
Abstract
Lipid droplet-associated hydrolase (LDAH) is a lipid hydrolase abundantly expressed in adipose and ovarian tissues and macrophages. However, LDAH’s functions in ovarian cancer are largely unknown. Analysis of publicly available patient datasets showed decreased LDAH expression in advanced stages of ovarian cancer, and low LDAH levels were associated with poor survival outcomes in ovarian cancer patients. Consistently, knockdown (KD) of LDAH in human ovarian cancer cell lines increased tumor cell proliferation but decreased endoplasmic reticulum (ER) stress and apoptosis upon cisplatin treatment. In addition, compared to scrambled control, LDAH KD ovarian cancer cells showed smaller lipid droplets (LDs), decreased triacylglycerol (TAG) content, and increased expression of adipose triglyceride lipase (ATGL), carnitine palmitoyltransferase 1 A (CPT1A), and phospho-NF-kB. Our xenograft studies also showed increased tumor growth, increased ATGL expression, and decreased apoptosis after cisplatin treatment in LDAH KD tumors. ATGL overexpression increased cisplatin resistance and expression of CPT1A and phospho-NF-kB, whereas treatment of LDAH KD cells with an ATGL inhibitor attenuated the phenotype. Lastly, we observed that high ATGL levels were associated with shorter survival in ovarian cancer patients. Collectively, our results suggest that ovarian cancer cells downregulate LDAH expression, leading to enhanced ATGL-mediated TAG hydrolysis and increased tumor growth and chemoresistance.
Similar content being viewed by others
Introduction
Ovarian cancer is the fifth leading cause of women’s cancer mortality and the most lethal gynecological cancer in the United States [1]. Acquired chemoresistance is the major challenge to effective ovarian cancer treatment [2, 3]. Dysregulated fatty acid metabolism has emerged as an important contributor to acquired chemoresistance in ovarian cancer cells [4,5,6]. Ovarian cancer cells frequently upregulate fatty acid uptake, de novo fatty acid synthesis, and mitochondrial fatty acid β-oxidation (FAO). FAO is a metabolic process of breakdown of free fatty acids (FFA) into acetyl-CoA, which can then be used to produce energy in the mitochondria to promote ovarian cancer growth, dissemination, and chemoresistance [6,7,8,9]. Ovarian cancer cells convert surplus fatty acids into triacylglycerol (TAG) and store them in lipid droplets (LDs), which allow them to regulate fatty acid metabolism and intracellular signaling by releasing free fatty acids and lipid mediators from LDs [10, 11]. However, it is still unclear how fatty acid metabolism promotes ovarian cancer chemoresistance and how fatty acid metabolism can be targeted to overcome ovarian cancer chemoresistance.
Lipid droplet-associated hydrolase (LDAH) is a ubiquitously expressed LD-associated protein with a lipase structure and a canonical lipase/esterase catalytic motif [12,13,14,15]. LDAH is localized in the cytosol in close association with the endoplasmic reticulum (ER) but readily translocalizes to LDs upon lipid loading [12, 16]. LDAH promotes the clustering and enlargement of LDs in Drosophila [16]. LDAH reportedly inhibits the activity of adipose triglyceride lipase (ATGL), a rate-limiting enzyme that initiates the breakdown of TAGs into diacylglycerol and FFAs, thus stimulating LD expansion in HEK293 cells treated with oleic acid [12]. The physiological role of LDAH in cancer is relatively unknown. It has been reported that LDAH levels are reduced in prostate tumor tissues, and LDAH suppression increases tumorigenicity in prostate cancer cell lines [15]. A recent study also reported that LDAH induces the activation of an anti-cancer drug lasonolide-A that accumulates in LDs by cleaving an ester bond in its side chain, which facilitates the release of a more active metabolite into the cytosol [17].
In this study, we found that LDAH levels are reduced in ovarian tumor tissues, and LDAH suppression leads to enhanced ATGL-mediated TAG hydrolysis, increased ovarian cancer proliferation, and chemoresistance. Overall, these studies stress the relevance of lipid metabolism to the progression and chemoresistance of ovarian cancer and the potential of targeting LD metabolic steps to mitigate the progression of the disease.
Results
Low levels of LDAH are associated with poor prognosis in ovarian cancer
To determine the clinical significance of LDAH in ovarian cancer patients, we analyzed the correlation between LDAH expression and survival outcomes of 1207 ovarian cancer patients using Kaplan–Meier Plotter. We found that ovarian cancer patients with low levels of LDAH showed shorter overall survival (OS; HR = 0.76 (0.64–0.91), log-rank P = 0.0024) (Fig. 1A) and shorter progression free survival after platinum-based chemotherapy (PFS; HR = 0.74 (0.56–0.97), log-rank P = 0.031) (Supplementary Fig. 1A). We also found that LDAH expression was significantly decreased in advanced stage ovarian cancer (Fig. 1B). Analysis of GEO datasets (GSE14407, GSE40595, GSE115939) revealed that LDAH mRNA expression levels were significantly lower in ovarian cancer tissues than in normal ovary tissues (Fig. 1C, D, Supplementary Fig. 1B).

A Kaplan-Meier survival curve of overall survival (OS) in 1207 serous ovarian cancer patients based on LDAH expression. Red indicates high LDAH expression. B Expression of LDAH in different stages of ovarian cancer (Stage IA, I, IIIA, IIIB, IV, IVA) in the GENT2 database. N = 272. Histological subtypes of samples are shown in Supplementary Table 3. C, D LDAH mRNA expression level in ovarian cancer and normal ovarian tissue of GEO datasets GSE14407 (normal vs serous ovarian cancer) (C) and GSE40595 (normal vs high grade serous ovarian cancer) (D). Students’s T-test. E Western blot of LDAH in scrambled control (Scr) and LDAH knockdown (ShLDAH#69, #71, #75) ES2 and A2780 cells. The quantification of band intensity normalized to GAPDH is shown on the right. One-way ANOVA followed by Tukey’s multiple test. F mRNA expression of LDAH in scrambled control (Scr) or LDAH knockdown (ShLDAH#69, #71, #75) ES2 and A2780 cells. Y axis, fold change compared to scrambled control. N = 3. Two-way ANOVA followed by Tukey’s test. The effect of LDAH knockdown (shLDAH#71, #75) on proliferation in ES2 (G) and A2780 cells (H) determined by MTT assay. N = 3. Student’s T-test. Error bars, SD. **, p < 0.01, ***, p < 0.001. ****, p < 0.0001.
