
Abstract
Cancer remains one of the leading health threats globally, with therapeutic resistance being a long-standing challenge across chemotherapy, radiotherapy, targeted therapy, and immunotherapy. In recent years, the association between epigenetic modification abnormalities and therapeutic resistance in tumors has garnered widespread attention, spurring interest in the development of approaches to target epigenetic factors. In this review, we explore the widespread dysregulation and crosstalk of various types of epigenetic modifications, including DNA methylation, histone modifications, and non-coding RNA changes, which interact through complex regulatory networks in tumors. Clinically, single-targeted therapy based on epigenetic modification usually has its limited effect against cancer. However, the combination of epigenetic drugs with other treatment modalities, such as chemotherapy, targeted therapy, or immunotherapy, shows potential for synergistically enhancing efficacy and reducing drug resistance. Therefore, we evaluate the possibility and potential mechanisms of targeting epigenetic modifications to overcome resistance in cancer therapy, and discuss the challenges and opportunities in moving epigenetic therapy into clinical practice. Moreover, the application of multi-omics technologies will aid in identifying core epigenetic factors from complex epigenetic networks, enabling precision treatment and overcoming therapeutic resistance in tumors. Furthermore, the development of spatial multi-omics technologies, by providing spatial coordinates of cellular and molecular heterogeneity, revolutionizes our understanding of the tumor microenvironment, offering new perspectives for precision therapy. In summary, the combined application of epigenetic therapies and the integration of multi-omics technologies herald a new direction for cancer treatment, holding the potential to achieve more effective personalized treatment strategies.
Similar content being viewed by others
Introduction
Cancer remains one of the leading causes of mortality worldwide, with the therapeutic resistance being a significant impediment to successful therapy.1 Despite advancements in chemotherapy, radiotherapy, immunotherapy, and targeted therapy, among all the possible reasons causing failure of anti-cancer treatments, development of therapeutic resistance accounts for up to 90% of cancer-associated deaths.2 The therapeutic resistance in cancer can be broadly classified into two categories: intrinsic (or de novo) and acquired resistance.3 Intrinsic resistance refers to the primary resistance exhibited by some cancers due to pre-existing genetic alterations or cellular states, which render them unresponsive to certain cytotoxic drugs and drug combinations from the outset. On the other hand, acquired resistance emerges during treatment as a result of an evolutionary process in which cancer cells adapt to survive therapeutic pressures. The acquired resistance can also arise through therapy-induced selection of pre-existing genetic alterations within the original malignancies, in which process epigenetic regulation plays a key role.4
The therapeutic resistance mechanism of cancer is multifaceted, involving genetic mutations, epigenetic alterations, cellular plasticity and so on. The mechanisms by which tumors develop resistance to various treatment modalities share both commonalities and differences. Almost all cancer hallmarks are closely related to tumor therapeutic resistance. Currently, a substantial amount of evidence indicates that there is an abnormal expression and activity of various epigenetic modifiers in tumors, leading to aberrant epigenetic modifications that are highly correlated with the malignant phenotype and therapeutic resistance of tumors.5 Epigenetic modifications, such as DNA methylation, histone modifications, and non-coding RNA regulation, are heritable changes in gene expression that do not involve alterations to the underlying DNA sequence. These modifications play a crucial role in the regulation of oncogenes and tumor suppressor genes expression and have been implicated in the development of cancer and resistance to therapy.6
In this review, we focus on the recent progress and provide an overview of the widespread epigenetic modification abnormalities in cancer. Then, the impact of epigenetic regulators on cancer therapeutic resistance will be discussed, exploring the mechanisms by which aberrant epigenetic regulations contribute to tumor resistance to treatment, and how targeting these regulators may offer a promising strategy to overcome the resistance. We will also highlight the roles of specific epigenetic modifiers (including their writers, erasers and readers), and their potential as therapeutic targets in cancer treatment. Furthermore, we underscore the importance of the interplay between different epigenetic modifications, which is a relatively unexplored area that could hold the key to understanding and overcoming therapy resistance. The future of epigenetic research in cancer therapy is promising but challenging. Among the various epigenetic modifications present in tumors, identifying which one is the core driver of malignant phenotypes is the most critical concern in clinical practice. Understanding the crosstalk between these epigenetic modifications will not only enhance our knowledge of cancer biology but also pave the way for the development of novel, targeted therapies that can effectively overcome resistance mechanisms. Furthermore, by leveraging multi-omics technologies, we can identify the core drivers among numerous epigenetic factors, which will enable a targeted approach to overcoming therapeutic resistance in cancer treatment, thus revolutionizing our ability to combat this complex disease. While the application of single-targeted epigenetic drugs alone in clinical oncology has not yet yielded the anticipated therapeutic outcomes, our review reveals the immense potential of combining epigenetic therapies with other treatment modalities to overcome therapeutic resistance. Rather than replicating several excellent reviews on the epigenetic modifications in relation to cancer, our aim here is to provide a novel perspective on the drivers of resistance in cancer therapy.
Epigenetic regulators in cancer
Epigenetics is fundamentally characterized by a diverse array of covalent modifications to histone proteins and nucleic acids, which collectively govern chromatin architecture and gene expression.7 These epigenetic modifications are reversible and subject to dynamic regulation, being initially established and later removed by specialized chromatin-modifying enzymes termed ‘writers’, ‘erasers’ and ‘readers'.8 Currently, the primary mechanisms of epigenetic regulation involve covalent modifications, including histone modification, DNA methylation, RNA modification, and non-coding RNAs9 (Fig. 1). These epigenetic patterns are intrinsically related to the occurrence, progression and treatment of tumors.10

Evolution of combination of epigenetic modifications and cancer therapy. It delineates the historical milestones in the combination of epigenetic regulation with cancer therapy. The progression of the combination of epigenetics and cancer treatment is depicted from four aspects: the association of different epigenetic mechanisms with cancer, the relevance of epigenetics to cancer treatment resistance, and the application of epigenetic inhibitors in preclinical research and clinical trials. Distinct epigenetic mechanisms are noted by different colors
Histone modifications
Histone modifications are key regulatory mechanisms in epigenetics that modulate chromatin structure and gene expression by adding or removing specific chemical groups on histones.11,12,13 These modifications mainly include acetylation, methylation, phosphorylation, and ubiquitination. Since the first isolation and discovery of histone acetylation in 1964,14 various forms of histone modifications, including such as methylation, phosphorylation, and ubiquitination have been gradually revealed (Fig. 1). In recent years, many novel histone modifications have been discovered, such as citrullination, crotonylation, succinylation, propionylation, butyrylation, 2-hydroxyisobutyrylation, and 2-hydroxybutyrylation.15,16,17,18,19,20,21,22 These histone modifications are essential for preserving the integrity of chromatin architecture, regulating DNA transcription, replication, repair, and recombination, have a close connection with the onset and development of various cancers23,24,25,26 (Fig. 2). In particular, representative histone modifications such as acetylation, methylation, phosphorylation, and ubiquitination have been extensively studied in the field of cancer therapy.27 These modifications are not only related to the development of cancer but also to therapeutic resistance in cancer treatment.28,29,30 Within this treatise, we will discuss the intricate role of histone modifications in oncogenesis and their pivotal impact on the resistance to a spectrum of therapeutic modalities.

Hallmarks of cancer regulated by epigenetics. It illustrates the impact of epigenetic mechanisms on the hallmarks of cancer. The epigenetic elements are presented sequentially in the order of chromosome, DNA, and RNA, with a concise description of their functions and regulatory roles. Furthermore, the figure emphasizes the principal hallmarks influenced by epigenetic modifications, specifically highlighting the dysregulation of tumor suppressors and oncogenes, the promotion of unlimited cell proliferation and resistance to apoptosis, microenvironmental remodeling, and metabolic reprogramming, which are mentioned in the review. DNMT DNA methyltransferase, TET ten-eleven translocation, HDAC histone deacetylase, KDM lysine demethylase, KMT lysine methyltransferase, KAT lysine acetyltransferase, MBD methyl-CpG binding domain
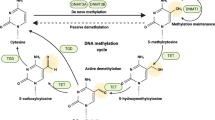
DNA methylation
DNA methylation represents an epigenetic modification, entailing the attachment of a methyl group to specific bases within the DNA molecule.31 This modification can trigger alterations in DNA conformation, DNA stability, chromatin structure, and the interplay between DNA and proteins, thereby exerting a regulatory influence on gene transcription. Specifically, DNA methylation serves as a physical barrier that hinders transcription proteins from binding to genes.32 Methyl-CpG-binding domain (MBD) proteins act as attractors, recruiting a variety of cofactors including histone deacetylases (HDACs) and chromatin reorganization proteins to the methylated DNA loci. Subsequently, compacted heterochromatin is formed, which functions to repress transcriptional activity.33. DNA methylation predominantly occurs at the fifth carbon of cytosine, forming 5-methylcytosine (5mC). In mammals, this modification is predominantly witnessed in CpG islands.34 Moreover, 5mC can be oxidized to yield derivatives such as 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5-fC), and 5-carboxylcytosine (5-caC). These 5mC derivatives have been found to be associated with active gene expression and contribute to cell development and the pathogenesis of diseases.35 Recent investigations have demonstrated that 5mC derivatives are closely related to the tumors development36 (Fig. 2). Apart from the aforementioned forms, there exist other manifestations of DNA methylation, including N6-methyladenine (N6-mA) and 4-methylcytosine (4-mC).37,38 Given its influence on regulation of DNA transcription, DNA methylation is integral to in a diverse array of biological processes including tumorigenesis, aging, and disease occurrence.39,40,41 Here, we will delve into the intricate and multifaceted role that DNA methylation plays in cancer progression and its implications regarding resistance to various therapeutic interventions.
RNA modifications
To date, over 100 distinct chemical modifications have been discovered on RNA within eukaryotic organisms. The study of RNA modifications can be traced back to the 1950s. Pseudouridine (Ψ) was the first type of RNA modification identified in 1950,42 and in 1965, the sequencing of yeast alanine tRNA confirmed 10 types of modifications. In 1975, RNA methylation at m6A was first observed43,44,45 (Fig. 1). Currently, RNA modifications mainly include N1-methyladenosine (m1A), 5-methylcytosine (m5C), N6-methyladenosine (m6A), 7-methylguanosine (m7G), pseudouridine (Ψ), and adenosine-to-inosine (A-to-I) editing.46 These modifications impact RNA stability, translation efficiency, and protein interactions, thereby influencing cell fate. Furthermore, it has been demonstrated that the abnormalities in RNA modifications are closely associated with the occurrence and development of various cancers (Fig. 2). For example, the m6A modification stands as the most prevalent form of methylation occurring in mRNA, and its abnormal changes are intimately connected with tumor proliferation, growth, invasion, and metastasis.47 m6A modification affects gene expression, transcription regulation, and immune evasion through influencing the structure and function of mRNA, thus becoming a new target for tumor treatment.48,49,50 In addition, m7G and m5C can also affect tumor occurrence and development through various mechanisms.51,52 RNA modifications exert significant regulatory influence on the occurrence and development of tumors, including affecting gene expression, cell cycle regulation, cell migration, and invasion capabilities. These revelations offer novel therapeutic targets and strategic avenues for oncological intervention.
Non-coding RNAs
It is widely acknowledged that ncRNAs are implicated in oncogenesis due to somatic genomic instability and the accumulation of mutations.53,54 Non-coding regions constitute over 97% of the entire genome. These regions encompass non-coding RNA genes, which possess the potential for transcription, as well as regulatory elements that are not transcribed. Besides, these regions also incorporate repetitive sequences and other segments that remain unelucidated.55 ncRNAs refer to RNA molecules which are not involved in protein - encoding and constitute an essential part of the transcriptome.56 They execute pivotal regulatory functions within a multitude of cellular processes via post-transcriptional mechanisms, exerting a profound influence on gene transcription and translation, cell proliferation, differentiation, senescence, apoptosis, and both genetic and epigenetic pathways57,58,59,60 (Fig. 2). The diversity of ncRNAs is extensive, including ribosomal RNA (rRNA), transfer RNA (tRNA), microRNA (miRNA), long non-coding RNA (lncRNA), and small interfering RNA (siRNA).61 With the continual advancements and implementation of high-throughput sequencing technologies, additional categories of non-coding RNAs, such as PIWI-interacting RNA (piRNA),62,63 tRNA-derived fragments (tRFs or tsRNA),64 and circular RNA (circRNA)65 have also been identified in recent years. Non-coding RNAs possess the inherent capacity to modulate cellular activities through interactions with DNA/chromosomes, other RNAs, and proteins, thereby forging a highly intricate ncRNA network.66 Virtually all epigenetic processes, including histone modifications, DNA modifications, and chromatin architecture, fall under the regulatory purview of ncRNAs. Consequently, ncRNAs have a vital impact on the interplay of epigenetic modifications. Our empirical study focuses on ncRNAs with regulatory functions in cancer, such as miRNAs, lncRNAs, piRNAs, and circRNAs, rather than the housekeeping ncRNAs customarily engaged in basic cellular functions.
Others
Chromatin remodeling
Chromatin remodeling stands as one of the significant mechanisms for regulating gene expression, exerting its influence on it by altering the configuration and composition of chromatin. Chromatin remodeling complexes (including SWI/SNF, BAF/PBAF, and others) harness the energy derived from ATP hydrolysis to translocate nucleosomes or modify the interaction between histones and DNA, thereby governing the accessibility and expression of genes.67 In the occurrence and development of tumors, chromatin remodeling plays a pivotal role. Studies have shown that mutations or dysregulation of chromatin remodeling factors can lead to abnormal gene expression, thereby promoting tumorigenesis and the subsequent advancement of malignancies.68 For instance, the SWI/SNF complex participates in tumor therapeutic resistance and progression, and mutations within its subunits can impact the responsiveness of tumor cells to treatment.69 In addition, chromatin remodeling is also related to tumor microenvironment composition and immune evasion, affecting the survival and metastasis of cancer cells.70,71 Chromatin remodeling is also involved in the regulation of DNA damage repair pathways in tumor cells, such as the chromatin modifications mediated by poly ADP-ribose polymerase 1 (PARP1) during DNA repair.72 In summary, chromatin remodeling plays a pivotal role in tumor initiation, progression, and therapeutic response. A deeper understanding of its mechanisms not only aids in elucidating the molecular foundations of tumorigenesis but also provides a theoretical basis and potential targets for the development of innovative anti-cancer therapies.
