Systems. 2017 Available at http://www.flir.com/G510/ (accessed June 2017).
Voiculescu I, McGill RA, Zaghloul ME et al. Micropreconcentrator for enhanced trace detection of explosives and chemical agents. IEEE Sensors Journal 2006; 6: 1094–1104.
Manginell RP, Adkins DR, Moorman MW et al. Mass sensitive microfabricated chemical preconcentrator. Journal of Microelectromechanical Systems. 2008; 17: 1396–1407.
Wong MY, Cheng WR, Liu MH et al. A preconcentrator chip employing μ-SPME array coated with in-situ synthesized carbon adsorbent film for VOCs analysis. Talanta 2012; 101: 307–313.
Camara EH, Breuil P, Briand D et al. A micro gas preconcentrator with improved performance for pollution monitoring and explosives detection. Analytica Chimica Acta 2011; 688: 175–182.
Lambertus G, Elstro A, Sensenig K et al. Design, fabrication and evaluation of microfabricated columns for gas chromatography. Analytical Chemistry. 2004; 76: 2629–2637.
Bhushan A, Yemane D, Trudell D et al. Fabrication of micro-gas chromatograph columns for fast chromatography. Microsystem Technologies 2007; 13: 361–368.
Reidy S, Lambertus G, Reece J et al. High-performance, static-coated silicon microfabricated columns for gas chromatography. Analytical Chemistry 2006; 78: 2623–2630.
Stadermann M, McBrady AD, Dick B et al. Ultrafast gas chromatography on single-wall carbon nanotube stationary phases in microfabricated channels. Analytical Chemistry. 2006; 78: 5639–5644.
Potkay JA, Lambertus GR, Sacks RD et al. A low power pressure and temperature-programmable micro gas chromatography column. Journal of Microelectromechanical Systems. 2006; 16: 1071–1079.
Radadia D, Salehi-Khojin A, Masel RI et al. The effect of microcolumn geometry on the performance of micro-gas chromatography columns for chip scale gas analyzers. Sensors and Actuators B: Chemical 2010; 150: 456–464.
Shakeel H, Agah M . High density semipacked separation columns with optimized atomic layer deposited phases. Sensors and Actuators B: Chemistry 2017; 242: 215–223.
Chen BX, Huang TY, Jian RS et al. A multidimensional micro gas chromatograph employing a parallel separation multi-column chip and stop-flow μGC×μGCs configuration. Lab on a Chip 2013; 13: 1333–1341.
Gaddes D, Westland J, Dorman FL et al. Improved micromachined column design and fluidic interconnects for programmed high-temperature gas chromatography separations. Journal of Chromatography A 2014; 1249: 96–104.
Archibald R, Datskos P, Devault G et al. Independent component analysis of nanomechanical responses of cantilever arrays. Analytica Chimica Acta 2007; 584: 101–105.
Li M, Myers E, Tang HX et al. Nanoelectromechanical resonator arrays for ultrafast, gas-phase chromatographic chemical analysis. Nano Letters. 2010; 10: 3899–3903.
Chen FY, Chang WC, Jian RS et al. Novel gas chromatographic detector utilizing the localized surface plasmon resonance of a gold nanoparticle mono layer inside a glass capillary. Analytical Chemistry 2014; 28: 5257–2564.
Zhu HB, Nidetz R, Zhou ML et al. Flow-through microfluidic photoionization detectors for rapid and highly sensitive vapor detection. Lab on a Chip 2015; 15: 3021–3029.
Sun J, Cui D, Chen X et al. Design, modeling, microfabrication and characterization of novel micro thermal conductivity detector. Sensors and Actuators B: Chemical 2011; 160: 936–941.
Akbar M, Shakeel H, Agah M . GC-on-chip: Integrated column and photoionization detector. Lab on a Chip 2015; 15: 1748–1758.
Liu J, Seo JH, Li YB et al. Smart multi-channel two-dimensional micro-gas chromatography for rapid workplace hazardous volatile organic compounds measurement. Lab on a Chip 2013; 13: 818–825.
Lee J, Zhou M, Zhu H et al. Fully automated portable comprehensive 2-dimensional gas chromatography device. Analytical Chemistry 2016; 88: 10266–10274.