To determine the effect of LDAH downregulation in ovarian cancer cells, we performed stable gene knockdown of LDAH using shRNA (shLDAH#69, shLDAH#71, and shLDAH#75) in ES2 and A2780 human ovarian cancer cell lines. The efficiency of LDAH knockdown was validated by western blotting and real-time PCR (Fig. 1E, F). The gene knockdown efficiency of shLDAH#75 was higher than shLDAH#69 and shLDAH#71 in ES2 and A2780 cells (Fig. 1E). Next, we determined the effect of LDAH knockdown (shLDAH#71, #75) on proliferation in ES2 and A2780 ovarian cancer cells and found that, compared to scrambled control, LDAH knockdown increased the proliferation of both cell lines (Fig. 1G, H). Collectively, our results showed that low LDAH expression was associated with poor prognosis in ovarian cancer patients, and LDAH downregulation promoted ovarian cancer cell proliferation.
LDAH knockdown promotes cisplatin resistance in ovarian cancer cells
Since low levels of LDAH are associated with shorter progression free survival (PFS) after platinum-based chemotherapy (Supplementary Fig. 1A), we sought to determine the effect of LDAH knockdown on cisplatin resistance. Our western blotting data showed that LDAH knockdown significantly decreased levels of cleaved caspase 3 after 16 µM cisplatin treatment compared to scrambled control in ES2 and A2780 cells (Fig. 2A, B). Similarly, MTT assay showed increased viability in LDAH knockdown ES2 and A2780 cells compared to scrambled control cells, both under basal conditions and under cisplatin treatment (Fig. 2C, D, Supplementary Fig. 2A, B). Consistently, Annexin V/7-AAD staining showed decreased percentages of apoptotic cells in LDAH knockdown ES2 and A2780 cells upon cisplatin treatment (Fig. 2E), indicating increased chemoresistance in LDAH knockdown cancer cells.

Western blot of LDAH in scrambled control or LDAH knockdown (shLDAH #69, #71 and #75) ES2 (A) and A2780 (B) ovarian cancer cell lines. The quantification of band intensity normalized to GAPDH is shown below. Two-way ANOVA followed by Tukey’s multiple comparison test. MTT assay to measure the effect of LDAH knockdown on proliferation in ES2 (C) and A2780 (D) cancer cells with or without cisplatin treatment. N = 3. Student’s T-test. E Annexin V/7-AAD apoptosis analysis of scrambled control or LDAH knockdown ES2 and A2780 cancer cells after cisplatin treatment. N = 3. Student’s T-test. F Representative images of excised tumors from xenograft mouse models. G Volume of excised tumors from (F). One-way ANOVA followed by Tukey’s multiple comparison test. Error bars, SD. *, p < 0.05, **, p < 0.01, ***, p < 0.001. ****, p < 0.0001. ns, not significant.
To further investigate the role of LDAH in chemoresistance in vivo, we established a xenograft model. We injected scrambled or LDAH-knockdown ES2 ovarian cancer cells subcutaneously into female nude mice. After palpable tumors were formed, mice were injected intraperitoneally with saline (vehicle) or 3 mg/kg cisplatin three times a week for 2 weeks (Fig. 2F; Supplementary Fig. 2C). In untreated mice, LDAH knockdown significantly increased tumor volume (Fig. 2F, G). Interestingly, LDAH knockdown also conferred resistance to cisplatin treatment, and while cisplatin significantly reduced the volume of scrambled tumors, it did not significantly affect the growth of tumors with LDAH knockdown (Fig. 2F, G). Taken together, our in vitro and in vivo studies suggest that LDAH knockdown enhances the viability of ovarian cancer cells and impacts their response to cisplatin treatment. Next, we sought to investigate the mechanisms.
LDAH knockdown inhibits ER stress-mediated apoptosis in ovarian cancer cells
LDAH is reported to be localized in the cytoplasm in close association with the endoplasmic reticulum (ER) in HEK293 cells in standard culture conditions [12]. To determine the localization of LDAH in ovarian cancer cells, we co-transfected ES2 cells with the ER marker DsRed2-ER5 [18] and LDAH-GFP [12]. Our results showed co-localization of LDAH-GFP and DsRed2-ER5 (Fig. 3A), suggesting LDAH expression in the ER of ovarian cancer cells. Since ER stress can promote apoptosis of tumor cells, we examined the effect of LDAH knockdown on ER stress. Western blotting data showed that LDAH knockdown ES2 and A2780 cancer cells had decreased expression of the ER stress markers (Ero1a, IREI, PERK, PDI) than scrambled control cells, with and without cisplatin treatment (Fig. 3B, Supplementary Fig. 3A). Similarly, immunofluorescence and western blotting data showed decreased PDI and CHOP in LDAH knockdown ES2 and A2780 cells with and without cisplatin treatment as compared to scrambled control cells (Fig. 3C, Supplementary Fig. 3B–D). These data suggests that LDAH knockdown inhibits ER stress in ovarian cancer cells.