Ribosomal RNA modifications
Ribosomal RNA modification constitutes a significant post-transcriptional modification mechanism within the biological landscape, being ubiquitously present across a diverse array of organisms. These modifications enhance the efficiency of protein translation by ribosomes through altering the local spatial structure of rRNA molecules.73 Three categories of chemical modifications are manifested in rRNA: ribose methylation (Nm), the isomerization of uridine to pseudouridine (Ψ), along with base modifications such as methylation (mN), acetylation (acN), and aminocarboxypropylation (acpN).74,75 These modifications have endured throughout the evolutionary process and are predominantly mediated by specific enzymes including Fibrillarin and small nucleolar RNAs (snoRNAs). rRNA modifications are of paramount importance for the functionality of ribosomes. They also participate in regulating the assembly of ribosomal subunits and translation functions.76 rRNA modifications not only affect the structure and function of ribosomes but may also be related to the occurrence of diseases. For instance, the methylation modification of rRNA engages in tumor growth by regulating ribosomal translation.77 In summary, rRNA modifications are integral to regulating translation efficiency, optimizing protein synthesis, and affecting the growth of organisms as well as the onset of diseases. Research into these modifications is conducive to deepening our comprehension of ribosomal function and its regulatory mechanisms.
Transcription factor binding site modifications
Transcription factor binding site modification refers to the chemical alterations that transpire when transcription factors (TFs) bind to specific sequences on DNA. These modifications may include DNA methylation, acetylation, phosphorylation, and various post-translational modifications of histones.78 Such chemical modifications possess the capacity to alter the interaction between DNA and histones, thereby exerting an impact on the binding affinity, stability, and efficiency of transcription factors in the modulation of gene expression.79 Modifications of transcription factor binding sites are of great significance in regulating gene expression. They are capable of transforming the chromatin structure, either facilitating or impeding the access of transcription factors to their target genes, thus enabling a precise regulation of gene activity. For example, histone acetylation is usually associated with gene activation, while methylation may be related to gene silencing.80,81 In the genesis and progression of tumors, alterations to transcription factor binding sites also exert a significant influence. Abnormal modification patterns may lead to gene expression dysregulation, thereby promoting tumor growth and spread. For example, abnormal methylation of certain transcription factors could cause the inactivation of tumor suppressor genes, while abnormal acetylation in some enhancer regions may promote the expression of oncogenes.80 In addition, modifications of transcription factor binding sites may also affect the process of chromatin remodeling, which is a dynamic change in the structure of chromatin within the cell nucleus, and is crucial for gene expression regulation, DNA replication, DNA repair, and cell division, among other nuclear activities.82 Overall, transcription factor binding site modification constitutes one of the crucial mechanisms for gene expression regulation, and its abnormalities may be linked to the emergence and progression of diverse diseases, including tumors. Consequently, delving into the patterns and regulatory mechanisms of these modifications is of paramount importance for comprehending the gene regulatory network and devising novel treatment strategies.
Abnormal epigenetic landscapes drive cancer therapeutic resistance
Alterations in the patterns of post-translational modifications (PTMs) have been extensively linked to cancer, whether considering the global level across the entire genome or the genetic information and functional status of cells.27,83 Over the past several decades, a multitude of histone modifications have been successively verified to bear a connection with the initiation and progression of cancer. Furthermore, during the last two decades, there has been an extensive and vigorous endeavor in the pharmacological targeting of these pathways for the purpose of intervening in cancer, which resulted in the occurrence of a number of novel cancer therapies84 (Fig. 1). Subsequently, the aberrant expression of histone modification regulators in diverse types of cancers will be expounded upon below (Fig. 3).

Abnormal epigenetic landscapes in various cancers. Aberrant expression of epigenetic regulators is presented across various cancer types. Distinct colors are used to denote different epigenetic pathways, while arrows indicate the direction of expression changes, either up-regulation or down-regulation
Histone post-translational modifications
Histone acetylation and deacetylation
Histone acetylation, a sophisticated epigenetic modification, plays a vital physiological role in the regulation of gene transcription by modulating the chromatin structure. Histone acetylation entails the addition of the acetyl groups (-COCH3) to lysine residues on histones. Acetylation can neutralize the positive charge of lysine residues, thereby diminishing the binding affinity between DNA and histones, leading to a relaxation of chromatin structure, thereby allowing transcriptional regulatory factors to bind and promoting gene expression.85 Histone acetylation is governed by two competing families of enzymes: histone lysine acetyltransferases (KATs), which add acetyl groups, and HDACs, which remove acetyl groups.86 The KAT family is categorized into two main types: type A and type B. Type A KATs are primarily located in the nucleus and can be further subdivided into three major families: the GNAT superfamily, the MYST family, and the CBP/p300 family.87 In contrast, type B KATs are mainly present in the cytoplasm and modify free histones.88 The HDAC family encompasses 18 distinct enzymes, categorized into four primary classes based on their sequence homology to yeast proteins. Class I, resembling the Rpd3-like enzymes, includes HDAC1, HDAC2, HDAC3, and HDAC8. Class II, akin to the Hda1-like enzymes, is bifurcated into two subclasses: Class IIa, which consists of HDAC4, HDAC5, HDAC6, HDAC7, and HDAC9, and Class IIb, comprising HDAC6 and HDAC10. Class III, the Sir2-like enzymes, encompasses SIRT1 through SIRT7. Lastly, Class IV is singular, containing only HDAC11.89
Histone acetylation is a crucial factor in the evolution and advancement of malignancies. The imbalance between HAT and HDAC activities leads to abnormal changes in histone acetylation levels, which may disrupt gene transcription regulation and participate during the appearance and growth of tumors.90 Specifically, in human and mouse tumor samples, the levels of H4K16 acetylation (H4K16ac) and H4K20 trimethylation (H4K20me3) have been significantly reduced, and these changes have been confirmed as biomarkers for tumor progression.91,92 Alongside the variations in histone acetylation levels, the expression of enzymes related to histone acetylation is also altered in cancer. These alterations are not only significant markers of cancer development but also serve as potential biomarkers and therapeutic targets in clinical practice. Specifically, general control of general nucleotide synthesis 5 (GCN5), which is one type of KATs, activation of this was detected in human glioma, colon cancer, breast cancer and lung carcinoma.93,94,95 Conversely, in solid tumors including ovarian, gastric, and esophageal cancer, the p300-CBP-associated factor (pCAF) is commonly diminished.83 Additionally, the aberrant expressions of KAT4, KAT5, MYST1, MYST3, MYST4, KAT2A, KAT2B, and p300 has been noted in colorectal cancer (CRC) as well. In contrast, overexpression of KAT2A, KAT2B, KAT4, and MOF is a characteristic feature of malignant kidney tumors.96,97 In contrast, KAT2B is downregulated in gastric cancer cells and appears to be positively correlated with the CDKN1A tumor suppressor mRNA levels.98 In addition, upregulation of lysine acetyltransferase 7 (KAT7) was observed in multiple breast cancer cell lines, which was found to enhance the PI3K/AKT signaling pathway and confer radioresistance by activating the transcription of PIK3CA99 (Fig. 3).
In addition to histone KATs, HDACs are also frequently dysregulated in tumors. For example, HDAC1 and HDAC2 expression levels are moderately elevated in papillary thyroid carcinoma tissues compared with normal tissues.100 Besides, HDAC1 is reported to be expressed in a significant proportion of cancers, such as gastric carcinoma, prostate cancer, lung cancer, breast cancer, and colon cancer101 (Table 1). However, HDAC2 is overexpressed in hepatocellular carcinoma (HCC), CRC and glioma.102,103 Furthermore, colon and breast cancers are notably characterized by elevated HDAC3 expression levels. In contrast, neuroblastoma cells are distinguished by a significant abundance of HDAC8.6 The activity of class III HDACs, specifically SIRT1, SIRT4, and SIRT7, is found to be heightened in myeloid leukemia, prostate and ovarian carcinoma, as well as non-melanoma skin cancers. Conversely, a reduction in SIRT2 expression has been documented in gliomas, gastric carcinomas, and melanomas104 (Fig. 3). Overall, aberrant histone acetylation landscape is intricately implicated in the pathogenesis of cancer and provide a novel way for cancer treatment.
Histone methylation and demethylation
Histone methylation is instrumental in the establishment and preservation of heterochromatin architecture, thereby mediating the repression of gene transcription. Histone methylation primarily occurs mainly on the arginine (Arg/R) and lysine (Lys/K) residues of histone H3 and H4. It is catalyzed by histone methyltransferases (HMTs), which transfer a methyl group from S-adenosylmethionine (SAM) to histones.105 HMTs primarily consist of histone lysine methyltransferases (HKMTs) and protein/histone arginine methyltransferases (PRMTs).106 The main KMTs include the SET1, SET2, MLL, Suv39, and EZH families. PRMTs can be categorized into three types based on their catalytic activity: Type I PRMTs (PRMT1, PRMT2, PRMT3, PRMT4, PRMT6, and PRMT8), Type II PRMTs (PRMT5 and PRMT9), and Type III PRMTs (PRMT7).107 Correspondingly, there were also histone demethylases have been identified, the enzymes of the histone-lysine demethylase group are divided into two main families: KDM1, which includes two members (LSD1/KDM1A and LSD2/KDM1B), and JmjC-containing HDMTs, which consist of several subfamilies with more than 30 proteins totally.108
Furthermore, a spectrum of studies has shown that alterations of histone methylation within various tumors are strongly linked to the prognosis and development of cancer. For example, H3K27me3 has been accessed as a prognostic indicator in patients with prostate, breast, ovarian, pancreatic and esophageal cancer.109 High levels of H3K27me3 correlate with poor prognosis in esophageal cancers.110 The variations in histone methylation levels, orchestrated by HMTs and demethylases, are frequently altered in tumors. Specifically, EZH2 was detected to be elevated in both primary and metastatic prostate cancers. Moreover, patients presenting with EZH2 overexpression showed a significantly lower survival rate compared to those having low EZH2 expression.111 KMT1C (G9a) is upregulated in breast cancer, colon cancer and gastric cancer,112,113,114 which is indicative of poor prognosis of various tumors. Besides, NSD1, NSD2, and NSD3 increase in glioblastoma bladder and prostate cancer.115,116,117 Histone demethylation enzymes are pivotal in tumor initiation and progression by modulating chromatin structure and gene expression. For instance, LSD1, a member of the histone-lysine demethylase family, exhibits overexpression in a wide range of human cancers, among which are leukemia and solid tumors. Similarly, JmjC domain-containing enzymes, another class of histone demethylases, are implicated in cancer pathogenesis. Overactivation of KDM4B has been noted in prostate cancer, while KDM4C overexpression was initially identified in esophageal, lung, and breast cancers.118,119,120 Additionally, the overexpression of KDM5B has been noted in prostate cancer. Interestingly, a single cancer type may exhibit dysregulation of multiple histone-lysine demethylases simultaneously. For example, in breast cancer, concurrent overactivation of HDMTs such as KDM5C, KDM5B, KDM4A, or KDM4B has been documented121 (Fig. 3). These results emphasize the elaborate relationship between histone methylation and cancer development, highlighting the potential of these enzymes as therapeutic targets for cancer treatment (Table 1).
Histone phosphorylation and dephosphorylation
In contrast to histone methylation, histone phosphorylation facilitates the unwinding of chromatin architecture and promotes gene transcription. In addition, histone phosphorylation is essential for chromosome condensation and segregation during mitosis. This dynamic and reversible modification occurs at serine, threonine, and tyrosine residues and is mediated by protein kinases. This process is distinct from the more stable methylation marks, as phosphorylation is transient, inducible, and often specific to particular pathways.122 Kinases such as PKA, PKC, AMPK, and JAK2 can add phosphate groups to proteins of histone, thereby influencing chromatin architecture and gene transcription. Conversely, histone dephosphorylation is carried out by phosphatases, including PP1, PP2A, PP2B (calcineurin), PP2C, PP4, PP5, PP6, and PP7. The readers of these phosphorylated histone marks primarily consist of 14-3-3 proteins and BRCT domain-containing proteins, which recognize and respond to the phosphorylation status of histones, further modulating cellular signaling and transcriptional regulation.123
The changes in histone phosphorylation in tumors mainly involve the H3S10 site and related enzymes. Elevated levels of H3S10ph, a phosphorylation marker at serine 10 of histone H3, have been identified in a wide range of cancers including invasive breast cancer, esophageal squamous cell carcinoma, gastric carcinoma, spongioblastoma, melanoma, and nasopharyngeal cancer. The presence of increased H3S10ph is not only more frequent in these cancers but also correlates with a worse prognosis, highlighting its potential as a clinical indicator.124,125,126,127 Besides, many H3S10 kinases, including MSK1/2, PIM1, CDK8, and AURORA kinases, are overexpressed in various types of cancer.128
Histone ubiquitination
As mentioned before, methylation, acetylation and phosphorylation modifications add small chemical groups to histones. In contrast, ubiquitination covalently attaches a larger 76-amino acid ubiquitin molecule to histone.129 The process of ubiquitination begins with the activation of ubiquitin by ubiquitin-activating enzymes (E1s), which transfer the activated ubiquitin to ubiquitin-conjugating enzymes (E2s). Finally, ubiquitin is transferred from E2 to the substrate by ubiquitin ligases (E3s).130 The E3 ligase include RNF168, RNF8, RING1A/1B, BMI1 and BRCA1/BARD1. And deubiquitinating enzymes contain USP3 USP44, BRCC36, BAP1 and MYSM1.131
In cancer, the process of histone ubiquitination often goes awry, which may result in alterations in the transcription of tumor suppressor genes and carcinogenic genes, thereby facilitating the proliferation and differentiation of cancer. For instance, the level of H2BK120ub1 is frequently reduced in tissues of breast cancer, lung cancer, and colorectal cancer compared to normal tissues.132,133 The widespread reduction of H2BK120ub1 is observed in approximately 70% of primary breast and colon cancer specimens, and is linked to a worse prognosis.134 Furthermore, the expression of enzymes associated with histone ubiquitination is also dysregulated in tumors. BMI1 exhibits elevated expression levels and facilitates the self-renewal capacity of cancer cells in acute myeloid leukemia (AML) and various solid tumor malignancies. Besides, RNF20 and RNF40 have been observed to be downregulated in seminoma, basal-like breast cancer, and colorectal cancer.135,136,137 And USP22 was found to overexpressed in prostate cancer.138 These investigations reveal that enzymes associated with histone ubiquitination are aberrantly regulated across diverse tumor types and exhibit a strong correlation with tumor proliferation, advancement, and metastatic dissemination. These observations underscore the intricate involvement of histone ubiquitination in oncogenesis and suggest its promising utility for therapeutic interventions and prognostic evaluation (Fig. 3).
