
Abstract
Human papillomaviruses, particularly high-risk human papillomaviruses, have been universally considered to be associated with the oncogenesis and progression of various cancers. The genome of human papillomaviruses is circular, double-stranded DNA that encodes early and late proteins. Each of the proteins is of crucial significance in infecting the epithelium of host cells persistently and supporting viral genome integrating into host cells. Notably, E6 and E7 proteins, classified as oncoproteins, trigger the incidence of cancers by fostering cell proliferation, hindering apoptosis, evading immune surveillance, promoting cell invasion, and disrupting the balance of cellular metabolism. Therefore, targeting human papillomaviruses and decoding molecular mechanisms by which human papillomaviruses drive carcinogenesis are of great necessity to better treat human papillomaviruses-related cancers. Human papillomaviruses have been applied clinically to different facets of human papillomavirus-related cancers, including prevention, screening, diagnosis, treatment, and prognosis. Several types of prophylactic vaccines have been publicly utilized worldwide and have greatly decreased the occurrence of human papillomavirus-related cancers, which have benefited numerous people. Although various therapeutic vaccines have been developed and tested clinically, none of them have been officially approved to date. Enhancing the efficacy of vaccines and searching for innovative technologies targeting human papillomaviruses remain critical challenges that warrant continuous research and attention in the future.
Similar content being viewed by others
Introduction
Human papillomaviruses (HPVs), recognized as one of the most common sexually transmitted viruses, can infect skin and mucosa.1,2 HPVs can be sorted into five genera: Alpha, Beta, Gamma, Mu, and Nu.3 HPVs from the Beta and Gamma genera can elicit inapparent infections of the skin. Alpha-HPVs are associated with various clinical conditions ranging from benign warts to cancers. For instance, HPV-6 and HPV-11 are connected with oral focal hyperplasia and anogenital warts. HPV-3, HPV-10, HPV-28 are closely linked with plane warts, whereas HPV-2, HPV-7, and HPV-40 are associated with common warts. Among Alpha-HPVs, only a small part of Alpha-HPVs is oncogenic, which is termed as high-risk HPVs (hr-HPVs) (Table 1). Most HPV infections are transient and asymptomatic without causing any health problems. Persistent infection with hr-HPVs can cause precancerous lesions, thus evolving into various cancers, including head and neck squamous cell carcinoma (HNSCC), cervical cancer (CC), anal cancer, as well as vulvar, penile, and vaginal cancers that are less prevalent.
In 2020, the World Health Organization (WHO) revised the classification of CC into two subtypes: HPV-associated and HPV-independent. In addition, HPV-associated adenocarcinoma has a better prognosis than HPV-independent adenocarcinoma in CC.4 Besides, the American Joint Committee on Cancer has set a specific staging system for HPV-positive oropharyngeal squamous cell carcinoma (OPSCC), which is a specific type of HNSCC. Research has confirmed that HPV-positive OPSCC is more sensitive to radiotherapy and chemotherapy, with a less aggressive phenotype and better prognosis.5,6 Moreover, HPV infection not only provides reliable biomarkers for the screening and early diagnosis of hr-HPV-related cancers, but also offers a series of potential therapeutic targets for treating hr-HPV-related cancers. Various vaccines targeting hr-HPVs have been developed to prevent and treat hr-HPV-related cancers, which will be narrated in detail in the following text.
The study of HPVs is time-honored (Fig. 1). In 1956, with the aid of the electron microscopic technique, the presence of HPV was identified and the feature of HPV was confirmed. Harald zur Hausen firstly proposed a hypothesis that HPV may be related with CC in 1974. The HPV genome was cloned into plasmids in 1976, providing methods for studying the underlying molecular mechanisms of HPV. And more and more research found the existence of HPV in lesions of cervix.7,8 Research on HPV saw significant advancement in the 1970s and 1980s, in which HPV-16 and HPV-18 were successfully cloned in 1983 and 1984, respectively.9 Moreover, HPV-16 and HPV-18 were classified as oncogenic in 1995. The characteristic of viral DNA such as immortalization was found and the oncogenic mechanisms of E6 and E7 proteins were further studied.10,11 Next, significant progress was witnessed in the 1990s and 2000s. Gardasil, Cervarix, and Gardasil-9 were developed and approved to prevent the occurrence of hr-HPV-related cancers. And the WHO has launched a worldwide strategy to eliminate CC in 2030. Nowadays, studies related to HPV include oncogenic signaling mechanisms, the development of various preventative and therapeutic vaccines, and different therapies targeting hr-HPV.

A timeline of HPV research. Since the presence was identified in 1956, study on HPV has continued to date. Apart from oncogenic mechanisms, vaccines targeting HPV have been developed. Moreover, WHO has launched strategies for worldwide CC elimination. This figure was created with BioRender (www.bioender.com)
Despite the availability of screening methods and various vaccines, the burden of hr-HPV-related malignancies still exists, which mainly detrimentally affects low- and meddle-income countries (LMICs) due to limited access to prevention and treatment. According to statistics, CC remains the most generally diagnosed cancers in women in 25 countries among 185 countries and CC is the main cause of cancer death in 37 countries especially in LMICs. Moreover, the incidence and mortality of CC ranks in top five in LMICs.12 In 2018, LMICs contribute to almost 90% of the mortality of CC. About 31% of LMICs have implemented HPV vaccination compared with 85% of high-income countries. The availability of HPV vaccine varies significantly among different regions (Fig. 2a).13 In addition, only 20% of women in LMICs have access to HPV screening, whereas more than 60% of women experience HPV screening in high-income countries.14,15

Global HPV vaccine availability and the association between hr-HPVs and cancers. a Percentage of HPV vaccine availability among different regions. b The association between hr-HPVs and cancers. Cervical cancer, oropharyngeal squamous cell carcinoma, anal cancer, vaginal cancer, vulvar cancer, and penile cancer are well recognized as closely associated with hr-HPVs. c Number of cancer cases caused by hr-HPV per year. This figure was created with BioRender (www.bioender.com)
The implications of this review extend beyond understanding the life cycle and oncogenic signaling pathways of hr-HPV infections. It also underscores the role of hr-HPV as a significant biomarker for cancer screening and early detection, as well as a therapeutic target for cancer treatment. As research progresses, these potential targets and biomarkers present opportunities to reduce the global burden of HPV-associated diseases through early intervention, personalized treatment strategies, and potentially curative therapies.
The epidemiology and current clinical management of hr-HPV-related cancers
It has been widely recognized that persistent hr-HPV infection contributes to certain types of cancers (Fig. 2b) and the number of cancer cases caused by hr-HPV has been estimated according to the Centers for Disease Control and Prevention (CDC) (Fig. 2c).16,17 Until now, 15 types of hr-HPVs have been found to be linked with the occurrence and development of CC, among which HPV-16 and HPV-18 contribute to almost 75% of CCs,18,19,20 while HPV-31, 33, 45, 52, and 58 contribute to the other 15–20% of CCs.21 CC, known as the fourth most common cancer in women around the world, had almost 604,000 cases and 342,000 deaths in 2020 according to the WHO.22 Due to the widespread application of screening and prevention, the incidence of CC has declined dramatically, and there will be an estimated 13,820 new cases in the United States in 2024.23 Meanwhile, compared with high-income countries, LMICs in Africa and southern Asia account for a large proportion of hr-HPV-related CCs due to the expensive costs of HPV screening and vaccination.24 Besides, a total of 891,453 cases of HNSCCs were estimated worldwide in 202212 and the incidence of HNSCCs is predicted to exceed that of CC in the United States and the UK. It’s also noteworthy that OPSCC has been found to be closely associated with hr-HPVs, with almost 70% of OPSCCs are caused by hr-HPVs, especially HPV-16.25 The incidence of hr-HPV-related OPSCC has increased dramatically over the past twenty years in the white population from high-income countries, especially in those with little or no tobacco exposure.26,27 Moreover, ~90% of anal cancers are also caused by persistent hr-HPV infection.23
ThinPrep cytologic test, HPV test, together with colposcopy has become valuable methods to detect pathological changes of the cervix, which has greatly contributed to the timely intervention of precancerous lesions and has benefited a large number of the female population. Radical hysterectomy with bilateral pelvic lymph node dissection is the standard treatment for early-stage CCs, whilst concurrent chemoradiation, based on drugs containing platinum is the first choice of advanced-stage CC. Moreover, pembrolizumab plus chemotherapy with or without bevacizumab is recommended as the first-line therapy for recurrent or metastatic CC.4
Notably, although HPV status has been included as an important staging factor in the 8th edition of the American Joint Committee on Cancer staging system for OPSCC, there are almost no differences in the treatment between hr-HPV-related OPSCC and hr-HPV-unrelated OPSCC.28 For early-stage OPSCC, surgery by a double transoral and transcervical approach or intensity-modulated radiotherapy is recommended, whilst cisplatin-based concurrent chemoradiation is the first-line therapy for advanced OPSCC. Meanwhile, nodal condition, tumor diameter, positive surgical margins, degree of differentiation and invasion pattern are also important factors to be considered in the treatment selection and prognostic prediction for OPSCC.6
Life cycle of hr-HPV during infection
HPV is a non-enveloped virus that owns an icosahedral capsid composed of 72 capsomers: 360 copies of the 55 kDa L1 protein and 12 copies of the 74 kDa L2 protein.29,30 The HPV genome is a circular, double-stranded DNA about 7–8 kb in size. The HPV genome has three characterized regions: the upstream regulatory region (URR) which is also called the long control region (LCR); the early (E) region; and the late (L) region. Owing to possessing binding sites for E1 and E2 proteins, and transcription elements such as papillomavirus enhancer factor-1, activating protein 1, octamer binding factor-1, transcription enhancer factor-1 (TEF-1) and TEF-2, transcription specificity protein 1, and nuclear factor-1,31,32,33 the URR is responsible for initiating the replication and transcription of HPV DNA. Besides, the URR contains the p97 promoter together with regulatory motifs and is recognized as the region that possesses the highest variation. According to this feature, analysis of URR can be applied to the classification of HPVs.29,34 The early region encodes E1, E2, E1^E4, E5, E6, E7, and E8^E2 proteins, which are closely associated with infection, viral replication, and oncogenesis.29 The late region encodes L1 and L2 proteins. In addition, the HPV genome contains early (PE) and late (PL) promoters, as well as early (pAE) and late (pAL) polyadenylation sites.
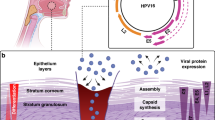
HPV particles enter the basal layer of the epithelium via minuscule abrasion or wound of epidermis. Then, HPV particles attach to heparan sulfate proteoglycans and secondary receptors such as α6-Integrin, which facilitate HPV particles entering into host cells. HPV infection undergoes four stages once entering the basal cells: initial, maintenance, vegetative amplification, as well as virus assembly and release (Fig. 3).

The life cycle of hr-HPVs. HPVs enter the basal layer of the epithelium and undergo different stages: infection, replication, capsid synthesis, virion assembly, and virion release. Early proteins are of great significance during HPV infection. Persistent infection results in hyperplasia and dysplasia. Without intervention, dysplasia has a great possibility to evolve into cancers. This figure was created with BioRender (www.bioender.com)
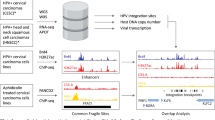
Through endocytosis, HPV particles have access to basal cells. Then, the L1 protein separates from the L2 protein. The L2 protein forms a complex with the HPV genome in the vesicle form, transports to the trans-Golgi and resides until HPV particles enter the nucleus when the nuclear envelope breaks down during mitosis. The promyelocytic leukemia (PML) protein is recruited to the surrounding viral genome prior to the genome entering the nucleus during early interphase. The L2 protein, together with the viral genome, remains at PML-nuclear bodies even in late interphase, which offers a protective environment and a possible position for the initial infection and low-copy number of viral genomes.35 Upon getting into the nucleus, the E1 and E2 proteins are expressed and cooperatively connect to the replication origin to initiate DNA replication. HPVs infect the proliferating basal cells of the epithelium to construct the maintenance stage, during which HPV genomes exist in the form of extrachromosomal plasmids36 and replicate along with the host genome in the S-phase. Notably, hr-HPVs have the ability to drive cell proliferation in the basal and parabasal layers, distinguishing them from other types of HPVs. DNA amplification occurs in the differentiated cells of the upper layers of the epithelium. Then the viral genome can be packaged and released from the surface of the epithelium. HPV triggers the DNA damage response (DDR) and employs the DDR mechanism to obtain materials necessary for viral DNA synthesis in the G2-like phase. DDR pathways, such as ataxia-telangiectasia mutated and Rad3-related pathways, are activated, and related factors are recruited to nuclear replication foci.35,37 These regions overlap fragile sites that are hard to replicate and susceptible to replication stress. It’s worth mentioning that HPV genome integration probably occurs in these fragile sites.35 Early proteins make a difference to maintaining the process of infection. The E1 and E2 proteins initiate the replication of HPV DNA and locate HPV particles to cellular chromosomes. E6 and E7 proteins maintain the life cycle of HPV replication. During the late phase of HPV replication, early proteins are increased to support the completion of this phase and facilitate the release of virions.38 The specific oncogenic mechanisms of early proteins of hr-HPVs are discussed in the following part.
Persistent hr-HPV infection can lead to the viral genome integrating with the host genome. Evidence has shown that the rate of genome integration is higher in HPV-16 and HPV-18 positive cancer samples than in non-cancer samples, and the integration facilitates intratumoral heterogeneity and clonal evolution.39,40,41 HPV integration mainly occurs in fragile sites that provide clonal selection advantages. HPV integration can be classified into two types. One copy is integrated in type 1 integration, while in type 2 integration, several tandem repeats are noticed. The viral genome connects to host cells mainly depend on the generation of E2 complex together with bromodomain protein 4 (Brd4), which can further activate the transcription of viral genome. In type 1 integration, despite the fact that the whole viral genome is integrated, high methylation of E2 binding sites in the p97 promoter region inhibits the expression of E2, thus reversing the inhibition of E6 and E7 and fostering the transcription of oncogenes. In type 2 integration, a combination of viral genome and Brd4 contributes to the transcription of E6 and E7. Moreover, genomic instability is caused via amplification of integration sites mediated by E1 during this process.42 The predominant mechanism of integration is the microhomologies-mediated DNA repair pathway. Research has demonstrated that enrichment of microhomologies is observed at or near integration breakpoints, especially when the length of microhomologies is over 4 bp. Meanwhile, the enrichment of 4-bp microhomologies remains noteworthy when the area of the flanking region extends.43 However, the specific progress of how microhomologies mediate the integration is still unknown. A new model has been proposed to elucidate the process of integration: synthesis‐dependent end‐joining, including junctional microhomologies, apparent blunt joining, and short insertion, which has been validated utilizing computational simulation in 4341 human-HPV junctional sequences.39 Notably, the hijacking of DDR to facilitate HPV DNA replication, the presence of epigenetic modification at integration sites, and the location of transcriptionally competent region within the host chromatin all contribute HPV integration, which further promotes oncogenesis.44
hr-HPV integration has numerous ways to alter the state of host cells, thereby promoting the initiation and development of cancers. hr-HPV integration results in the formation of fusion transcripts, which have been found in hr-HPV-related CCs. Fusion transcripts elevate the mRNA expression levels of E6 and E7 and inhibit the E2 expression, which serves as a transcriptional inhibitor of E6 and E7.45 The overexpression of E6 and E7 facilitates oncogenesis by disrupting the transcription of tumor suppressor genes and triggering flanking gene amplification. The subsequent overexpression of oncogenes in host cells contributes to oncogenesis.46 The fusion gene E1C, a representative product of HPV integration, has been confirmed to facilitate the malignant transformation of cervical cells.47 HPV integration contributes to the generation of extrachromosomal DNA (ecDNA), which enables cells to proliferate unlimitedly, making them promising drivers of clonal evolution.48 ecDNA keeps the integrity of E6 and E7 genes, which can encode oncogenic proteins and specific oncogenic signaling pathways are detailed in the following text. Moreover, together with epigenetic modification, the dysregulation of cellular genes mediated by HPV integration can cause oncogenesis as well.49,50 HPV integration induces chromosomal instability, rendering host cells more susceptible to cancers. Upon integration, the HPV breakpoint-induced cellular super-enhancers (BP-cSEs) are generated. BP-cSEs can interact with chromosomes and induce genomic rearrangements. Acting as ecDNA, BP-cSEs dysregulate chromosomal genes, indicating the role of HPV integration in oncogenesis.51 Moreover, HPV integration leads to chromosomal translocation and genomic variation, promoting clonal evolution and heterogeneity in cancers, mainly through the form of ecDNA.40 Research has also observed that HPV integration dysregulates local chromatin and transcriptome in HPV-positive cancers. This disruption upregulates the expression of oncogenes, thereby promoting oncogenesis by altering gene splicing, modifying the epigenome, and affecting transcriptional regulation.52
Moreover, dysregulated epigenetic modification, including DNA methylation, histone modification, noncoding RNA (ncRNA) regulation, and chromatin regulation, plays a key role in the occurrence and progression of cancers. Aberrant methylation of the host cell genome and HPV DNA causes the dysfunction of tumor suppressor genes, thereby accelerating the progression of cancers. WID-qCIN test, based on DNA methylation, has been developed to detect cervical intraepithelial neoplasia (CIN) and predict the development of diseases in hr-HPV-positive women.53 Histone modifications, like acetylation, methylation, and phosphorylation, are vital to the development of cancers. The E6 of hr-HPVs can suppress SET7-mediated methylation of p53 and downregulate histone methylation mediated by p53 coactivators, decreasing the binding of p53 to chromatin and transactivation of p53.54 E6 inhibits acetylation of p53 and nucleosomal core histones mediated by p300, which disrupts the activation of genes targeted by p53 and causes the development of cancers.55,56
HPV has various ways to evade host immune responses. Transporting viral DNA in the form of vesicles can protect HPV particles from the identification of pattern recognition receptors (PRRs), which is crucial to the persistent infection of HPVs. Early proteins of hr-HPVs can inhibit viral DNA detection by the cyclic GMP-AMP synthase -stimulator of interferon genes (cGAS-STING) pathway, thus impairing the transcription of interferons (IFNs) and IFN-stimulated genes (ISGs) by inhibiting the transcription of IFN-κ. E6 and E7 proteins of hr-HPVs also work cooperatively to suppress the generation of ISGs, thereby evading the antiviral responses of host cells. In addition, E6 and E7 proteins of hr-HPVs can dysregulate the cGAS-STING pathway and Toll-like receptor (TLR) signaling, which ultimately leads to immune evasion and persistent infection.57,58 Apart from innate immunity, early proteins of hr-HPVs, especially E6 and E7 proteins, affect adaptive immunity as well. hr-HPVs can impair specific human leukocyte antigen (HLA) molecules and recruit regulatory T cells to inhibit the amplification of effector T cells, therefore impeding antitumor responses.57 Meanwhile, the emergence of dysfunctional and exhausted T-cell subsets has been observed in HPV-positive malignancies. HPV-positive malignant cells can also generate tenascin C to interact with stroma to suppress the proliferation and activation of T cells, thus aiding hr-HPVs to evade immune responses.59
Oncogenic signaling pathways of hr-HPV
Given that persistent hr-HPV infection is a key factor in the initiation and development of cancers, oncogenic signaling pathways of hr-HPVs need to be discussed. In this review, we focus on the early proteins that hr-HPVs encode and their roles in initiating and developing cancers (Fig. 4).