A Immunofluorescence images of DsRed2-ER5 (red) in LDAH-GFP expressing ES2 cells. Blue, DAPI. Scale bars, 20 µm. B Western blot of ER stress markers (Ero 1a, IREI, PDI, PERK) in scrambled control and LDAH knockdown ES2 cells, with or without cisplatin treatment. The quantification of band intensity normalized to GAPDH is shown on the right. Two-way ANOVA followed by Tukey’s multiple comparison test. C Immunofluorescence images of PDI (red) in scrambled control and LDAH knockdown ES2, with or without cisplatin treatment. Blue, DAPI. Scale bars, 50 µm. D Western blot of Bak1 in scrambled control and LDAH knockdown ES2cells, with or without cisplatin treatment. The quantification of band intensity normalized to GAPDH is shown on the right. One-way ANOVA followed by Tukey’s multiple comparison test. E, F qPCR analysis showing mRNA expression of Bak1 (E; pro-apoptotic) and BIRC2 (F; anti-apoptotic) in scrambled control and LDAH knockdown ES2, with and without cisplatin treatment. One-way ANOVA followed by Tukey’s multiple comparison test. G Immunofluorescence images of LDAH and PERK in scrambled control and LDAH overexpressing ES2 cancer cells. Scale bars, 20 µm. Quantification of PERK signal is shown on the right. Student’s T-test. H Western blot of cleaved caspase 3, p-PERK, PERK, and LDAH in scrambled and LDAH overexpressing cells after treatment with vehicle, cisplatin alone, or in combination with a PERK inhibitor GSK2606414. GAPDH, a loading control. The quantification of band intensity normalized to GAPDH is shown on the right. One-way ANOVA followed by Tukey’s multiple comparison test. I mRNA expression level of Bak1 (pro-apoptosis) in LDAH overexpressing cells after treatment with vehicle, cisplatin alone or combination of in combination with a PERK inhibitor GSK2606414. One-way ANOVA followed by Tukey’s multiple comparison test. Error bars, SD. *, p < 0.05, **, p < 0.01, ***, p < 0.001. ****, p < 0.0001. ns, not significant.
To further investigate the impact of LDAH knockdown in apoptosis, we measured the expression levels of pro-apoptotic (BAK1) and anti-apoptotic (BIRC2) proteins in scrambled and LDAH knockdown cells. The protein levels of the BAK1 were lower in LDAH knockdown ES2 and A2780 cancer cells than in scrambled controls, with and without cisplatin treatment (Fig. 3D, Supplementary Fig. 3E). qPCR analyses also showed lower BAK1 and higher BIRC2 mRNA levels in LDAH knockdown in ES2 and A2780 cells, with and without cisplatin treatment (Fig. 3E, F, Supplementary Fig. 3F, G).
Based on the results under LDAH knockdown, we asked whether LDAH upregulation would enhance cisplatin-induced apoptosis through increasing ER stress. We overexpressed LDAH in ES2 cells and found that LDAH overexpression significantly increased the immunofluorescence intensity of an ER stress marker PERK (Fig. 3G, Supplementary Fig. 3H). Next, we treated LDAH overexpressing ES2 cells with cisplatin alone or in combination with a PERK inhibitor GSK260641,4 and measured the levels of p-PERK and cleaved caspase 3. Compared to scrambled cells, LDAH-overexpressing ES2 cancer cells showed a marked increase in cleaved caspase 3 after cisplatin treatment, which was attenuated by GSK2606414 (Fig. 3H). Similarly, LDAH overexpressing ES2 cells showed increased BAK1 mRNA levels upon cisplatin treatment, which was also abrogated by GSK2602414 treatment (Fig. 3I). Altogether, studies on LDAH downregulation and overexpression suggest that LDAH knockdown mitigates ER stress and increases resistance to cisplatin-induced apoptosis.
LDAH knockdown increases TAG hydrolysis in ovarian cancer cells
Ovarian cancer cells often upregulate de novo fatty acid synthesis and store excess fatty acids in the form of triglyceride (TAG) in lipid droplets (LDs) [10, 11, 19,20,21]. Since LDAH enhances LD accumulation in HEK293 cells loaded with oleic acid [12], we asked if LD formation was altered in scrambled, LDAH overexpressing, and LDAH knockdown ovarian cancer cells. Similar to the results from HEK293 cells [12], we observed that, compared to controls, LDAH overexpression and knockdown in ES2 cells resulted in accumulation of larger and smaller LDs, respectively (Fig. 4A, Supplementary Fig. 4A). Consistently, LDAH knockdown cancer cells showed decreased TAG levels while LDAH overexpressing cancer cells showed increased TAG levels than scrambled control (Fig. 4B).

A BODIPY 493/503 staining (green) of lipid droplets in scrambled, LDAH knockdown (shLDAH), and LDAH overexpressing (OE) ES2 cancer cells. Blue, DAPI. Scale bars, 10 µm. Quantification of BODIPY signals is shown on the right. One-way ANOVA followed by Tukey’s multiple comparison test. B TAG levels (mg/dl) in scrambled, LDAH knockdown (shLDAH), and LDAH overexpressing (OE) ES2 cancer cells. One-way ANOVA followed by Tukey’s multiple comparison test. Western blot of CPT1A in scrambled control and LDAH knockdown ES2 (C) and A2780 (D) cancer cells with or without cisplatin treatment. The quantification of band intensity normalized to GAPDH is shown below. One-way ANOVA followed by Tukey’s multiple comparison test. Western blot of p-NF-kB and total NF-kB in scrambled control and LDAH knockdown ES2 (E) and A2780 (F) cancer cells with or without cisplatin treatment. The quantification of band intensity normalized to GAPDH is shown below. One-way ANOVA followed by Tukey’s multiple comparison test. Error bars, SD. *, p < 0.05, **, p < 0.01, ***, p < 0.001, ****, p < 0.0001.
The differences in LD size and TAG content in LDAH knockdown cells could be due to changes in TAG biogenesis, hydrolysis, or both. To address this question, we synchronized ES2 scrambled or shLDAH cancer cells by incubating them with charcoal stripped lipid-free medium for 24 h, followed by treatment with Triacsin C, an inhibitor of TAG synthesis. After 16 h of treatment with Triascin C, we incubated the cells with lipid-containing FBS medium, and cells were collected and fixed at different time points (4 h, 8 h, 12 h, 24 h) for BODIPY staining and TAG analysis. Our results showed no difference in BODIPY staining and TAG level (mg/dl) up to 12 h. We observed a significant decrease in BODIPY staining and TAG levels in shLDAH cells after 24 h (Supplementary Fig. 4B, C). Furthermore, western blotting data showed no difference in expression of diacylglycerol O-acyltransferase 1 (DGAT1; a key enzyme involved in TAG biogenesis) in Scr and shLDAH cells in ES2 and A2780 cell lines (Supplementary Fig. 4D). These results suggest that LDAH does not affect TAG biogenesis, and reduced size of LDs in shLDAH cancer cells is due to increased TAG hydrolysis.
