
Abstract
Background
Sepsis is a leading global health burden in children, and its unavoidable heterogeneity has hindered providing therapies beyond antibiotics and supportive care. Recently, we identified four computable phenotypes showing distinct cytokine profiles, clinical outcomes, and therapeutic response characteristics (PedSep-A, B, C, and D) in a multicenter pediatric sepsis cohort.
Methods
In the cohort data, we collected whole-exome sequencing data and identified rare variants associated with PedSep-D phenotype by conducting a gene-based analysis in an aggregated fashion.
Results
As a result, one whole-exome significant gene (LTBP4) and two suggestive significant genes (PLA2G4E, CCDC157) showed association with PedSep-D, the phenotype characterized by the most severe outcomes and highest inflammation. The associated variants in LTBP4 were enriched for predicted deleterious effects based on established functional prediction metrics. All three associated genes are implicated in inflammation and immune cell activation based on existing gene function and expression data. Although the circulating cytokine profiles were overlapping between the rare variant carriers, we also identified gene-specific cytokine changes.
Conclusion
Altogether, our study provides valuable insights into the genetic architecture of a pediatric sepsis phenotype with the highest inflammation level and the most severe outcomes, highlighting potential candidate genes and pathways for further biomarker and therapeutic studies.
Impact
-
Pediatric sepsis exhibits substantial heterogeneity, with genetic variation contributing to this variability. Rare variants in LTBP4 are significantly associated with the most severe pediatric sepsis phenotype (PedSep-D), while variants in PLA2G4E and CCDC157 show associations with this phenotype in suggestive significance.
-
Expands on the concept of sepsis phenotypes (PedSep-A, B, C, D) by incorporating genetic insights, moving beyond clinical and cytokine profiles to uncover molecular drivers.
-
Opens new avenues for mechanistic studies to understand the genetic underpinnings of severe inflammation and immune activation in sepsis.
Similar content being viewed by others

Introduction
Pediatric sepsis is a life-threatening condition associated with organ failure in children due to a dysregulated host immune response to infection. It is a recognized global public health problem that affects 20.3 million children and causes 2.9 million deaths in those under five years old every year.1 Despite global efforts to improve clinical outcomes for pediatric sepsis, its phenotypic heterogeneity remains a significant barrier to therapeutic advancement.2 Several recent studies have advanced efforts to characterize pediatric sepsis phenotypes using data-driven approaches, including genome-wide expression profiling,3,4 dynamic modeling of organ dysfunction trajectories,5 and supervised classification of inflammation-based subtypes.6 While these contributions are important, key limitations remain. Studies, such as Wong et al.3 and Sweeney et al.4 primarily rely on transcriptomic data, which, although informative at the molecular level, may lack direct applicability at the bedside. While Sanchez-Pinto et al.5 utilized subscores of the pediatric Sequential Organ Failure Assessment (pSOFA) score to define multiple organ dysfunction syndrome (MODS) phenotypes, these phenotypes may not fully reflect the broader complexity and heterogeneity of pediatric sepsis, particularly features not directly manifested as organ failure. Although Carcillo et al.6 included a broader range of clinical features, their supervised approach introduces the potential for bias due to dependence on predefined categories or outcomes. In contrast, our approach aims to address these limitations by leveraging a broader and more granular set of clinical data in an unsupervised framework that emphasizes both statistical robustness and bedside applicability. Recently, by applying machine learning approaches to 25 first-day bedside clinical variables of 404 pediatric sepsis patients with organ dysfunction enrolled as part of a multicenter cohort, PHENOtyping sepsis-induced Multiple organ failure Study (PHENOMS), between 2015 to 2017, we derived four computable phenotypes PedSep-A, B, C, and D with differences in infection source, cytokine profiles, organ failure, outcomes, and treatment responses.7
Several studies suggested that host genetic factors contribute to the heterogeneity of pediatric sepsis. While early family studies8 and targeted candidate gene analyses9,10,11 have supported this notion, discovery efforts have been limited to discover novel functional sepsis-related genes. Alternatively, researchers have conducted several genome-wide association studies (GWAS) on adult and pediatric populations to identify common variants underlying sepsis susceptibility and outcomes.12,13,14 However, although common variants have been used to understand the genetic basis of clinical outcomes (e.g., survival from sepsis or hospital admissions), they are limited in elucidating the genetic architecture for computable pediatric sepsis phenotypes with poor outcomes. Since common variants usually have small effects on complex traits, their systematic study requires a prohibitively large sample size and their small effects are of limited clinical benefit in a large fraction of the population, which is not feasible to study in pediatric sepsis.15,16 Additionally, to the best of our knowledge, studies investigating rare variants (low penetrance, low allele frequency (AF)) in pediatric critical illness are limited,17 with no prior rare variant analyses specifically focusing on sepsis subtypes. This gap underscores the need for more targeted genomic investigations to identify genetic contributors to pediatric sepsis severity and heterogeneity. These findings underscore the need for more comprehensive and functionally focused genomic studies, especially in understudied pediatric populations. Without involving further functional validation, the large fraction of findings in non-coding regions challenges the interpretation of the sepsis GWAS results.