Histone lactylation, citrullination and crotonylation
Histone lactylation attenuates the chromatin compaction facilitating the attachment of transcription factors and enhancing gene transcription, thereby modulating a series of physiological activities such as embryogenesis, cell metabolism and signal transduction. Histone lactylation refers to the process by which lactic acid modifies the lysine residues on histones, a post-translational modification known as lysine lactylation (Kla or Klac). Histone lactylation is linked to numerous physiological and pathological processes, including pulmonary fibrosis, tumors, cardiovascular diseases. We will elucidate the role of histone lactylation in tumorigenesis. Recently, alanyl-tRNA synthetase (AARS1) was found to function as a lactate sensor that modulates global lactylation and increases the lactylation of p53, contributing to tumorigenesis.139 Moreover, there is a study indicated that extent of histone lactylation is connected to an unfavorable prognosis for those suffering from clear cell renal cell carcinoma (ccRCC).140 Studies have shown that an increase in histone lactylation promotes liver metastasis of colorectal cancer cells. Furthermore, the level of histone lactylation is also increased in prostate cancer and lung adenocarcinoma.21,141 In ocular melanoma, the increase in histone lactylation leads to enhanced proliferation and migration of ocular melanoma cells by promoting the transcription of the m6A-modified recognition protein YTHDF.141 Histone lactylation not only promotes cancer but also acts as tumor suppressors in some contexts. In non-small cell lung cancer (NSCLC), the increased level of histone lactylation leads to the inhibition of glucose uptake and glycolysis, as well as the reduction of cell proliferation and migration. In uveal melanoma, histone H3K18la modifies nuclear enlargement and induces cell cycle arrest.142 The progression of UM was further inhibited. As for histone lactylation-related enzymes, recent studies have shown that SIRT2, a deacetylase, can inhibit the proliferation and migration of neuroblastoma cells.143 Besides, SIRT3 has the ability to impede the proliferation of HCC cells by modulating the level of Cyclin E2 lactate modification144 (Fig. 3).
Histone citrullination modulates gene expression by diminishing the hydrogen bond complement within chromatin, leading to chromatin decondensation, and it exerts a pivotal influence on cellular division, programmed cell death and tumor progression. Moreover, histone citrullination a post-translational modification mediated by the peptidyl-arginine deiminase (PAD) enzyme family, entails the conversion of arginine residues within histones to citrulline.145,146 This modification involves the genesis of neutrophil extracellular traps (NETs) and is closely related to tumors. In cancer, histone citrullination is linked to the tumor microenvironment, proliferation, metastasis, and drug resistance of cancer cells. PAD2, PAD4 and citrullinated histones are highly expressed in prolactinomas. PAD4 exhibits substantial expression in a range of malignant tissues, however, it is notably absent or present at significantly lower levels in both normal tissues and benign tumors.147 Furthermore, heightened levels of PAD4 have been detected in a multitude of solid malignancies and concomitantly noted to be upregulated in the peripheral blood of individuals afflicted with lung carcinoma148 (Fig. 3).
Significantly, histone crotonylation induces a heightened relaxation of chromatin architecture and exerts a more potent stimulatory effect on gene expression compared to acetylation. Furthermore, the equilibrium between histone crotonylation and acetylation, as well as other acylation modifications, imparts functional implications on gene expression. Thereby, histone crotonylation plays a pivotal role in the developmental trajectory of embryonic stem cells, cellular metabolism, the DNA damage response, and tumor progression. Histone crotonylation refers to the addition of a crotonyl group to a lysine residue. Histone crotonylation was discovered in 2011.149 The function of histone crotonylation has been thoroughly investigated in the last decades and is closely related to the transcription and replication of genes.150,151,152 This modification plays a role in a variety of biological processes, including gene expression regulation, cell signaling and is related to the occurrence and development of a variety of diseases, especially cancer.153,154,155 Histone crotonylation is intricately linked to the etiology, progression, metastasis, and therapeutic response of tumors.156,157 Recently, the p300-regulated lysine crotonylome was characterized by a quantitative proteomics study, which also showed that p300-targeted Kcr substrates are potentially linked to cancer.158 This implies that crotonylation could serve as an oncogenic factor that advances tumor progression. In the EDRN database, 4.5% of tumor biomarkers have been identified as crotonylated, and 32 crotonylated proteins are connected with tumor genes.159 Histone crotonylation is also associated with metabolic regulation in diverse types of tumors (Fig. 3). For instance, levels of histone crotonylation are decreased in liver, gastric, and renal cancers, while they are increased in thyroid, esophageal, pancreatic, and lung cancers. Elevated levels of crotonylation can inhibit the motility and proliferation of liver cancer cells.160 Collectively, the aberrant expression of histone post-translational modifications mentioned above in various cancers holds significant impact on tumor progression. As the research spectrum broadens, the biological functions of these novel modifications and their roles in tumors are gradually being revealed, providing novel perspectives and potential candidates for tumor diagnosis and treatment (Table 1).
DNA methylation
5-mC
5mC represents a chemical modification on DNA and constitutes the most prevalent form of DNA methylation.161 The process of DNA methylation is catalyzed by a family of DNA methyltransferases (DNMTs), with these enzymes transferring a methyl group from SAM to the fifth carbon of a cytosine, thereby giving rise to 5mC.162 DNMT3A and DNMT3B have the ability to form a new methylation pattern to unmodified DNA and are hence known as de novo Dnmt.163 In mammals, DNMT3A and DNMT3B are the enzymes responsible for de novo methylation, whereas DNMT1 primarily maintains the methylation pattern during the process of DNA replication.164 The proteins involved in erasing DNA methylation primarily encompass the TET family proteins (TET1, TET2, and TET3).165 Meanwhile, “readers” refer to proteins capable of recognizing and binding to methylated DNA, such as the MBD protein family, the UHRF (ubiquitin-like with PHD and RING finger domains) protein family, and proteins containing zinc finger domains. These proteins contribute to the regulation of gene expression by binding to methylated DNA.166 In the following text, we will delve into the specific details regarding the functions and roles of these enzymes and proteins involved in DNA methylation, as well as their implications in cancer development.
DNMT1
DNMT1 serves as a crucial enzyme in the maintenance of the genome’s methylation patterns, and its dysregulated expression is intimately linked to the occurrence and development of a diverse range of malignancies. In numerous types of cancer, including breast cancer, lung cancer, colorectal cancer, pancreatic cancer, gastric cancer, and cervical cancer, the expression of DNMT1 is frequently upregulated,167 and this upregulation is associated with the enhancement of tumor cell proliferation, migration, and invasive potential. For example, in breast cancer, DNMT1 promotes the occurrence and development of tumors by suppressing the expression of tumor suppressor genes through methylation.168 In lung cancer, the inhibition of DNMT1 can reduce cell proliferation and increase apoptosis, and its activity is related to the invasiveness and metastatic potential of tumors.169 In CRC, the overexpression of DNMT1 is related to the progression of tumors and may be associated with the invasiveness and metastatic potential of tumors.170 The inhibition or downregulation of DNMT1 exerts a significant anti-cancer effect in certain tumors. Overall, DNMT1 plays a multifaceted and intricate role on the pathogenesis and progression of malignancies, including processes such as cellar proliferation, apoptosis, invasion, and metastasis. Crucially, the dysregulation of DNMT1 expression can confer resistance to various treatment modalities by promoting cancer stem cell properties, such as the aforementioned malignant phenotypes. For instance, DNMT1 downregulates FOXO3a, thereby enhancing the chemoresistance of breast cancer stem cells.168 In conclusion, DNMT1 conspicuously emerges as a pivotal factor in tumorigenesis and tumor progression, with its profound and far-reaching impacts on multiple aspects of cancer biology. Given its capacity to confer treatment resistance as well, further in-depth investigations into DNMT1 are both necessary and justified to explore potential therapeutic strategies for more effectively managing and combating various cancers (Fig. 3).
DNMT3A
DNMT3A, a highly significant enzyme, assumes a crucial role in the regulation of gene expression. It undertakes the responsibility of appending methylation marks onto DNA molecules, thereby exerting a regulatory influence on gene activity. DNMT3A occupies a central position in the origin and progression of a broad spectrum of cancers, with its impact being particularly pronounced in AML. In the context of AML, DNMT3A manifests a notably high mutation frequency, which is closely intertwined with an unfavorable prognosis and resistance to therapeutic interventions.171 Abnormal expression of DNMT3A in tumors is usually characterized by upregulation, which is associated with enhanced abilities of tumor cells to proliferate, migrate, and invade. For example, in AML, mutations in DNMT3A can result in altered de novo DNA methylation patterns, affecting gene expression and promoting the occurrence and development of leukemia.172 In NSCLC, increased expression of DNMT3A is associated with the tumor’s potential for invasion and metastasis, and its inhibition can reduce cell proliferation and increase apoptosis.173 In colorectal cancer, overexpression of DNMT3A is associated with the activation of MEK/ERK signaling pathway, leading to malignant characteristics such as high invasiveness and mobility of the tumor.174 The abnormal activity of DNMT3A can also lead to resistance to cancer treatment.175 Given its role in tumors, DNMT3A has emerged as a prime target for cancer treatment. Treatment strategies centered around DNMT3A, such as the application of DNMT3A inhibitors, may well constitute a novel direction for future cancer therapy.
DNMT3B
Similar to DNMT3A, DNMT3B also plays a significant part in the de novo synthesis of DNA methylation.176 The deviant expression of DNMT3B is related to the progression and resistance phenotype of various malignant tumors (Fig. 3). In a variety of tumors, such as breast cancer, lung cancer, colorectal cancer, pancreatic cancer, gastric cancer, and cervical cancer, the expression of DNMT3B is often upregulated,177 and this upregulation is associated with enhanced tumor cell proliferation, migration, and invasive capacity.178 For example, in AML, mutations in DNMT3B can lead to changes in de novo DNA methylation patterns, affecting gene expression and boosting the occurrence and development of leukemia.179 However, in some cases, the downregulation or loss of function of DNMT3B is also related to the occurrence of tumors. I In a mouse model with a knockout of the Dnmt3b gene, the absence of DNMT3B accelerates the development of lymphoma, indicating that DNMT3B may act as a tumor suppressor gene in normal cells, inhibiting tumor development by stabilizing DNA methylation patterns.180 It is worth highlighting that the abnormal expression of DNMT3B varies among different types of cancer, which implies that both its overexpression and silencing can exert an impact on gene expression. In summary, DNMT3B plays a multifaceted and complex role in the occurrence and development of tumors, and interventions targeting DNMT3B may potentially constitute a novel direction for tumor treatment.
TETs
TET proteins constitute a category of enzymes that play a crucial role in oxidizing 5mC to 5-hydroxymethylcytosine (5hmC) and subsequent oxidation products, thereby indirectly facilitating DNA demethylation. Aberrations in DNA methylation patterns represent a defining characteristic of cancer. The activity and expression of TET enzymes, which is involved in removing this epigenetic mark, has also emerged as an important tumor suppressor mechanism in cancer.181 The functionality and expression of TET enzymes, which play a pivotal role in the removal of these epigenetic modifications, have also been recognized as a crucial tumor-suppressive mechanism in oncogenesis. Diminished expression of TET proteins and decreased levels of 5hmC are prevalent features across number cancer types, encompassing gastric carcinoma, prostate cancer, hepatocellular carcinoma, pulmonary neoplasms, and breast cancer, as well as glioblastoma multiforme and cutaneous melanoma.182,183,184,185 For example, mutations in the TET2 gene are associated with a poor prognosis for AML, and low expression or inactivation of TET2 is common in a variety of tumors, while activation of TET2 can suppress tumor development.186 Besides, under certain circumstances, high expression of TET proteins may increase chemoresistance. For instance, in ovarian cancer, high expression of TET proteins may enhance chemoresistance, and elevated expression levels of TET1 contribute to chemoresistance, possibly by modulating the DNA damage response and repair systems.187 Interestingly, tumors can acquire resistance to DNMT inhibitors by modulating the expression of TET proteins. Deletion of the DNMT1 gene rendered cancer cells susceptible to TET2 upregulation following exposure to DNMT inhibitors. This elevation in TET2 expression coincides with the development of resistance to DNMT inhibitors under conditions of DNMT1 deficiency.188 The aforementioned results suggest that mutations in TET proteins and alterations in their expression can lead to the epigenetic disruption of 5hmC and 5mC patterns. Nevertheless, the exact influence of altered TET activity on the initiation, advancement, and sustenance of these malignancies remains predominantly enigmatic and represents a domain that is presently under vigorous scientific investigation.
MBD proteins
The MBD proteins family stands out as primary contenders for deciphering DNA methylation, as they can recruit chromatin remodelers, HDACs, and methylases to methylated DNA, which participates in gene repression.189 The MBD protein family includes multiple members, such as MBD2 and MBD3, which affect gene expression by regulating DNA methylation and histone modifications to, thereby participating in the formation and progression of tumors.190 MBD2 exhibits significant differences in expression levels and functions across different types of cancer (Fig. 3). In lung and breast cancer, MBD2 is highly expressed and closely related to tumor progression and metastasis.191 In gastric cancer, however, MBD2 expression is reduced, and its downexpression may affect the biological characteristics of the tumor.192 Additionally, MBD3 plays a significant role in the occurrence and development of cancer, with higher expression in liver cancer tissue compared to adjacent normal liver tissue, and its expression level is negatively correlated with patient prognosis, such as overall survival, disease-free survival, and metastasis-free survival.193 This suggests that high expression of MBD3 may be associated with the development of liver cancer and a worse prognosis. Mutations in the MBD4 gene heighten the risk and intricacy of various cancers by affecting DNA repair mechanisms, increasing mutational burden, and promoting the formation of specific mutational spectra.194 Given the important role of the MBD protein family in tumor development, targeting MBD proteins for treatment may represent a new direction for future cancer therapy.166 Through in-depth investigations into the expression patterns and functions of MBD proteins in different cancer types, scientists can devise more precise diagnostic tools and treatment approaches aimed at curbing tumor growth and dissemination by capitalizing on the specific mechanisms of these proteins.195 Furthermore, comprehending how MBD proteins impact DNA repair and mutational spectra can assist us in gaining a better understanding of the genetic underpinnings of tumors and furnishing new strategies for cancer prevention and treatment.