Based on this reduction in TAG levels, we speculated that LDAH could affect fatty acid oxidation (FAO), a major metabolic pathway that breaks down free fatty acids and helps cancer cells meet their energetic and metabolic needs [9]. Consistent with decreased TAG levels, the expression of CPT1A, a rate-limiting enzyme of FAO, was increased in LDAH knockdown ES2 and A2780 cells compared to scrambled cells (Fig. 4C, D). When TAGs are hydrolyzed to free fatty acids, it can trigger the activation of NF-κB, a crucial transcription factor which promotes tumor growth and chemoresistance [22]. Our results showed increased p-NF-kB (indicative of NF-kB activation) in LDAH knockdown ES2 and A2780 cells compared to scrambled control, irrespective of cisplatin treatment (Fig. 4E, F). Collectively, our data suggests that LDAH knockdown cancer cells exhibit reduced TAG levels and increased FAO and NF-ĸB activity.
ATGL expression is increased in LDAH knockdown cells and is linked to poor prognosis
It has been reported that LDAH promotes LD accumulation by increasing proteasomal degradation of ATGL (the primary enzyme responsible for TAG hydrolysis) in HEK293 cells [12]. Since TAG levels were reduced in LDAH knockdown cells, we asked whether these cells have increased ATGL expression. Confirming this, western blotting data showed increased ATGL expression in LDAH knockdown ES2 and A2780 cancer cells compared to scrambled control (Fig. 5A, Supplementary Fig. 5A) with or without cisplatin treatment. Furthermore, MG132 (a proteasome inhibitor) treatment increased ATGL expression in both ES2 scr and shLDAH cells (Supplementary Fig. 5B), suggesting that LDAH decreases ATGL expression in ovarian cancer cells potentially via proteasomal degradation.

A Western blot of ATGL in scrambled and LDAH knockdown ES2 cancer cells with or without cisplatin treatment. GAPDH, a loading control. B Cells viability of scrambled control (SCR), LDAH knockdown (shLDAH), and ATGL overexpressing (ATGL OE) ES2 cancer cells. Student’s T-test. TAG levels (mg/dl) (C) and Western blot of CPT1A, ATGL, pNF-ĸB, and NF-ĸB (D) in scrambled control and ATGL-overexpressing ES2 cells. Student’s T-test. E Immunofluorescence images of LDAH, ATGL, and cleaved caspase 3 in excised subcutaneous tumors from Fig. 2F. Scale bars, 100 µm (LDAH/ATGL IF), and 50 µm (cleaved caspase 3 IF). Quantification of fluorescence intensity of ATGL and cleaved caspase 3 are shown on the right. One-way ANOVA followed by Tukey’s multiple comparison test. F Cell viability of scrambled, LDAH knockdown (shLDAH), and atglistatin (50 µM)- treated shLDAH ES2 cells after 8 µM cisplatin treatment for 3 days. Student’s T-test. G BODIPY 493/503 staining (green) of lipid droplets in scrambled, LDAH knockdown (shLDAH), and atglistatin (50 µM)-treated shLDAH ES2 cancer cells. Blue, DAPI. Scale bars, 10 µm. H Western blot of CPT1A, pNFĸB, and NFĸB in scrambled, LDAH knockdown (shLDAH), and atglistatin (50 µM)-treated shLDAH ES2 cancer cells. I Kaplan-Meier survival curve of overall survival (OS) in 212 ovarian cancer patients based on ATGL expression. Red indicates high ATGL expression. *, p < 0.05, ***, p < 0.001. ****, p < 0.0001.
Next, we asked whether ATGL upregulation causes similar phenotypes as LDAH knockdown in ovarian cancer cells. We overexpressed ATGL in ES2 and A2780 cells (Supplementary Fig. 5C) and examined cancer cell viability after cisplatin treatment, TAG levels, FAO, and NF-ĸB activation. Our results showed that ATGL overexpression increased cancer cell viability upon cisplatin treatment (Fig. 5B). Similar to LDAH knockdown cancer cells, ATGL overexpressing cancer cells also showed decreased TAG levels and increased expression of CPT1A and p-NF-kB (Fig. 5C, D, Supplementary Fig. 5D, E).
Consistent with our in vitro data, both immunohistochemistry and immunofluorescence staining of ATGL in scrambled control or LDAH knockdown tumors (Fig. 2F) showed increased ATGL expression in LDAH knockdown tumors than control tumors (Fig. 5E, Supplementary Fig. 6). Importantly, cleaved caspase 3 staining significantly decreased in LDAH knockdown tumors compared to scrambled tumors upon cisplatin treatment (Fig. 5E), showing inverse relationship between ATGL expression and cleaved caspase 3 levels in tumors.
To determine if ATGL inhibition can attenuate the effects of LDAH knockdown in ovarian cancer cells, we utilized atglistatin, a pharmacological inhibitor of ATGL. Cell viability of cisplatin-treated LDAH knockdown ES2 cancer cells was significantly attenuated by atglistatin treatment (Fig. 5F). BODIPY staining showed that atglistatin treatment increased LD size in LDAH knockdown ES2 cancer cells (Fig. 5G). Furthermore, atglistatin-treated LDAH knockdown cancer cells also showed decreased expression of CPT1A and pNF-kB (Fig. 5H). Finally, we sought to determine the clinical significance of ATGL in ovarian cancer patients. Analysis of the TCGA dataset revealed that high levels of ATGL were associated with shorter overall survival in ovarian cancer patients (Fig. 5I).