Dziuban JA, Mroz J, Szczygielska M et al. Portable gas chromatograph with integrated components. Sensors and Actuators A: Physical 2004; 115: 318–330.
Manginell RP, Bauer JM, Moorman MW et al. A Monolithically-integrated μGC chemical sensor system. Sensors 2011; 11: 6517–6532.
Qin YT, Gianchandani YB . A fully electronic microfabricated gas chromatography with complementary capactitive detectors for indoor pollutants. Microsystems and Nanoengineering 2016; 2: 15049.
Akbar M, Restaino M, Agah M . Chip-scale gas chromatography: From injection through detection. Microsystems and Nanoengineering 2015; 1: 15039.
Lewis PR, Manginell RP, Adkins DR et al. Recent advancements in the gas-phase μChem Lab. IEEE Sensors Journal 2006; 6: 784–795.
Zampolli S, Elmi I, Mancarella F et al. Real-time monitoring of sub-ppb concentrations of aromatic volatiles with a MEMS-enabled miniaturized gas-chromatograph. Sensors and Actuators, B: Chemical 2009; 141: 322–328.
Garg A, Akbar M, Vejerano E et al. Zebra GC: A mini gas chromatography system for trace-level determination of hazardous pollutants. Sensors and Actuators, B: Chemical 2015; 212: 145–154.
Tzeng TH, Kuo CY, Wang SY et al. A portable micro gas chromatography system for lung cancer associated volatile organic compound detection. IEEE Journal of Solid-State Circuits 2016; 51: 259–272.
Tian WC, Pang S, Lu CJ et al. Microfabricated preconcentrator-focuser for a microscale gas chromatograph. Journal of Microelectromechanical Systems 2003; 12: 264–272.
Sukaew T, Zellers ET . Evaluating the dynamic retention capacities of microfabricated vapor preconcentrators as a function of flow rate. Sensors and Actuators B: Chemical 2013; 183: 163–171.
Bryant-Genevier J, Zellers ET . Toward a microfabricated preconcentrator-focuser for a wearable micro-scale gas chromatograph. Journal of Chromatography A 2015; 1422: 299–309.
Serrano G, Reidy S, Zellers ET . Assessing the reliability of wall-coated microfabricated gas chromatographic separation columns. Sensors and Actuators B: Chemical 2009; 141: 217–226.
Zhong Q, Steinecker W, Zellers ET . Characterization of a high-performance portable GC with a chemiresistor array detector. Analyst 2009; 134: 283–293.
Hsieh MD, Zellers ET . Limits of recognition for simple vapor mixtures determined with a microsensor array. Analytical Chemistry 2004; 76: 1885–1895.
Jin C, Kurzawski P, Hierlemann A et al. Evaluation of multitransducer arrays for the determination of organic vapor mixtures. Analytical Chemistry 2008; 80: 227–236.
Kim SJ, Serrano G, Wise KD et al. Evaluation of a microfabricated thermal modulator for comprehensive two-dimensional gas chromatography. Analytical Chemistry 2011; 83: 5556–5562.
Collin WR, Bondy A, Paul D et al. μGC×μGC: comprehensive two-dimensional gas chromatographic separations with microfabricated components. Analytical Chemistry 2015; 87: 1630–1637.
Scholten K, Collin WR, Fan X et al. Nanoparticle-coated micro-optofluidic ring resonator as a detector for microscale gas chromatographic vapor analysis. Nanoscale 2015; 7: 9282–9289.
Serrano G, Sukaew T, Zellers ET . Hybrid preconcentrator/focuser module for determinations of TNT marker compounds with a micro-scale gas chromatograph. Journal of Chromatography A 2013; 1279: 76–85.
Collin WR, Nunovero N, Paul D et al. Comprehensive two-dimensional gas chromatographic separations with a temperature programmed microfabricated thermal modulator. Journal of Chromatography A 2016; 1444: 114–122.
Collin WR, Scholten KW, Fan X et al. Polymer-coated micro-optofluidic ring resonator detector for a comprehensive two-dimensional gas chromatographic microsystem: μGC×μGC- μOFRR. Analyst 2016; 141: 261–269.
Lu CJ, Steinecker WH, Tian WC et al. First-generation hybrid MEMS gas chromatograph. Lab on a Chip 2005; 5: 1123–1131 1151.