Oncogenic mechanisms of hr-HPVs and therapeutic strategies for hr-HPV-related cancers. hr-HPVs contribute to the oncogenesis through various ways, including evading immune responses, promoting cell proliferation, resisting apoptosis, inducing cell immortality, fostering angiogenesis, and promoting cell invasion. Different types of therapies treating hr-HPV-related cancers have been widely applied in clinic, aiming to gain better efficacy and prognosis. This figure was created with BioRender (www.bioender.com)
E1 and E2
The E1 protein is an adenosine triphosphate-dependent helicase with conserved structure. The E1 protein binds to the replication origin and unwinds the viral DNA to initiate DNA replication. The E1 protein consists of an N-terminal domain, a DNA binding domain, an oligomerization domain, and a C-terminal helicase domain. The N-terminal domain is responsible for transporting the E1 protein between the nucleus and the cytoplasm. The DNA binding domain is capable of identifying particular sequences and binding to the helicase domain around viral DNA. The E2 protein, existing in the form of a dimer, includes an N-terminal domain responsible for the transcriptional activation of DNA replication and a C-terminal domain that serves to bind with specific DNA. In order to initiate DNA replication, the E2 protein binds to E1 to form a complex at the replication origin. Then, the E2 protein is extracted, and the E1 protein is recruited to form double trimers. At last, the double hexamer E1 complex unwinds DNA and recruits replication factors such as topoisomerase I and DNA polymerase α-primase to initiate DNA synthesis.60
E1 can modulate HPV transcription epigenetically via binding to nucleosomes that are essential for regulating HPV transcription.61 In addition, E1 affects cell functions by dysregulating the expression of different genes. Research has verified that the overexpression of HPV-16 E1 and HPV-31 E1 suppresses the proliferation of cells by means of inducing cell cycle arrest, necrosis, or apoptosis.62 E1 dysregulates the expression of genes and proteins involved in transcription, apoptosis, DNA damage pathways, and immune pathways,63 which ultimately promotes oncogenesis. E1C, composed of an integration of the HPV-58 E1 and CMAHP gene, fosters colony formation and malignant transformation of cervical cells. Analyzed by RNA sequencing, E1C upregulates gene expression such as S1PR3, NCL, and HNRNPD, high levels of which indicate a poor prognosis of CC. Meanwhile, E1C downregulates the expression of genes, including MTRNR2L8, KRT80, and CLDN4, and the decreased expression of these genes also suggests a poor prognosis.47 Besides, E1 has been shown to inhibit the expression of IFNs, including IFNβ1 and IFNλ1, and activate the nuclear factor kappa-B (NF-κB) pathway, which indicates the role of E1 in immune regulation.60,64
Apart from being able to initiate DNA synthesis, E2 can inhibit the expression of E6 and E7 by binding to specific sites in the LCR region. Binding to Brd4, TIP60, and EP400 contributes to E2-mediated transcriptional repression of the LCR region. Demethylation of histone H3 lysine 4 is induced by the interaction between E2 and SMCX; therefore, the transcriptional activity of the LCR region is suppressed, which finally contributes to the inhibited expression of E6 and E7.65 E2 also blocks p97 and p105 promoters to inhibit the expression of E6 and E7.66,67 Disruption of E2 leads to the aberrant expression of E6 and E7, which is a precursor of oncogenesis. What’s more, E2, E6 and E7 increase the generation and secretion of interleukin 10 (IL-10) that can aid cancer cells to escape immunologic surveillance. IL-10 increases the expression of E6 and E7, which results in a vicious cycle. The overexpression of IL-10 can lead to the transformation of squamous intraepithelial lesions (SIL) to CC.68
E4
The E4 open reading frame (ORF) exists in the E2 ORF. The E4 protein is generated as an E1^E4 fusion protein through mRNA splicing.20 The E4 protein works in the late stage of the viral life cycle. The E4 protein owns a leucine cluster motif near the N-terminus, which is necessary for its interaction with keratin. The C-terminus of the protein aids to regulate keratin, vital for viral assembly and transportation. The E4 protein can inhibit the phosphorylation of SPRK1 to modulate the distribution of E2.69 Research has demonstrated that the expression and accumulation of E4 can lead to G2-phase arrest, which prevents nuclear translocation and phosphorylation of proteins participating in the process of mitosis. The viral vegetative amplification is ultimately promoted, fostering persistent HPV infection.70
E5
The E5 protein, a small hydrophobic protein, comprised of three regions, an N-terminal domain, a C-terminal domain, and a central domain located between them,20 is characterized as sequence heterologous in different types of HPVs.71 Notably, only alpha-HPVs can encode the E5 protein.72 E5 proteins such as those derived from HPV-16 and HPV-18 exist in the endoplasmic reticulum, Golgi, endosomes, nuclear membrane, and plasma membrane.73,74,75 E5 is recognized as one of the oncoproteins and can reinforce the function of E6 and E7 in immortalizing keratinocytes.76 E5 is of great significance in modulating cell growth, viral replication, and various signaling pathways to initiate oncogenesis (Fig. 5a).

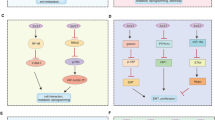
Oncogenic signaling pathways of hr-HPVs. a Oncogenic signaling pathways of E5. E5 can activate epidermal growth factor receptor (EGFR) signaling. Through EGFR, E5 activates the PI3K/AKT and MAPK/ERK pathways to promote uncontrolled cell proliferation. Moreover, E5 facilitates cell migration and inhibits apoptosis and immune responses, which ultimately leads to the generation of cancers. b Oncogenic signaling pathways of E6. E6 is well-known for binding with and degrading p53 in a ubiquitin–proteasome way. Degradation of p53 promotes cell proliferation and inhibits apoptosis. E6 is able to inhibit autophagy and immune responses and facilitate cell immortalization and invasion independent of p53 degradation as well. In addition, E6 promotes glycolysis and lipid synthesis to support cell growth. c Oncogenic signaling pathways of E7. E7 binds with pRb to release E2F, promoting cells entering the S-phase. E7 can increase the expression of cyclins to enable cells to alter from the G1-phase to S-phase unrestrictedly, resulting in uncontrolled cell proliferation. Moreover, E7 boosts cell migration and hinders immune responses to further stimulate the occurrence of cancers. This figure was created with BioRender (www.bioender.com)
By means of diverse pathways, E5 is capable of modulating the proliferation, differentiation, and transformation of cells. E5 inhibits the transforming growth factor (TGF-β)/SMAD signaling, which modulates immune responses and uncontrolled cell proliferation.77 Moreover, E5 can interact with the epidermal growth factor receptor (EGFR) and activate pathways associated with EGFR.78 Through the interaction between E5 and the vacuolar ATPase (v-ATPase), the v-ATPase-mediated acidification of endosomes is disrupted, facilitating the recycling of EGFR.79 In addition, E5 suppresses the activity of casitas B-lineage lymphoma that can degrade EGFR via ubiquitination.78,80 Therefore, EGFR can persist longer. EGFR activates the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) and the mitogen-activated protein kinase (MAPK)/extracellular regulated protein kinases (ERK) pathways. EGFR activates PI3K and mitogen-activated extracellular signal-regulated kinase (MEK) 1/2, which can phosphorate Akt and ERK1/2 respectively. Vascular endothelial growth factor (VEGF), a pivotal factor in angiogenesis, is finally upregulated.81 Interestingly, research has shown that the expression level of the E5 protein has no influence on the transcription of EGFR or VEGF in patients with HPV-16 or HPV-18 positive CC.82 Activation of MAPK sustains the expression of cyclin D1, which promotes cell proliferation. Activator protein 1 (AP-1), a dimer composed of the Fos and Jun proteins,83 can be activated by MAPK and possess the constitutive activity. AP-1 is highly significant for initiating and maintaining the expression of E6 and E7, and the viral expression is subsequently enhanced.84 Research has confirmed that the high expression of c-Jun is associated with poor prognosis in patients with HPV-16 and HPV-18 positive CC.85 Activated AKT phosphorylates GSK3β, while phosphorylated GSK3β contributes to the accumulation of cyclin D1. Moreover, activation of AKT leads to the phosphorylation of p21 and p27. Phosphorylated p21 and p27 accumulate in the cytoplasm and lose the ability to inhibit the activity of cyclin-dependent kinase (CDK) 4/6-cyclin D1, thus promoting cells entering the S-phase from the G-phase.86 In addition, the elevated expression of CDK4/6-cyclin D1 promotes E2F releasing from the RB/E2F complex, facilitating the progression of the cell cycle.87 Ultimately, the S-phase is prolonged, and the proliferation of basal layer cells and replication of viral genome is enhanced. Apart from regulating cell proliferation, E5 regulates the differentiation of keratinocytes located in the upper layer via keratinocyte growth factor receptor/fibroblast growth factor receptor 2b (KGFR/FGFR2b). E5 downregulates the expression of KGFR/FGFR2b and inhibits KGFR transporting to the endocytic degradative pathway, resulting in the inhibition of differentiation and regulation of suprabasal keratinocytes.88 Moreover, by means of downregulating the expression of KGFR/FGFR2b, E5 represses autophagy, which may cause oncogenesis.89
E5 can inhibit apoptosis to facilitate the survival and replication of hr-HPVs.90 E5 mainly induces the ubiquitination and degradation of death receptors to inhibit apoptosis.91 The extrinsic apoptotic pathway is initiated by the activation of cell surface death receptors such as tumor necrosis factor (TNF) receptor, TNF-related apoptosis-inducing ligand (TRAIL) receptors, and Fas. Once these receptors bind with corresponding ligands, trimerization occurs at the cell surface. Subsequently, the Fas-associated protein with death domain (FADD), pro-caspase 8, and/or pro-caspase 10 are recruited to form the death-inducing signaling complex (DISC). Pro-caspase 8 is activated and altered to caspases 3, 6, and 7.92,93 Apart from downregulating the expression of Fas to inhibit the recruitment of FADD, HPV-16 E5 also disrupts the formation of the TRAIL DISC and the cracking of pro-caspase 8,3, and poly ADP-ribose polymerase.94 HPV-16 E5 decreases the expression of Bax and Bak through increasing the proteasome-ubiquitin degradation of Bax. Meanwhile, E5 increases the expression of Bcl-2.95 As a result, apoptosis is suppressed.
E5 plays an essential role in evading immune responses. E5 is able to downregulate the expression of major histocompatibility complex (MHC) I, which is important for antigen presentation and subsequent induction of immune responses. Via inhibiting compartment acidification, E5 mediates the retention of MHC I molecules in the Golgi and decreases the expression of MHC I on the membrane surface. Moreover, by inhibiting MHC II transporting to the cell surface, E5 disturbs the expression and stability of MHC II. Through binding with MHC molecules and further suppressing their transportation to the plasma membrane, E5 hinders antigen processing and promotes immune evasion.96,97 CD1d, which presents antigens to natural killer T cells, is downregulated by E5 and is incapable of being transported to the surface of cells, thus successfully evading immunologic surveillance.98,99 E5 directly binds with mitochondrial antiviral-signaling protein and STING to inhibit the cGAS-STING and retinoic acid-inducible gene-1 (RIG-1)/melanoma differentiation-associated gene 5 pathways, ultimately eliminating immunologic surveillance.100
E5 upregulates the expression of paxillin and downregulates the expression of A/C protein; thereby altering cell migration.101,102 What’s more, E5 may modulate cell adhesion and cell migration via matrix-metalloproteinases (MMPs). MMPs pave the way for cell migration due to the ability to degrade the basement and extracellular matrix and separate cells from the extracellular matrix and adjacent cells.103 HPV-16 E5 promotes cell migration, and accelerates the transition to a malignant phenotype by downregulating the expression of E-cadherin.104 Nevertheless, the detailed underlying mechanisms need to be further studied.
E5 is involved in metabolic reprogramming and aims to provide energy and environment conducive to the proliferation of cancer cells. Elevated expression of EGFR activates the MAPK/ERK, Janus kinase (JAK)/signal transducer and activator of transcription (STAT), PI3K/Akt, and protein kinase C pathways, contributing to increased glycolysis, lipid synthesis, and inhibited glycogen synthesis.105
E6
E6, composed of around 150 amino acids, includes two zinc finger-binding domains flanked by four Cys–X–X–Cys (CXXC) motifs. Possessing a C-terminal PDZ (PSD-95/DLG/ZO-1) binding motif (PBM) is a unique characteristic of hr-HPVs. PBM is essential for the oncogenesis of the E6 protein of hr-HPVs.106 This motif facilitates the binding of E6 to proteins containing PDZ domains, thus influencing the generation of cancers. E6-associated protein (E6AP), which owns a PDZ structure, is well-known for interacting with E6 and forming a heterodimer. The interaction between E6 and the LXXLL motif of E6AP is a prerequisite for the fabrication of E6/E6AP/P53 complex.107 Degradation of p53 in a ubiquitin–proteasome way, a marker that indicates the activity of E6, causes genomic instability,108,109 which is closely linked with oncogenesis. Here, we summarize mechanisms through which E6 induces oncogenesis (Fig. 5b).