To address these limitations, we performed a gene-based exome-wide rare variant analysis using data from the PHENOMS study of severe pediatric sepsis. Using whole-exome sequencing data from 319 children (Fig. 1) with sepsis and organ dysfunction in the PHENOMS study, we focused our analysis on the highest-risk phenotype, PedSep-D, with the aim of identifying biologically impactful variants and informing future therapeutic strategies. Altogether, we present the first rare variant burden test in the computable phenotype with the worst outcomes in pediatric sepsis.

CONSORT (Consolidated Standards of Reporting Trials) diagram for the study.
Methods and Materials
Consent statement
The study was approved by the Institutional Review Board at University of Utah Central IRB # 70976.
Cohort and phenotyping
The phenotyping data and blood samples were obtained from pediatric sepsis patients of a multicenter cohort, PHENOMS.6 The cohort enrolled pediatric patients from 2015 to 2017 with written informed consent from at least one of the guardians. Children were qualified for enrollment if they met all four criteria: (1) at the ages of 44 weeks to 18 years old; (2) were suspected of infection meeting two or more SIRS (systemic inflammatory response) criteria;18 (3) presented one or more organ failures; and (4) had an indwelling arterial or central venous catheter. Patients without a commitment to aggressive care or lack of blood samples were further excluded from the enrollment.
The data-driven phenotyping approach and results were described in previous work.7 Briefly, four phenotypes named PedSep-A, B, C, and D were identified by applying consensus k-means clustering on 25 day-one bedside variables. As WES data was available for only a subset of the complete cohort, we confirmed the phenotyping analysis using this subset of patients with available genetic data to ensure that phenotype assignment was robust in this reduced sample size. In the following genetic analysis, we performed a case-control study between children in the computable phenotype subgroup with the highest severity of illness and mortality (PedSep-D) and the others (PedSepA,B, and C) to identify the genetic factors exclusively associated with increased sepsis severity susceptibility.
DNA extraction and genotyping
Out of 404 pediatric patients enrolled in the cohort, a total of 381 parents of the children provided WES consent, and 2 mL of whole blood was collected for DNA extraction using standard methods. Whole-exome sequencing was successfully completed on 332 patients from 2018 to 2020 by the University of Pittsburgh Genomics Research Core performed on the Ion Torrent platform. Libraries were constructed by the Ampliseq Exome RDY (Thermo Fisher Scientific) with 100 × target coverage. FASTQ files were aligned to Homo sapiens reference sequence GRCh37/hg19 to generate VCF files. Variant calling was performed by GATK (Genome Analysis Toolkit).19
Quality control
Two levels of quality control were conducted on 332 samples with completed whole-exome sequencing data, patient-level, and variant-level. At the patient level, we excluded nine individuals without phenotype information. Four pairs of individuals were identified as relatives based on IBD (identity by descent). In each IBD pair, the individual with the higher missingness was removed from the analysis. In terms of variant-level quality control, we filtered sites with SOR (Strand Odds Ratio) > 3, MQ (root mean square Mapping Quality) < 40, QD (variant confidence normalized by depth) < 2.0, average GQ (Genotyping confidence) < 20, average DP (Depth) < 10, missingness > 0.05, HWE (Hardy-Weinberg equilibrium p) < 1e-06, and those located in sex chromosomes. No imputation of missing genotypes was performed due to concerns for potentially low imputation quality of rare variants in datasets with small sample sizes. Quality control was performed by software bcftools (v1.9),20 VCFtools (v0.1.16),21 and PLINK (v1.9).22 Then variant function was annotated by ANNOVAR.23
Principal Component (PC) derivation
To account for potential population stratification and other confounders in the statistical model, we derived 10 principal components (PCs) based on common SNPs following linkage disequilibrium (LD) pruning. LD pruning was performed using PLINK (version 1.07) with the argument “–indep-pairwise 50 5 0.2”. This procedure involves considering a sliding window of 50 SNPs, calculating LD between each pair of SNPs within this window, and removing one SNP from any pair exhibiting an LD greater than 0.2. After pruning within a window, the window is shifted forward by 5 SNPs, and the pruning process is repeated until the entire dataset is processed.
Gene-based analysis
Variants that passed quality control were included if they were in hg19 annotated exon regions and had a MAF (minor AF) lower than 1%. Genes with less than three qualified variants were excluded from the analysis. The final number of genes tested was 3846. Therefore, the p-value threshold for declaring whole-exome level significance was 0.05/3846 = 1.3e-05.
Then, we aggregately examined the relationships between the rare variants and the binary indicator of phenotype membership by gene-based association test SKAT (Sequence Kernel Association Test).24 SKAT is a widely-employed method to test the association between a group of variants and the trait, which increases the power to detect rare variant associations by pooling rare variants across a given region of interest, such as chromosome region or gene. In running the SKAT test, a single null model was fitted containing only the covariates to be adjusted (i.e., age, sex, and the first four ancestry PCs constructed from common LD-pruned SNPs). Then the effect of SNPs from each gene was tested by variance-component score tests in a mixed model, and their statistics were aggregated with weights through a kernel matrix to form a gene-level statistic. Compared to other gene-based tests, such as the Burden Test and SKAT-O, one advantage of applying SKAT in our analysis is that it makes few assumptions about rare-variant effects and retains statistical power when variants within a gene have different directions and magnitude of effects.25 This property aligns with the study design that contrasts one phenotype with others and allows us to better account for potential heterogeneity in phenotypes.