RNA modifications
N6-methyladenosine (m6A)
In RNA molecules, m6A, a crucial epigenetic modification, involves the addition of a methyl group to the nitrogen atom at the sixth position of adenosine. This modification is prevalent among diverse various RNA species, particularly in the 3’-untranslated regions (3’-UTRs) and near mRNA stop codons.196 The core methyltransferase complex, often referred to as the “writer,” is composed of methyltransferase-like 3 (METTL3) and methyltransferase-like 14 (METTL14), with METTL3 acting as the catalytic subunit. This complex is supported by additional proteins, including Wilms tumor 1-associated protein (WTAP), VIR-like m6A methyltransferase associated (VIRMA), RNA-binding motif protein 15 (RBM15), and zinc finger CCCH-type containing 13 (ZC3H13), which contribute to the co-transcriptional deposition of m6A on nascent pre-mRNAs. Demethylation, the reversal process, is carried out by enzymes known as “erasers,” such as Fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5).197 The expression of m6A regulators is frequently dysregulated in cancer, and they exert significant influence on cancer development and progression.
METTL3
Undoubtedly, METTL3 is the most studied type of m6A writer enzyme, which is active in complex with METTL14 and the splicing regulator WTAP.198 Emerging research has demonstrated that METTL3 enhances tumor proliferation and metastasis. METTL3 mRNA and protein exhibit substantial expression levels in acute myeloid leukemia.199 In bladder cancer, METTL3 is significantly overexpressed in patient-derived samples and facilitates the advancement of bladder cancer through the AFF4/NF-κB/MYC signaling cascade.200 In addition, Studies have documented that METTL3 is markedly elevated in HCC and correlates with reduced overall survival in HCC patients.201 Moreover, evidence indicates that elevated METTL3 expression is linked to advanced pathological stages in pancreatic ductal adenocarcinoma (PDAC).202 And its function as a tumor suppressor has also been established in distinct subgroups of breast and colorectal cancers as well as glioblastomas.203,204 In summary, the expression of METTL3 in tumors is complex, and it may play different roles in various types of cancers (Fig. 3), acting as an oncogene to promote tumor development in some cases, and as a tumor suppressor in others. These observations imply that METTL3 represents a promising therapeutic target for oncological interventions, but its exact role and mechanisms in cancer treatment require further research and exploration.
METTL14
The expression of METTL14 patterns closely mirror those of METTL3, and it has been implicated in playing dual roles as both an oncogene and a tumor suppressor.198 More studies have demonstrated the role of METTL14 as a tumor suppressor.205,206,207 Downregulation of METTL14 promotes cancer cell growth, invasion, migration and forecasts poor prognosis in patients with HCC and CRC contributes to progression and metastasis.208,209,210 Besides, the expression of METTL14 was observed to be elevated in CRC tissues, and survival analysis revealed that the METTL14 expression level had a significantly link to the improved prognosis of CRC. Similarly, METTL14 has been proposed a potential indicator for the diagnosis and prognosis of endometrial cancer.49,211 In the tissue samples of gastric cancer (Fig. 3), METTL14 was found to be under-expressed. This low expression of METTL14 played a role as a prognostic factor associated with poor survival in those suffering from gastric cancer.210 Moreover, the knockout of METTL14 is capable of triggering the Wnt and PI3K - Akt signaling, which in turn promotes the growth and invasion of gastric cancer cells.212 On the other hand, METTL14 plays an oncogenic role in stimulating the development and progression of tumors in some cases.213,214,215 The overexpression of METTL14 decreases PERP mRNA and protein levels and enhances tumor cell migration and colony formation.214 In addition, METTL14 mediates the expression of downstream genes CXCR4 and CYP1B1, thus facilitating tumor growth and development.216 The expression of METTL4 in tumors is complex, contributing to tumor development, metastasis, and metabolism, and it may become a potential target for cancer therapy.
FTO
The tumorigenic function of FTO in malignancies r was initially established in research on melanoma, wherein particular FTO variants were linked to an elevated risk of developing melanoma.217 Over the past few years, the involvement of FTO in oncogenesis has also been progressively explored. The upregulation of FTO is connected to enhanced tumor proliferation and progression, which is attributed to its capacity to diminish m6A methylation in oncogenes. This reduction in m6A residues stabilizes oncogenes, thereby promoting the translation of proteins that contribute to tumorigenesis.218 Recent studies on gastric cancer have shown that FTO was also highly expressed in the tumor region and promotes the occurrence of gastric cancer by promoting the proliferation, migration and lymph node metastasis of gastric cancer cells. Decreased expression of FTO protein is related to poor clinical outcomes in gastric cancer patients, suggesting that FTO exerts a regulatory influence on the progression and metastatic dissemination of gastric cancer.219 FTO is elevated expression in NSCLC tissues and cellular models, while m6A content is reduced.220,221 On the other hand, the antitumor effects of FTO have been gradually explored. Expression of FTO is reduced in melanoma.222 And upregulation of FTO overexpression inhibits CSC proliferation and tumorigenic potential in vitro cultures and in vivo xenograft murine models.223,224,225 Besides, in ovarian cancer, FTO is downregulated and promotes tumorigenesis and self-renewal by affecting the m6A modification levels of specific genes.223 The role of FTO may vary in different tumors, and additional investigations are required to delineate its specific mechanisms of action in tumor development.
ALKBH5
The dual function of ALKBH5 in modulating tumor growth is evident in colon cancer, lung cancer, renal cell carcinoma, and osteosarcoma, highlighting the complex nature of each m6A regulator in disease onset and progression.203,226 ALKBH5 exhibits high expression levels in hepatocellular carcinoma (HCC), gastric cancer and breast cancer (Fig. 3). Moreover, the upregulation of ALKBH5 is associated with a worse prognosis in patients with these types of cancer. Additionally, ALKBH5 contributes to poor survival rates in glioblastoma multiforme (GBM) by regulating ADAM19 and the transcription factor FOXM1.227,228 Besides, in bladder cancer, osteosarcoma, and multiple myeloma, the expression of ALKBH5 is also upregulated, primarily promoting the development and progression of tumors by affecting m6A modification.229,230,231 Oppositely, ALKBH5 is downregulated in prostate cancer and is pivotal in regulating the Wnt signaling pathway. It achieves this by decreasing the RNA methylation of Wnt inhibitory factor 1 (WIF-1), thereby inhibiting the tumorigenesis of PC.232,233 Moreover, reduced levels of ALKBH5 are correlated with an unfavorable prognosis in PDAC.234 In summary, ALKBH5 plays a multifaceted role in cancer, with its expression levels and impact on tumorigenesis varying across different cancer types. Its ability to modulate the m6A modification pathway and influence key signaling mechanisms, such as the Wnt pathway, underscores the complexity of its function in both promoting and suppressing tumor growth, depending on the cancer context. The prognostic significance of ALKBH5 is further highlighted by its association with patient outcomes in various malignancies.
YTHDF1
YTHDF1 has been identified as an oncogenic factor in various types of cancer. Comprehensive analysis from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases, along with immunohistochemical studies, have revealed that YTHDF1 is consistently overexpressed in liver and breast cancers, correlating with poorer overall survival rates and advanced pathological stages.235,236 Recent research has extended these findings, demonstrating upregulation of YTHDF1 in melanoma and ovarian cancer as well.237,238,239 This protein stands out as an independent prognostic marker, being of vital importance in the regulation of cell cycle progression and metabolic pathways in liver cancer.240 The primary mechanism through which YTHDF1 fosters tumorigenesis involves facilitating the translation for m6A-modified mRNAs that are crucial for cell proliferation and the epithelial-mesenchymal transition (EMT).241 In the context of CRC, YTHDF1 has been shown to enhance the initiation and progression of the disease by activating the Wnt/β-catenin signaling pathway.237,242,243 These insights underscore the value of YTHDF1 as a probable therapeutic target in cancer treatment strategies.
YTHDF2
YTHDF2 exhibits a complex and context-dependent role in cancer biology, with its function varying across different types of research (Fig. 3). In the context of colon cancer, high levels of YTHDF2 are associated with increased malignancy, as it enhances the expression and translation of mRNAs that drive cell proliferation and invasion.241 In HCC, YTHDF2 expression is elevated and closely linked to the severity of the disease.244 Besides, in lung cancer, YTHDF2 interacts with the m6A modification site on the 3’-untranslated region (3’-UTR) of 6-phosphogluconate dehydrogenase (6PGD) mRNA, thereby promoting 6PGD mRNA translation and contributing to the proliferation of lung cancer cells.245 Conversely, in esophageal cancer, YTHDF2 is typically under expressed. Interestingly, when YTHDF2 is upregulated in esophageal and gastric cancers, it suppresses neoplastic cell proliferation and triggers programmed cell death, indicating a protective role against these malignancies.246,247 These findings highlight the dual nature of YTHDF2 in cancer, where its role as a promoter or suppressor of cancer progression is profoundly contingent upon the specific tumor subtype and cellular milieu. Understanding these nuances is crucial for formulating targeted strategies that can exploit the differential effects of YTHDF2 in cancer treatment.
YTHDF3
YTHDF3 is of significance in promoting the translation of oncogenic genes within diverse cancers. In CRC, YTHDF3 is notably overexpressed and has been linked to poorer patient survival outcomes.236,248 Its function in breast cancer is characterized by the m6A-dependent promotion of oncogene translation. Additionally, YTHDF3 also enhances the stability of m6A-modified ZEB1 mRNA, thereby facilitating metastasis of liver cancer.244 Moreover, YTHDF3 is frequently overexpressed in HCC, with higher expression levels correlating with an increased risk of cancer recurrence in patients.249 Furthermore, recent research has implicated YTHDF3 in tumorigenesis. It has been revealed to promote the translation of Integrin subunit α 6 (ITGA6) mRNA, which is instrumental in the development of bladder cancer.250,251 In the context of pancreatic cancer, YTHDF3’s role is further underscored by its interaction with ZDHHC20, which mediates S-palmitoylation of YTHDF3. This post-translational modification stabilizes MYC mRNA, thereby contributing to the progression of pancreatic cancer.252 These findings highlight YTHDF3 as a key factor in the regulation of cancer development and progression, underscoring its potential as a therapeutic target.
N1-methyladenosine(m1A)
The significance of N1-methyladenosine (m1A) modifications in cancer progression and prognosis has garnered increasing attention in recent research. The regulators associated with m1A modifications encompass a range of proteins, including TRMT6/TRMT61A, ALKBH1, ALKBH3, and the YTH-domain family proteins.253 Emerging evidence suggests that the levels of m1A methylation and the expression of m1A-related RNAs could serve as innovative biomarkers for predicting cancer prognosis. In CRC, the content of m1A is notably higher in patients compared to healthy individuals, indicating its potential diagnostic and prognostic value.254 To assess the m1A modification profile personalized patient contexts, the m1A score has been developed. In cervical cancer and oral squamous cell carcinoma (OSCC), patients exhibiting a high m1A score have been observed to increased lymphatic invasion, reduced survival, and a poorer response to immunotherapy.255 The m1A level has demonstrated the potential to predict the prognosis of various cancers. Above results highlight the importance of m1A modifications within the complex landscape for cancer biology and their prospective utility as therapeutic targets and prognostic indicators.
TRMT6/TRMT61A
The TRMT6/61A complex is of great significance in a variety of biological systems, especially in the context of cancer. Current research indicates that TRMT6/61A is upregulated in bladder cancer and is associated with an increase in m1A modification levels.256 Additionally, TRMT6/61A has been found to enhance the proliferation of gastrointestinal and breast cancer cells, exerting an oncogenic effect.257,258,259,260 TRMT6 exhibits an upregulated trend in certain types of cancer, particularly bladder cancer, and is associated with the degree of malignancy and the unfolded protein response. These findings highlight the potential role of TRMT6 in tumor development and suggest that it may serve as a target for future cancer treatments.
ALKBH1
When exploring the role of ALKBH1 across different cancers, we have found that expression levels of ALKBH1 have a significant correlation with patient prognosis. There is a significant negative correlation between the overexpression of ALKBH1 and overall survival rates in CRC. Recent research has highlighted that ALKBH1 is not only overexpressed in CRC but also plays a pivotal role in cancer metastasis.261 Besides, ALKBH1 typically acts as an oncogene, promoting tumorigenesis, as evidenced in glioma and gastric cancer.261 Furthermore, in lung cancer, the upregulation of ALKBH1 in both tissue and cellular contexts has been shown to enhance cell invasion and migration.262 However, in the context of pancreatic cancer, low levels of ALKBH1 expression are associated with a particularly poor prognosis263 (Fig. 3). As our comprehension of the mechanisms through which ALKBH1 operates in diverse cancers deepens, new therapeutic strategies may be developed in the future to target the expression and function of ALKBH1, thereby improving patient treatment outcomes (Table 1).
ALKBH3
Recent progress has revealed ALKBH3’s function as a cancer-promoting factor in various cancers. In prostate cancer, ALKBH3 serves as a critical biomarker for early detection and histopathological classification. Numerous studies have demonstrated that ALKBH3 expression level in prostate cancer are significantly elevated.264,265 Besides, ALKBH3 has been identified as a facilitator of neoplastic cell proliferation in gastrointestinal (GI) malignancies.257,258 Heightened the expression of ALKBH3 in lung adenocarcinoma (LUAD) has been correlated with post-recurrence survival.266 ALKBH3 enhances the glycolytic activity of neoplastic cells by modulating the expression of m1A-modified ATP5D mRNA.267 ALKBH3 has been documented to modulate the cell cycle.268 Moreover, Silencing of ALKBH3 induces the senescence and halts the cell cycle in lung cancer and urothelial carcinomas by upregulating the expression of cell cycle arrest proteins p27 and p21.266 ALKBH3 could produce tRNA-derived small RNAs (tDRs) through demethylation of m1A-tRNA thereby enhancing the growth and invasive potential of cancer cells.269 In summary, the multifunctionality of ALKBH3 in cancer development suggests that it may become a potential target for future cancer therapies, particularly in strategies that target its expression and function.