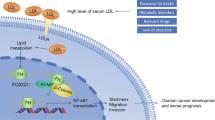
Collectively, these data suggest that LDAH expression is downregulated in ovarian cancer cells, which leads to increased tumor growth and chemoresistance due to the upregulation of ATGL-mediated TAG hydrolysis. Our results provide novel insights on how ovarian cancer cells regulate ER stress and TAG hydrolysis through LDAH downregulation to promote tumor growth and chemoresistance.
Discussion
Ovarian cancer cells undergo alterations in fatty acid metabolism to meet the high energy demand for persistent tumorigenesis [19, 23, 24]. However, the cellular and molecular mechanism which regulates alterations in fatty acid metabolism in ovarian cancer cells have not been fully elucidated. LDAH is abundantly expressed in white and brown adipose tissues and ovarian tissues as well. Here, we found that LDAH is downregulated in ovarian cancer, and loss of LDAH promotes cisplatin resistance by suppressing ER stress and promoting TAG hydrolysis and FAO. Our results are consistent with previous reports [12, 16] that LDAH promotes TAG accumulation and enlargement of LDs while loss of LDAH promotes TAG hydrolysis [12, 16].
Cisplatin resistance is mediated by multiple mechanisms, such as increased drug efflux, decreased drug accumulation, increased drug metabolism, increased DNA repair, or decreased ER stress-induced apoptosis [3, 25, 26]. The ER is the major site for fatty acid metabolism as most enzymes associated with fatty acid synthesis and modification are located in the ER. Excess fatty acids by increased fatty uptake, synthesis, or LD hydrolysis, can overwhelm the ER’s capacity to fold and transport proteins and lead to the accumulation of misfolded proteins in the ER lumen, triggering ER stress [27, 28]. The ER then sequesters excess fatty acids and forms LDs. LDAH and ATGL are initially located in the ER but transported from the ER to newly synthesized LDs [12, 14]. Although induction of ER stress is primarily an ER repair mechanism, CHOP triggers apoptosis in the absence of a functional cell repair. Our results showed decreased expression of ER stress markers including CHOP in LDAH knockdown ovarian cancer cells, suggesting a role of LDAH in inducing ER stress. The ER promotes homeostasis in lipid metabolism, and unmitigated ER stress can dysregulate lipid metabolism, thus interfere with fatty acid oxidation. Further research is needed to understand the mechanism on how LDAH induces ER stress in ovarian cancer cells.
Lipid droplets (LDs) are ubiquitous cytoplasmic organelles which act as energy storage by compartmentalizing TAGs and sterols within cancer cells. Cancer cells store excess fatty acids in LDs to prevent the cell from lipotoxicity and subsequently fine-tune the release of free fatty acids in the cytoplasm based on metabolic needs of cancer cells [10, 29]. Accumulation of LDs also predict poor prognosis in ovarian cancer patients [30, 31]. It has been shown that LDAH overexpression promotes clustering of LDs while knockdown of LDAH/CG9186 decreases TAG storage in 6-day-old fly [16]. Similarly, Goo et al. reported that LDAH knockdown promotes TAG hydrolysis in HEK 293 cells by inducing ATGL expression [12]. Consistent with these reports, we also showed increased TAG hydrolysis in LDAH KD ovarian cancer cells. However, two previous studies showed the effect of LDAH on LD clustering and TAG hydrolysis in the presence of oleic acid (OA), which facilitates LD formation in cells [12, 16]. Strikingly, we found that ES2 and A2780 ovarian cancer cells showed the presence of LDs without supplementation with OA. Since de novo fatty acid synthesis is upregulated in ovarian cancer cells, the presence of glucose in the culture medium might induce LD formation in ovarian cancer cells without OA supplementation [32,33,34]. Another possibility is that FBS in the culture medium might serve as an additional source of fatty acids for cancer cells. Although our study is the first to report the function of LDAH without OA supplementation, it helped us to distinctly understand the effects of LDAH on fatty acid metabolism or LD function per se in cancer cells. We showed that LDAH downregulation increases ATGL expression and TAG hydrolysis, leading to FAO upregulation and activation of NF-kB signaling to promote cisplatin resistance in ovarian cancer cells.
ATGL is a rate-limiting enzyme that catalyzes the first step of TAG hydrolysis which readily gets degraded by ubiquitin-proteosome pathway [35,36,37]. Goo et al. showed that LDAH increases polyubiquitination of ATGL, thereby degrading ATGL in HEK293 cells [12]. We found the same results in ovarian cancer cells where LDAH knockdown increased the levels of ATGL due to decreased proteasomal degradation of ATGL. While ATGL overexpression phenocopies LDAH knockdown, treatment of LDAH knockdown cancer cells with an ATGL inhibitor attenuated cisplatin resistance and TAG hydrolysis, suggesting ATGL as a major contributory factor for the phenotype observed in LDAH knockdown cancer cells. Importantly, high levels of ATGL are associated with poor survival outcomes in ovarian cancer patients. Monoacylglycerol lipase (MAGL), an enzyme that catalyzes the last step of TAG hydrolysis, is reported to regulate fatty acid metabolism to promote ovarian cancer migration, invasion, survival, and in vivo tumor growth [38]. Thus, it is possible that TAG hydrolysis enhanced by LDAH downregulation might promote ovarian cancer progression and chemoresistance. However, we cannot exclude the possibility of involvement of other proteins or signaling pathways in promoting tumor progression and chemoresistance in LDAH knockdown ovarian cancer cells.
In conclusion, our study showed that LDAH is downregulated in ovarian cancer, which promotes ovarian cancer progression and cisplatin resistance. We established LDAH as a novel therapeutic target, especially in cancer with dysregulated lipid metabolism. Our study provides strong evidence for further studies to identify small molecules that can restore LDAH expression in ovarian cancer cells as anti-cancer drugs.