Genes showing whole-exome level significance and suggestive significance were further investigated to query the gene function (GeneCards),26 common variant evidence from previous GWAS analysis (GWAS Catalog),27 gene enrichment in GO biological process (FUMA, Enrichr),28,29 and gene expression level in the GTEx database.30 Rare variants that contributed to gene significance were annotated with four different types of score (CADD,31 GERP,32 SIFT,33 Polyphen234) to indicate the effect of each variant.
Comparison of cytokine profiles between rare variant carriers and non-carriers
To further investigate the effect of variations on inflammation, levels of the 33 pre-collected biomarkers of the rare variant carriers were further visualized and compared with non-carriers. The cytokine heatmap was used to present the log ratio of the median biomarker values of the host response. The red color represents a greater value for the group compared to the entire cohort, while the blue color represents a lower value for the group compared to the entire cohort. Hierarchical clustering was used to visualize the similarity of cytokine patterns between rare variant carriers. Additionally, we calculated p-values from a pairwise t-test comparing cytokine values of rare variant carriers and non-carriers.
Mediation Analysis
To further explore potential biological mechanisms, we conducted a causal mediation analysis to evaluate whether biomarkers mediate the relationship between rare variant gene burden and PedSep-D membership. Mediation analysis requires three conditions to be met: 1) A significant association between gene burden and PedSep-D membership (which was established by SKAT results); 2) A significant association between gene burden and the candidate biomarker; and 3) A significant association between the biomarker and PedSep-D membership.
When all three conditions were satisfied, we used the mediation package in R,35 applying 1000 bootstrap iterations, to estimate the indirect effect (via the biomarker), direct effect, total effect, and proportion mediated for each gene–biomarker pair.
Sensitivity Analysis
In order to further validate the top genes identified from the gene-based analysis, we investigated the influence of the ancestry information on the genes with a sensitivity analysis that was performed as follows. Briefly, we randomly swap phenotype labels between pairs of individuals with the same reported ancestry information to keep the ancestry makeup of the groups the same and run 100,000 iterations of SKAT gene-based analysis to calculate how many times the test statistic value is greater than the test statistic value from observed data. Thus, we generated an empirical p-value for each gene.
Pathway-based analysis
In addition to identifying association signals between individual genes and the sepsis phenotype of interest, we also sought to identify associations between genetic variation at the pathway level, using Gene set analysis Association Using Sparse Signals (GAUSS).36 GAUSS is constructed with gene-based test results and calculates a gene set level p-value by identifying a subset of genes (i.e., core genes) to maximize the association signal. Using the GAUSS method, we aggregated the SKAT test statistics of individual genes into groups based on GO biological process (GOBP) pathway annotations and examined the associations between 7482 GOBP pathways and phenotype of interest. This was performed for the PedSep-D phenotype (Table 1). We also detected the active genes driving the pathway-trait associations to facilitate the interpretation of test results.
Results
PedSep-D phenotype has the highest mortality with unique clinical presentation and immune system profile
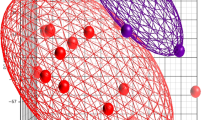
Using the 319 pediatric patients from the parent cohort who passed quality control both in bedside features and whole exome sequencing (WES) data (Fig. 1), we assigned them to one of four established phenotypes (PedSep-A, B, C, D) as previously determined by the consensus k-means clustering of 25 first-day bedside features.7 Among these 319 patients, the sample sizes of PedSep-A, B, C, D are 116 (36%), 86 (27%), 77 (24%), and 40 (13%), respectively (Table 2, Supplemental Tables 1–3). The proportions of patients in each phenotype are close to our original study, in which PedSep-A, B, C, D contained 34, 25, 27, and 14 percent of the 404 patients, respectively. To estimate the homogeneity of each phenotype, we projected them on the t-SNE plot of all 25 features and observed good separation between each phenotype and the other phenotypes (average Euclidean distance=6.4, Fig. 2). Specifically, the PedSep-D phenotype was distinct from PedSep-A, B, and C patients (Euclidean distance = 8.9).

Samples in two-dimensional space were labeled by PedSep phenotype membership. Euclidean distance was measured between centroids of two groups to quantitatively compare the difference between one phenotype versus the remaining phenotypes.
Our data, which is sampled from the original PHENOMS cohort, confirmed distinct clinical (Table 2; Supplemental Tables 1–3) and biomarker (Supplemental Tables 4–7) profiles across phenotypes. PedSep-A showed the mildest presentation (Supplemental Tables 1 and 4) and PedSep-D had the most severe profile with multi-organ failure and highest mortality (Tables 2–4), where PedSep-B and -C are in bet-ween (Supplemental Tables 2 and 6). These patterns recapitulate the importance of analyzing PedSep-D as the highest-risk group (Supplemental Table 8).
Gene-based test associates LTBP4, PLA2G4E, and CCDC157 with PedSep-D
To detect genetic factors associated with the sepsis phenotypes, we performed a whole exome-wide rare variant analysis. To increase power in detecting associations, we aggregated the rare variant association signals by gene and estimated the significance in the following steps (see Methods). First, we performed quality control and selected a total of 3864 genes that had more than three variants for the association between rare variants and the phenotype of interest. Then, we ran SKAT on the WES data separately for each of the four PedSep phenotypes (PedSep-A, B, C, D) versus any of the other three phenotypes while adjusting for age, sex, and ancestry based upon the first four PCs constructed based on common variants. (Fig. 3, Supplemental Figs. S1–3, Tables 2, 3, Supplemental Table 9).