5-Methylcytosine (m5C)
In the context of cancer, the m5C modification is intricately linked to the proliferation, migration, invasion, and therapeutic resistance of tumor cells.270,271 The biological function of m5C is closely associated with the proteins that modulate their presence: the writers, erasers, and readers. M5C writers, such as DNMT2 and the NSUN family proteins, are responsible for the establishment of methylation marks. Readers, including ALYREF and YBX1, are proteins that identify and attach to these methylation sites. Conversely, m5C erasers, such as the TET family proteins and ALKBH1, are involved in the removal of these methylation marks, creating a dynamic equilibrium between the two opposing processes.272 The role of m5C RNA modification in gastric cancer is predominantly oncogenic, with elevated m5C levels being indicative of a poor prognosis and reduced overall survival rate.273 The oncogenic impact of m5C modulation may also extend to immune suppression, as gastric cancer patients with lower levels of m5C modulation levels have been observed to exhibit higher immune activity, along with longer progression-free survival and overall survival.273 These insights highlight the complex interaction between m5C modification and cancer biology, highlighting its potential as a therapeutic target and prognostic marker. Additionally, absence of DNMT2 is associated with alterations in mRNA expression and methylation patterns and the suppression of cell proliferation and migration.274
M5C-related enzymes are frequently dysregulated in tumors, and the NSUN family proteins, which consists of NSUN1-7, plays a significant role in this context.275 Recent investigations have demonstrated that NSUN3 expression is significantly elevated in individuals diagnosed with low-grade glioma and head and neck squamous cell carcinoma (HNSCC). Moreover, it has been elucidated that NSUN3-facilitated m5C methylation of tRNA potentiates metastatic progression by augmenting the translational efficiency of mitochondrial mRNA.276 The immune cell infiltration associated with NSUN3 predominantly encompasses CD8+ T lymphocytes and M2-polarized macrophages,273,277,278 which suggests its role in modulating the tumor microenvironment. High levels of m5C were negatively related to prognosis of patients with glioma.279,280 Besides, overexpression of NSUN5 has been associated with tumorigenesis in HCC.281 Research has suggested that NSUN6 functions as a protective agent against triple-negative breast cancer (TNBC), pancreatic carcinoma, testicular cancer, thyroid malignancies, and ovarian cancer, while serving as a risk factor for CRC.282,283,284,285 These findings underscore the complex and context-dependent roles of NSUN family proteins in the biology of various cancers.
The m5C demethylase identified to date include the TET family proteins and ALKBH1. TET2 is primarily responsible for catalyzing the conversion of m5C to hm5C, thereby facilitating the removal of m5C modifications in RNA.286 Studies have measured TET2 expression across a spectrum of cancer types, revealing its upregulation in patients with low-grade glioma.287 In contrast, TET2 expression is reduced in clear cell renal cell carcinoma (ccRCC), ovarian cancer, and prostate adenocarcinoma.288,289 The specific role of TET3 in mediating m5C elimination remains to be fully elucidated. Nonetheless, some research has suggested that elevated TET3 expression in prostate cancer may correlate with a poorer prognosis, emphasizing the intricate and context-dependent roles of TET enzymes in the realm of cancer biology.290 Additionally, research has indicated that ALKBH1 is upregulated in a variety of cancers, including gastric, head and neck, and liver cancers. High expression levels of ALKBH1 have been associated with poor prognosis in multiple tumor types. Moreover, ALKBH1 is markedly overexpressed in advanced tumors that are high metastatic, and exhibit high malignancy, underscoring its potential role as a biomarker for aggressive cancer behavior.291
Readers, or proteins that bind to m5C sites, include ALYREF and YBX1. ALYREF has been identified as a significant oncogenic factor, correlating with poor prognosis in patients across various cancer types, such as HCC, glioblastoma, glioma, and neuroblastoma.292,293,294 In HCC patients, increased levels of ALYREF are associated with the upregulation of eIF4A3 expression, as well as disruptions in the cell cycle and mitosis.295 Within the context of lung adenocarcinoma, ALYREF, in conjunction with NSUN2, enhances the m5C modification of Yes-Associated Protein (YAP) mRNA, a factor whose high expression in tumors is linked to increased invasiveness and metastatic potential.296,297,298,299 YBX1, another m5C reader, plays a multifaceted role in cancer development, with its oncogenic effects observed in gastric cancer, bladder cancer, glioblastoma, CRC, cholangiocarcinoma, prostate cancer, epithelial ovarian cancer, and cervical cancer.272,297,300,301,302,303 In summary, the expression of m5C and its related enzymes in tumors is closely related to the occurrence, development, and prognosis of tumors, providing new strategies for the diagnosis and treatment of tumors.
7-Methylguanosine (m7G)
The m7G modification occurs in a specific subset of tRNAs, serves to stabilize these modified tRNAs and is essential for the efficient translation of mRNA.304 The methylation process of m7G modification is primarily catalyzed by methyltransferases such as METTL1 and RNMT, which are writer proteins that add m7G modifications to RNA molecules, including tRNA and mRNA. Reader proteins that recognize m7G modifications include eIF4E, QKI, and NCBP2, which are involved in regulating RNA maturation, nuclear export, and translation processes. However, no demethylase for m7G has been identified to date.305 A growing body of evidence implicates m7G in the development of various human diseases, particularly cancer.306 Disruptions in m7G levels are intricately linked to the onset and advancement of malignancies, as they regulate the transcription of numerous oncogenes and tumor suppressor genes.307 Abnormalities in m7G modification may lead to changes in tumor cell proliferation, migration and invasion.308
METTL1
The writer of the m7G modification is primarily METTL1, which forms a complex with WDR4. METTL1 and WRD4 are upregulated in diverse kinds of cancer, including esophageal, liver, colorectal, lung, and nasopharyngeal carcinomas.309,310,311 In bladder cancer cells and lung cancer cells, METTL1 notably enhances cellular proliferation, migration, and invasion.299 Besides, the expression of WDR4 is significantly correlated with advanced-stage prostate cancer.312 And elevated expression of METTL1 or WDR4 indicates a less favorable prognosis in patients with osteosarcoma.313 The above results highlight their potential as prognostic biomarkers and therapeutic targets in cancer.
Quaking proteins (QKI)
Recent research has revealed that the QKI protein, which is downregulated in a spectrum of malignant tumors such as lung, gastric, and CRC,314,315,316,317 and serves as a tumor suppressor in various human cancers, encompassing oral, colon, gastrointestinal, and prostate cancer318,319 (Fig. 3). Specifically, in NSCLC, QKI-6 has been shown to inhibit EMT processes by modulating the EGFR/SRC/STAT3 signaling pathway, thereby upregulating the expression of AGR2.320 QKI is suppressed in a variety of tumors, and its expression levels are strongly correlated with the aggressiveness, metastatic potential, and clinical outcomes of these tumors. In summary, functioning as intrinsic m7G-recognizing proteins within mRNA, QKIs orchestrate the regulation of target mRNA metabolism and modulate cellular chemoresistance, making them as promising candidates for therapeutic intervention.321
Nuclear cap binding protein subunit 2 (NCBP2)
NCBP2 exhibited significant upregulation in pancreatic cancer tissues compared with normal tissues. Overexpression of NCBP2 was associated with poor prognosis, especially in early-stage patients with pancreatic cancer.322 There is a study indicated that the expression of NCBP2 is upregulated in various types of malignant tumors, including OSCC. It has been confirmed that NCBP2 indeed suppresses the migration, invasion, and proliferation of OSCC cells.323 However, the specific mechanisms of NCBP2 in tumorigenesis still require further exploration.
Non-coding RNAs
LncRNAs
Long non-coding RNAs (lncRNAs) represent a category of non-protein-coding transcripts exceeding 200 nucleotides in length. This extensive definition encompasses a diverse and highly variable array of transcripts that vary in their biogenesis, genomic origin, and mechanisms of action.324 LncRNAs play essential regulatory roles in numerous cellular processes, such as gene expression, cell differentiation, and development.325 They are capable of exerting an impact on DNA, RNA, and proteins to modulate gene expression by alterations in chromatin structure, transcription, and post-transcriptional processing.326,327
LncRNAs are increasingly recognized as pivotal regulators that are implicated in gene expression as well as a wide array of physiological and pathological processes.328,329 In the context of cancer, they are capable of regulating the growth, differentiation, invasiveness, and metastasis of cancer cells.330 Recent research has demonstrated that lncRNAs display elevated levels of expression and are frequently linked to diverse varieties of tumors. The dysregulation and mutations of these lncRNAs are significantly correlated with tumorigenesis, metastasis, and tumor progression.331 Furthermore, lncRNAs demonstrate specific expression patterns in certain cancer types and can be detected in circulating blood and/or urine.327 As such, lncRNAs represent a novel class of potential molecular indicators and therapeutic targets for oncological interventions.
Genome-wide RNA sequencing (RNA-Seq) analysis has identified numerous lncRNAs that are either upregulated or downregulated in breast cancer. LncRNAs implicated in breast cancer include HOTAIR, ANRIL, ZFAS1, HOTAIRM1, PVT1, MALAT1, and LNP1, among others.332 HOTAIR suppresses tumor suppressor genes like PGR, PCDH10, PCDHB5, and JAM2, promoting breast cancer development, and is overexpressed in colorectal, hepatocellular, gastrointestinal, and NSCLC.333,334,335 Besides, ANRIL is also upregulated in breast cancer. In lung cancer, lncRNAs such as MALAT1, CCAT2, HOTAIR, AK126698, HNF1A-AS1, SOX2-OT, MEG3, ANRIL, H19, CARLo-5, MVIH, PVT1, EVADR, SPRY4-IT1, GAS5, PANDAR, BANCR, and TUG1 are involved.336,337 MALAT1 is overexpressed in lung cancer, enhancing cell proliferation, EMT, and angiogenesis.338 In addition, CCAT2 is also overexpressed in NSCLC, increasing its invasiveness.339 MEG3 is downregulated in NSCLC, influencing immunity and autophagy via the miR-543/IDO pathway.340 Besides, In HCC, several lncRNAs, including MALAT1, HULC, HEIH, DILC, and HOTAIR, are upregulated, with HULC promoting HCC growth metastasis and drug resistance.341,342 However, lncRNA DILC has been identified as a tumor suppressor gene that can suppress the stemness of tumor cells.343 A vast number of lncRNAs have been identified in various other types of cancers, and more details are presented in figures and tables. Many lncRNAs are abnormally expressed in different tumors (Fig. 3), with some being cancer-specific. They are stable in body fluids and can be detected in the plasma and urine of cancer patients, reflecting the severity of the disease. These characteristics render lncRNAs promising non-invasive biomarkers and therapeutic targets for cancer treatment, although challenges and the need for validation still exist for their clinical application.
miRNAs
miRNAs are small non-coding RNAs with a length of approximately 22 nucleotides (nt), which are widely recognized to play a significant role in the post-transcriptional regulation of mRNA.344 The typical biogenesis process of miRNA involves three distinct stages: primary miRNA (pri-miRNA), precursor miRNA (pre-miRNA), and miRNA duplex formation.345,346 Subsequently, one strand of the miRNA duplex is integrated into the RNA-induced silencing complex (RISC), which triggers the decay of mRNA and translational suppression by interacting with the complementary sequences in the 3’-untranslated region (3’-UTR) of target gene mRNA.347,348 Notably, miRNA-mediated gene expression control is critical for the cellular response to the environmental stresses, like starvation, hypoxia, oxidative stress, and DNA damage.346,349
Undoubtedly, miRNAs stand out as the most intensively studied category of ncRNAs, especially within the context of cancer research. In 2002, the first evidence showing the role of miRNAs in human disease was reported350 (Fig. 1). Since then, a substantial amount of research has indicated that aberrant regulation of miRNA expression is an intricately linked to the progression of tumors.351,352 The underlying mechanisms encompass chromosomal aberrations (such as the amplification or deletion of miRNA genes), alterations in transcriptional regulation, epigenetic modifications, and flaws in the miRNA biogenesis machinery.353 Over the past two decades, the association between miRNAs and diverse types of cancers has been the subject of extensive investigation. Drawing upon the evidence regarding miRNAs, a multitude of potential cancer biomarkers for both diagnosis and prognosis have been proposed, thereby offering a novel vantage point for cancer screening.
In 2002, the deletion and low-expression of miR-15 and miR-16 cluster in chronic lymphocytic leukemia were demonstrated, which initially shed light on the role of miRNAs in the progression of cancers.354 Concurrently, a decrease in these two miRNAs was noted in cancerous tissues compared to normal tissues. Over the past years, miRNAs have been reported to be implicated in almost all known cancer processes. miR-21 demonstrates a potential oncogenic function and targets tumor inhibitor proteins in almost all types of cancer, including glioblastoma, head and neck cancer, ovarian cancer, B-cell lymphoma, HCC, cervical cancer, and lung cancer.355 Likewise, miR145 is also highly expressed in numerous malignancies and plays a profound role in cancer initiation. miR-145 is overexpressed in colon cancer, ovarian cancer and so on356,357 (Fig. 3). Conversely, miRNA-34 exhibits tumor-suppressive effects in various types of cancer, including gastric cancer, CRC, prostate cancer, breast cancer. miR-34 is epigenetically downregulated or silenced in colorectal cancer tissues and cell lines, miR-34 regulates several different target genes and signaling pathways, inducing apoptosis, senescence, and cell cycle arrest and repressing gastric cancer cell proliferation, migration and metastasis, thus contributing to the suppression of carcinogenesis and cancer progression.358
Additionally, miR-155 has been identified as being dysregulated in several types of human tumors, and it is regarded as functioning either as an oncogene or a tumor suppressor, depending on tumor system. high expression of miR-155 has been shown in B-cell lymphomas, colon cancer and lung cancer as an oncogenes,359,360 whereas low expression in ovarian cancer and melanoma as an oncosuppressor-miR.360,361 There are many miRNAs are also dysregulated during the development and progression of cancer. For example, miR-148a and miR-152 are downregulated in gastric cancer tissues362; miR-128 is significantly downregulated in breast and lung cancer tissues363,364; and miR-375 is significant overexpressed in lung cancer.365 The information presented in Fig. 3 and Table 1 further enriches our understanding of these miRNA-mediated cancer-related phenomena, emphasizing the need for continued research in this significant area.