Materials and methods
Cell culture
A2780 human ovarian endometrioid adenocarcinoma cell line was purchased from SIGMA (St. Louis, MO, USA). ES2 human ovarian clear cell carcinoma line was purchased from ATCC (Manassas, VA, USA). Lenti-X 293 T cell line was purchased from Takara Bio (formerly Clontech; Kusatsu, Shiga, Japan). A2780 cells were cultured in RPMI (Gibco Life Technologies; Waltham, MA, USA), and ES2 and Lenti-X 293 T cell lines were cultured in DMEM (Gibco Life Technologies, Waltham, MA, USA) supplemented with 10% FBS (Sigma-Aldrich, St. Louis, MO, USA) and 1x penicillin/streptomycin (Gibco Life Technologies; Waltham, MA, USA). All cells were cultured at 37 °C with 5% CO2. All cell lines were tested negative for mycoplasma contamination and authenticated by short tandem repeat (STR) profiling before experimentation.
Lentiviral shRNA knockdown
Stable gene knockdown was performed using short hairpin RNAs (shRNAs) as described earlier [5, 6]. Lentiviral constructs containing different shRNAs against human LDAH were purchased from SIGMA (Product number: SHCLNV, clone ID: TRCN0000150271, TRCN0000149469, TRCN0000127675). pCMV-dR8.2 dvpr and CMV-VSV-G were gifts from Bob Weinberg (Addgene plasmids # 8455 and 8454). Lipofectamine 2000 (Life Technologies; Carlsbad, CA, USA) was used to co-transfect pCMV-dR 8.2 dvpr, pCMV-VSV-G, and a lentiviral construct containing shRNA into Lenti-X 293 T cells. Forty-eight hours after transfection, lentivirus-containing media was harvested and filtered through a 0.45 µM PVDF low protein-binding membrane filter (Celltreat; Pepperell, MA, USA). Lentivirus-containing medium was concentrated using Lenti-X concentrator (Takara Bio; Kusatsu, Shiga, Japan). Cancer cells were incubated in the lentivirus-containing medium with 8 µg/mL polybrene (EMD Millipore; Burlington, MA, USA; #TR-1003-G) for 72 h at 37 °C with 5% CO2 followed by 5 µg/mL puromycin (ThermoFisher, Waltham, MA, USA; #A1113803). Cancer cells infected with lentiviruses harboring pLKO.1 non-target shRNA control plasmid DNA (SIGMA, catalog no. SHC106) were used as the scrambled control.
Plasmid DNA transfection
FLAG-LDAH, pEGFP-LDAH, and FLAG-ATGL constructs were previously described [12]. DsRed2-ER5 was a gift from Michael Davidson (Addgene plasmid # 55836). Lipofectamine 2000 (Life Technologies; Carlsbad, CA, USA) was used to transfect the constructs into ES2 and A2780 human ovarian cancer cells according to the manufacturer’s protocols. Expression of LDAH and ATGL constructs were validated by western blotting. Expression of GFP-LDAH was determined by immunofluorescence imaging in ZOETM Fluorescent Cell Imager (Bio-Rad; Hercules, CA, USA).
Western blotting
Western blotting was performed as described previously [5, 6]. 25 µg of protein were run on 4–20% Mini-PROTEAN® TGX Stain-Free™ Protein Gels (BioRad; Hercules, CA, USA) and transferred onto PVDF membranes. Membranes were blocked with 2% BSA in PBST for 1 h at room temperature and incubated with primary antibodies (in 5% BSA/PBST) overnight at 4 °C. Next day, membranes were washed and incubated with HRP-conjugated secondary antibodies for 1 h at room temperature and imaged. A list of antibodies is provided in Supplementary Table 1. Quantification of bands was done by performing densitometry analysis using Image J Software.
Cell viability assays
MTT assay (SIGMA; St. Louis, MO, USA) was performed to evaluate cell viability using a Glomax Explorer Microplate Reader (Promega), according to the manufacturer’s protocols. For the apoptosis assay, FITC Annexin V Apoptosis Detection Kit I was purchased from BD Biosciences (Franklin Lakes, NJ, USA; #556547), and Annexin V/7-AAD staining was performed according to the manufacturer’s instructions. Cells were incubated with FITC Annexin V in a buffer containing 7-AAD for 15 min at room temperature and analyzed by flow cytometry using ZE5 Cell Analyzer (BioRad; Hercules, CA, USA).
RT-qPCR
The qRT-PCR reaction was performed using a CFX96 real-time PCR detection system (BioRad; Hercules, CA, USA) as described before [5, 6]. A total of 1 µg of RNA was reverse transcribed into cDNA using the Quantitect Reverse Transcription Kit (Germantown, MD, USA; # 205311). Then, 50 ng of cDNA was amplified using the relevant human gene primers and iQSYBR-Green Supermix (Bio-Rad; Hercules, CA, USA; #1708880). The primers used in this study are listed in Supplementary Table 2. The data were analyzed by the 2−ΔΔC(T) method.
Mouse experiments
All animal experiments were approved by the Albany Medical College’s Institutional Animal Care and Use Committee. Six- to eight-week-old female NCr nude mice were purchased from Taconic Biosciences (Germantown, NY, USA) and housed in pathogen-free environment at animal resource facility of Albany Medical College. Five million ES2 scrambled or LDAH KD cancer cells in serum-free DMEM media were mixed with Matrigel (BD Biosciences) in 1:1 ratio and injected into the left and right flank of NCr nude female mice. Two weeks after tumor cell injection, mice were randomly distributed into two groups. One group was given 0.1 ml of vehicle (0.9% saline), and the other group was given cisplatin (3 mg/kg) three times a week for 2 weeks by intraperitoneal injection. All animals were monitored daily, and all mice were euthanized at 3 weeks by CO2 asphyxiation followed by cervical dislocation. Subcutaneous tumors were dissected, and the volume (V) of tumors was calculated using a formula V = ½ (Length × Width2) [39, 40, 41].
Immunohistochemistry
Immunohistochemistry was performed as previously described [42]. Tissues were incubated overnight at 4 °C with primary antibodies (1:200) diluted in wash buffer containing 1% BSA, followed by incubations with goat anti-rabbit or goat anti-mouse secondary HRP-conjugated antibodies (1: 600). Antibodies used in this study were listed in Supplementary Table 1. Immunolabelling was visualized by using DAB staining (Abcam, Cambridge, UK; #64238) followed by hemoxylin staining (Abcam Cambridge, UK; #220365). Images were acquired at 4x, 10x, and 20x magnifications on an Olympus BX61 upright microscope with a PCO.EDGE 4.2 scientific CMOS camera and analyzed with MetaMorph software (version 7.10.2, Molecular Device).