E6 is capable of triggering uncontrolled cell proliferation. Research has verified that the proliferation of HPV-positive cells can be inhibited via downregulating the expression of E6.110,111 p53 suppresses cyclin B, leading to G2/M-phase arrest and cell cycle regulation.112 By degrading p53, the mentioned functions can be reversed. HPV-16 E6 inhibits the coactivation of p300/CBP, leading to uncontrolled cell proliferation.113 E6 impairs cell cycle checkpoints by downregulating the expression of CDKs and cyclin inhibitor kinases such as p21 and p27, thus causing uncontrolled proliferation.114 E6 inhibits p21 and p27 probably by degrading p53 and upregulating c-Myc, which needs to be further studied.115 E6 activates the PI3K/Akt/mammalian target of rapamycin (mTOR) pathway. The mTOR complex (mTORC) 2 phosphorylates Akt, which induces the activation of mTORC1. mTORC1 promotes viral replication and cell proliferation, and inhibits autophagy.116,117 Research has verified that inhibiting the PI3K/Akt/mTOR pathway can further inhibit cell proliferation.118,119 E6 inhibits the expression and nuclear import of STAT1 to promote the amplification of the viral genome, thereby maintaining the low-level replication of the viral genome.120,121,122,123 E6 further enhances STAT3 signaling by inducing IL-6 expression through the Ras-related C3 botulinum toxin substrate 1/NF-κB pathway. STAT3 can in turn promote the expression of E6.124,125 Activated STAT3 is able to promote the transcription of genes involved in cell proliferation.126 STAT3 colocalizes with AP-1 components and interacts with AP-1, which further influences the cell cycle and proliferation of cells.85 Nevertheless, recent research has found that there’s no obvious difference in the proliferation rates of cells between STAT3 KO CC cells and their parental counterparts. Although ruxolitinib effectively inhibits the phosphorylation of STAT3, it shows no effect in the proliferation or growth of cells,127 which needs further research. Mastermind like transcriptional coactivator 1 (MAML1), a coactivator of NOTCH, interacts with E6 and stabilizes E6. A synergistic effect between E6AP and MAML1 fosters cell proliferation.128 Research has confirmed that E6 increases the expression of NOTCH1 and elevates NOTCH signaling to promote cell proliferation.129 However, the distinct mechanism still needs to be studied. Na+ /H+ exchanger regulatory factor-1 (NHERF1) can be degraded by E6 in an E6AP-dependent proteasome manner. The degradation of NHERF1 activates the canonical Wnt/β-catenin pathway.130 E6 induces the expression of MZF1 and the subsequent transcription of NKX2-1 and FOXM1. FOXM1 enhances the transcription and translocation of β-catenin.131 Besides, HPV-18 E6 increases the transcription of β-catenin and T-cell factor 4, which further activates the Wnt/β-catenin pathway and promotes the expression of targeted genes such as c-Myc and cyclin D1.132 The activated Wnt/β-catenin pathway cooperatively works with E6 to enhance cell proliferation.133 Notably, extracellular vesicular Wnt7b, upregulated by E6 in HPV-16 and HPV-18 positive cells, can activate β-catenin signaling to promote angiogenesis and cancer invasion.134 Via increasing SNAT1, a glutamine transporter, E6 further elevates intracellular levels of glutamine and glutamate, which provides abundant energy to support uncontrolled cell proliferation.135 RBM15, which determines N6 methyladenine (m6A) modification, is upregulated and accumulated in CC cells through inhibition of E6-mediated degradation. RBM15 promotes the proliferation and growth of CC cells through m6A modification. Furthermore, RBM15 increases the expression of c-Myc, which promotes cell proliferation and oncogenesis.136 Upregulated c-Myc suppresses miR-146a-5p promoter to inhibit the expression of miR-146a-5p. Meanwhile, KDM2B is upregulated by E6, which in turn elevates the expression of E6. Decreased miR-146a-5p and increased KDM2B work synergistically to promote cell proliferation and the progression of cell cycle.137 Moreover, p53 degradation contributes to the downregulation of miR-22 and miR-34a, which subsequently promotes cell proliferation and migration.138
The presence of PBM region is a unique feature of E6-induced oncogenesis. E6 regulates tumor invasion via binding to proteins containing the PDZ motif.106 Research has confirmed that PBM of HPV-16 contributes to the migration and invasion of cancer cells.139 The Scribble complex, the Crumbs complex, and the Par complex, all of which possess a PDZ motif, play pivotal roles in maintaining cellular polarity.140 Membrane-associated guanylate kinase 1 (MAGI1) can serve as a tumor suppressor in the progression of cancer. E6 targets and degrades MAGI1, disrupting cell adhesion and thereby promoting the invasion and metastasis of cancer cells.141,142 Once binding with E6, the mentioned complexes fail to maintain contact between cells and cellular polarity. Loss of polarity may lead to abnormalities in signal transportation, cell transformation, the occurrence of epithelial to mesenchymal transition (EMT), and ultimately the progression and invasion of cancer.106 NHERF1 is downregulated by HPV-16 E6, leading to the upregulation of ACTN4 and F-actin. Actin cytoskeleton assembly and cancer cell migration are thus promoted.139 EMT is well recognized to be of vital importance in cancer metastasis. E6 has been found to upregulate EMT levels and stemness of HPV-positive CC cells. As prominent markers of EMT, the mRNA expressions of N-cadherin and vimentin are significantly elevated in C33A cells. The overexpression of E6 can increase the sphere-forming activity of C33A cells and mRNA expression of OCT4, Nanog, SOX4, and other stemness-related transcription factors.143 Consequently, E6 promotes the invasion and migration of cancer cells, resulting in malignant phenotypes.
E6 is able to inhibit apoptosis in various ways. p53 is well-known to trigger apoptosis in cells with DNA damage and interacts with pro-apoptotic genes such as Puma, Bax, and Noxa. Moreover, p53 interacts with and suppresses anti-apoptotic genes such as BCL-2 and BCL-xL, inducing mitochondrial outer membrane permeabilization, thereby causing mitochondrial apoptosis in a transcription-independent manner.144,145 E6 reverses the function of p53 by binding to and degrading p53. E6 degrades FADD and pro-caspase 8 to inhibit the formation of DISC and the subsequent activation of caspases 8, 3, and 7. Therefore, TRAIL-mediated apoptosis is inhibited independently of p53.146
Through interacting with TYK2, E6 inhibits the phosphorylation of STAT1 and STAT2 and the subsequent transcription of ISGs. The impaired ISGs signaling disrupt the production of chemokines and antigen-presenting molecules such as MHC, leading to inadequate antiviral responses and the initiation of cancers.147 E6-mediated STAT3 upregulation promotes tumor growth by suppressing immune responses that are partially dependent on the NF-κB pathway. Studies have shown that inhibition of STAT3 leads to increased phosphorylation of p65 and NF-κB, as well as enhanced infiltration of T cells, resulting in impaired tumor growth and robust immune responses.148 Owing to the phosphorylation, transcriptional activity, and translocation ability of interferon regulatory factor 3 being inhibited by its binding with E6, the downstream expression of IFNβ is suppressed, which facilitates HPV to evade immune responses.149
Immortalization of cells is a hallmark of HPV-induced cancer progression. E6 activates human telomerase reverse transcriptase (hTERT) via several mechanisms: promoting the expression of c-Myc, recruiting the transcriptional factor Sp1, downregulating NFX1-91, and increasing telomerase activity, which collectively promote the proliferation and immortalization of cells.150,151,152 NFX1-123, highly expressed in CC, interacts with HPV-16 E6 and synergistically increases the activity of hTERT and telomerase.153 E6 increases the expression of AurB, and the interaction between AurB and E6 further enhances the level of hTERT, which results in cell immortalization.154
Degradation of p53 decreases the expression of TP53-induced glycolysis and apoptosis regulators, which leads to increased glucose transporters-1 (GLUT-1), GLUT-2, and glycolysis. Degraded p53 increases the level of hypoxia-inducible factor-1α (HIF-1α), promoting glycolysis, lipid uptake, and synthesis.155 Besides, elevated expression of c-Myc upregulates enzymes involved in glycolysis and fosters glycolysis.156 Meanwhile, E6 activates the PI3K/Akt/mTOR pathway to facilitate glycolysis and lipid synthesis.105,157,158
E7
E7, a small protein of around 100 amino acids, is composed of conserved regions 1/2/3 (CR1/2/3). The sequences of a tiny part of CR1 and almost total CR2 are similar to the adenovirus E1A protein and a large T antigen of SV40.159 Positioned at the C-terminal end, CR3 consists of conserved sequences and encodes a zinc finger domain, which includes two CXXC motifs separated by 29 amino acid residues.160 E7 disturbs DNA damage repair, resulting in the accumulation of DNA lesions and the occurrence of homologous recombination deficiency.161 E7 directly interacts with and regulates RING finger protein 168, which is essential for viral replication. Through the interaction, E7 suppresses DNA double-strand break signaling, promoting the occurrence of genomic instability and the development of cancers.162 Due to its ability to trigger DNA damage and genomic instability, E7 is considered as an oncoprotein that promotes the generation and progression of cancers (Fig. 5c).
E7 is widely known for binding with pRb to disturb the cell cycle and promote uncontrolled cell proliferation. Serving as a cell cycle regulator, pRb can interact with E2F to strictly modulate the transition of cells into the S-phase. In late G1-phase, cyclin E-CDK2 phosphorylates pRb to dissociate from E2F, thereby facilitating the transition of the cell cycle into the S-phase.163,164 Cyclin Y-CDK4 and cyclin D-CDK4/6 phosphorylate pRb to release E2F, enhance the transcriptional activity of E2F, and promote the G1/S-phase alternation.165,166 E7 competitively binds with and degrades pRb, as well as related proteins p107 and p130, resulting in the release and increased activity of E2F. Interestingly, a study has revealed that inhibition of HPV-18 E7 mainly leads to the elevated expression of pRb in HeLa cells, while the expression of pRb differs insignificantly in EC109, EC9706, and Tca83 cell lines, which indicates that E7 tends to interact with different targets in diverse cancer cells.167 The expression of cyclin A and cyclin E is subsequently increased and promotes the transition into the S-phase.168 E7 interacts with p21 and p27 to reverse the inhibition of CDK2, thus facilitating the transition into the S-phase.169,170 Moreover, E7 targets p21 and p27 to inhibit apoptosis.171 E7 can directly bind with E2F6, a member of the E2F family that can repress transcription, causing the extension of the S-phase.172 The DREAM complex, composed of dimerization partner, Rb-like, E2F, and MuvB, functions as a transcriptional repressor. By dissociating MuvB from p130, E7 disrupts the assembly of the DREAM complex and promotes the formation of the Myb-MuvB (MMB) complex, which is necessary for G2/M-phase gene expression. Genes targeted by the DREAM and MMB complex are upregulated, and cell proliferation is thus boosted.173 E7 induces the expression of H3K27-specific demethylases KDM6A and KDM6B and promotes cell growth in HPV-16 positive cells. Moreover, p16, a marker of hr-HPVs, is induced by KDM6A and KDM6B.174 E7 upregulates and activates miR-106a in HPV-16-positive CC cells. miR-106a further regulates the AMPK/mTOR pathway and increases the expression of phosphorylated mTOR via targeting liver kinase B1, which ultimately leads to uncontrolled cell proliferation and inhibited autophagy.175 E7 cooperates with E6 to upregulate miR-18a, which inhibits the STK4-Hippo pathway and facilitates cell proliferation in HPV-positive cells.176 HPV-16 E7 decreases the phosphorylation level of AKT, p70 S6K, and 4E-BP1, accompanied by increased internal ribosomal entry site-dependent translation, which increases the expression of c-Myc and BAX, leading to cell proliferation and cancer progression.177
Cell invasion and metastasis play pivotal roles in the progression of cancer. MMPs can degrade the basal membrane and extracellular matrix to promote cell invasion. Research has confirmed that high expression of MMPs, especially MMP-2, and tissue inhibitors of metalloproteinase-2 (TIMP-2) is closely associated with high aggressiveness and poor prognosis.178 HPV-16 E7 upregulates the activity of MMP-9 and downregulates proteins, including cysteine-rich proteins with kazal motif gene and TIMP-2, which inhibits the activity of MMP-9, thus contributing to cell invasion and tumor metastasis.179 HPV-18 E7 synergistically enhances the expression of MMPs in conjunction with H-rasV12, mediated by the MEK/ERK pathway. Therefore, the upregulated MMPs enable cells to migrate and invade in a manner unrelated to the degradation of pRb.180 HPV-16 E7 and E6 synergistically induce the accumulation of β-catenin in the nucleus, increase the expression of c-Myc and transcription factors, and promote the occurrence of EMT.181 In addition, E7 also induces the activity of fibroblast growth factors, which enhances the invasive ability of cancer cells.182 By interacting with gelsolin, HPV-16 E7 promotes EMT via cytoskeletal actin remodeling. Moreover, the interaction between E7 and gelsolin leads to actin polymerization and accumulation of phosphorylated YAP in the cytoplasm, which may contribute to EMT.183 By increasing c-Myc, HPV-16 E7 and E6 upregulate SNHG12, which can further upregulate the expression of Slug, a transcriptional repressor of E-cadherin, and ultimately contribute to the occurrence of EMT.184 E7 can directly upregulate the expression of Slug, which decreases the expression of E-cadherin and increases vimentin expression simultaneously, contributing to EMT and migration.185 Through activation of the PI3K/AKT1 pathway, HPV-16 E7 overexpresses pirin, which promotes cell migration and EMT in CC and oral cancer cells.186,187 Moreover, research has confirmed that HPV-16 E7 and E6 work cooperatively to decrease the expression of miR-23b-3p, thereby enhancing the occurrence of EMT and promoting the invasion and migration of CC cells.188
E7 upregulates H3K9-specific methyltransferase and SUV39H1, which leads to the formation of heterochromatin at the promoter regions of RIG-1, cGAS, and STING. The activation of the RIG-1 and cGAS-STING pathways is thus suppressed, and the synthesis and release of IFNs are inhibited as well, causing immune evasion from the host immune system.189 HPV-16 E6 and E7 downregulate the expression of TLR9 at mRNA and protein levels,190 inhibiting the generation of IFNs mediated by NF-κB and IRF7.191 HPV-16 and HPV-18 E7 downregulate the expression of miR-142-5p that directly targets PD-L1. PD-L1 is thus enhanced, and immune responses are suppressed.192 HPV-16 E7 negatively regulates the expression of MHC I, inhibits antigen processing, and simultaneously hinders T-cell-mediated immune responses.193,194 HPV-16 E7 impedes the formation and translocation of interferon-stimulated gene factor 3, which induces the transcription of ISGs. Therefore, further synthesis of IFNs is suppressed.184
Telomerase activity, together with the inactivation of pRb and p16, results in the immortalization of cells.195 hr-HPV E7 is capable of degrading PTPN14, contributing to the limited differentiation and increased immortalization of keratinocytes.196 HPV-16 E7 is capable of inhibiting the activity of TNFα-induced NF-κB due to its integrity of pRb binding sites. The inhibition of NF-κB promotes the immortalization of cells, especially in the cervical transformation zone.177,197
E7 is able to alter the metabolism of cells to facilitate the development of cancers as well. HPV-16 and HPV-18 E7 and E6 upregulate IGF2BP2 to increase the expression of c-Myc by identifying m6A-modified sites, which can increase aerobic glycolysis in CC cells.156 Besides, E7 activates the PI3K/Akt/mTOR pathway and upregulates the expression of HIF-1α, causing enhanced glycolysis and lipid synthesis.105
hr-HPV as a target used in the clinic
Since hr-HPV plays an irreplaceable role in the generation and progression of cancers, the detailed applications of hr-HPV in screening, diagnosis, and treatment need to be discussed. Currently, the application of hr-HPV, especially in CC, is widely recognized and has benefited a large number of individuals suffering from persistent hr-HPV infection.
Value of hr-HPV in prevention and screening
Unless detected and treated as early as possible, cells that experience persistent hr-HPV infection can progress from low-grade SIL (LSIL) into high-grade SIL (HSIL), which are prone to oncogenesis. Hence, prevention and timely screening are of vital significance.
The cervical HPV test has 90% sensitivity for detecting precancer. HPV screening combined with cytology test is recommended once every 5 years for individuals aged 30–65 years.198 The possibility of evolving into CC will be less than 0.15% within 5 years if the HPV test result is negative.199 If the HPV test result is negative, re-examination is recommended once every 3 or 5 years. Considering the positive HPV test result together with LSIL for cytology test, annual re-examination is recommended, along with a colposcopy. If the HPV test result is positive and the cytology result is HSIL, colposcopy is required and subsequent treatment, such as ablative treatment, should be considered. Additionally, expedited treatment is highly recommended if the risk of CIN3 is greater than 60% (e.g., HPV-16-positive accompanied by HSIL).199,200 Apart from testing genotypes of HPV, the p16/Ki67 dual-stain test is also applied to further predict the risk of precancer and confirm the necessity of colposcopy.201 HPV DNA combined with DNA methylation detection, which can indicate CIN2/3 and CC, shows promise for the future of CC screening. Research has shown that the result of HPV DNA test together with DNA methylation test from urine is highly consistent with the result from cervical scrapes.202 Due to the easy access to urine samples, this approach may be more beneficial for the promotion of CC screening.