CircRNAs
CircRNAs fall within the classification of ncRNA molecules, which were initially identified in pathogens back in the 1970s.366 Typically, circRNAs are generated from precursor mRNA (pre-mRNA) through the back-splicing process, also referred to as alternative splicing.367 CircRNA can be categorized into three types: exonic circular RNA (ecircRNA), which consists solely exons, ciRNA, which originates from intron lariat 3 and exon-intron circRNA (EI-ciRNA), which is made up of both exon and intron sequences.368 CircRNAs represents a single-stranded RNA that differentiates itself from linear RNA on account of its covalently closed structure. Lacking 5’ caps and 3’ poly(A) tails, this unique structural feature endows circRNAs with enhanced stability and a remarkable resistance to RNase R. This unique form grants circRNAs greater stability and resistance to RNase R. They possess the ability to bind miRNAs, functioning as sponges, and thereby partake in a diverse range of physiological functions, such as cell cycle regulation, intercellular communication, as well as transcriptional and translational regulation.369,370
In the past, circRNAs were regarded as by-products resulting from aberrant splicing. CircRNAs were extensively discovered in mammalian transcriptomes in 2012 (Fig. 1), which subsequently led to their attracting substantial attention in the realm of cancer research.371,372 They are crucial in tumorigenesis, invasion, metastasis, and chemoresistance, and may serve as novel diagnostic biomarkers and anticancer targets. Research indicates that circRNAs are differentially expressed in various tumors, including colon, ovarian, gastric, esophageal cancers, and gliomas.373,374 They regulate the expression of oncogenes and tumor suppressor genes through various mechanisms, including acting as miRNA sponges to adsorb miRNAs or binding to RNA-binding proteins (RBPs) to form complexes, thereby affecting gene transcription.375 CircRNAs have been associated with a range of physiological states and cellular attributes, such as stemness and pluripotency, and are thus potentially involved in initiating and perpetuating oncogenesis. Furthermore, circRNAs have been linked to various clinical parameters, including tumor grade, size, metastatic stage, and malignancy aggressiveness.
Recent studies have revealed that abnormal expression of circRNAs are widespread across nearly all types of cancer and play crucial roles in cancer pathogenesis (Fig. 3), functioning either as tumor suppressors or oncogenes.373 For example, circMTO1 is downregulated in HCC and suppress HCC progression by sponging oncogenic miR-9 to promote p21 expression.376 A similar ceRNA mechanism also applies to circTRIM33-12, which is also down-regulated in HCC tissues and cell lines.377 Conversely, circRHOT1 is significantly upregulated in HCC, which promote proliferation and metastasis.378 Additionally, multiple circRNAs are found to be dysregulated in gastric cancer. For the past several years, circRNAs were regarded as potential targets in the clinical treatments of gastric cancer. There were studies indicated that circCACTIN and circNHSL1 are upregulated in gastric cancer, which promoted cell migration and invasion,379,380 and circFGD4 is downregulated in gastric cancer.381 In addition, recent studies have indicated that a variety of circRNAs are involved in the progression of breast cancer, primarily by acting as miRNA sponges,382 circCDYL and circHSDL2 were found upregulated the breast cancer cell lines and clinical tissues,383,384 which indicate a potential prognostic marker for breast cancer. Besides, circRNAs also play regulatory roles in the progression and metabolism of lung cancers. Another study has identified a novel exon-derived circRNA, circSLC25A16, which has the ability to accelerate the glycolysis and proliferation of NSCLC cells.385 The circTP63 and cercaria were also found to be overexpressed in lung cancer.386,387 Dysregulation of circRNAs has also been found in other types of cancer. circLPAR3 and hsa_circRNA6448-14 are upregulated in esophageal cancer.388,389 CircFOXK2 and circBFAR were found to be overexpressed in pancreatic cancer (PC).390,391 These circRNAs have emerged as hot topics in current cancer research due to their significant roles in tumor development, as well as their potential as biomarkers and therapeutic targets. As research continues to advance, an increasing number of circRNAs will be identified to play a crucial part in the diagnosis and treatment of cancer.
piRNAs
piRNAs constitute a distinct class of small silencing RNAs in the animal kingdom, distinguishing themselves from miRNAs and siRNAs. They possess 2ʹ-O-methyl-modified 3ʹ ends and form complexes with PIWI proteins, in contrast to miRNAs and siRNAs which pair with AGO proteins.392,393,394 The PIWI/piRNA complex is known for regulating transposon silencing and reproductive development by controlling gene expression at the transcriptional or post-transcriptional level.395,396 PIWI protein/piRNA deletion or disruption can reactivate transposons, potentially leading to germ cell tumors. In contrast, PIWI proteins are highly expressed in various other tumors, including gastric, colon, liver, glioma, and bladder cancers.397,398,399,400 piRNAs regulate gene expression in cancer analogous to normal cells, dependent on PIWI proteins, and can also exert epigenetic control by interacting with regulatory factors or through direct regulating their expression.401 Additionally, piRNA can also bind to target genes, thereby degrading mRNA, altering its stability, or inhibition of its translation.402,403
Around 2010, it was initially reported that piRNAs exhibited abnormal expressions in cancer (e.g. upregulation of piR-651 in several cancer cell lines404). Current studies indicated that piRNA and PIWI are significantly abnormally expressed in gastric, breast, kidney, colon, and lung cancers, and are involved in the initiation, progression, metastasis and therapy resistance of cancers, which may be the potential diagnostic tools, prognostic markers, and therapeutic targets for cancers.402 A study has analyzed the expression patterns of piRNAs in tumor tissues by means of whole-transcriptome piRNA sequencing or PIWI-interacting/bound RNA sequencing, which has unveiled an abnormal expression profile of piRNAs/PIWI between tumor tissues and normal tissues.405
Among these piRNAs, abnormal expression of pir-651 and pir-823 is the most widely associated with various cancer types.406 It has been proved that in gastric cancer, the expression of piR-651 is significantly higher than in adjacent normal tissue,404 while the expression of piR-823 is markedly reduced.407 Concurrently, the expression of PIWIL1 is upregulated in gastric cancer cells. Moreover, these aberrant changes are associated with the progression of gastric cancer and poor prognosis. Similarly, it has been reported that piR-651 was overexpressed in breast cancer,408 and it has the ability to boost the proliferation and migration, while suppressing apoptosis of breast cancer cells by facilitating DNMT1-mediated PTEN promoter methylation.408 In addition, other piRNAs such as piR-4987, piR-20365, piR-20485, piR-20582,409 piR-021285410 and piR-932411 are highly expressed in breast cancer, while the expression of piR-36712 is significantly lower in breast cancer patients compared to normal tissue.412 Notably, piR-021285 mediates the hypermethylation of AATF, ARHGAP11A, PIP4K2B, THAP10, and other related oncogenes in breast cancer tissue, thereby promoting the progression of breast cancer, highlighting the importance of crosstalk between various epigenetic modifications.410 In various types of tumors, the abnormal expression of piRNAs has been widely reported, such as piR-823, piR-54265, piR-18849, and piR-19521 in colorectal cancer,413,414 and piR-651 and piR-55490 in lung cancer.415,416 For more details, please refer to (Fig. 3) and the Table 1. It is worth highlighting that circulating piRNAs in the blood of tumor patients are regarded as potential cancer biomarkers for detection and prognosis. Furthermore, machine learning-based diagnostic methods for CRC utilizing piRNAs have already been developed.417 As we further investigate the intricate regulatory mechanisms and functions of piRNAs in the context of cancer, it is expected that they will offer more valuable insights and possibilities for improving cancer diagnosis, prognosis, and treatment strategies in the future, highlighting the importance of further in-depth investigations in this promising area.
Mechanisms of cancer therapy resistance by epigenetic networks
Cancer therapy resistance (or therapeutic resistance) pertains to the diminished sensitivity of tumor cells towards various treatment modalities, including chemotherapy, radiotherapy, targeted drugs, and immunotherapy.418 This resistance is one of the main reasons for treatment failure in cancer patients, leading to diminished responsiveness, rapid disease progression, relapse, metastasis, and ultimately, patient mortality.419 Therapy resistance can be roughly categorized into intrinsic and acquired resistance.2 The intrinsic resistance is mediated by the inherent or endogenous characteristics that are exist in tumor cells or tissues prior to initial treatment, such as effective DNA damage repair mechanisms, tumor growth kinetics and stem cell like properties, that offer cancer cells survival benefits and ability to adapt to primary therapeutic stress.420,421 This can manifest as an unresponsive initial reaction to therapies, even at elevated doses. Differently, acquired resistance refers to the development of resistance to cancer therapies after an initial response, mainly based on factors such as genomic instability (mutations), tumor cells heterogeneity, cellular plasticity, and adaptive responses, which can manifest as local tumor recurrence or distal metastases following clinical remission.422
In recent years, the mechanisms by which cancer cells develop resistance to various treatment methods have been gradually revealed, and it has been comprehensively summarized in several reviews.418,423,424 Additionally, various forms of epigenetic regulations have been successively verified to be associated with cancer treatment resistance (Figs. 4–7). In general, the decreased responsiveness of cancer cells are related to diverse mechanisms, which typically entail the interactions of genetic factors, nongenetic elements and microenvironment.423 It is widely acknowledged that genetic factors play a predominant role in the development of therapeutic resistance in cancer cells.2 One of the hallmark characteristics of cancer cells is genomic instability, which means that as cancer cells proliferate, various mutations will accumulate in genome, thereby inducing a range of phenotypes to swiftly adjust their transcriptional and/or metabolic program to cope with and endure the therapeutic pressure.425 However, accumulative evidence also supports those nongenetic factors, including epigenetic regulation, also play a significant role in the therapeutic resistance of cancer. The notion that a single cancer genome is capable of producing a multitude of phenotypic states, and cancer cells can shift between these states without fundamental genomic alterations is gaining greater acknowledgement.426

Mechanisms of epigenetics impacting on radiotherapy resistance. It delineates the mechanisms by which epigenetic factors contribute to radiotherapy resistance, primarily through the modulation of DNA damage repair processes and tumor-related phenotypes. DNA damage repair pathways play a role in radiotherapy resistance. Among them the DNA double-strand break repair pathways, specifically homologous recombination repair and nonhomologous end joining, are crucial in conferring radioresistance. Furthermore, the activation of these DNA damage repair pathways enhances resistance to radiotherapy. Besides, tumor-associated phenotypes, including cell cycle regulation, apoptosis, autophagy, EMT, and cell proliferation, may serve as potential targets for enhancing radiosensitivity through epigenetic modifications. HR homologous recombination repair, NHEJ nonhomologous end joining, BER base excision repair, MMR mismatch repair, EMT epithelial-mesenchymal transition

Mechanisms of epigenetics impacting on chemotherapy resistance. It depicts the pathways of epigenetic regulation of chemotherapy resistance, which can be summarized into the following four aspects: dysregulation of tumor suppressor gene and oncogene, metabolic reprogramming, unlimited cell proliferation and resistance to cell death, and microenvironment remodeling

Mechanisms of epigenetics impacting on immunotherapy resistance. It illustrates the mechanisms by which epigenetic modifications influence resistance to immunotherapy. Epigenetic processes predominantly modulate the activity of immune cells and the expression of the immune checkpoint proteins, specifically, PD-1 and PD-L1. Particularly, the activity of CD8+ T cells, Tregs, and MDSCs, along with the expression of PD-1, are principal targets for epigenetic regulation in the context of immunotherapy resistance. Tregs regulatory T cells, MDSCs myeloid-derived suppressor cells, NK nature killer cell, DC dendritic cell, M1 M1 macrophages, M2 M2 macrophages

Mechanisms of epigenetics impacting on targeted therapy resistance. It delineates the mechanisms by which epigenetic factors contribute to targeted resistance. The factors could be summarized into the following four aspects: tumor growth, tumor metastasis, tumor death and other tumor-related phenotypes including glycolysis, cancer stemness and drug transport
When cancer cells are subjected to different therapeutic pressures such as chemotherapeutic drugs, targeted agents, ionizing radiation, and monoclonal antibodies, they can gradually evolve different mechanisms of resistance.3,427,428 Due to the diverse mechanisms of action of various chemotherapeutic drugs, the mechanisms of chemotherapeutic resistance in cancer are quite complex. Tumor cells primarily achieve resistance to traditional chemotherapy drugs through mechanisms such as alteration of drug metabolism, reduction of drug absorption, increased drug pumping out, efficient DNA damage repair, resistance to cell death, EMT, cancer stem cells, and the tumor microenvironment.429 In addition to the aforementioned factors, tumor cells can gradually acquire resistance to targeted drugs through heterogeneity, plasticity, and adaptability.427 Unlike chemotherapy, the mechanism of radiotherapy is relatively straightforward, mainly relying on the destruction of biomolecules such as DNA by ionizing radiation. Therefore, tumor cells can mitigate the damage of ionizing radiation through chromatin remodeling, and achieve resistance to radiotherapy by relying on efficient DNA damage repair and growth dynamics (cell cycle regulation, cell death resistance and metabolic reprogramming).420 The resistance of tumor cells to immunotherapy mainly arises from the tumor’s regaining of immune evasion capabilities through various mechanisms, coupled with the suppression of immune cells within the microenvironment.430 The factors mentioned above are all intricately regulated by epigenetic modifications. In this context, we will focus on the contribution of epigenetic modifications to tumor treatment resistance.
Radiosensitivity
Radiotherapy is a pivotal modality in cancer treatment.431 However, the resistance exhibited by numerous tumor cells significantly diminishes its therapeutic efficacy.432,433 It is well established that radiotherapy exerts a substantial cytotoxic effect on proliferating cells. Nonetheless, tumor cells that endure radiation exposure concurrently activate a variety of pro-survival signaling pathways, such as ATM, ATR, AKT, ERK and NF-κB-mediated DNA damage checkpoints, DNA damage repair, inhibition of apoptosis, and cancer stem cell-related stem-like pathways. The configuration of compact chromatin enhances the protection of tumor cells against radiotherapy. Notably, radioresistant pathways are subject to epigenetic regulation.434,435,436 In recent years, epigenetic mechanisms in the cancer cells radioresistance have received increasing attention. Moreover, the integration of radiotherapy with epigenetic pharmacological agents has been implemented in clinical treatment.434,437
Histone modifications in radiosensitivity
Histone modifications have a vital function in controlling gene expression and chromatin structure, which modulate DNA damage response and repair pathways that are crucial to radioresistance. Currently, evidence shows that histone acetylation is closely related to radiotherapy resistance.438 The level of histone acetylation can affect DNA damage repair and thereby regulate radioresistance. For example, treatment with the HDAC inhibitor PCI-24781 leads to increased acetylation levels, which reduce the accuracy of DSB damage repair in SiHa cervical cancer and WiDr colon cancer cells, thereby reducing radiotherapy resistance.439 Conversely, decreased acetylation levels promote radiotherapy resistance. In addition to affecting DNA damage repair, histone acetylation levels also influence tumor angiogenesis and migration processes. HDAC inhibitors can suppress the expression of MMP14 in GBM, and the inhibition of MMP14 helps to reduce tumor angiogenesis, inflammation, cancer cell invasion, and metastasis, thereby promoting tumor radiotherapy resistance.440,441 Furthermore, histone acetylation affects DNA damage repair in cancer cells by regulating chromatin remodeling. Targeting BRG1 chromatin remodeling enzymes may enhance the radiosensitivity of cancer cells. BRG1-BRD increases the sensitivity of cancer cells to radiotherapy by disrupting the γ-H2AX and 53BP1 pathways, leading to reduced DNA repair efficiency, G2-M checkpoint defects, and increased apoptosis.442 Additionally, acetylation of the chromatin remodeling protein MORC family CW-type zinc finger 2 (MORC2) can activate the G2 checkpoint and induce DNA damage, thereby improving the radiosensitivity of breast cancer cells443 (Fig. 4).