Red line: whole-exome wide significant –log10(P) value; Blue line: suggested significant –log10(P) value.
While our primary analyses centered on PedSep-D, we included results from the other phenotype comparisons to provide broader context and to highlight the distinctiveness of PedSep-D-specific genetic associations. For PedSep-A or C versus the remaining phenotypes, no significantly associated genes were detected. For PedSep-B, the PLXNA2 gene presented a suggestive association (Supplemental Fig. S2). However, the QQ plot (S. Fig. 4B) showed genomic inflation, suggesting that the potential association of PLXNA2 has a high risk of being false positive. In contrast, for PedSep-D, variation in LTBP4 was significantly associated with phenotype development at the exome-wide level (p-value = 1.069E-05, 6 [15%] carriers in case group, 2 [0.7%] carriers in control group), while variations in PLA2G4E (4 [10%] carriers in case group, 2 [0.7%] carriers in control group) and CCDC157 (7 [17.5%] carriers in case group, 10 [3.6%] carriers in control group) were suggestively associated with phenotype development. Four, 4, and 8 rare variants contributed to the significance of LTBP4, PLA2G4E, and CCDC157, respectively (Table 5). All variants encode missense variants except one in the LTBP4 gene. However, this silent variant, rs370696272, replaces a common leucine code (CTG, 0.361) with a less common codon (TTG, 0.134) based on the CoCoPUT database,37 explaining its high CADD score (17.55). Most variants in three genes were predicted to be deleterious based on their CADD score (12 out of 16 with CADD > 10), among which SNP rs573310430 in LTBP4 had the highest CADD score of 34, ranked over the top 0.1% in terms of deleteriousness among variants across the whole genome. This variant creates an unpaired cysteine in the 14th calcium-binding epidermal growth factor-like (cbEGF) domain of LTBP4, a domain stabilized by three pairs of cysteines forming intradomain disulfide bonds particularly sensitive to removal or addition of cysteine residues.38
We observed well-calibrated test statistics and little evidence of inflation (Supplemental Fig. S4, lambda = 0.98) for PedSep-D, suggesting that these associations are true signals. To explore the AF of variants contributing to significant and suggestive genes, we compared the AF of all variants across three populations (Black, White, and Asian) in the gnomAD database (Supplemental Table 9). No large difference was observed between the AFs across populations, indicating that the top signals are less likely related to ancestry distinctions. To further investigate if there is an ancestry difference driving the top signals, we conducted a sensitivity analysis by randomly swapping the phenotype labels between pairs of individuals with similar ancestry information to keep the ancestry makeup of the groups the same while generating a meaningful empirical p-value. With 100,000 iterations of permutation for each of the three genes, we observed 0 times that permutated statistics were larger than the previously estimated statistic. This indicates the significance of the three genes is not likely driven by ancestry differences.
LTBP4, PLA2G4E, and CCDC157 underlie distinct cytokine patterns in patients
To explore the genes’ association with inflammation status, we grouped patients based on whether they carried rare variants in one of the three genes of interest, generated a heatmap showing the normalized levels of 33 cytokines (Fig. 4), and statistically tested the group-wise differences (Supplemental Tables 10, 11). Comparison between the rare variants carriers and non-carriers indicated some similarity shared by carriers groups. For example, a higher level of IL-6 is significantly related to both LTBP4 and CCDC157 rare variant carriers (p-value = 0.032 and 0.043, respectively), and higher level of M-CSF is significantly related to both PLA2G4E and CCDC157 rare variant carriers (p-value = 0.013 and 0.011 separately). Simultaneously, several cytokines important in regulating inflammation showed distinct patterns when comparing carriers to non-carriers with rare variants in LTBP4, PLA2G4E, and CCDC157 (Supplemental Table 13). For instance, IL-4 is significantly higher in LTBP4 rare variant carriers (p-value = 0.025) but is significantly lower in CCDC157 rare variant carriers (p-value = 0.035). Compared to non-carriers, PLA2G4E rare variant carriers presented significantly higher levels of IL-16, and SCF, but significantly lower levels of CRPH (p-value = 0.034, 0.005, and 0.022, separately), while LTBP4 and CCDC157 rare variant carrier groups showed no significant difference with non-carriers for these biomarkers. The ferritin level is uniquely higher in LTBP4 rare variant carriers (p-value = 0.023). These results imply that the three genes might be involved in different pathological mechanisms driving the phenotype.

The log ratio of the median values of 33 inflammatory biomarkers by rare variants carriers and non-carriers. Red represents a greater median biomarker value for that group compared with the median for the entire study cohort, whereas blue represents a lower median biomarker value compared with the median for the entire study cohort.
Mediation Analysis was further performed to evaluate whether biomarkers mediate the relationship between rare variant gene burden and PedSep-D membership. The results (Supplemental Table 12) highlight three gene–biomarker pairs that showed significant mediation effects. For example, the effect of LTBP4 burden on PedSep-D membership was partially mediated by ADAMTS13 levels, with 16% of the total effect explained by this biomarker. Similarly, IL-16 and IL-8 were significant mediators for PLA2G4E and CCDC157, respectively, with mediation proportions ranging from 16 to 41%. These findings suggest that biomarkers may serve as functional intermediates linking rare variant burden to sepsis subtype PedSep-D and highlight potential targets for mechanistic validation.