HNSCC tends to be diagnosed late, so new methods to timely detect HNSCC need to be developed. hr-HPV serological tests have been widely studied. Predictive models incorporating hr-HPV serological tests are of great convenience when tumor tissue is unavailable and of predominant sensitivity, specificity, and accuracy. A new predictive model for HNSCC using hr-HPV serostatus, genetic factors, and risk factors has been developed in Europe. The accuracy of prediction for OPSCC is over 90% when hr-HPV serostatus is included.203 A newly clinico-serological predictive model has been developed and has shown its superiority in predicting hr-HPV status and prognosis in OPSCC compared with p16 tissue standard. Moreover, researchers have found that HPV-16-related OPSCC tends to match the model best.204 However, hr-HPV serological tests have not been widely applied clinically. In terms of feasibility, specificity, sensitivity, and accuracy, models incorporating hr-HPV serological tests are promising in the screening of HNSCC.
Despite the fact that HPV DNA test has been widely applied for cervical cancer screening in clinic, limitations still exist. Although the HPV DNA test is considered to be a sensitive screening tool, it also produces substantial false-negative results. There are several possible explanations for these false-negative results: on one hand, it may result from the small lesion size with a few abnormal cells or the presence of genotypes not covered by HPV test; on the other hand, the integration of the virus inside the host cell could significantly hinder HPV detection by reducing the viral load at the surface of lesions.205,206,207 Besides, in LMICs with limited number of well-trained health workers and financial resources, people may have no access to HPV screening, which greatly limits its generalizability. Moreover, variations in populations and testing methods may lead to bias in the sensitivity and specificity of HPV DNA test results.208 Therefore, making these techniques more convenient, cheaper and stable to benefit people around the world may be the future focus.
Prophylactic vaccines have been developed and can prevent the occurrence of CC, HNSCC, anal cancer, penile cancer, vulvar cancer, and vaginal cancer to a certain extent. Data has shown that the effectiveness of vaccination decreases with increasing age,209 and the incidence of cancers remarkedly decreases with the widespread access to HPV vaccines.210 Prophylactic vaccines are based on virus-like particles derived from the L1 protein and can prevent infection through the generation of L1-specific neutralizing antibodies.211 Nowadays, different types of prophylactic vaccines have been applied since 2006 (Table 2). Gardasil, approved by the Food and Drug Administration (FDA) for use in women aged 9–26 in 2006, is the first generation of vaccines targeting HPV-16/18/6/11. Cervarix is a bivalent vaccine targeting HPV-16/18, approved by the FDA for use in women aged 9–25 in 2009. Gardasil-9 is a nonavalent vaccine targeting HPV-16/18/31/33/45/52/58/6/11, approved in 2014.212 Xiamen Innovax Biotech has developed a bivalent vaccine targeting HPV-16/18, Cecolin, which has been approved in China in 2019. It can be applied to women aged 9–45. Based on Cecolin, a new nonavalent vaccine named Cecolin-9 has been developed, and a phase II clinical trial has been conducted to validate its effectiveness (NCT03935204).213 The Serum Institute of India has developed a quadrivalent HPV vaccine called Cervavac targeting HPV-16/18/6/11. The vaccine is suitable for people aged 9–26 regardless of gender. The vaccine has completed its phase III clinical trial and has been approved by India in 2022.214 A bivalent vaccine targeting HPV-16/18 developed by Shanghai Zerun, which has completed a phase III clinical trial (NCT01548118), has been approved by China in 2022. Despite the fact that vaccines based on L2 show lower immunogenicity and lower levels of neutralizing antibodies than L1-based vaccines, L2-based prophylactic vaccines are still promising. Chimeric L1-L2 virus-like particles and vaccines combining L2 with E6 and/or E7 proteins may be wonderful strategies to enhance the prophylactic efficacy of L2-based vaccines.215,216 Prophylactic vaccines have been validated to be effective in clinical trials and nearly have no serious adverse events (SAEs).217,218 Cervarix has been confirmed to be effective and titers of antibodies remain high 9.4 years after the first vaccination. In various studies, Gardasil has been verified to be effective against HPV-16/18-related CIN2/3 or worse lesions and titers of antibodies remain high especially against HPV-16 5 years postvaccination.219 Notably, 5 years after vaccination, compared with Cervarix, Gardasil has lower levels of geometric mean titers (GMTs) against HPV-16 and HPV-18. Meanwhile, in terms of geometric mean number of CD4+ T cells against HPV-16 and HPV-18 induced by vaccination, Cervarix is better than Gardasil.220 Gardasil-9 has been confirmed to be effective against CIN2/3 8 years after vaccination.221 However, almost 20% of recipients have a loss of anti-HPV-18 titers and 15% of recipients have a loss of anti-HPV-45 titers 2 years after vaccination of Gardasil-9. Besides, anti-HPV-31, 33, 52, and 58 titers can decline 2 years after vaccination.212 Since there are difficulties in the completion of three doses of vaccines, the WHO has recommended a single dose to replace multiple doses in order to better promote the spread of vaccines. A meta-analysis has observed that compared with 3 doses of Gardasil, 2 doses of Gardasil have inferior antibody responses against HPV-18 in 18 months. While two doses of Gardasil-9 have higher peak GMTs at 12-month interval and final protection is irrelevant to the time interval of the second Gardasil-9 dose, which makes vaccination more practical in clinic.212 Moreover, research has confirmed that a single dose of quadrivalent vaccine still has high titers of antibodies ten years after vaccination.222 Given the convenience and lower cost, a single dose of vaccine may be a trend. Due to the high price of vaccines, a large number of people from LMICs have no access to vaccines. Therefore, the incidence of hr-HPV-related cancers remains high in LMICs. More investment and lower costs of vaccines are urgently needed to be addressed in order to facilitate the spread of vaccines worldwide.25,223
Therapeutic advances in vaccines
Conventional therapy, such as surgery, radiotherapy, and chemotherapy, has been widely utilized in clinic. Given the essential role of hr-HPV in the initiation and progression of cancers, various vaccines targeting hr-HPV, expected to induce intense immune responses, have been developed to treat cancers. To date, four types of vaccines, including vector-based vaccines, peptide- or protein-based vaccines, whole-cell vaccines, and nucleic acid vaccines, have been developed and tested in clinic (Table 3).
Vector-based vaccines
Usually, bacterial and viral vectors are employed as vector platforms to deliver hr-HPV-specific antigens. Vector-based vaccines possess intense immunogenicity and are capable of inducing innate and adaptive immune responses. However, high immunogenicity may be harmful for patients with low immunity. Furthermore, immune responses targeting vectors may exceed responses targeting hr-HPV-specific antigens, which is an obstacle to the use of vector-based vaccines.
Among bacterial vectors, live-attenuated listeria monocytogenes (Lm) is considered as an effective vector due to its ability to present antigens, induce antigen-presenting cell maturation, and trigger immune responses. ADXS11-001, a vaccine composed of HPV-16 E7 fused with non-hemolytic listeriolysin O, utilizing Lm as a carrier, is a therapeutic vaccine that is expected to induce both CD4+ and CD8+ T-cell immune responses. A phase I clinical trial was conducted in 2009. In 2014, 110 patients with recurrent or refractory CC following chemotherapy and/or radiotherapy were recruited, and a phase II clinical trial was conducted to study the effectiveness of ADXS11-001 with or without cisplatin. The 12-month overall survival (OR) rate was 34.9%, and the 18-month OR rate was 24.8%. Three SAEs related to drugs were reported.224 The results were promising. To explore the effects of ADXS11-001 in patients at high risk of recurrence, a phase III clinical trial was conducted, and the disease-free survival rate was focused. Although the trial was completed, the results of the trial have not been issued yet. GLBL101c, a vaccine targeting HPV-16 E7 based on heat-attenuated recombinant Lactobacillus casei, has been developed to treat patients diagnosed with CIN2. A phase IIB clinical trial was conducted to validate the effectiveness of the vaccine. Unfortunately, no significant differences were observed in the complete response rate between placebo group and GLBL101c group.225 Owing to the unsatisfactory result, a new vaccine, IGMKK16E7, based on GLBL101c, has been developed, which combines HPV-16-derived mutated E7Rb proteins with an anchor protein, cA, derived from Lactococcus lactis.226 A phase I/II clinical trial has shown that the complete response rate in the high-dose group is greater than that in the placebo group only in HPV-16 positive patients, but there’s no difference in the objective response rate between two groups.227
Viral vectors like adenoviruses (Ad), adeno-associated viruses, vaccinia viruses, and alphaviruses have been widely used in the production of vaccines because of the capability of transducing genetic information to the host. Vaccines such as MVA-E2, TA-HPV, TG4001, Vvax001, and PRGN-2009 have been validated to be effective in suppressing tumor growth clinically. Moreover, a phase II clinical trial is now recruiting patients with recurrent or metastatic CC who are pembrolizumab resistant, aiming at comparing the effectiveness of pembrolizumab with that of PRGN-2009 in combination with pembrolizumab. Ad26/35-based vaccines targeting E2, E6, and E7 of HPV-16/18 have been developed and shown great therapeutic efficacy in the TC-1 model.228 An Ad19a/64/5-based vaccine targeting E1, E2, E6, and E7 of HPV-16 shows superb effects in the C3 model as well.229 However, no Ad-based vaccines have been tested in clinical trials till now.
Peptide- or protein-based vaccines
Peptides and proteins can be processed by dendritic cells (DCs) to activate MHC I or MHC II, which ultimately induces CD8+ or CD4+ T-cell responses. Although vaccines based on peptides and proteins are safe and easy to manufacture, they lack high immunogenicity and may need to be combined with adjuvants to enhance immune responses. Short peptide-based vaccines are MHC-specific and need to match specific HLA, while synthetic long peptide and protein vaccines have no limitations of MHC restriction on account of the enrichment of CD8+ and CD4+ T-cell epitopes.
Peptide- or protein-based vaccines tend to be combined with other drugs in the clinic to treat hr-HPV-related cancers, especially hr-HPV-related HNSCC. ISA101 is a long peptide vaccine composed of 9 overlapping E6 peptides and 4 overlapping E7 peptides complemented by Montanide ISA-51.230 In a phase II trial, ISA101 together with nivolumab was tested in patients with uncurable HPV-16-positive cancer. Compared with nivolumab alone, the overall response rate (ORR) is 33%, and the median OR is 17.5 months, both of which are promising.231 A phase II trial is ready for recruitment to evaluate the effectiveness of ISA101 together with cemiplimab in patients with recurrent or metastatic HPV-16 positive CC. PepCan, containing 4 HPV-16 E6 synthetic peptides and adjuvant Candin, was tested in patients with HSIL.232 Due to the effectiveness of PepCan, a phase II trial has been conducted in patients with HSIL. The efficacy of TVGV-1 was verified in patients with HSIL in a phase IIA trial, whose results remain unknown. TVGV-1 together with the adjuvant CpG or GPI-0100 can induce tumor regression and prolong OR time in mouse models.233 A phase II trial is ready to be conducted to assess the efficacy and safety of TVGV-1 compared to those of the adjuvant GPI-0100 in patients with HSIL induced by HPV. PDS0101 has been tested clinically alone or in combination with pembrolizumab to evaluate its efficacy in patients with hr-HPV-associated OPSCC.
Notably, nanoparticles, which can be easily engulfed by DCs and induce immune responses, are recognized as powerful delivery platforms for peptide antigens.234 Polymeric nanoparticles, liposome nanoparticles and virus-like particles can be physically mixed or incorporated with antigens via encapsulation, self-assembly, absorption, and chemical conjugation. A new poly(lactide-co-glycolide)-acid nanoparticle vaccine, encapsulating HPV-16 E744-62 and using ATP as an adjuvant, can inhibit tumor growth in the TC-1 model.235 A nanoparticle peptide vaccine composed of a pentapeptide FF-Amp-FF and its derivatives AmpFE7 (AmpFE749–57 and AmpFE744–57) has demonstrated to be capable of suppressing tumor growth and inducing the proliferation of T cells in the TC-1 model.236 Liposomes composed of 1,2-dioleoyl-3-trimethylammonium propane and HPV-16 E749-57 can induce immune responses and tumor regression.237 An HPV-16 L1-based virus-like particle vaccine containing HPV-16 E6 and E7 epitopes can inhibit tumor growth in the TC-1 model.238 Interestingly, a new vaccine consisting of HPV-16 E744-62 enclosed by bacterial outer membrane vesicles has shown efficacy in suppressing tumor growth and inducing antigen-specific immune responses.239
Whole-cell vaccines
DCs are considered as the most effective antigen-presenting cells in vivo and can provoke strong immune responses. DCs can also serve as adjuvants.240 DCs pulsed with HPV-18 E7 in vitro were subcutaneously injected into a patient with metastatic CC, which could suppress disease progression, although it couldn’t entirely cure the disease.241 A phase I trial has been conducted in patients with hr-HPV-positive or recurrent CC, which has shown that DC-based vaccines are promising in treating CC.242 DCs pulsed with HPV-16 E7 have been intended to be evaluated in patients with recurrent CC in a phase I trial. Unfortunately, the result of the trial is still unknown. DCs pulsed with HPV-16/18 E7 and keyhole limpet hemocyanin have been confirmed that humoral and cellular immunity are enhanced after immunized with the vaccine.243 However, difficulty in manufacturing is a limiting factor in the promotion of DC-based vaccines.
Engineered T-cell therapy can efficiently generate antigen-specific T cells in vitro and then repatriate T cells to patients to maximally induce immune responses. T-cell receptors (TCRs) can identify and target peptides derived from intracellular or extracellular proteins. The weaknesses of TCR therapy include HLA restriction and immune escape due to the deficiency of HLA elements or ingredients of antigen-processing mechanisms.244 Targeting E7 with TCR-T cells contributes to tumor regression in hr-HPV-related cancers.235 A phase II trial, which aims to evaluate the efficacy and safety of TCR gene therapy targeting HPV-16 E7 in patients diagnosed with hr-HPV-related cancers, is now recruiting participants. Coincidently, another phase I/II of TCR gene therapy targeting HPV-16 E7 in patients with CIN, tumors in situ, HPV infection, and vulvar neoplasms is recruiting participants as well.