Immunofluorescence staining
Cells were fixed in 4% paraformaldehyde, followed by permeabilization (1X PBS/2% goat serum/0.4% Triton X-100 buffer) and blocking (1X PBS/10% goat serum/0.2% Tween 20 buffer). Cells were incubated overnight at 4 °C with primary antibodies diluted in wash buffer containing 1% BSA, followed by incubations with goat anti-rabbit or goat anti-mouse secondary Alexa fluor labelled antibodies. Antibodies are listed in Supplementary Table 1. Lipid droplets were visualized by BODIPY 493/503 (ThermoFisher, Waltham, MA, USA; #D3922), and nucleus was visualized by DAPI staining (ThermoFisher, Waltham, MA, USA #62248). Images were taken with Olympus BX61 upright microscope with a PCO.EDGE 4.2 scientific CMOS camera and analyzed with MetaMorph software (version 7.10.2, Molecular Device).
Measurement of TAG levels
TAG levels were detected by Triglyceride colorimetric assay kit (Cayman chemicals, Ann Arbor, Michigan, USA; #10010303). Briefly, cells were harvested, and the pellet was resuspended in standard diluent buffer. Equal amounts of protein (1 mg) from each sample were used for experiments. The hydrolysis reaction was initiated by adding the enzyme mixture followed by incubation at room temperature for 60 min. Absorbance was measured at 530–560 nm using a Glomax Explorer Microplate Reader (Promega; Madison, WI, USA). The amount of TAG (mg/dl) was calculated by using the standard curve prepared according to manufacturer’s protocol.
Patient data analysis
LDAH Kaplan–Meier plots were generated by the KM plotter (https://www.kmplot.com) using the overall survival data of 1207 serous ovarian cancer patients expressing high or low mean expression of LDAH (Affymetrix ID: 222193_at) with an auto select best cutoff option. LDAH expression data in different stages and grades of ovarian cancer (Supplementary Table 3) was downloaded from the GENT2 database (http://gent2.appex.kr/gent2/), and the boxplot with samples (n = 272) was generated using GraphPad PRISM. GSE14407, GSE40595, and GSE115939 datasets were used to analyze the expression level of LDAH (C2orf43; Affymetrix ID: 222192_s_at, 222193_at) in normal ovarian tissues as compared to ovarian cancer tissues. ATGL Kaplan–Meier plots were generated by the GEPIA (http://gepia.cancer-pku.cn) using TCGA and GTEx datasets with high cut-off 75% and low cut-off 25%.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software version 7.0 (GraphPad Software; San Diego, CA, USA). A Student’s t-test was used to compare the means of two groups, and a one-way ANOVA was applied to compare the mean difference of three or more groups. Data were plotted as mean ± standard deviation (SD). p < 0.05 was considered statistically significant.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary information files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Chien J, Kuang R, Landen C, Shridhar V. Platinum-sensitive recurrence in ovarian cancer: the role of tumor microenvironment. Front Oncol. 2013;3.
Pokhriyal R, Hariprasad R, Kumar L, Hariprasad G. Chemotherapy resistance in advanced ovarian cancer patients. Biomark Cancer. 2019;11:1179299X1986081.
Suh DH, Kim HS, Kim B, Song YS. Metabolic orchestration between cancer cells and tumor microenvironment as a co-evolutionary source of chemoresistance in ovarian cancer: a therapeutic implication. 92, p. 43–54. Biochemical Pharmacology; Elsevier Inc.: 2014.
Rada M, Nallanthighal S, Cha J, Ryan K, Sage J, Eldred C, et al. Inhibitor of apoptosis proteins (IAPs) mediate collagen type XI alpha 1-driven cisplatin resistance in ovarian cancer. Oncogene. 2018;37:4809–20.
Nallanthighal S, Rada M, Heiserman JP, Cha J, Sage J, Zhou B, et al. Inhibition of collagen XI alpha 1-induced fatty acid oxidation triggers apoptotic cell death in cisplatin-resistant ovarian cancer. Cell Death Dis. 2020;11:258.
Lin H, Patel S, Affeck VS, Wilson I, Turnbull DM, Joshi AR, et al. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol. 2017;19:43–54.
Houten SM, Wanders RJA. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherit Metab Dis. 2010;33:469–77.
Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227–32.
Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. 20, Nature Reviews Molecular Cell Biology. Nature Publishing Group; 2019. p. 137–55.
Cruz ALS, Barreto E de A, Fazolini NPB, Viola JPB, Bozza PT. Lipid droplets: platforms with multiple functions in cancer hallmarks. 11, Cell Death and Disease. Springer Nature; 2020.
Goo YH, Son SH, Paul A. Lipid droplet-associated hydrolase promotes lipid droplet fusion and enhances ATGL degradation and triglyceride accumulation. Sci Rep. 2017;7:2743.
Goo YH, Son SH, Kreienberg PB, Paul A. Novel lipid droplet-associated serine hydrolase regulates macrophage cholesterol mobilization. Arterioscler Thromb Vasc Biol. 2014;34:386–96.
Kory N, Grond S, Kamat SS, Li Z, Krahmer N, Chitraju C, et al. Mice lacking lipid droplet-associated hydrolase, a gene linked to human prostate cancer, have normal cholesterol ester metabolism. J Lipid Res. 2017;58:226–35.
Currall BB, Chen M, Sallari RC, Cotter M, Wong KE, Robertson NG, et al. Loss of LDAH associated with prostate cancer and hearing loss. Hum Mol Genet. 2018;27:4194–203.
Thiel K, Heier C, Haberl V, Thul PJ, Oberer M, Lass A, et al. The evolutionarily conserved protein CG9186 is associated with lipid droplets, required for their positioning and for fat storage. J Cell Sci. 2013;126:2198–212.