In addition, we accessed tissue-specific gene expression data from the GTEx database. Given that GTEx compiles transcriptomic profiles across a wide range of human tissues, we observed that all three genes are expressed in multiple tissue types (Fig. 5a). LTBP4 is highly expressed in multiple tissues, including the lung, kidney, stomach, skin, and others. PLA2G4E is specifically expressed in the skin. CCDC157 is specifically expressed in testis. In terms of cell-type specific expression of three genes, all of them displayed expression in immune cells of the cardiovascular and pulmonary systems, both of which are highly affected by severe sepsis or septic shock (Fig. 5b).

a Heat map of tissue-specific log2 transformed average expression level for three genes based on GTEx v8 RNA data. Red color indicates a higher expression within a tissue compared to other tissues, whereas blue color indicates a lower expression within a tissue compared to other tissues. b Dot plot of cell-type-specific expression level for three genes in two sepsis-related tissues (GTEx Single Cell data). The dot color reports the mean expression value with each cell type. The dot size reports the fraction of cells in which a gene is detected. Black dot size reports the total detected level of all three genes.
Discussion
In this study, rare variants in LTBP4 were significantly associated with the development of the previously reported high-mortality PedSep-D phenotype, with additional suggestions of associations with rare variations in PLA2G4E and CCDC157. To our knowledge, this is the first time a rare variant burden test has been applied to pediatric sepsis with deep phenotyping.
The top signal found in our study, LTBP4, a member of the latent transforming growth factor β binding protein family, shares structural homology with fibrillin and is moderately expressed in plasma cells and immune cells.39 Mutations in LTBP4 have been associated with autosomal recessive cutis laxa type 1C,40,41,42 Duchenne Muscular Dystrophy (DMD),43 fibrosis-related disorders,44 cancer,45 pulmonary disorders, and cardiovascular disorders.46 PedSep-D patients had the most severe kidney involvement, whereas LTBP4 was found to protect against tubular interstitial fibrosis by strengthening angiogenesis, downregulating inflammatory gene expression, and facilitating the maintenance of mitochondrial structure in tubular epithelial cells.47 Common variants in LTBP4 have previously been reported in GWASs to be associated with several traits, including lung function (FEV1/FVC),48 peak expiratory flow,49 hematocrit and hemoglobin,50 eosinophil counts,51 carotid intima-media thickness,52 and diastolic blood pressure.53 The precise molecular mechanism by which deleterious LTBP4 alleles may contribute to the PedSep-D phenotype awaits future functional studies. It remains to be determined whether known activities of LTBP4, elastic fiber organization and regulation transforming growth factor β (TGF-β) signaling54 or as yet undiscovered functions play a role in sepsis pathogenesis. TGF-β remains an attractive candidate given its potent anti-inflammatory effects.55 However, our finding that ADAMTS13 levels mediate part of the LTBP4 effect provide evidence for previously unknown molecular interactions between these proteins in sepsis. Because another ADAMTS protease family member, ADAMTS7 is known to cleave LTBP4,56 LTBP4 may serve either as a substrate or a competitive inhibitor of ADAMTS13. In the field of sepsis and trauma, Bergmann et al. have postulated the role of TGF-β and connected it, as well as other immunosuppressive cytokines, with the high mortality rate of patients discharged from ICU.55
As one of the two genes with suggestive significance, the PLA2G4E gene encodes a member of the cytosolic phospholipase A2 group IV family involved in membrane tubule-mediated transport regulation. It plays an important role in trafficking through the clathrin-independent endocytic pathway.57 PLA2G4E was also up-regulated in Alzheimer’s disease APP-PS1 transgenic mice lacking CD8 T cells compared to the control group.58 Common variants in PLA2G4E have been reported in previous GWASs to be associated with several sepsis clinical prognostic factors, such as neutrophil count,59,60 white blood cell count,61 and mean platelet volume.51
As another gene displaying suggestive significance, CCDC157 encodes a protein coiled-coil domain containing 157. Common variants in CCDC157 have been reported in previous GWASs to be associated with sepsis risk factors, such as hematocrit,50 pulse pressure,62 and calcium levels.59 No clear function of immune dysregulation has been reported for CCDC157 to date.