Moreover, given that whole-tumor cell lysates can include total potential antigens, whole-tumor cell lysate-based vaccines have been developed.245 Heat shock protein 27 and HPV-16 E7 are transduced into TC-1 cells to develop a tumor cell lysate-based vaccine, which has shown potential for suppressing tumor growth and secreting cytokines such as IFN-γ and TNF-α.246
Nucleic acid vaccines
Nucleic acid vaccines directly introduce a DNA plasmid or mRNA sequence encoding antigens, with or without costimulatory molecules. Notably, nucleic acid vaccines induce both cellular and humoral immunity and deliver antigens in a non-MHC-restricted way.223
DNA-based vaccines, dependent on bacterial plasmids encoding antigens, must be delivered into the nucleus to elicit the expression of antigens and immune responses. DNA-based vaccines are featured as stable, safe, and easy to manufacture. The disadvantages of these vaccines are poor immunogenicity and inability to amplify. VGX-3100, composed of E6 and E7 proteins from HPV-16/18, has finished a phase II trial, which has shown that VGX-3100 can efficiently induce viral elimination and histopathological regression, especially in patients with CIN2/3.247 Moreover, high levels of IFN-γ can still be detected even 18 months after the last immunization248 A phase III trial has been conducted to further confirm the efficacy. About 27.6% of patients in the experimental group show complete viral elimination and histopathological regression, while the ratio in the placebo group is only 8.7%. Histologic regression is observed in 3 out of 9 patients with CIN2/3 immunized with high doses of pNGVL4a-Sig/E7(detox)/HSP70, although the result is not satisfactory.249 To better relieve patients from CIN2/3, the efficacy of pNGVL4a-Sig/E7(detox)/HSP70 combined with TA-HPV has been validated in the TC-1 model, which has shown that more robust immune responses are elicited with the combination of the two vaccines.250 Moreover, a phase I trial has been conducted to further confirm the effectiveness and safety of the combined therapy in patients with CIN3. A phase I trial is now recruiting patients with HPV-16-positive ISIL or tumors in situ to validate the efficacy of different doses of pNGVL4aCRTE6E7L2 with or without TA-CIN. After chemoradiation, patients with CC receive the immunization with INO-3112, which results in humoral and cellular responses targeting HPV and the elimination of HPV DNA.251 Besides, the efficacy of INO-3112 combined with durvalumab has been proven in patients with recurrent or metastatic HPV-associated cancers. The ORR is 21%. However, unfortunately, the study is interrupted due to the low ORR in patients with CC.252 GX188E, proven to be effective in treating patients with HPV-16/18-associated cancers, has been combined with pembrolizumab to treat patients suffering from HPV-16 or 18 positive advanced CC. The ORR is 42%, and the combined therapy is relatively safe for patients.253,254
RNA-based vaccines are based on RNA sequences of the antigen. When taken up by antigen-presenting cells and other target cells, vaccines induce expression of antigenic proteins. Owing to the instability and inability to spread intercellularly, RNA-based vaccines are always packaged into vectors, including cationic lipids and polymer materials. HPV RNA-lipoplex (RNA-LPX) can induce robust T-cell responses via intravenous injection and can persistently suppress tumor growth in the TC-1 model.255 Unmodified or nucleoside-modified non-replicating mRNA as well as self-amplifying mRNA vaccines, which encode HPV-16 E7 and herpes simplex virus type 1 glycoprotein D, are encapsulated by lipid nanoparticles. The vaccine has shown that it can suppress tumor growth, provide prolonged antitumor protection, and elicit strong immune responses in both subcutaneous and orthotopic tumor models.256 mHTV, an mRNA vaccine composed of E6 and E7 proteins from HPV-16/18, has displayed the ability to suppress tumor growth, modulate the tumor microenvironment, and offer continuous protection in subcutaneous and orthotopic tumor models in mice, especially in those that receive immunization via subcutaneous injection. Moreover, the vaccine can provoke antigen-specific immune responses in rhesus monkeys.257 The mRNA-LNP vaccine, which encodes HPV-16 E7, can induce robust HPV-specific CD8+ T-cell responses and IFNγ secretion in an hr-HPV-related OPSCC mouse model. In addition, mRNA-LNP vaccine combined with immune checkpoint blockade can better promote tumor regression.258
Significance of hr-HPV in prognosis
According to a meta-analysis, hr-HPV positivity is associated with a lower cancer stage, squamous cell carcinoma histology, and better disease-free survival and OR rate. Nevertheless, it has no connection with the reoccurrence of CC.259 Moreover, compared with HPV-16 positivity, HPV-18 positivity is associated with a poor prognosis, especially for early-stage cancers.260 hr-HPV positivity indicating better prognosis of cancers can be applied to clinic in the future. Research has confirmed that hr-HPV-positive patients have a better OR rate than hr-HPV-negative patients in OPSCC.261 Since hr-HPV contributes to the epigenetic reprogramming of host cells, hr-HPV-related DNA methylation has also been applied to clinic. hr-HPV-related DNA methylation is explored to predict the prognosis of patients diagnosed with CC via gene set enrichment analysis, Cox regression analysis, and Kaplan–Meier survival analysis.262 Utilizing E7 sequences, a marker for circulating hr-HPV DNA, to predict the prognosis of patients diagnosed with CC is a new method identified in a prospective cohort of CC. In this study, researchers have found that remaining circulating hr-HPV DNA after treatment is closely related to poor prognosis of patients suffering from CC and can predict the reoccurrence of CC.263
Owing to the widespread use of hr-HPV screening and prophylactic vaccines, the occurrence of hr-HPV-related cancers, especially CC, has declined significantly. Unlike CC, the detection of hr-HPV in HNSCC depends on the original sites of tumors. hr-HPV has become a potent biomarker in OPSCC, which indicates a favorable prognosis and assists in staging of cancers. Nowadays, detection of HPV can adopt p16 immunohistochemistry, PCR, RNA in situ hybridization, and single-versus multimodality HPV analysis.264 Circulating hr-HPV DNA is correlated with tumor burden and prognosis. Low levels of hr-HPV DNA imply low hr-HPV copy numbers and hr-HPV integration.265 Therefore, circulating hr-HPV DNA may be a new method for detecting hr-HPV-related cancers in the future.
Conclusion and perspective
In this review, we summarize the detailed oncogenic signaling pathways of hr-HPV and its indispensable role in clinic, including screening, diagnosis, prevention, and treatment of cancers. hr-HPV infection results in the occurrence of cancers mainly dependent on early proteins, among which E6 and E7 are recognized as main oncoproteins in the process of oncogenesis. They can disturb the cell cycle, affect cell proliferation, inhibit apoptosis, and alter cellular metabolism, leading to the occurrence and development of cancers.75
Several types of vaccines have been developed and popularized to prevent hr-HPV-related cancers, from which large quantities of people have benefited. Given the popularization of prophylactic vaccines, the incidence of hr-HPV-related cancers has declined dramatically. A majority of therapeutic vaccines targeting E6 and E7 proteins have been developed. Moreover, vaccines tend to be utilized in combination with other therapies such as radiotherapy, chemotherapy, and immunotherapy. To exert the antitumor effects of vaccines to the largest extent, an increasing number of clinical trials have been conducted to validate the safety and efficacy of combined therapy for treating hr-HPV-related cancers.
Apart from vaccines targeting hr-HPVs, other therapies have been applied clinically as well (Fig. 4). Immunotherapy, such as immune checkpoint blockade therapy and adoptive T-cell therapy, aiming to reverse the immunosuppressive environment, have been widely studied. Immune checkpoint blockades, such as nivolumab, pembrolizumab, tremelimumab, and durvalumab, have been tested in clinic alone or in combination with standard therapies in hr-HPV-related cancers to verify their efficacy. Research has proved that inhibition of CD96 can enhance the blockade of PD-1 and boost the function of CD8+ T cells, which provides inspiration for therapy.266 Adoptive cell therapy, including tumor-infiltrating lymphocytes, genetically modified TCRs, and T cells with chimeric antigen receptors, also offers a prospective therapy for hr-HPV-related cancers. Although numerous clinical trials have been conducted to verify the efficacy and safety of these therapies,267 their clinical application remains controversial. In addition to immunotherapy, various drugs have been applied as well. Bevacizumab, which belongs to anti-VEGF antibodies, has been widely applied, particularly to patients diagnosed with recurrent or metastatic cancers. REBACIN has shown its great potential in eliminating HPV infection via suppressing E6 and E7 genes.268 Nocardia rubra cell wall skeleton, recognized as National Category II New Drug in China, can impair T-cell exhaustion and enhance local immune responses in patients undergoing HPV infection or diagnosed with CIN.269 Imiquimod, considered as an agonist of TLR7, can activate DCs and promote the secretion of IFNγ and TNFα.270 A phase II clinical trial has witnessed its efficacy in eliminating HPV infection and treating HSIL, especially in combination with 9-valent HPV vaccines.271 Gene silencing and editing, including meganucleases, transcription activator-like effector nucleases, zinc finger nucleases, and clustered regularly interspaced short palindromic repeat (CRISPR)-associated Cas9 has provided new insight into treating hr-HPV-related cancers. Research has found that shRNA or siRNA targeting E6 and/or E7 dramatically downregulates the expression of E6 and/or E7 and upregulates the expression of p53 and pRb simultaneously in hr-HPV-positive HNSCC cells. CRISPR-Cas9 targeting E7 delivered by nanoparticles can interfere with viral genome constantly and demonstrate its efficacy in eliminating tumor in a CC xenograft mouse model.272 Though CRISPR-Cas9 shows potential in treating hr-HPV-related cancers, it is limited by low efficiency of editing. Enhancing the gene editing efficiency and specificity needs to be considered in the future.273 Physical therapies such as radiotherapy and hyperthermia have been noticed. A clinical trial has been conducted to compare the efficacy of radiotherapy with that of hyperthermia plus radiotherapy. Hyperthermia plus radiotherapy has the better local control efficacy in locally advanced pelvic tumors, especially in CC.274 In addition, hyperthermia downregulates E6 and contributes to the accumulation of p53, triggering the occurrence of p53-mediated apoptosis and G2-phase arrest in HPV-positive cells, which may benefit patients and decrease the normal tissue damage maximally.275 Research has found that the stability of IGFBP1 can be destroyed and the expression of E7 can thus be downregulated by heat treatment, which provides a new strategy to treat hr-HPV-related cancers on the basis of heat treatment.276
Considering that a high proportion of cancers, such as CC and HNSCC, are caused by persistent infection with hr-HPVs and that a large number of people suffer from cancers, deeper research on hr-HPV is of necessity. Despite the fact that various therapeutic vaccines are produced, none of them have been widely used due to the safety and adverse events. Enhancing the potency and safety of vaccines to benefit people suffering from hr-HPV-related cancers may be the focus of future studies.
References
Głowienka-Stodolak, M. et al. Human papillomavirus infections and the role played by cervical and cervico-vaginal microbiota-evidence from next-generation sequencing studies. Cancers 16, 399 (2024).
Haręża, D. A., Wilczyński, J. R. & Paradowska, E. Human papillomaviruses as infectious agents in gynecological cancers. oncogenic properties of viral proteins. Int. J. Mol. Sci. 23, 1818 (2022).
de Villiers, E. M., Fauquet, C., Broker, T. R., Bernard, H. U. & zur Hausen, H. Classification of papillomaviruses. Virology 324, 17–27 (2004).
Abu-Rustum, N. R. et al. NCCN Guidelines® insights: cervical cancer, version 1.2024. J. Natl. Compr. Cancer Netw. 21, 1224–1233 (2023).
Antonioli, M. et al. HPV sensitizes OPSCC cells to cisplatin-induced apoptosis by inhibiting autophagy through E7-mediated degradation of AMBRA1. Autophagy 17, 2842–2855 (2021).
Bozec, A., Culié, D., Poissonnet, G., Demard, F. & Dassonville, O. Current therapeutic strategies in patients with oropharyngeal squamous cell carcinoma: impact of the tumor HPV status. Cancers 13, 5456 (2021).
Meisels, A. et al. Human papillomavirus infection of the cervix: the atypical condyloma. Acta Cytol. 25, 7–16 (1981).
Della Torre, G., Pilotti, S., de Palo, G. & Rilke, F. Viral particles in cervical condylomatous lesions. Tumori J. 64, 549–553 (1978).
Dürst, M., Gissmann, L., Ikenberg, H. & zur Hausen, H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA 80, 3812–3815 (1983).
Dürst, M., Dzarlieva-Petrusevska, R. T., Boukamp, P., Fusenig, N. E. & Gissmann, L. Molecular and cytogenetic analysis of immortalized human primary keratinocytes obtained after transfection with human papillomavirus type 16 DNA. Oncogene 1, 251–256 (1987).
Schwarz, E. et al. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 314, 111–114 (1985).
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Bruni, L. et al. HPV vaccination introduction worldwide and WHO and UNICEF estimates of national HPV immunization coverage 2010-2019. Prev. Med. 144, 106399 (2021).
Abbas, K., Yoo, K. J., Prem, K. & Jit, M. Equity impact of HPV vaccination on lifetime projections of cervical cancer burden among cohorts in 84 countries by global, regional, and income levels, 2010-22: a modelling study. EClinicalMedicine 70, 102524 (2024).
Brisson, M. et al. Impact of HPV vaccination and cervical screening on cervical cancer elimination: a comparative modelling analysis in 78 low-income and lower-middle-income countries. Lancet 395, 575–590 (2020).
Jensen, J. E., Becker, G. L., Jackson, J. B. & Rysavy, M. B. Human papillomavirus and associated cancers: a review. Viruses 16, 680 (2024).
Saraiya, M. et al. US assessment of HPV types in cancers: implications for current and 9-valent HPV vaccines. J. Natl Cancer Inst. 107, djv086 (2015).
Chua, B. W. B., Ma, V. Y., Alcántar-Fernández, J. & Wee, H. L. Is it time to genotype beyond HPV16 and HPV18 for cervical cancer screening? Int. J. Public Health 67, 1604621 (2022).
Tian, R. et al. Gene knock-out chain reaction enables high disruption efficiency of HPV18 E6/E7 genes in cervical cancer cells. Mol. Ther. Oncolytics 24, 171–179 (2022).
Bhattacharjee, R. et al. Mechanistic role of HPV-associated early proteins in cervical cancer: molecular pathways and targeted therapeutic strategies. Crit. Rev. Oncol. Hematol. 174, 103675 (2022).
Wei, F., Georges, D., Man, I., Baussano, I. & Clifford, G. M. Causal attribution of human papillomavirus genotypes to invasive cervical cancer worldwide: a systematic analysis of the global literature. Lancet 404, 435–444 (2024).
Lee, Y. M., Lee, B., Cho, N. H. & Park, J. H. Beyond the microscope: a technological overture for cervical cancer detection. Diagnostics 13, 3079 (2023).
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 74, 12–49 (2024).
de Martel, C., Plummer, M., Vignat, J. & Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 141, 664–670 (2017).
Williamson, A. L. Recent developments in human papillomavirus (HPV) vaccinology. Viruses 15, 1440 (2023).
Garolla, A., Graziani, A., Grande, G., Ortolani, C. & Ferlin, A. HPV-related diseases in male patients: an underestimated conundrum. J. Endocrinol. Investig. 47, 261–274 (2024).
Lechner, M., Liu, J., Masterson, L. & Fenton, T. R. HPV-associated oropharyngeal cancer: epidemiology, molecular biology and clinical management. Nat. Rev. Clin. Oncol. 19, 306–327 (2022).
Amin, M. B. et al. The Eighth Edition AJCC Cancer Staging Manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 67, 93–99 (2017).
Burd, E. M. Human papillomavirus and cervical cancer. Clin. Microbiol. Rev. 16, 1–17 (2003).
Pereira, R., Hitzeroth, I. I. & Rybicki, E. P. Insights into the role and function of L2, the minor capsid protein of papillomaviruses. Arch. Virol. 154, 187–197 (2009).
Chong, T., Apt, D., Gloss, B., Isa, M. & Bernard, H. U. The enhancer of human papillomavirus type 16: binding sites for the ubiquitous transcription factors oct-1, NFA, TEF-2, NF1, and AP-1 participate in epithelial cell-specific transcription. J. Virol. 65, 5933–5943 (1991).
Yu, L., Majerciak, V. & Zheng, Z. M. HPV16 and HPV18 genome structure, expression, and post-transcriptional regulation. Int. J. Mol. Sci. 23, 4943 (2022).
O’Connor, M. & Bernard, H. U. Oct-1 activates the epithelial-specific enhancer of human papillomavirus type 16 via a synergistic interaction with NFI at a conserved composite regulatory element. Virology 207, 77–88 (1995).
Bletsa, G. et al. Genetic variability of the HPV16 early genes and LCR. Present and future perspectives. Expert Rev. Mol. Med. 23, e19 (2021).
Warburton, A., Della Fera, A. N. & McBride, A. A. Dangerous liaisons: long-term replication with an extrachromosomal HPV genome. Viruses 13, 1846 (2021).
Coursey, T. L. & McBride, A. A. Development of keratinocyte cell lines containing extrachromosomal human papillomavirus genomes. Curr. Protoc. 1, e235 (2021).
Della Fera, A. N., Warburton, A., Coursey, T. L., Khurana, S. & McBride, A. A. Persistent human papillomavirus infection. Viruses 13, 321 (2021).
Graham, S. V. The human papillomavirus replication cycle, and its links to cancer progression: a comprehensive review. Clin. Sci. 131, 2201–2221 (2017).
Tian, R. et al. Genome-wide virus-integration analysis reveals a common insertional mechanism of HPV, HBV and EBV. Clin. Transl. Med. 12, e971 (2022).
Akagi, K. et al. Intratumoral heterogeneity and clonal evolution induced by HPV integration. Cancer Discov. 13, 910–927 (2023).
Zhou, L. et al. Long-read sequencing unveils high-resolution HPV integration and its oncogenic progression in cervical cancer. Nat. Commun. 13, 2563 (2022).
Catalán-Castorena, O. et al. The role of HR-HPV integration in the progression of premalignant lesions into different cancer types. Heliyon 10, e34999 (2024).
Hu, Z. et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 47, 158–163 (2015).
McBride, A. A. & Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 13, e1006211 (2017).
Liu, H., Liang, H., Li, D., Wang, M. & Li, Y. Association of cervical dysbacteriosis, HPV oncogene expression, and cervical lesion progression. Microbiol. Spectr. 10, e0015122 (2022).
Johnson, D. E. et al. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 6, 92 (2020).
Liu, M. et al. Long-read sequencing reveals oncogenic mechanism of HPV-human fusion transcripts in cervical cancer. Transl. Res. 253, 80–94 (2023).
McBride, A. A. & White, E. A. HPV integration can drive the formation of virus-host extrachromosomal DNA in tumors. Cancer Discov. 13, 814–816 (2023).
Lim, Y. X., Mierzwa, M. L., Sartor, M. A. & D’Silva, N. J. Clinical, morphologic and molecular heterogeneity of HPV-associated oropharyngeal cancer. Oncogene 42, 2939–2955 (2023).
Tabatabaeian, H., Bai, Y., Huang, R., Chaurasia, A. & Darido, C. Navigating therapeutic strategies: HPV classification in head and neck cancer. Br. J. Cancer. 131, 220–230 (2024).