Contrarily to histone acetylation, increased level of histone methylation leads to chromatin condensation, resulting in gene silencing.444 Specifically, the methylation of H3K36 and H3K27me3 may participate in the cellular response to radiation through regulation of the DNA repair pathways of homologous recombination (HR) and nonhomologous end joining (NHEJ).445,446,447 For instance, the trimethylation of H3K36 mediated by SETD2 is crucial for the activation of the ATM kinase and the recruitment of 53BP1 to DNA double-strand breaks (DSBs). Furthermore, overexpression of JMJD2A leads to a decrease in H3K36 methylation, reducing HR repair efficiency, which may weaken the radiation resistance of tumors.448,449 On the other hand, Metnase, a methyltransferase for H3K36, can promote NHEJ repair, thereby enhancing the radiation resistance of tumor cells.450 In addition, the dynamics of H3K27me3 are also related to radiotherapy sensitivity. Studies have shown that the H3K27 demethylase inhibitor GSKJ4 can disrupt radiation-induced DSB repair and reduce the radiation resistance of tumor cells. Meanwhile, the histone demethylase UTX has been found to enhance radiotherapy resistance444 (Fig. 4). Overall, histone methylation contributes significantly to modulate the repair of DNA damage and DSBs, thereby affecting the radiation resistance of cancer cells.
DNA methylation in radiosensitivity
DNA methylation exerts a substantial influence on the radioresistance of cancer cells. Researches have revealed that the DNA methylation status within tumors is intimately linked to radiosensitivity, and aberrant alterations in DNA methylation can impact the efficacy of tumors to radiotherapy. Precisely, the hypomethylation state of certain genes may facilitate the radioresistance of tumor cells by inducing specific signaling pathways. For example, in CRC, the hypomethylation of the DSTN gene potentiates the resistance for cancer to radiotherapy though activating the Wnt/β-Catenin signaling pathway.451 Moreover, DNA methylation can also affect the radiosensitivity of tumor cells by influencing genes related to DNA damage repair. For genes that promote DNA damage repair (e.g., ATM/ERCC1), DNA methylation can increase the sensitivity of tumors to radiation, while demethylation leads to radiotherapy resistance in tumor cells452,453 (Fig. 4). For genes that inhibit DNA repair (e.g., RASSF1A), DNA methylation results in radiotherapy resistance in tumor cells.454 DNA methylation profile of aggressiveness‐associated genes modulates the radiosensitivity in distinct manners. The elevated expression of tumor proliferation-suppressing genes serves to restrain the proliferation of tumor cells and consequently trigger radiosensitivity in the context of radiotherapy. The SERPINB5 and HIC1 genes were observed to be hypermethylated within radioresistant cells,455,456 whereas the TM4SF4 and miR24 genes manifested hypomethylation in these same radioresistant cells.457 DNA methylation can also affect cell cycle-related genes, thereby altering radiotherapy sensitivity. Radiosensitivity varies across different phases of the cell division cycle. It is worth noting that the radiosensitivity varies across different cell division cycles. Cells in the S phase exhibit resistance to irradiation, while those in the M and G2 phases display sensitivity to it. When radiotherapy is applied, cells in the sensitive phases like M or G2 are selectively eradicated.458 Therefore, hypermethylation of genes that promote cell entry into the G2/M phase can lead to radioresistance, such as hypermethylation of promoter site of the RPRM, which promotes radioresistance in nasopharyngeal carcinoma (NPC) cells.459 CCND2 is hypermethylated in radioresistant cell lines of HNSCC, and hypomethylated in radiosensitive cells.460 Besides, in nasopharyngeal carcinoma, hypermethylation of p53 and p21 can inhibit cell cycle arrest and reduce apoptosis, thereby leading to radiotherapy resistance.461 Furthermore, certain genes may also impact changes in related pathways, leading to radiotherapy resistance. The methylation-induced silencing of the tumor suppressor genes RASSF1 and RASSF2A, coupled with the subsequent activation of the Ras/PI3K/Akt signaling axis, constitutes the underlying mechanism of radioresistance in individuals with OSCC.461 Hypomethylation of the TM4SF4 upregulates its expression and the secretion of IGF, thereby activating the IGF1R/Akt/PI3K and NF-κB signaling cascades to drive cell proliferation, migration, and invasion in lung adenocarcinoma cells exhibiting intrinsic radioresistance.462 Besides, hypermethylation of TIMP3 activates the STAT1/FOXO1 pathway, leading to radiotherapy resistance463 (Fig. 4). In summary, DNA methylation wields a profound and intricate influence on the radioresistance of cancer cells through multiple dimensions, such as impinging on signaling pathways, DNA damage repair genes, cell proliferation and cycle-related genes, as well as related pathways. A profound comprehension of these mechanisms holds substantial promise for devising more targeted strategies to surmount radioresistance and enhance the therapeutic effectiveness of radiotherapy in oncological interventions.
RNA modifications in radiosensitivity
During radiotherapy, RNA modifications play a crucial role in regulating DNA damage repair, cancer stemness, apoptosis and G2/M arrest, all of which are relevant to radiosensitivity of cancer cells.464,465 Ionizing radiation can alter the levels of RNA modifications and the activity of related enzymes, which in turn may modulate the sensitivity of cancer cells to radiation.466,467
Research has demonstrated that the m6A methyltransferase METTL3 serves as a critical element in enhancing radioresistance by promoting DNA damage repair and inhibiting cell death pathways. In the context of GBM, exposure to IR has been shown to upregulate METTL3 expression. Notably, overexpressed METTL3 interacts with SOX2 transcripts, stabilizing them and thereby contributing to heightened DNA damage repair capabilities, which in turn bolsters the resistance of glioma stem-like cells to γ-irradiation.468 Additionally, METTL3-mediated m6A modification of mRNA impedes the degradation of H2A histone family member X (H2AX) mRNA, leading to increased H2AX expression and facilitating both DNA damage repair and cell survival, and then promote radiation resistance.469 In pharyngeal squamous cell carcinoma, METTL3 has been identified as a key inducer of radioresistance. It achieves this by enhancing the stability of circCUX1, which in turn binds to caspase1 mRNA and suppresses its expression. This inhibition of caspase1 leads to a decrease in programmed cell death, thereby conferring radioresistance.469,470 Additionally, in lung adenocarcinoma, METTL3 expression is upregulated, contributing to the stabilization of VANGL1. This upregulation activates the BRAF/TP53BP1/RAD51 pathway, reducing DNA damage and fostering radioresistance.471 Conversely, METTL3 also enhances radioresistance by modulating the invasiveness and migratory capabilities of cancer cells. In gastric cancer, the upregulation of WTAP, a subunit of the METTL3 complex, boosts TGF-β expression. This increase in TGF-β promotes EMT and the migration of gastric cancer cells, ultimately leading to radioresistance in gastric cancer472 (Fig. 4).
RNA demethylases also function as a central component in radiation sensitivity. Recent studies have shed light on the distinct roles of m6A demethylase ALKBH5 in radioresistance compared to METTL3. Contrary to METTL3, ALKBH5 does not have an opposing effect; instead, it enhances radiotherapy resistance in GBM stem cells by modulating HR.469,473 Furthermore, the transcription factor FOXM1 has been implicated in DNA damage repair. The upregulation of FOXM1 induced by IR is mitigated by ALKBH5 inhibition, suggesting an alternative pathway through which ALKBH5 may mediate radioresistance in GBM.474,475,476 In addition, the m6A demethylase FTO has been implicated in the development of radioresistance. This enzyme is found to be upregulated in radioresistant NPC, where it enhances the radioresistance of the cancer cells,477 This enhancement is achieved by promoting the activity of the deubiquitinase OTUB1, which in turn mediates anti-ferroptosis, a process that stabilizes the expression of SLC7A11 and is crucial in human cancer.478 Beyond its role in DNA DSB repair, excision repair also contributes to radioresistance. FTO is capable of demethylating β-catenin mRNA in cervical squamous cell carcinoma, thereby stabilizing β-catenin expression and activating the excision repair gene, ERCC1, which is a key factor in conferring resistance to radiotherapy.479 The biological functions of m6A modification extend beyond its “writing” (writers) and “erasing” (erasers) processes; its “reading” (readers) proteins also play a crucial role in regulating radiation sensitivity. YTHDF3, as an m6A reader, can recognize m6A modifications and promote the m6A-dependent translation of the DNA repair protein RAD51 paralog 4 (RAD51D), thereby affecting the role of hepatocyte nuclear factor 1-α (HNF1α) in promoting radioresistance in cervical cancer cells480 (Fig. 4). This discovery reveals the significant role of m6A in the regulation of radiation therapy sensitivity, providing new molecular mechanisms and promising anticancer targets of radiotherapy.
Beyond the realm of m6A, both N4-methylcytosine (m5C) and N7-methylguanosine (m7G) have been implicated in their association with radiosensitivity. The expression of the m5C writer NSUN6 has been found to be clinically correlated with radioresistance and unfavorable prognosis in cervical cancer.481 NSUN6 facilitates the m5C modification of NDRG1 mRNA, thereby enhancing its stability and activating the HR pathway, which in turn augments the radioresistance of cervical cancer.481 On the other hand, the m7G writer METTL1 is crucial for nonhomologous end-joining repair and plays a role in the radioresistance of HCC by modulating the DNA-dependent protein kinase catalytic subunit or DNA ligase IV.482 Additionally, METTL1 expression in tumor tissue is significantly associated with a poor prognosis for HCC patients undergoing radiotherapy. In the context of HCC, gene networks related to m7G and radiotherapy resistance, as well as prognostic models, have been established.483 In summary, RNA modifications play a crucial role in the occurrence, development, and the formation of radiotherapy resistance in cancer (Fig. 4). By precisely regulating RNA modifications, we can reduce the resistance of tumors to radiotherapy and thereby enhance the effectiveness of cancer treatment. This regulatory strategy not only targets the biological characteristics of cancer but also may offer new perspectives and approaches for cancer therapy.
Non-coding RNAs in radiosensitivity
LncRNAs
The role of lncRNAs in cancer drug resistance has now been firmly established.484 Emerging evidence suggests the involvement of lncRNAs in radiation therapy response. LncRNAs alter the radiosensitivity of cancer cells by regulating radiation-related pathways, including DNA damage repair, cell cycle, cancer stem cell phenotype, and apoptosis.485 For example, the downregulation of the lncRNA XIST expression inhibits the viability and survival of NSCLC cells, facilitates apoptosis, and enhances sensitivity to ionizing radiation. Conversely, XIST promotes the radioresistance of NSCLC cells by modulating the expression of miR-16-5p and WEE1. Therefore, it may serves as a novel target for NSCLC radiotherapy,486,487 Additionally, lncRNA SBF2-AS1 and lncRNA FAM201A also promote radiotherapy resistance in NSCLC by reducing cellular apoptosis488 (Fig. 4).
Furthermore, lncRNAs are capable of regulating the stemness of cancer cells, thereby impacting radiosensitivity. LncRNA HOTAIR can uphold the stemness of liver cancer stem cells (LCSCs) and further intensify their radioresistance through the JMJD6-BRD4-HOTAIR-LSD1-ERK2 (MAPK1) axis.489 LncRNA HOTAIR also exerts a pivotal influence in DNA damage repair by promoting recruitment of EZH2 to the MYC promoter and upregulating the transcription of DDR molecules such as KU70, KU80, DNA-PKs, and ATM, which increases DNA repair in breast cancer and leads to radioresistance.490 In addition to targeting ATM, lncRNA HOTAIR can also interact with ATR to reduce DNA damage and promote radiotherapy resistance in CRC334 (Fig. 4).
LncRNAs modulate intracellular autophagy levels, thereby affecting the radiosensitivity of cancer cells. There was a study indicated that LINC-RA1 maintained the stability of H2B K120 monoubiquitination (H2BK120ub1) by interacting with H2B and suppressing the engagement between H2Bub1 and ubiquitin-specific protease 44 (USP44), thereby suppressing autophagy and consequently fostering radioresistance in glioma.491 Another crucial function of lncRNAs is to act as a sponge for miRNAs, exerting pivotal functions in radioresistance through the modulation of genes or proteins at both the transcriptional and post-translational levels. LncRNA SP100-AS1 induces radioresistance in CRC through sponging miR-622 and enhancing the stability of ATG3.492 LncRNA FGD5-AS1 promotes the radiotherapy resistance in breast cancer cells by upregulating MACC1 expression through competitive binding with miR-497-5p.493 Besides, lncRNA also induce radioresistance via activating multiple signaling pathway, including PI3K/AKT/mTOR494 Wnt /β-Catenin495 YAP1/AKT.496
In summary, the mechanisms through which lncRNAs contribute to radiotherapy resistance are complex and diverse, presenting potential targets for adjunctive radiotherapy treatments in the future. Continued exploration of these molecular mechanisms is indispensable for devising novel strategies to enhance the efficacy of radiotherapy in cancer patients.
miRNAs
miRNAs play an intricate and pivotal role in tumor radiotherapy resistance. They exert an impact on the sensitivity of tumor cells towards radiotherapy by regulating a diverse array of molecular mechanisms and signaling pathways. Specifically, miRNAs modulate the DNA damage repair process, either promoting or inhibiting radiotherapy resistance. For instance, miR-200c-3p is downregulated in radiotherapy-resistant prostate cancer cells, where it promotes DNA damage repair by targeting HP1α, reducing cell death, and thus contributing to radiotherapy resistance.497 Additionally, miRNAs are involved in regulating the activation of cell cycle checkpoints, autophagy processes, and apoptosis, thereby affecting the efficacy of radiotherapy. miR-450a-5p inhibits autophagy to enhance the sensitivity of esophageal squamous cell carcinoma to radiotherapy.498 In head and neck cancer, miR-630 induces anti-apoptotic effects through the Nrf2-GPX2 molecular axis, enhancing radiotherapy resistance.499 miRNAs can also foster radiotherapy resistance by targeting specific signaling pathways, such as miR-193b-3p in nasopharyngeal carcinoma, which accelerates tumor-associated macrophages (TAM) activation by directly repressing mitogen-activated protein/ERK kinase kinase 3 (MEKK3), promoting the invasion and radiotherapy resistance of nasopharyngeal cancer cells.500 In breast cancer, miR-21 causes G2/M phase arrest and reduces apoptosis in cancer cells, which is related to radiotherapy resistance.501 Moreover, miRNAs also induce EMT in tumor cells, such as miR-6855-5p in pancreatic cancer, which promotes radiotherapy resistance by inducing EMT through the suppression of FOXA1.502 It is widely acknowledged that the level of reactive oxygen species (ROS) constitutes an important factor in radiosensitivity. Recent studies have demonstrated that miRNA could modulate ROS production.503 In glioma stem cells, the downregulation of miR-153 leads to the upregulation of its putative target Nrf2, resulting in the accumulation of glutathione peroxidase 1 (GPx1). This antioxidant enzyme increases radiation resistance by downregulating ROS levels.504 miRNAs exhibit a multifaceted and intricate role in the development of resistance to radiotherapy (Fig. 4). A comprehensive investigation into the mechanisms through which miRNAs influence radiotherapy resistance is crucial for the advancing novel therapeutic strategies and enhancing radiotherapy efficacy.