Although none of the pathways are significantly associated with PedSep-D phenotype, top-ranked pathways involve important biological processes related to sepsis development. For example, the high rank of N-acylphosphatidylethanolamine metabolic process supported previous investigations revealing evaluated fatty acids as candidate biomarkers of sepsis.63 The high rank of growth hormone (GH) secretion was supported by previous studies suggesting GH level is higher in septic shock patients compared to sepsis patients and is also higher in sepsis non-survivors.64 Other top pathways involve endocytic recycling that plays a role in infection-host interaction65 and regulation of cilium beat frequency which is related to respiratory disease and airway infection.66
There are several limitations in this study. First, the tested sample size is small, limiting the statistical power to detect associations. As such, larger independent cohorts are needed for validation and meta-analysis. Second, the signals from rare variants may be caused by local ancestry differences, in which situation the number of alleles derived from distinct ancestral populations at a given locus is different. Therefore, although we account for global ancestry by adjusting for top PCs in the association test and conducting sensitivity analysis, it is crucial to further perform local ancestry inference and examine the results in diverse populations separately to validate our findings. Third, we acknowledge the potential impact of residual LD on our association findings. Although we implemented LD pruning, it is challenging to fully resolve LD structure, particularly in regions with complex haplotypes or in diverse populations where reference panels may be limited. As such, some observed associations may reflect correlated signals rather than direct causal effects. Fourth, while our current analysis focused on individual variant and gene-level associations, future studies leveraging polygenic risk scores or burden scores that integrate both common and rare variants may offer a more continuous and potentially powerful framework for assessing genetic contributions to disease severity and subtype classification in sepsis. For example, Rautanen et al. identified genetic variants associated with 28-day survival in sepsis, providing a foundation for polygenic modeling of sepsis outcomes.12 The application of PRS in this context is an emerging area and will benefit from larger datasets and phenotype-specific GWAS to enable accurate score derivation. As a secondary analysis, we examined GTEx-based tissue-specific expression profiles to provide biological context for the potential regulatory function of the identified variants, particularly in tissues implicated in sepsis pathophysiology. However, we recognize that GTEx may not fully reflect gene expression dynamics in children with sepsis. To address this limitation and strengthen the biological relevance of our findings, future studies incorporating transcriptomic or proteomic data from pediatric sepsis patients and age-matched healthy controls will be essential for validation. Finally, given the nonsignificant findings from the GAUSS method, interpretation of the pathway analysis results should be approached with caution, and the results require further validation in a larger cohort. In summary, our study identified rare variants that, if found to have functional effects in future studies, might play a role in pediatric sepsis outcomes, providing evidence for a genetic contribution to disease heterogeneity.
Data availability
Sepsis exome data is available upon request. However, as the data is governed by IRB regulations, requesters must ensure that the IRB policies at their institution are compatible with the regulations under which the data is protected.
References
World Health Organisation. Global Report on the Epidemiology and Burden of Sepsis: Current Evidence, Identifying Gaps and Future Directions. World Health Organization (WHO, 2020).
Cavaillon, J. M., Singer, M. & Skirecki, T. Sepsis therapies: learning from 30 years of failure of translational research to propose new leads. EMBO Mol. Med. 12, e10128 (2020).
Wong, H. R. et al. Identification of pediatric septic shock subclasses based on genome-wide expression profiling. BMC Med. 7, 34 (2009).
Sweeney, T. E. et al. Unsupervised analysis of transcriptomics in bacterial sepsis across multiple datasets reveals three robust clusters. Crit. Care Med. 46, 915–925 (2018).
Sanchez-Pinto, L. N., Stroup, E. K., Pendergrast, T., Pinto, N. & Luo, Y. Derivation and validation of novel phenotypes of multiple organ dysfunction syndrome in critically Ill children. JAMA Netw. Open 3, e209271 (2020).
Carcillo, J. A. et al. A multicenter network assessment of three inflammation phenotypes in pediatric sepsis-induced multiple organ failure. Pediatr. Crit. Care Med. 20, 1137–1146 (2019).
Qin, Y. et al. Machine learning derivation of four computable 24-h pediatric sepsis phenotypes to facilitate enrollment in early personalized anti-inflammatory clinical trials. Crit. Care 26, 128 (2022).
Sørensen, T. I. A., Nielsen, G. G., Andersen, P. K. & Teasdale, T. W. Genetic and environmental influences on premature death in adult adoptees. N. Engl. J. Med. 318, 727–732 (1988).
Borghesi, A. et al. Whole-exome sequencing for the identification of rare variants in primary immunodeficiency genes in children with sepsis: a prospective, population-based cohort study. Clin. Infect. Dis. 71, e614–e623 (2020).
Asgari, S. et al. Exome sequencing reveals primary immunodeficiencies in children with community-acquired Pseudomonas aeruginosa sepsis. Front. Immunol. 7, 357 (2016).
Kernan, K. F. et al. Prevalence of pathogenic and potentially pathogenic inborn error of immunity associated variants in children with severe sepsis. J. Clin. Immunol. 42, 350–364 (2022).
Rautanen, A. et al. Genome-wide association study of survival from sepsis due to pneumonia: an observational cohort study. Lancet Respir. Med. 3, 53–60 (2015).
Hernandez-Beeftink, T. et al. A genome-wide association study of survival in patients with sepsis. Crit. Care 26, 341 (2022).
Butler-Laporte, G., Harroud, A., Forgetta, V. & Richards, J. B. Elevated body mass index is associated with an increased risk of infectious disease admissions and mortality: a mendelian randomization study. Clin. Microbiol. Infect. 27, 710–716 (2021).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Burnham, K. L. et al. eQTLs identify regulatory networks and drivers of variation in the individual response to sepsis. Cell Genomics 4, 100587 (2024).
Motelow, J. E. et al. Risk variants in the exomes of children with critical illness. JAMA Netw. Open 5, e2239122 (2022).
Singer, M.et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA - Journal of the American Medical Association. 315, 801 (2016).
Auwera, vander, G., O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. O’Reilly Media: Sebastopol, CA, 2020.
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575 (2007).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Wu, M. C. et al. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93 (2011).