Tian, R. et al. HPV integration generates a cellular super-enhancer which functions as ecDNA to regulate genome-wide transcription. Nucleic Acids Res. 51, 4237–4251 (2023).
Karimzadeh, M. et al. Human papillomavirus integration transforms chromatin to drive oncogenesis. Genome Biol. 24, 142 (2023).
Schreiberhuber, L. et al. Cervical cancer screening using DNA methylation triage in a real-world population. Nat. Med. 30, 2251–2257 (2024).
Hsu, C. H. et al. The HPV E6 oncoprotein targets histone methyltransferases for modulating specific gene transcription. Oncogene 31, 2335–2349 (2012).
Thomas, M. C. & Chiang, C. M. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol. Cell 17, 251–264 (2005).
Liu, H., Ma, H., Li, Y. & Zhao, H. Advances in epigenetic modifications and cervical cancer research. Biochim. Biophys. Acta Rev. Cancer 1878, 188894 (2023).
Lo Cigno, I., Calati, F., Girone, C., Catozzo, M. & Gariglio, M. High-risk HPV oncoproteins E6 and E7 and their interplay with the innate immune response: uncovering mechanisms of immune evasion and therapeutic prospects. J. Med. Virol. 96, e29685 (2024).
Moody, C. A. Regulation of the innate immune response during the human papillomavirus life cycle. Viruses 14, 1797 (2022).
Fan, J. et al. Multi-omics characterization of silent and productive HPV integration in cervical cancer. Cell Genom. 3, 100211 (2023).
Baedyananda, F., Sasivimolrattana, T., Chaiwongkot, A., Varadarajan, S. & Bhattarakosol, P. Role of HPV16 E1 in cervical carcinogenesis. Front. Cell Infect. Microbiol. 12, 955847 (2022).
Burley, M., Roberts, S. & Parish, J. L. Epigenetic regulation of human papillomavirus transcription in the productive virus life cycle. Semin Immunopathol. 42, 159–171 (2020).
Fradet-Turcotte, A., Moody, C., Laimins, L. A. & Archambault, J. Nuclear export of human papillomavirus type 31 E1 is regulated by Cdk2 phosphorylation and required for viral genome maintenance. J. Virol. 84, 11747–11760 (2010).
Baedyananda, F., Chaiwongkot, A., Varadarajan, S. & Bhattarakosol, P. HPV16 E1 dysregulated cellular genes involved in cell proliferation and host DNA damage: a possible role in cervical carcinogenesis. PLoS ONE 16, e0260841 (2021).
Castro-Muñoz, L. J. et al. The human papillomavirus (HPV) E1 protein regulates the expression of cellular genes involved in immune response. Sci. Rep. 9, 13620 (2019).
Smith, J. A. et al. SMCX and components of the TIP60 complex contribute to E2 regulation of the HPV E6/E7 promoter. Virology 468-470, 311–321 (2014).
Ribeiro, A. L., Caodaglio, A. S. & Sichero, L. Regulation of HPV transcription. Clinics 73, e486s (2018).
Evande, R., Rana, A., Biswas-Fiss, E. E. & Biswas, S. B. Protein-DNA interactions regulate human papillomavirus DNA replication, transcription, and oncogenesis. Int. J. Mol. Sci. 24, 8493 (2023).
Berti, F. C. B., Pereira, A. P. L., Cebinelli, G. C. M., Trugilo, K. P. & Brajão de Oliveira, K. The role of interleukin 10 in human papilloma virus infection and progression to cervical carcinoma. Cytokine Growth Factor Rev. 34, 1–13 (2017).
Prescott, E. L. et al. Human papillomavirus type 1 E1^E4 protein is a potent inhibitor of the serine-arginine (SR) protein kinase SRPK1 and inhibits phosphorylation of host SR proteins and of the viral transcription and replication regulator E2. J. Virol. 88, 12599–12611 (2014).
Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 445, 80–98 (2013).
Sudarshan, S. R., Schlegel, R. & Liu, X. Two conserved amino acids differentiate the biology of high-risk and low-risk HPV E5 proteins. J. Med. Virol. 94, 4565–4575 (2022).
Doorbar, J., Egawa, N., Griffin, H., Kranjec, C. & Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 25, 2–23 (2015).
Disbrow, G. L., Sunitha, I., Baker, C. C., Hanover, J. & Schlegel, R. Codon optimization of the HPV-16 E5 gene enhances protein expression. Virology 311, 105–114 (2003).
Hu, L. & Ceresa, B. P. Characterization of the plasma membrane localization and orientation of HPV16 E5 for cell-cell fusion. Virology 393, 135–143 (2009).
Basukala, O. & Banks, L. The not-so-good, the bad and the ugly: HPV E5, E6 and E7 oncoproteins in the orchestration of carcinogenesis. Viruses 13, 1892 (2021).
Stöppler, M. C., Straight, S. W., Tsao, G., Schlegel, R. & McCance, D. J. The E5 gene of HPV-16 enhances keratinocyte immortalization by full-length DNA. Virology 223, 251–254 (1996).
de Freitas, A. C., de Oliveira, T. H. A., Barros, M. R. Jr. & Venuti, A. hrHPV E5 oncoprotein: immune evasion and related immunotherapies. J. Exp. Clin. Cancer Res. 36, 71 (2017).
Ilahi, N. E. & Bhatti, A. Impact of HPV E5 on viral life cycle via EGFR signaling. Micro. Pathog. 139, 103923 (2020).
DiMaio, D. & Petti, L. M. The E5 proteins. Virology 445, 99–114 (2013).
Zhang, B., Srirangam, A., Potter, D. A. & Roman, A. HPV16 E5 protein disrupts the c-Cbl-EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene 24, 2585–2588 (2005).
Kim, S. H. et al. Human papillomavirus 16 E5 up-regulates the expression of vascular endothelial growth factor through the activation of epidermal growth factor receptor, MEK/ ERK1,2 and PI3K/Akt. Cell Mol. Life Sci. 63, 930–938 (2006).
Basto, D. L. et al. The papillomavirus E5 gene does not affect EGFR transcription and overall survival in cervical cancer. J. Med Virol. 92, 1283–1289 (2020).
Bejjani, F., Evanno, E., Zibara, K., Piechaczyk, M. & Jariel-Encontre, I. The AP-1 transcriptional complex: local switch or remote command? Biochim Biophys. Acta Rev. Cancer 1872, 11–23 (2019).
Mahata, S. et al. Berberine modulates AP-1 activity to suppress HPV transcription and downstream signaling to induce growth arrest and apoptosis in cervical cancer cells. Mol. Cancer 10, 39 (2011).
Thakur, K. et al. Physical interaction between STAT3 and AP1 in cervical carcinogenesis: Implications in HPV transcription control. Biochim Biophys. Acta Mol. Basis Dis. 1869, 166817 (2023).
Yang, Z. et al. RNA N6-methyladenosine reader IGF2BP3 regulates cell cycle and angiogenesis in colon cancer. J. Exp. Clin. Cancer Res. 39, 203 (2020).
Gutierrez-Xicotencatl, L. et al. Cellular functions of HPV16 E5 oncoprotein during oncogenic transformation. Mol. Cancer Res. 19, 167–179 (2021).
Belleudi, F. et al. HPV16 E5 affects the KGFR/FGFR2b-mediated epithelial growth through alteration of the receptor expression, signaling and endocytic traffic. Oncogene 30, 4963–4976 (2011).
Aranda-Rivera, A. K., Cruz-Gregorio, A., Briones-Herrera, A. & Pedraza-Chaverri, J. Regulation of autophagy by high- and low-risk human papillomaviruses. Rev. Med. Virol. 31, e2169 (2021).
Zamaraev, A. V., Zhivotovsky, B. & Kopeina, G. S. Viral infections: negative regulators of apoptosis and oncogenic factors. Biochemistry 85, 1191–1201 (2020).
Khalil, M. I. et al. HPV upregulates MARCHF8 ubiquitin ligase and inhibits apoptosis by degrading the death receptors in head and neck cancer. PLoS Pathog. 19, e1011171 (2023).
Bertheloot, D., Latz, E. & Franklin, B. S. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol. Immunol. 18, 1106–1121 (2021).
Carneiro, B. A. & El-Deiry, W. S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17, 395–417 (2020).
Kabsch, K. et al. The HPV-16 E5 protein inhibits TRAIL- and FasL-mediated apoptosis in human keratinocyte raft cultures. Intervirology 47, 48–56 (2004).
Oh, J. M. et al. Human papillomavirus type 16 E5 protein inhibits hydrogen-peroxide-induced apoptosis by stimulating ubiquitin-proteasome-mediated degradation of Bax in human cervical cancer cells. Carcinogenesis 31, 402–410 (2010).
Ashrafi, G. H., Brown, D. R., Fife, K. H. & Campo, M. S. Down-regulation of MHC class I is a property common to papillomavirus E5 proteins. Virus Res. 120, 208–211 (2006).
Jain, M. et al. Epidemiology, molecular pathogenesis, immuno-pathogenesis, immune escape mechanisms and vaccine evaluation for HPV-associated carcinogenesis. Pathogens 12, 1380 (2023).
Miura, S. et al. CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: a possible mechanism for immune evasion by HPV. J. Virol. 84, 11614–11623 (2010).
Scarth, J. A., Patterson, M. R., Morgan, E. L. & Macdonald, A. The human papillomavirus oncoproteins: a review of the host pathways targeted on the road to transformation. J. Gen. Virol. 102, 001540 (2021).
Miyauchi, S. et al. Human papillomavirus E5 suppresses immunity via inhibition of the immunoproteasome and STING pathway. Cell Rep. 42, 112508 (2023).
Kivi, N., Greco, D., Auvinen, P. & Auvinen, E. Genes involved in cell adhesion, cell motility and mitogenic signaling are altered due to HPV 16 E5 protein expression. Oncogene 27, 2532–2541 (2008).
Liao, S. et al. Human papillomavirus 16/18 E5 promotes cervical cancer cell proliferation, migration and invasion in vitro and accelerates tumor growth in vivo. Oncol. Rep. 29, 95–102 (2013).
Barillari, G., Monini, P., Sgadari, C. & Ensoli, B. The impact of human papilloma viruses, matrix metallo-proteinases and HIV protease inhibitors on the onset and progression of uterine cervix epithelial tumors: a review of preclinical and clinical studies. Int. J. Mol. Sci. 19, 1418 (2018).
Ranieri, D., Belleudi, F., Magenta, A. & Torrisi, M. R. HPV16 E5 expression induces switching from FGFR2b to FGFR2c and epithelial-mesenchymal transition. Int. J. Cancer 137, 61–72 (2015).
Sitarz, K., Czamara, K., Szostek, S. & Kaczor, A. The impact of HPV infection on human glycogen and lipid metabolism—a review. Biochim Biophys. Acta Rev. Cancer 1877, 188646 (2022).
Ganti, K. et al. The human papillomavirus E6 PDZ binding motif: from life cycle to malignancy. Viruses 7, 3530–3551 (2015).
Conrady, M. C. et al. Structure of high-risk papillomavirus 31 E6 oncogenic protein and characterization of E6/E6AP/p53 complex formation. J. Virol. 95, 10–1128 (2020).
Martinez-Zapien, D. et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 529, 541–545 (2016).
Duensing, S. et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc. Natl. Acad. Sci. USA 97, 10002–10007 (2000).
Díaz, L. et al. The phytochemical α-mangostin inhibits cervical cancer cell proliferation and tumor growth by downregulating E6/E7-HPV oncogenes and KCNH1 gene expression. Int. J. Mol. Sci. 24, 3055 (2023).
Zhao, X. et al. Curcumin suppressed the proliferation and apoptosis of HPV-positive cervical cancer cells by directly targeting the E6 protein. Phytother Res. 38, 4967–4981 (2023).
Fischer, M., Quaas, M., Steiner, L. & Engeland, K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 44, 164–174 (2016).
Patel, D., Huang, S. M., Baglia, L. A. & McCance, D. J. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 18, 5061–5072 (1999).
Tavakolian, S., Goudarzi, H. & Faghihloo, E. Cyclin-dependent kinases and CDK inhibitors in virus-associated cancers. Infect. Agent Cancer 15, 27 (2020).
Bretones, G., Delgado, M. D. & León, J. Myc and cell cycle control. Biochim Biophys. Acta 1849, 506–516 (2015).
Bossler, F., Hoppe-Seyler, K. & Hoppe-Seyler, F. PI3K/AKT/mTOR signaling regulates the virus/host cell crosstalk in HPV-positive cervical cancer cells. Int. J. Mol. Sci. 20, 2188 (2019).
Zhang, L., Wu, J., Ling, M. T., Zhao, L. & Zhao, K. N. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol. Cancer 14, 87 (2015).
Zheng, Y. et al. Effects of miR-202-5p silencing PIK3CA gene expression on proliferation, invasion, and epithelial-mesenchymal transition of cervical cancer SiHa cells through inhibiting PI3K/Akt/mTOR signaling pathway activation. Mol. Cell Biochem. 476, 4031–4044 (2021).
Jin, Y., Li, Y., Wang, X. & Yang, Y. Secretory leukocyte protease inhibitor suppresses HPV E6-expressing HNSCC progression by mediating NF-κB and Akt pathways. Cancer Cell Int. 19, 220 (2019).
Hong, S., Mehta, K. P. & Laimins, L. A. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J. Virol. 85, 9486–9494 (2011).
Wang, Y. et al. Orthogonal ubiquitin transfer reveals human papillomavirus E6 downregulates nuclear transport to disarm interferon-γ dependent apoptosis of cervical cancer cells. FASEB J. 35, e21986 (2021).
Bordignon, V. et al. How human papillomavirus replication and immune evasion strategies take advantage of the host DNA damage repair machinery. Viruses 9, 390 (2017).
Zhang, M. et al. Fra-1 inhibits cell growth and the Warburg effect in cervical cancer cells via STAT1 regulation of the p53 signaling pathway. Front. Cell Dev. Biol. 8, 579629 (2020).
Morgan, E. L. & Macdonald, A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1-NFκB-IL-6 signalling axis. PLoS Pathog. 15, e1007835 (2019).
Shukla, S. et al. Functional regulatory role of STAT3 in HPV16-mediated cervical carcinogenesis. PLoS ONE 8, e67849 (2013).
Johnson, D. E., O’Keefe, R. A. & Grandis, J. R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 15, 234–248 (2018).
Strobel, T. D. et al. Revisiting the role of endogenous STAT3 in HPV-positive cervical cancer cells. J. Med Virol. 95, e29230 (2023).
Skelin, J. et al. MAML1-induced HPV E6 oncoprotein stability is required for cellular proliferation and migration of cervical tumor-derived cells. J. Med. Virol. 95, e28624 (2023).
Weijzen, S., Zlobin, A., Braid, M., Miele, L. & Kast, W. M. HPV16 E6 and E7 oncoproteins regulate Notch-1 expression and cooperate to induce transformation. J. Cell Physiol. 194, 356–362 (2003).
Drews, C. M., Case, S. & Vande Pol, S. B. E6 proteins from high-risk HPV, low-risk HPV, and animal papillomaviruses activate the Wnt/β-catenin pathway through E6AP-dependent degradation of NHERF1. PLoS Pathog. 15, e1007575 (2019).
Bello, J. O. et al. Regulation of the Wnt/β-catenin signaling pathway by human papillomavirus E6 and E7 oncoproteins. Viruses 7, 4734–4755 (2015).
Muñoz-Bello, J. O. et al. HPV-18 E6 oncoprotein and its spliced isoform E6*I regulate the Wnt/β-catenin cell signaling pathway through the TCF-4 transcriptional factor. Int. J. Mol. Sci. 19, 3153 (2018).
Skelin, J., Luk, H. Y., Butorac, D., Boon, S. S. & Tomaić, V. The effects of HPV oncoproteins on host communication networks: therapeutic connotations. J. Med. Virol. 95, e29315 (2023).
Qiu, J. J., Sun, S. G., Tang, X. Y., Lin, Y. Y. & Hua, K. Q. Extracellular vesicular Wnt7b mediates HPV E6-induced cervical cancer angiogenesis by activating the β-catenin signaling pathway. J. Exp. Clin. Cancer Res. 39, 260 (2020).
Ortiz-Pedraza, Y. et al. HPV16 E6 and E7 oncoproteins stimulate the glutamine pathway maintaining cell proliferation in a SNAT1-dependent fashion. Viruses 15, 324 (2023).
Nie, G. et al. HPV E6 promotes cell proliferation of cervical cancer cell by accelerating accumulation of RBM15 dependently of autophagy inhibition. Cell Biol. Int. 47, 1327–1343 (2023).