Kim KS, Chang H, Zellers ET . Microfabricated gas chromatography for the selective determination of trichloroethylene vapor at sub-parts-per-billion concentration in complex mixtures. Analytical Chemistry 2011; 83: 7198–7206.
Kim SK, Burris DR, Chang H et al. Microfabricated gas chromatograph for on-site determinations of trichloroethylene in indoor air arising from vapor intrusion, part 1: field evaluation. Environmental science & technology 2012; 46: 6065–6072.
Collin WR, Serrano G, Wright LK et al. Microfabricated gas chromatograph for rapid, trace-level determinations of gas phase explosive marker compounds. Analytical Chemistry 2014; 86: 655–663.
Wright LK, Zellers ET . A nanoparticle-coated chemiresistor array as a microscale gas chromatograph detector for explosive marker compounds: flow rate and temperature effects. Analyst 2013; 138: 6860–6868.
Lin Z, Nuñovero N, Wang J et al. A zone-heated gas chromatographic microcolumn: energy efficiency. Sensors and Actuator B: Chemical 2018; 254: 561–572.
ACGIH. Threshold Limit Values for Chemical Substances and Physical Agents & Biological Exposure Indices. 2016 ed. Cincinnati: ACGIH. 2016.
Rowe MP, Plass KE, Kim K et al. Single-phase synthesis of functionalized gold nanoparticles. Chemistry of materials 2004; 16: 3513–3517.
Jian RS, Huang RX, Lu CJ . A micro GC detector array based on chemiresistors employing various surface functionalized monolayer-protected gold nanoparticles. Talanta 2008; 88: 160–167.
Bohrer FI, Covington E, Kurdak C et al. Characterization of dense arrays of chemiresistor vapor sensors with sub-micron features and patterned nanoparticle interface layers. Analytical Chemistry 2011; 83: 3687–3695.
NIH U.S. National Library of Medicine. TOXNET Databases: Hazardous Substances Data Bank (HSDB). Bethesda: U.S. National Library of Medicine. 2017 Available at https://toxnet.nlm.nih.gov/newtoxnet/hsdb.htm (accessed April 2017).
Bryant-Genevier J, Scholten K, Kim SK et al. Multivariate curve resolution of co-eluting vapors from a gas chromatograph with microsensor array detector. Sensors and Actuator B: Chemical 2014; 202: 167–176.
Grob RL, Barry EF . Modern Practice of Gas Chromatography4th ednHoboken: Wiley Interscience. 2004, 114–115.
Krasteva N, Besnard I, Guse B et al. Self-assembled gold nanoparticle/dendrimer composite films for vapor sensing applications. Nano Letters 2002; 2: 551–555.
Acknowledgements
We sincerely thank Dr Will Collin for technical assistance with various critical aspects of prototype development and Professor Katsuo Kurabayashi for advice on thermal modeling. Devices were constructed in the Lurie Nanofabrication Facility. Funding was provided by the National Institute for Occupational Safety and Health of the Centers for Disease Control and Prevention (Grant. R01-OH-010297). We are solely responsible for the content of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Microsystems & Nanoengineering website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, J., Bryant-Genevier, J., Nuñovero, N. et al. Compact prototype microfabricated gas chromatographic analyzer for autonomous determinations of VOC mixtures at typical workplace concentrations. Microsyst Nanoeng 4, 17101 (2018). https://doi.org/10.1038/micronano.2017.101
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/micronano.2017.101
This article is cited by
-
Exhaled volatile organic compounds as novel biomarkers for early detection of COPD, asthma, and PRISm: a cross-sectional study
Respiratory Research (2025)
-
Review on micro-gas chromatography system for analysis of multiple low-concentration volatile organic compounds: preconcentration, separation, detection, integration, and challenges
Micro and Nano Systems Letters (2024)
-
Investigation of sorptive interactions between volatile organic compounds and supramolecules at dynamic oscillation using bulk acoustic wave resonator virtual sensor arrays
Microsystems & Nanoengineering (2024)
-
Portable comprehensive two-dimensional micro-gas chromatography using an integrated flow-restricted pneumatic modulator
Microsystems & Nanoengineering (2022)
-
Built-in Chamber-Coupled Electronic Pressure Control of Ambient Air as the Carrier Gas for a Cylinder-Free Portable Gas Chromatograph
Chromatographia (2022)