Lee, S. et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet 91, 224–237 (2012).
Stelzer, G. et al. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinf. 54, 1–33 (2016).
Sollis, E. et al. The NHGRI-EBI GWAS catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51, D977–D985 (2023).
Watanabe, K., Taskesen, E., Van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Chen, E. Y. et al. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinforma. 14, 128 (2013).
Lonsdale, J. et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47, D886–D894 (2019).
Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6, e1001025 (2010).
Ng, P. C. & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874 (2001).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Tingley, D., Yamamoto, T., Hirose, K., Keele, L. & Imai, K. Mediation: R package for causal mediation analysis. J.Stat. Softw. 59, 1–38 (2014).
Dutta, D. et al. A powerful subset-based method identifies gene set associations and improves interpretation in UK Biobank. Am. J. Hum. Genet 108, 669–681 (2021).
Alexaki, A. et al. Codon and Codon-pair usage tables (CoCoPUTs): facilitating genetic variation analyses and recombinant gene design. J. Mol. Biol. 431, 2434–2441 (2019).
Downing, A. K. et al. Solution structure of a pair of calcium-binding epidermal growth factor-like domains: implications for the Marfan syndrome and other genetic disorders. Cell 85, 597–605 (1996).
Su, C. T. & Urban, Z. Ltbp4 in health and disease. Genes 12, 795 (2021).
Zhang, Q. et al. Two novel compound heterozygous variants of LTBP4 in a Chinese infant with cutis laxa type IC and a review of the related literature. BMC Med Genomics 13, 183 (2020).
Mazaheri, M., Jahantigh, H. R., Yavari, M., Mirjalili, S. R. & Vahidnezhad, H. Autosomal recessive cutis laxa type 1C with a homozygous LTBP4 splicing variant: a case report and update of literature. Mol. Biol. Rep. 49, 4135–4140 (2022).
Urban, Z. et al. Mutations in LTBP4 cause a syndrome of impaired pulmonary, gastrointestinal, genitourinary, musculoskeletal, and dermal development. Am. J. Hum. Genet 85, 593–605 (2009).
Kosac, A. et al. LTBP4, SPP1, and CD40 variants: genetic modifiers of duchenne muscular dystrophy analyzed in serbian patients. Genes 13, 1385 (2022).
Lu, J. et al. Increased expression of latent TGF-β-binding protein 4 affects the fibrotic process in scleroderma by TGF-β/SMAD signaling. Lab. Investig. 97, 1121–1601 (2017).
Li, Y. et al. Integrated genomic characterization of the human immunome in cancer. Cancer Res. 80, 4854–4867 (2020).
Rocchiccioli, S. et al. Hypothesis-free secretome analysis of thoracic aortic aneurysm reinforces the central role of TGF-β cascade in patients with bicuspid aortic valve. J. Cardiol. 69, 570–576 (2017).
Su, C. T. et al. LTBP4 affects renal fibrosis by influencing angiogenesis and altering mitochondrial structure. Cell Death Dis. 12, 943 (2021).
Shrine, N. et al. Multi-ancestry genome-wide association analyses improve resolution of genes and pathways influencing lung function and chronic obstructive pulmonary disease risk. Nat. Genet. 55, 410–422 (2023).
Shrine, N. et al. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat. Genet 51, 481–493 (2019).
Vuckovic, D. et al. The polygenic and monogenic basis of blood traits and diseases. Cell 182, 1214–1231 (2020).
Chen, M. H. et al. Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell 182, 1198–1213.e14 (2020).
Yeung, M. W. et al. Twenty-five novel loci for carotid intima-media thickness: a genome-wide association study in >45 000 individuals and meta-analysis of >100 000 individuals. Arterioscler Thromb. Vasc. Biol. 42, 484–501 (2022).
Plotnikov, D. et al. High blood pressure and intraocular pressure: a mendelian randomization study. Invest. Ophthalmol. Vis. Sci. 63, 29 (2022).
Rifkin, D. et al. The role of LTBPs in TGF beta signaling. Developmental Dynamics 251, 75–84 (2022).
Bergmann, C. B. et al. Potential targets to mitigate trauma- or sepsis-induced immune suppression. Front. Immunol. 12, 622601 (2021).
Colige, A., Monseur, C., Crawley, J. T. B., Santamaria, S. & De Groot, R. Proteomic discovery of substrates of the cardiovascular protease ADAMTS7. J. Biol. Chem. 294, 8037–8045 (2019).
Capestrano, M. et al. Cytosolic phospholipase A2ε drives recycling through the clathrin-independent endocytic route. J. Cell Sci. 127, 977–993 (2014).
Pérez-González, M. et al. PLA2G4E, a candidate gene for resilience in Alzheimer´s disease and a new target for dementia treatment. Prog. Neurobiol. 191, 101818 (2020).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
Kachuri, L. et al. Genetic determinants of blood-cell traits influence susceptibility to childhood acute lymphoblastic leukemia. Am. J. Hum. Genet 108, 1823–1835 (2021).
Astle, W. J. et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167, 1415–1429.e19 (2016).
Evangelou, E. et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet 50, 1755 (2018).
Amunugama, K., Pike, D. P. & Ford, D. A. The lipid biology of sepsis. J. Lipid Res 62, 100090 (2021).