Peta, E. et al. HPV16 E6 and E7 upregulate the histone lysine demethylase KDM2B through the c-MYC/miR-146a-5p axys. Oncogene 37, 1654–1668 (2018).
Choi, P. W. et al. The dysregulation of microRNAs in the development of cervical pre-cancer-an update. Int. J. Mol. Sci. 23, 7126 (2022).
Wang, Q. et al. HPV16 E6 promotes cervical cancer cell migration and invasion by downregulation of NHERF1. Int. J. Cancer 144, 1619–1632 (2019).
Buckley, C. E. & St Johnston, D. Apical-basal polarity and the control of epithelial form and function. Nat. Rev. Mol. Cell Biol. 23, 559–577 (2022).
Wörthmüller, J. & Rüegg, C. MAGI1, a scaffold protein with tumor suppressive and vascular functions. Cells 10, 1494 (2021).
Glaunsinger, B. A., Lee, S. S., Thomas, M., Banks, L. & Javier, R. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene 19, 5270–5280 (2000).
Lu, Y. et al. HPV16 E6 promotes cell proliferation, migration, and invasion of human cervical cancer cells by elevating both EMT and stemness characteristics. Cell Biol. Int. 46, 599–610 (2022).
Wei, H. et al. Structural insight into the molecular mechanism of p53-mediated mitochondrial apoptosis. Nat. Commun. 12, 2280 (2021).
Wang, H., Guo, M., Wei, H. & Chen, Y. Targeting p53 pathways: mechanisms, structures, and advances in therapy. Signal Transduct. Target Ther. 8, 92 (2023).
Garnett, T. O., Filippova, M. & Duerksen-Hughes, P. J. Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16 E6 impairs TRAIL-mediated apoptosis. Cell Death Differ. 13, 1915–1926 (2006).
Ivashkiv, L. B. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 18, 545–558 (2018).
Rossetti, R. A. M. et al. Local and systemic STAT3 and p65 NF-KappaB expression as progression markers and functional targets for patients with cervical cancer. Front. Oncol. 10, 587132 (2020).
Poirson, J. et al. High-risk mucosal human papillomavirus 16 (HPV16) E6 protein and cutaneous HPV5 and HPV8 E6 proteins employ distinct strategies to interfere with interferon regulatory factor 3-mediated beta interferon expression. J. Virol. 96, e0187521 (2022).
Liu, X. et al. HPV E6 protein interacts physically and functionally with the cellular telomerase complex. Proc. Natl. Acad. Sci. USA 106, 18780–18785 (2009).
Gewin, L., Myers, H., Kiyono, T. & Galloway, D. A. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 18, 2269–2282 (2004).
Wang, A. et al. Overexpression of the telomerase holoenzyme induces EMT and tumorigenesis of HPV-immortalized keratinocytes. J. Med. Virol. 95, e28681 (2023).
Vliet-Gregg, P. A., Robinson, K. L., Levan, J., Matsumoto, L. R. & Katzenellenbogen, R. A. NFX1-123 is highly expressed in cervical cancer and increases growth and telomerase activity in HPV 16E6 expressing cells. Cancer Lett. 449, 106–113 (2019).
Boon, S. S. et al. Interaction between human papillomavirus-encoded E6 protein and AurB induces cell immortalization and proliferation—a potential target of intervention. Cancers 15, 2465 (2023).
Priego-Hernández, V. D. et al. Expression of HIF-1α and genes involved in glucose metabolism is increased in cervical cancer and HPV-16-positive cell lines. Pathogens 12, 33 (2022).
Hu, C. et al. HPV E6/E7 promotes aerobic glycolysis in cervical cancer by regulating IGF2BP2 to stabilize m(6)A-MYC expression. Int. J. Biol. Sci. 18, 507–521 (2022).
Xie, Y. et al. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (review). Mol. Med. Rep. 19, 783–791 (2019).
Cruz-Gregorio, A., Aranda-Rivera, A. K., Ortega-Lozano, A. J., Pedraza-Chaverri, J. & Mendoza-Hoffmann, F. Lipid metabolism and oxidative stress in HPV-related cancers. Free Radic. Biol. Med. 172, 226–236 (2021).
Phelps, W. C., Yee, C. L., Münger, K. & Howley, P. M. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell 53, 539–547 (1988).
McIntyre, M. C., Frattini, M. G., Grossman, S. R. & Laimins, L. A. Human papillomavirus type 18 E7 protein requires intact Cys-X-X-Cys motifs for zinc binding, dimerization, and transformation but not for Rb binding. J. Virol. 67, 3142–3150 (1993).
He, S. et al. Human papillomavirus E7 protein induces homologous recombination defects and PARPi sensitivity. J. Cancer Res. Clin. Oncol. 150, 27 (2024).
Sitz, J. et al. Human papillomavirus E7 oncoprotein targets RNF168 to hijack the host DNA damage response. Proc. Natl. Acad. Sci. USA 116, 19552–19562 (2019).
Ezhevsky, S. A., Ho, A., Becker-Hapak, M., Davis, P. K. & Dowdy, S. F. Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin-dependent kinase complexes in vivo. Mol. Cell Biol. 21, 4773–4784 (2001).
Zhou, L. et al. Post-translational modifications on the retinoblastoma protein. J. Biomed. Sci. 29, 33 (2022).
Topacio, B. R. et al. Cyclin D-Cdk4,6 drives cell-cycle progression via the retinoblastoma protein’s C-terminal helix. Mol. Cell 74, 758–770.e754 (2019).
Chen, L. et al. Cyclin Y binds and activates CDK4 to promote the G1/S phase transition in hepatocellular carcinoma cells via Rb signaling. Biochem. Biophys. Res. Commun. 533, 1162–1169 (2020).
Boon, S. S. et al. Human papillomavirus type 18 oncoproteins exert their oncogenicity in esophageal and tongue squamous cell carcinoma cell lines distinctly. BMC Cancer 19, 1211 (2019).
Cheng, S., Schmidt-Grimminger, D. C., Murant, T., Broker, T. R. & Chow, L. T. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev. 9, 2335–2349 (1995).
Funk, J. O. et al. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 11, 2090–2100 (1997).
Zerfass-Thome, K. et al. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene 13, 2323–2330 (1996).
Yadav, C. et al. Overview of genetic and epigenetic regulation of human papillomavirus and apoptosis in cervical cancer. Apoptosis 28, 683–701 (2023).
McLaughlin-Drubin, M. E., Huh, K. W. & Münger, K. Human papillomavirus type 16 E7 oncoprotein associates with E2F6. J. Virol. 82, 8695–8705 (2008).
James, C. D. et al. Restoring the DREAM complex inhibits the proliferation of high-risk HPV positive human cells. Cancers 13, 489 (2021).
McLaughlin-Drubin, M. E., Crum, C. P. & Münger, K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. USA 108, 2130–2135 (2011).
Cui, X. et al. miR-106a regulates cell proliferation and autophagy by targeting LKB1 in HPV-16-associated cervical cancer. Mol. Cancer Res. 18, 1129–1141 (2020).
Morgan, E. L. et al. MicroRNA-18a targeting of the STK4/MST1 tumour suppressor is necessary for transformation in HPV positive cervical cancer. PLoS Pathog. 16, e1008624 (2020).
Peng, Q. et al. HPV E6/E7: insights into their regulatory role and mechanism in signaling pathways in HPV-associated tumor. Cancer Gene Ther. 31, 9–17 (2024).
Azevedo Martins, J. M., Rabelo-Santos, S. H., do Amaral Westin, M. C. & Zeferino, L. C. Tumoral and stromal expression of MMP-2, MMP-9, MMP-14, TIMP-1, TIMP-2, and VEGF-A in cervical cancer patient survival: a competing risk analysis. BMC Cancer 20, 660 (2020).
Cardeal, L. B. et al. HPV16 oncoproteins induce MMPs/RECK-TIMP-2 imbalance in primary keratinocytes: possible implications in cervical carcinogenesis. PLoS ONE 7, e33585 (2012).
Yoshida, S., Kajitani, N., Satsuka, A., Nakamura, H. & Sakai, H. Ras modifies proliferation and invasiveness of cells expressing human papillomavirus oncoproteins. J. Virol. 82, 8820–8827 (2008).
Chakraborti, S. et al. Induction of epithelial to mesenchymal transition in HPV16 E6/E7 oncogene transfected C33A cell line. Tissue Cell 82, 102041 (2023).
Cheng, Y. M., Chou, C. Y., Hsu, Y. C., Chen, M. J. & Wing, L. Y. The role of human papillomavirus type 16 E6/E7 oncoproteins in cervical epithelial-mesenchymal transition and carcinogenesis. Oncol. Lett. 3, 667–671 (2012).
Matarrese, P., Vona, R., Ascione, B., Paggi, M. G. & Mileo, A. M. Physical interaction between HPV16E7 and the actin-binding protein gelsolin regulates epithelial-mesenchymal transition via HIPPO-YAP axis. Cancers 13, 353 (2021).
Gutiérrez-Hoya, A. & Soto-Cruz, I. Role of the JAK/STAT pathway in cervical cancer: its relationship with HPV E6/E7 oncoproteins. Cells 9, 2297 (2020).
Srivastava, K. et al. ΔNp63γ/SRC/Slug signaling axis promotes epithelial-to-mesenchymal transition in squamous cancers. Clin. Cancer Res. 24, 3917–3927 (2018).
Carrillo-Beltrán, D. et al. Human papillomavirus 16 E7 promotes EGFR/PI3K/AKT1/NRF2 signaling pathway contributing to PIR/NF-κB activation in oral cancer cells. Cancers 12, 1904 (2020).
Carrillo, D. et al. Upregulation of PIR gene expression induced by human papillomavirus E6 and E7 in epithelial oral and cervical cells. Open Biol. 7, 170111 (2017).
Hu, J. et al. The HPV16 E6, E7/miR-23b-3p/ICAT signaling axis promotes proliferation, migration, invasion and EMT of cervical cancer cells. Carcinogenesis 44, 221–231 (2023).
Lo Cigno, I. et al. Human papillomavirus E7 oncoprotein subverts host innate immunity via SUV39H1-mediated epigenetic silencing of immune sensor genes. J. Virol. 94, 10–1128 (2020).
Hasan, U. A. et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J. Immunol. 178, 3186–3197 (2007).
Lo Cigno, I., Calati, F., Albertini, S. & Gariglio, M. Subversion of host innate immunity by human papillomavirus oncoproteins. Pathogens 9, 292 (2020).
Ling, J. et al. Human papillomavirus 16 E6/E7 contributes to immune escape and progression of cervical cancer by regulating miR-142-5p/PD-L1 axis. Arch. Biochem. Biophys. 731, 109449 (2022).
Zhou, F., Chen, J. & Zhao, K. N. Human papillomavirus 16-encoded E7 protein inhibits IFN-γ-mediated MHC class I antigen presentation and CTL-induced lysis by blocking IRF-1 expression in mouse keratinocytes. J. Gen. Virol. 94, 2504–2514 (2013).
Bottley, G. et al. High-risk human papillomavirus E7 expression reduces cell-surface MHC class I molecules and increases susceptibility to natural killer cells. Oncogene 27, 1794–1799 (2008).
Kiyono, T. et al. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 396, 84–88 (1998).
Hatterschide, J. et al. PTPN14 degradation by high-risk human papillomavirus E7 limits keratinocyte differentiation and contributes to HPV-mediated oncogenesis. Proc. Natl. Acad. Sci. USA 116, 7033–7042 (2019).
Vandermark, E. R. et al. Human papillomavirus type 16 E6 and E 7 proteins alter NF-kB in cultured cervical epithelial cells and inhibition of NF-kB promotes cell growth and immortalization. Virology 425, 53–60 (2012).
Curry, S. J. et al. Screening for cervical cancer: US Preventive Services Task Force Recommendation Statement. Jama 320, 674–686 (2018).
Perkins, R. B., Wentzensen, N., Guido, R. S. & Schiffman, M. Cervical cancer screening: a review. JAMA 330, 547–558 (2023).
Egemen, D. et al. Risk estimates supporting the 2019 ASCCP risk-based management consensus guidelines. J. Low. Genit. Trac. Dis. 24, 132–143 (2020).
Wentzensen, N. et al. Clinical evaluation of human papillomavirus screening with p16/Ki-67 dual stain triage in a large organized cervical cancer screening program. JAMA Intern. Med. 179, 881–888 (2019).
van den Helder, R. et al. HPV and DNA methylation testing in urine for cervical intraepithelial neoplasia and cervical cancer detection. Clin. Cancer Res. 28, 2061–2068 (2022).
Budhathoki, S. et al. A risk prediction model for head and neck cancers incorporating lifestyle factors, HPV serology and genetic markers. Int. J. Cancer 152, 2069–2080 (2023).
Ren, J. et al. Multiple imputation and clinico-serological models to predict human papillomavirus status in oropharyngeal carcinoma: an alternative when tissue is unavailable. Int. J. Cancer 146, 2166–2174 (2020).
Naryshkin, S. & Austin, R. M. Limitations of widely used high-risk human papillomavirus laboratory-developed testing in cervical cancer screening. Drug Health. Patient Saf. 4, 167–172 (2012).
Igidbashian, S. et al. Tissue genotyping of 37 in situ and invasive cervical cancer with a concomitant negative HC2 HPV DNA test. J. Low. Genit. Trac. Dis. 18, 87–91 (2014).
Coquillard, G., Palao, B. & Patterson, B. K. Quantification of intracellular HPV E6/E7 mRNA expression increases the specificity and positive predictive value of cervical cancer screening compared to HPV DNA. Gynecol. Oncol. 120, 89–93 (2011).
Sy, F. et al. Accuracy of HPV testing on self-collected and clinician-collected samples for different screening strategies in African settings: a systematic review and meta-analysis. Gynecol. Oncol. 166, 358–368 (2022).
Laprise, J. F. et al. Effectiveness and cost-effectiveness of human papillomavirus vaccination through age 45 years in the United States. Ann. Intern. Med. 172, 22–29 (2020).
Mix, J. M., Van Dyne, E. A., Saraiya, M., Hallowell, B. D. & Thomas, C. C. Assessing impact of HPV vaccination on cervical cancer incidence among women aged 15-29 years in the United States, 1999-2017: an ecologic study. Cancer Epidemiol. Biomark. Prev. 30, 30–37 (2021).
Garbuglia, A. R., Lapa, D., Sias, C., Capobianchi, M. R. & Del Porto, P. The use of both therapeutic and prophylactic vaccines in the therapy of papillomavirus disease. Front. Immunol. 11, 188 (2020).
Harper, D. M. & DeMars, L. R. HPV vaccines—a review of the first decade. Gynecol. Oncol. 146, 196–204 (2017).
Hu, Y. M. et al. Immunogenicity and safety of an Escherichia coli-produced human papillomavirus (types 6/11/16/18/31/33/45/52/58) L1 virus-like-particle vaccine: a phase 2 double-blind, randomized, controlled trial. Sci. Bull. 68, 2448–2455 (2023).
Sharma, H. et al. Immunogenicity and safety of a new quadrivalent HPV vaccine in girls and boys aged 9-14 years versus an established quadrivalent HPV vaccine in women aged 15-26 years in India: a randomised, active-controlled, multicentre, phase 2/3 trial. Lancet Oncol. 24, 1321–1333 (2023).
Schellenbacher, C., Roden, R. & Kirnbauer, R. Chimeric L1-L2 virus-like particles as potential broad-spectrum human papillomavirus vaccines. J. Virol. 83, 10085–10095 (2009).
Yousefi, Z. et al. An update on human papilloma virus vaccines: history, types, protection, and efficacy. Front. Immunol. 12, 805695 (2021).
Saeki, Y. et al. Effectiveness of prophylactic HPV vaccines against cervical abnormalities and HPV infection in Japan: The J-HERS 2021 multicenter study. J. Med. Virol. 96, e29413 (2024).
Arbyn, M. & Xu, L. Efficacy and safety of prophylactic HPV vaccines. A Cochrane review of randomized trials. Expert Rev. Vaccines 17, 1085–1091 (2018).
De Vincenzo, R., Conte, C., Ricci, C., Scambia, G. & Capelli, G. Long-term efficacy and safety of human papillomavirus vaccination. Int. J. Women’s Health 6, 999–1010 (2014).
Godi, A., Bissett, S. L., Miller, E. & Beddows, S. Relationship between humoral immune responses against HPV16, HPV18, HPV31 and HPV45 in 12-15 year old girls receiving Cervarix® or Gardasil® vaccine. PLoS ONE 10, e0140926 (2015).
Zhao, C. et al. Opportunities and challenges for human papillomavirus vaccination in China. Hum. Vaccin Immunother. 20, 2329450 (2024).