Dubey R, Stivala CE, Nguyen HQ, Goo YH, Paul A, Carette JE, et al. Lipid droplets can promote drug accumulation and activation. Nat Chem Biol. 2020;16:206–13.
Day RN, Davidson MW. The fluorescent protein palette: tools for cellular imaging [published correction appears in Chem Soc Rev. 2011 Dec;40(12):5923]. Chem Soc Rev. 2009;38:2887–921.
Tania M, Khan MA, Song Y. Association of lipid metabolism with ovarian cancer. Curr Oncol. 17;2010:6–11.
Garcia G, Zhang H, Moreno S, Kimberly Tsui C, Webster BM, Higuchi-Sanabria R, et al. Lipid homeostasis is essential for a maximal ER stress response. Elife. 2023;12:e83884.
Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, et al. HIF2α-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 2016;5:653–67.
Zhang C, Liao Y, Liu P, Du Q, Liang Y, Ooi S, et al. FABP5 promotes lymph node metastasis in cervical cancer by reprogramming fatty acid metabolism. Theranostics. 2020;10:6561–80.
Molendijk J, Robinson H, Djuric Z, Hill MM. Lipid mechanisms in hallmarks of cancer. Mol Omics. 2020;16:6-18.
Zhao G, Cardenas H, Matei D. Ovarian cancer—why lipids matter. Cancers. 2019;11:1870.
Jiang C, Shen C, Ni M, Huang L, Hu H, Dai Q, et al. Molecular mechanisms of cisplatin resistance in ovarian cancer. Genes Dis. 2024;11:101063.
Zoń A, Bednarek I. Cisplatin in ovarian cancer treatment—known limitations in therapy force new solutions. Int J Mol Sci. 2023;24:7585.
Liu KT, Yeh IJ, Chou SK, Yen MC, Kuo PL. Regulatory mechanism of fatty acid-CoA metabolic enzymes under endoplasmic reticulum stress in lung cancer. Oncol Rep. 2018;40:2674–2682.30.
Zhu Y, Li Y, Bai B, Shang C, Fang J, Cong J, et al. Effects of apoptin-induced endoplasmic reticulum stress on lipid metabolism, migration, and invasion of HepG-2 cells. Front Oncol. 2021;11:614082.
Jarc E, Petan T. Lipid droplets and the management of cellular stress. Yale J Biol Med. 2019;92:435–52.
Iwahashi N, Ikezaki M, Fujimoto M, Komohara Y, Fujiwara Y, Yamamoto M, et al. Lipid droplet accumulation independently predicts poor clinical prognosis in high-grade serous ovarian carcinoma. Cancers. 2021;13:5251.
Jin Y, Tan Y, Wu J, Ren Z. Lipid droplets: a cellular organelle vital in cancer cells. Cell Death Discov. 2023;9:254.
Mashima T, Seimiya H, Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer. 2009;100:1369–72.
Grunt TW, Wagner R, Ries A, Berghoff AS, Preusser M, Grusch M, et al. Targeting endogenous fatty acid synthesis stimulates the migration of ovarian cancer cells to adipocytes and promotes the transport of fatty acids from adipocytes to cancer cells. Int J Oncol. 2024;64:24.
Lin L, Li X, Wu AJ, Xiu JB, Gan YZ, Yang XM, et al. TRPV4 enhances the synthesis of fatty acids to drive the progression of ovarian cancer through the calcium-mTORC1/SREBP1 signaling pathway. iScience. 2023;26:108226.
Zhang R, Meng J, Yang S, Liu W, Shi L, Zeng J, et al. Recent advances on the role of ATGL in cancer. Front Oncol. 2022;12:944025.
Olzmann JA, Richter CM, Kopito RR. Spatial regulation of UBXD8 and p97/VCP controls ATGL-mediated lipid droplet turnover. Proc Natl Acad Sci USA. 2013;110:1345–50.
Dai Z, Qi W, Li C, Lu J, Mao Y, Yao Y, et al. Dual regulation of adipose triglyceride lipase by pigment epithelium-derived factor: a novel mechanistic insight into progressive obesity. Mol Cell Endocrinol. 2013;377:123–34.
Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140:49–61.
Roby KF, Taylor CC. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 21;2000:585–91.
Hamilton TC, Young RC, Louie KG, Behrens BC, Mckoy WM, Grotzinger KR, et al. Characterization of a xenograft model of human ovarian carcinoma which produces ascites and intraabdominal carcinomatosis in mice [Internet]. Cancer Res. 1984;44:5286–90.
Charan J, Kantharia N. How to calculate sample size in animal studies? J Pharm Pharmacother. 2013;4:303–6.
Heiserman JP, Nallanthighal S, Gifford CC, Graham K, Samarakoon R, Gao C, et al. Heat shock protein 27, a novel downstream target of collagen type XI alpha 1, synergizes with fatty acid oxidation to confer cisplatin resistance in ovarian cancer cells. Cancers. 2021;13:4855.
Acknowledgements
This work was supported by the Department of Defense Ovarian Cancer Research Program Pilot Award (W81XWH-21-1-0256) to DC and YG and the Caring Together Research Fund to DC.
Author information
Authors and Affiliations
Contributions
Conceptualization: BD, YG, DC; methodology: BD, SN; validation: BD, YG, AP, DC; formal analysis: BD, YG, AP, DC; investigation: BD, SN, DC.; resources: YG, AP, DC; data curation: BD, SN, EN, ZM, DC; writing—original draft preparation: BD and DC.; writing—review and editing: BD, SN, EN, ZM, YG, AP, DC; visualization: BD; supervision: YG, AP, DC; funding acquisition: YG, DC. All authors have read and agreed to the published version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. All animal experiments were approved by the Albany Medical College’s Institutional Animal Care and Use Committee (ACUP-24-06001).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Deswal, B., Nallanthighal, S., Nikpayam, E. et al. Lipid droplet-associated hydrolase (LDAH) knockdown enhances TAG hydrolysis and promotes ovarian cancer progression and chemoresistance. Oncogenesis 14, 22 (2025). https://doi.org/10.1038/s41389-025-00566-1
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41389-025-00566-1