Wasyluk, W., Wasyluk, M. & Zwolak, A. Sepsis as a pan-endocrine illness—endocrine disorders in septic patients. Journal of Clinical Medicine 10, 2075 (2021).
Vale-Costa, S. & Amorim, M. Recycling endosomes and viral infection. Viruses 8, 64 (2016).
Tilley, A. E., Walters, M. S., Shaykhiev, R. & Crystal, R. G. Cilia dysfunction in lung disease. Annu Rev. Physiol. 77, 379–406 (2015).
Acknowledgements
Clinical Research Investigation and Systems Modeling of Acute illness center: Ali Smith, BS; Octavia Palmer, MD; Vanessa Jackson, AA; Renee Anderko, BS, MS. Children’s Hospital of Pittsburgh: Jennifer Jones, RN; Luther Springs. Children’s Hospital of Philadelphia: Carolanne Twelves, RN, BSN, CCRC; Mary Ann Diliberto, BS, RN, CCRC; Martha Sisko, BSN, RN, CCRC, MS; Pamela Diehl, BSN, RN; Janice Prodell, RN, BSN, CCRC; Jenny Bush, RNC, BSN; Kathryn Graham, BA; Kerry Costlow, BS; Sara Sanchez. Children’s National Hospital: Elyse Tomanio, BSN, RN; Diane Hession, MSN, RN; Katherine Burke, BS. Children’s Hospital of Michigan, Central Michigan University: Ann Pawluszka, RN, BSN; Melanie Lulic, BS. Nationwide Children’s Hospital: Lisa Steele, RN, CCRC; Andrew R. Yates, MD; Josey Hensley, RN; Janet Cihla, RN; Jill Popelka, RN; Lisa Hanson-Huber, BS. Children’s Hospital Los Angeles and Mattel Children’s Hospital: Jeni Kwok, JD; Amy Yamakawa, BS. Children’s Hospital of Washington University of Saint Louis: Michelle Eaton, RN. Mott Children’s Hospital: Frank Moler, MD; Chaandini Jayachandran, MS, CCRP. University of Utah Data Coordinating Center: Teresa Liu, MPH, CCRP; Jeri Burr, MS, RN-BC, CCRC, FACRP; Missy Ringwood, BS, CMC; Nael Abdelsamad, MD, CCRC; Whit Coleman, MSRA, BSN, RN, CCRC.
Funding
Funding was supported, in part, by grant R01GM108618 (to Dr Carcillo PI, HJ Park CoI) from the National Institutes of General Medical Sciences, by 5U01HD049934-10S1 (to Dr Carcillo) and K12HD047349 (to Dr Kernan) from the Eunice Kennedy Shriver National Institutes of Child Health and Human Development, National Institutes of Health, Department of Health and Human Services, and the following cooperative agreements: U10HD049983, U10HD050096, U10HD049981, U10HD063108, U10HD63106, U10HD063114, U10HD050012, and U01HD049934.
Author information
Authors and Affiliations
Contributions
Y.Q., K.K., Y.B., J.R.S., Z.U., S.C., M.P., K.M., C.N., T.S., R.E.H., M.H., J.A.C. and H.J.P. designed the project, edited the manuscript, and supervised the study. All authors have approved the final version of this paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors have no competing interest but did have the following and sources of funding. Drs. Carcillo’s, Berg’s, Wessel’s, Pollack’s, Meert’s, Hall’s, Doctor’s, Cornell’s, Harrison’s, Zuppa’s, Reeder’s, Banks’s, and Holubkov’s institutions received funding from the National Institutes of Health (NIH). Drs. Carcillo’s, Newth’s, Shanley’s, and Dean’s institutions received funding from the National Institutes of Child Health and Human Development. Drs. Carcillo, Berg, Wessel, Polack, Meert, Hall, Newth, Doctor, Shanley, Cornell, Harrison, Zuppa, Reeder, Banks, Holubkov, Notterman, and Dean received support for article research from the NIH. Dr. Carcillo’s institution also received funding from the National Institutes of General Medical Sciences. Dr. Pollack disclosed that his research is supported by philanthropy from Mallinckrodt Pharmaceuticals. Dr. Hall received funding from Bristol Myers-Squibb (for service on an advisory board) and LaJolla Pharmaceuticals (service as a consultant), both unrelated to the current submission. Dr. Newth received funding from Philips Research North America. Dr. Doctor’s institution received funding from the Department of Defense and Kalocyte. Dr. Shanley received funding from Springer publishing, International Pediatric Research Foundation, and Pediatric Academic Societies. Dr. Cornell disclosed he is co-founder of Pre-Dixon Bio. Dr. Holubkov received funding from Pfizer (Data Safety Monitoring Board [DSMB] member), Medimmune (DSMB member), Physicians Committee for Responsible Medicine (biostatistical consulting), DURECT Corporation (biostatistical consulting), Armaron Bio (DSMB past member), and St Jude Medical (DSMB past member). The remaining authors have disclosed that they do not have any potential competing interests.
Consent statement
The study was approved by the Institutional Review Board at University of Utah Central IRB # 70976.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qin, Y., Kernan, K.F., Bai, Y. et al. Deleterious variants in LTBP4 are associated with severe pediatric sepsis. Pediatr Res (2025). https://doi.org/10.1038/s41390-025-04420-3
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41390-025-04420-3