Joshi, S. et al. Evaluation of immune response to single dose of quadrivalent HPV vaccine at 10-year post-vaccination. Vaccine 41, 236–245 (2023).
Skolnik, J. M. & Morrow, M. P. Vaccines for HPV-associated diseases. Mol. Asp. Med. 94, 101224 (2023).
Basu, P. et al. A randomized phase 2 study of ADXS11-001 Listeria monocytogenes-Listeriolysin O immunotherapy with or without cisplatin in treatment of advanced cervical cancer. Int. J. Gynecol. Cancer 28, 764–772 (2018).
Ikeda, Y. et al. A placebo-controlled, double-blind randomized (Phase IIB) trial of oral administration with HPV16 E7-expressing Lactobacillus, GLBL101c, for the treatment of cervical intraepithelial neoplasia grade 2 (CIN2). Vaccines 9, 329 (2021).
Komatsu, A., Igimi, S. & Kawana, K. Optimization of human papillomavirus (HPV) type 16 E7-expressing lactobacillus-based vaccine for induction of mucosal E7-specific IFNγ-producing cells. Vaccine 36, 3423–3426 (2018).
Kawana, K. et al. Phase I and II randomized clinical trial of an oral therapeutic vaccine targeting human papillomavirus for treatment of cervical intraepithelial neoplasia 2 and 3. JNCI Cancer Spectr. 7, pkad101 (2023).
Khan, S. et al. Development of a replication-deficient adenoviral vector-based vaccine candidate for the interception of HPV16- and HPV18-induced infections and disease. Int. J. Cancer 141, 393–404 (2017).
Boilesen, D. R. et al. Efficacy and synergy with cisplatin of an adenovirus vectored therapeutic E1E2E6E7 vaccine against HPV genome-positive C3 cancers in mice. Cancer Immunol. Res. 11, 261–275 (2023).
Kenter, G. G. et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. New Engl. J. Med. 361, 1838–1847 (2009).
Massarelli, E. et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol. 5, 67–73 (2019).
Coleman, H. N. et al. Human papillomavirus type 16 viral load is decreased following a therapeutic vaccination. Cancer Immunol. Immunother. 65, 563–573 (2016).
Da Silva, D. M. et al. Therapeutic efficacy of a human papillomavirus type 16 E7 bacterial exotoxin fusion protein adjuvanted with CpG or GPI-0100 in a preclinical mouse model for HPV-associated disease. Vaccine 37, 2915–2924 (2019).
Zhang, J. et al. Peptide-based nanovaccines in the treatment of cervical cancer: a review of recent advances. Int. J. Nanomed. 17, 869–900 (2022).
Zhang, Q. et al. Employing ATP as a new adjuvant promotes the induction of robust antitumor cellular immunity by a PLGA nanoparticle vaccine. ACS Appl. Mater. Interfaces 12, 54399–54414 (2020).
Song, Y. et al. Precisely shaped self-adjuvanting peptide vaccines with enhanced immune responses for HPV-associated cancer therapy. ACS Appl. Mater. Interfaces 13, 49737–49753 (2021).
Chen, W. & Huang, L. Induction of cytotoxic T-lymphocytes and antitumor activity by a liposomal lipopeptide vaccine. Mol. Pharm. 5, 464–471 (2008).
Monroy-García, A. et al. Immunization with an HPV-16 L1-based chimeric virus-like particle containing HPV-16 E6 and E7 epitopes elicits long-lasting prophylactic and therapeutic efficacy in an HPV-16 tumor mice model. Arch. Virol. 159, 291–305 (2014).
Chen, H. et al. Peptide-based therapeutic HPV cancer vaccine synthesized via bacterial outer membrane vesicles. Int. J. Nanomed. 18, 4541–4554 (2023).
Santin, A. D. et al. Therapeutic vaccines for cervical cancer: dendritic cell-based immunotherapy. Curr. Pharm. Des. 11, 3485–3500 (2005).
Santin, A. D., Bellone, S., Gokden, M., Cannon, M. J. & Parham, G. P. Vaccination with HPV-18 E7-pulsed dendritic cells in a patient with metastatic cervical cancer. New Engl. J. Med. 346, 1752–1753 (2002).
Ramanathan, P., Ganeshrajah, S., Raghanvan, R. K., Singh, S. S. & Thangarajan, R. Development and clinical evaluation of dendritic cell vaccines for HPV related cervical cancer-a feasibility study. Asian Pac. J. Cancer Prev. 15, 5909–5916 (2014).
Santin, A. D. et al. Human papillomavirus type 16 and 18 E7-pulsed dendritic cell vaccination of stage IB or IIA cervical cancer patients: a phase I escalating-dose trial. J. Virol. 82, 1968–1979 (2008).
Norberg, S. M. & Hinrichs, C. S. Engineered T cell therapy for viral and non-viral epithelial cancers. Cancer Cell 41, 58–69 (2023).
Tagliamonte, M., Petrizzo, A., Tornesello, M. L., Buonaguro, F. M. & Buonaguro, L. Antigen-specific vaccines for cancer treatment. Hum. Vaccin Immunother. 10, 3332–3346 (2014).
Rezaei, F. et al. Development of novel HPV therapeutic vaccine constructs based on engineered exosomes and tumor cell lysates. Life Sci. 340, 122456 (2024).
Trimble, C. L. et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet 386, 2078–2088 (2015).
Bhuyan, P. K. et al. Durability of response to VGX-3100 treatment of HPV16/18 positive cervical HSIL. Hum. Vaccin Immunother. 17, 1288–1293 (2021).
Trimble, C. L. et al. A phase I trial of a human papillomavirus DNA vaccine for HPV16+ cervical intraepithelial neoplasia 2/3. Clin. Cancer Res. 15, 361–367 (2009).
Sun, Y. Y. et al. Local HPV recombinant vaccinia boost following priming with an HPV DNA vaccine enhances local HPV-specific CD8+ T-cell-mediated tumor control in the genital tract. Clin. Cancer Res. 22, 657–669 (2016).
Hasan, Y. et al. A phase 1 trial assessing the safety and tolerability of a therapeutic DNA vaccination against HPV16 and HPV18 E6/E7 oncogenes after chemoradiation for cervical cancer. Int. J. Radiat. Oncol. Biol. Phys. 107, 487–498 (2020).
Morris, V. K. et al. Phase II trial of MEDI0457 and durvalumab for patients with recurrent/metastatic human papillomavirus-associated cancers. Oncologist 28, 618–623 (2023).
Youn, J. W. et al. Pembrolizumab plus GX-188E therapeutic DNA vaccine in patients with HPV-16-positive or HPV-18-positive advanced cervical cancer: interim results of a single-arm, phase 2 trial. Lancet Oncol. 21, 1653–1660 (2020).
Choi, Y. J. et al. A phase II, prospective, randomized, multicenter, open-label study of GX-188E, an HPV DNA vaccine, in patients with cervical intraepithelial neoplasia 3. Clin. Cancer Res. 26, 1616–1623 (2020).
Grunwitz, C. et al. HPV16 RNA-LPX vaccine mediates complete regression of aggressively growing HPV-positive mouse tumors and establishes protective T cell memory. Oncoimmunology 8, e1629259 (2019).
Ramos da Silva, J. et al. Single immunizations of self-amplifying or non-replicating mRNA-LNP vaccines control HPV-associated tumors in mice. Sci. Transl. Med. 15, eabn3464 (2023).
Lee, S. et al. mRNA-HPV vaccine encoding E6 and E7 improves therapeutic potential for HPV-mediated cancers via subcutaneous immunization. J. Med. Virol. 95, e29309 (2023).
Qiu, K. et al. mRNA-LNP vaccination-based immunotherapy augments CD8(+) T cell responses against HPV-positive oropharyngeal cancer. NPJ Vaccines 8, 144 (2023).
Volesky-Avellaneda, K. D., Laurie, C., Tsyruk-Romano, O., El-Zein, M. & Franco, E. L. Human papillomavirus detectability and cervical cancer prognosis: a systematic review and meta-analysis. Obstet. Gynecol. 142, 1055–1067 (2023).
Lei, J. et al. Human papillomavirus infection determines prognosis in cervical cancer. J. Clin. Oncol. 40, 1522–1528 (2022).
Shi, J. et al. The effect of HPV DNA and p16 status on the prognosis of patients with hypopharyngeal carcinoma: a meta-analysis. BMC Cancer 22, 658 (2022).
Yang, S. et al. HPV-related methylation-based reclassification and risk stratification of cervical cancer. Mol. Oncol. 14, 2124–2141 (2020).
Jeannot, E. et al. Circulating HPV DNA as a marker for early detection of relapse in patients with cervical cancer. Clin. Cancer Res. 27, 5869–5877 (2021).
Ferris, R. L. & Westra, W. Oropharyngeal carcinoma with a special focus on HPV-related squamous cell carcinoma. Annu. Rev. Pathol. 18, 515–535 (2023).
Chera, B. S. et al. Rapid clearance profile of plasma circulating tumor HPV type 16 DNA during chemoradiotherapy correlates with disease control in HPV-associated oropharyngeal cancer. Clin. Cancer Res. 25, 4682–4690 (2019).
Wang, Y. et al. Targeting CD96 overcomes PD-1 blockade resistance by enhancing CD8+ TIL function in cervical cancer. J. Immunother. Cancer 10, (2022).
Shamseddine, A. A., Burman, B., Lee, N. Y., Zamarin, D. & Riaz, N. Tumor immunity and immunotherapy for HPV-related cancers. Cancer Discov. 11, 1896–1912 (2021).
Yang, Y. et al. REBACIN® as a noninvasive clinical intervention for high-risk human papillomavirus persistent infection. Int. J. Cancer 145, 2712–2719 (2019).
Chen, W. et al. Nocardia rubra cell wall skeleton up-regulates T cell subsets and inhibits PD-1/PD-L1 pathway to promote local immune status of patients with high-risk human papillomavirus infection and cervical intraepithelial neoplasia. Front. Immunol. 11, 612547 (2020).
Hamar, B. et al. Imiquimod is effective in reducing cervical intraepithelial neoplasia: a systematic review and meta-analysis. Cancers 16, 1610 (2024).
Sheth, S. S. et al. Randomized phase II trial of imiquimod with or without 9-valent HPV vaccine versus observation in patients with high-grade pre-neoplastic cervical lesions (NCT02864147). Clin. Cancer Res. 30, 1768–1777 (2024).
Jubair, L., Lam, A. K., Fallaha, S. & McMillan, N. A. J. CRISPR/Cas9-loaded stealth liposomes effectively cleared established HPV16-driven tumours in syngeneic mice. PLoS ONE 16, e0223288 (2021).
Ye, J., Zheng, L., He, Y. & Qi, X. Human papillomavirus associated cervical lesion: pathogenesis and therapeutic interventions. MedComm 4, e368 (2023).
van der Zee, J. et al. Comparison of radiotherapy alone with radiotherapy plus hyperthermia in locally advanced pelvic tumours: a prospective, randomised, multicentre trial. Dutch Deep Hyperthermia Group. Lancet 355, 1119–1125 (2000).
Oei, A. L. et al. Hyperthermia selectively targets human papillomavirus in cervical tumors via p53-dependent apoptosis. Cancer Res. 75, 5120–5129 (2015).
Wang, L. et al. m(6)A modification confers thermal vulnerability to HPV E7 oncotranscripts via reverse regulation of its reader protein IGF2BP1 upon heat stress. Cell Rep. 41, 111546 (2022).
Zhao, F. H. et al. Efficacy, safety, and immunogenicity of an Escherichia coli-produced Human Papillomavirus (16 and 18) L1 virus-like-particle vaccine: end-of-study analysis of a phase 3, double-blind, randomised, controlled trial. Lancet Infect. Dis. 22, 1756–1768 (2022).
Li, J. et al. Safety and immunogenicity of a pichia pastoris-expressed bivalent human papillomavirus (types 16 and 18) L1 virus-like particle vaccine in healthy Chinese women aged 9-45 years: a randomized, double-blind, placebo-controlled phase 1 clinical trial. Vaccine 41, 3141–3149 (2023).
Rosales, R. et al. Regression of human papillomavirus intraepithelial lesions is induced by MVA E2 therapeutic vaccine. Hum. Gene Ther. 25, 1035–1049 (2014).
Baldwin, P. J. et al. Vaccinia-expressed human papillomavirus 16 and 18 e6 and e7 as a therapeutic vaccination for vulval and vaginal intraepithelial neoplasia. Clin. Cancer Res. 9, 5205–5213 (2003).
Brun, J. L. et al. Regression of high-grade cervical intraepithelial neoplasia with TG4001 targeted immunotherapy. Am. J. Obstet. Gynecol. 204, 169.e161–168 (2011).
Borcoman, E. et al. Phase Ib/II trial of tipapkinogene sovacivec, a therapeutic human papillomavirus16-vaccine, in combination with avelumab in patients with advanced human papillomavirus16-positive cancers. Eur. J. Cancer 191, 112981 (2023).
Komdeur, F. L. et al. First-in-human phase I clinical trial of an SFV-based RNA replicon cancer vaccine against HPV-induced cancers. Mol. Ther. 29, 611–625 (2021).
Pellom, S. T. et al. Characterization of recombinant gorilla adenovirus HPV therapeutic vaccine PRGN-2009. JCI Insight 6, (2021).
Lauterbach, H. et al. Development and characterization of a novel non-lytic cancer immunotherapy using a recombinant arenavirus vector platform. Front. Oncol. 11, 732166 (2021).
de Jong, A. et al. Enhancement of human papillomavirus (HPV) type 16 E6 and E7-specific T-cell immunity in healthy volunteers through vaccination with TA-CIN, an HPV16 L2E7E6 fusion protein vaccine. Vaccine 20, 3456–3464 (2002).
Smalley Rumfield, C., Pellom, S. T., Morillon Ii, Y. M., Schlom, J. & Jochems, C. Immunomodulation to enhance the efficacy of an HPV therapeutic vaccine. J. Immunother. Cancer 8, (2020).
Karkada, M., Quinton, T., Blackman, R. & Mansour, M. Tumor inhibition by DepoVax-based cancer vaccine is accompanied by reduced regulatory/suppressor cell proliferation and tumor infiltration. ISRN Oncol. 2013, 753427 (2013).
Alvarez, R. D. et al. A pilot study of pNGVL4a-CRT/E7(detox) for the treatment of patients with HPV16+ cervical intraepithelial neoplasia 2/3 (CIN2/3). Gynecol. Oncol. 140, 245–252 (2016).
Aggarwal, C. et al. Safety and efficacy of MEDI0457 plus durvalumab in patients with human papillomavirus-associated recurrent/metastatic head and neck squamous cell carcinoma. Clin. Cancer Res. 29, 560–570 (2023).
Kim, T. J. et al. Clearance of persistent HPV infection and cervical lesion by therapeutic DNA vaccine in CIN3 patients. Nat. Commun. 5, 5317 (2014).
Chandra, J. et al. A phase 1, single centre, open label, escalating dose study to assess the safety, tolerability and immunogenicity of a therapeutic human papillomavirus (HPV) DNA vaccine (AMV002) for HPV-associated head and neck cancer (HNC). Cancer Immunol. Immunother. 70, 743–753 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (J.J.R., 82272777); The Science and Technology Department of Sichuan Province [K.Q., 2024NSFSC1510; J.J.R., 2024YFHZ0335]; Postdoctor Research Fund of West China Hospital, Sichuan University [K.Q., 2024HXBH111]; Sichuan University Interdisciplinary Innovation Fund [K.Q.].
Author information
Authors and Affiliations
Contributions
Jianjun Ren, Yu Zhao, and Ping Cheng offered main direction and significant guidance of this manuscript. Yu Zhang and Ke Qiu drafted the manuscript and illustrated figures and tables. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Qiu, K., Ren, J. et al. Roles of human papillomavirus in cancers: oncogenic mechanisms and clinical use. Sig Transduct Target Ther 10, 44 (2025). https://doi.org/10.1038/s41392-024-02083-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41392-024-02083-w
This article is cited by
-
Association of EGFR and EGF gene polymorphisms with cervical cancer in a case–control study and cross-cancer meta-analysis
Scientific Reports (2026)
-
Unravelling the molecular crosstalk between Epithelial-Mesenchymal transition and human papillomavirus in oral cancer
Molecular Biology Reports (2026)
-
Human papillomavirus (HPV) genotypes extended prevalence in the female population from a city in Northern Chile
BMC Women's Health (2025)
-
Human papillomavirus and vaccine knowledge, willingness, and uptake among university students: a systematic review and meta-analysis
BMC Public Health (2025)
-
Human papilloma virus genotypes associated with non-cervical HPV positive cancer development in UK and Ireland cohorts: a systematic review
BMC Infectious Diseases (2025)