CircRNAs
CircRNAs play a role in the regulation of cancer resistance of radiotherapy via diverse mechanisms, among which functioning as miRNA sponges to modulate radiosensitivity is a prominent aspect. In gliomas, circ_0008344 can function as a molecular for miR-433-3p and promote the transcription of RNF2, thereby promoting radiotherapy resistance.505 In esophageal squamous cell carcinoma, circVRK1 positively regulates PTEN by operating as a molecular sink for miR-624-3p, and the upregulated PTEN suppresses the functionality of the PI3K/AKT signaling cascade, leading to increased radiotherapy sensitivity in esophageal squamous cell carcinoma.506 Additionally, circRNAs affect tumor radioresistance by influencing glycolysis. For example, circ-PITX1 promotes glycolysis in gliomas and induces radiotherapy resistance.507 CircRNAs also affect tumor radiotherapy resistance by influencing apoptosis, autophagy and EMT. In NSCLC, upregulated circ-0086720 inhibits cell survival and reduces apoptosis, thereby increasing its radiotherapy resistance.508 In CRC, circ-ZNF609 promotes advancement and radioresistance of prostatic carcinoma cells by accelerating the glycolytic action of the miR-501-3p/HK2 axis.509 In CRC, the upregulation of circ-0055625 promotes cell migration, proliferation, migration, and further induces radiotherapy resistance in CRC.510 CircRNAs also induce radiotherapy resistance by binding to related molecules. In hypopharyngeal squamous cell carcinoma (HPSCC), circCUX1 binds to caspase1 and inhibits its expression, leading to reduced release of inflammatory factors and the development of resistance to radiotherapy.470 During cell development, different cell cycles exhibit varying sensitivities to radiation, Specifically, the G2/M phase exhibits the highest sensitivity to radiation, whereas the S phase demonstrates radioresistance. CircRNAs can also regulate cell cycles and thereby influence radiotherapy sensitivity. In nasopharyngeal carcinoma, low expression of circFIP1L1 leads to an increased proportion of the S phase during cell development, resulting in radiotherapy resistance in nasopharyngeal carcinoma.511 Currently, research on the role of circRNAs in tumor radiotherapy remains in its nascent stage, and numerous aspects of their regulatory mechanisms remain obscure. It is evident that the majority of circRNAs act as miRNA sponges to activate or inhibit signaling pathways. We believe that further research could place greater emphasis on other molecular mechanisms, such as post-transcriptional modifications and translation mechanisms (Fig. 4). A thorough understanding of the molecular mechanisms of circRNAs will aid in identifying novel and effective diagnostic and therapeutic targets.
Chemoresistance
Chemotherapy is one of the cornerstones of cancer treatment, however, its effectiveness is frequently undermined by the phenomenon of chemoresistance, which diminishes the therapeutic outcomes for cancer patients.512 Chemoresistance is primarily characterized by the survival and sustained proliferation of tumor cells following multiple rounds of chemotherapy drugs, leading to tumor recurrence and metastasis, ultimately affecting patients’ survival and quality of life. Frequently employed chemotherapy agents encompass alkylating agents (cyclophosphamide), antimetabolites (5-fluorouracil and cytarabine), DNA crosslinking agents (cisplatin and carboplatin), anthracycline antibiotics (including doxorubicin, idarubicin, and mitoxantrone), antimicrotubular agents (paclitaxel and docetaxel), topoisomerase inhibitors (etoposide), nucleoside analogs (gemcitabine), DNA methyltransferase inhibitors (5-azacytidine), and proteasome inhibitors (bortezomib, melphalan, and carfilzomib).513 The development of chemoresistance is not due to a single factor but involves the complex and interwoven effects of multiple factors, including oncogene activation, impaired DNA repair function, hypoxia in the tumor microenvironment, and changes in cellular metabolism.514 Epigenetic modifications are notably significant in the context of chemoresistance. These modifications operate through distinct mechanisms, allowing for the control of gene expression and cellular phenotype without altering the fundamental DNA sequence, thus influencing the effectiveness of chemotherapeutic agents. For instance, epigenetic alterations such as DNA methylation and histone modification can lead to the silencing of tumor suppressor genes, thereby facilitating the development of chemoresistance.515 Furthermore, during chemotherapy, epigenetic reprogramming may transform the originally transient transcriptional state into a stable drug-resistant state.516 In the ensuing discussion, we will elaborate on the specific impacts of the epigenetic modifications on chemoresistance.
Histone modifications in chemoresistance
Recently, a multitude of researches underscored the significant role of histone PTMs in shaping chemoresistance to cancer therapy, influencing processes such as apoptosis, EMT, DDR, and the intricate dynamics of cancer stem cell behavior.517,518
In the realm of histone acetylation, HDACs have emerged as pivotal factors in chemotherapy resistance. HDACs mitigate cancer chemotherapy resistance by inhibiting the proliferation of cancer stem cells though regulating oncogene promoters. Domatinostat (a histone deacetylase inhibitors (HDACi)) have been shown to sensitize pancreatic cancer to gemcitabine/taxol by targeting the cancer stem cell compartment through the modulation of FOXM1.519 Additionally, inhibitors of HDAC11 are found to attenuate the self-renewal capacity of lung adenocarcinoma stem cells and overcome resistance to chemotherapy agents by downregulating Sox2, thereby offering novel pathways for enhancing the efficacy of cancer treatments520 (Fig. 5).
The correlation between histone methylation and resistance to cancer chemotherapy has been thoroughly investigated, revealing its influence on chemotherapy resistance through the modulation of DSBs repair and the induction of apoptosis. For instance, the overexpression of EZH2, a histone methyltransferase, has been shown to stimulate chemoresistance in glioblastoma, small-cell lung cancer, and HNSCC.521,522 Regarding the underlying mechanisms, EZH2-mediated gene silencing of SLFN11, a crucial factor in DNA damage repair, via H3K27 hypermethylation, leads to chemoresistance in small-cell lung cancer cells.521 Additionally, histone methylation alterations modulate tumor cell apoptosis and proliferation to influence chemotherapy resistance. Reports indicate that targeting EZH2 can modulate the H3K27me3 level of HMGA2 to inhibit PI3K/Akt phosphorylation, thereby suppressing cancer cell proliferation and reducing the resistance of CRC to oxaliplatin.523 In addition, some histone demethylases are also implicated in the development of radiation resistance. The level of G9a is positively correlated with cisplatin resistance, as it inhibits apoptosis by modulating glutathione levels, thereby increasing cisplatin resistance.524 Furthermore, KDM2B and GSK can enhance the chemoresistance of glioblastoma by promoting DNA damage repair.525 The upregulation of KDM6 can also increase the chemoresistance of lymphoma by upregulating the expression of BCL-6, which reduces apoptosis.526,527 Similarly, a Jumonji inhibitor, JIB-04, may serve as a potential therapeutic agent for chemo-resistant NSCLC refractory to taxane and platinum-based chemotherapy, through upregulation of pro-apoptotic genes528 (Fig. 5).
In addition to histone acetylation and methylation, other histone modifications also affect chemoresistance. It has been reported that reduced levels of histone H2AK119ub1 in ovarian cancer inhibit apoptosis, leading to chemoresistance.529 Deubiquitinating enzymes also affect the sensitivity of cancer cells to chemotherapeutic drugs through various mechanisms. For instance, USP7 contributes to chemoresistance in cervical cancer by maintaining the stability of the Chk1 protein and reducing DNA damage responses.530 Additionally, USP22 induces resistance to cisplatin in lung adenocarcinoma by regulating DNA damage repair mediated by γH2AX and apoptosis mediated by Ku70/Bax.531 Histone lactylation has also been found to affect chemoresistance. H3K9 lactylation activates LUC7L2 transcription, which reduces the expression of MutL homolog 1 (MLH1), thereby inhibiting mismatch repair (MMR), and ultimately leading to resistance to temozolomide (TMZ) in GBM532 (Fig. 5). Overall, histone modifications are closely related to chemoresistance, which has been well demonstrated in preclinical studies. Furthermore, the combination of histone modification inhibitors with traditional chemotherapeutic drugs has been used to improve therapeutic outcomes.
DNA methylation in chemoresistance
DNA methylation constitutes a crucial mechanism within the domain of epigenetics, which exerts its regulatory function on gene expression through the attachment of methyl groups to DNA molecules. The role of DNA methylation in cancer chemotherapy resistance has been widely studied and concerned. DNA methylation has the potential to influence the expression of specific genes, thereby contributing to the development of drug resistance in tumor cells. In bladder cancer, merely 35% of metastatic bladder cancer patients initially show a response to cisplatin chemotherapy, and in the end, most bladder cancer patients who are sensitive to cisplatin chemotherapy develop resistance.533 High methylation of TLX3, TAp73, and HOXA9 promotes the anti-apoptotic effects of bladder cancer cells, while high methylation of SOCS3, STST3 and SOX2 promotes the development of cancer stem cells, leading to cisplatin resistance.534,535 Hypermethylation of the DNA repair protein MLH1 hypermethylation results in platinum resistance in ovarian cancer through reduced MLH1/c-Abl apoptotic signaling.536 Additionally, MLH1 hypermethylation causes an increase in drug resistance in CRC and is responsible for enhanced resistance by means of the loss of DNA mismatch repair capability, which causes genomic instability537 (Fig. 5). Besides apoptosis, DNA methylation plays a significant role in causing drug resistance in cancer cells through the modulation of the expression of transporters and metabolism-related genes. In HCC, the hypomethylation of drug delivery gene (ABCB1, ABCC2, ABCC5 and ABCG2) promoters can enhance the expression of these genes. This, in turn, boosts the cells’ ability to pump out drugs, consequently reducing the concentration of sorafenib within tumor cells and attenuating the cytotoxicity of sorafenib and hypomethylation and subsequent upregulation of ABCB1 and ABCG2 promote paclitaxel resistance in ovarian cancer.538 Beside, DNA hypomethylation of around the transcriptional start site of human organic cation transporter-1 (hOCT1, gene SLC22A1) decreases the uptake of quinine-inhibitable sorafenib by hepatoma cells, ultimately leading to sorafenib resistance in liver cancer cells539 (Fig. 5). Overall, DNA methylation plays a multifaceted role in various cancers by impacting gene expression patterns and contributing to the development of drug resistance. This underlines its critical importance in the pursuit of understanding and potentially surmounting the challenges posed by chemotherapy.
RNA modifications in chemoresistance
Epigenetic modifications of RNA are intricately linked to the sensitivity to various chemotherapy drugs.540 In particular, m6A modification is pivotal in modulating apoptosis, cancer cell migration, and invasion, thereby affecting chemotherapy resistance. In recent years, the connection between m6A modification regulators and chemoresistance has garnered significant attention and progress.476,541,542,543 HNRNPA2B1 is an RNA-binding protein that is upregulated in gastric cancer that has developed resistance to chemotherapy drugs. This protein stabilizes lncRNA NEAT1 through m6A modification, thereby activating the Wnt/β-catenin signaling pathway. The activation of this pathway endows gastric cancer cells with stem cell characteristics and enhances their resistance to 5-fluorouracil (5-FU).544 METTL3, an m6A “writer,” affects chemotherapy resistance by regulating autophagic degradation. LncRNA ARHGAP5-AS1 can recruit METTL3 to promote m6A modification of ARHGAP5 mRNA, maintaining its stability in the cytoplasm and preventing it from being degraded by autophagy, which leads to resistance to cisplatin.545 Additionally, METTL3 can induce chemotherapy resistance by triggering DNA damage repair. In breast cancer cell lines MCF-7 and MDA-MB-231, METTL3 augments resistance to doxorubicin (ADR) by improving the efficiency of HR and alleviating ADR-induced DNA damage.546 In MCF-7 cells that have developed resistance to ADR, the expression level of METTL3 is elevated. Furthermore, METTL3 also promotes the maturation of pri-miR-221-3p through m6A modification, which is associated with ADR resistance.547,548 In addition to m6A “writers,” “erasers” such as ALKBH5 also play an important role in chemotherapy resistance. ALKBH5 promotes the demethylation of WIF-1 mRNA, enhances its transcription, and inhibits the Wnt signaling pathway in AsPC-1 and PANC-1 cells, thereby increasing the sensitivity of pancreatic cancer cells to gemcitabine.549 Moreover, ALKBH5 affects chemotherapy resistance by regulating the stemness of cancer cells. Through m6A demethylation, ALKBH5 also stabilizes FOXO1 mRNA, leading to elevated levels of FOXO1 protein. This upregulation increases the expression of superoxide dismutase 2 (SOD2), diminishing intracellular ROS, and preserving the stemness of cancer cells as well as resistance to doxorubicin.228 In the treatment of ovarian cancer, elevated levels of ALKBH5 were identified to enhance the multiplication of epithelial cells and tumor growth, leading to resistance to cisplatin.550 Furthermore, the interaction between ALKBH5 and the homeobox A10 can inhibit the degradation of the JAK2/STAT3 signaling pathway mediated by YTHDF2, a key factor in the development of cisplatin resistance.550 Concurrently, another demethylase, FTO, through its interaction with lncRNAs adjacent to X-inactive specific transcript, facilitates the demethylation process of phosphoinositide-dependent kinase-1, which not only promotes aerobic glycolysis in glioma cells but also strengthens their resistance to temozolomide.551 The role of m6A reader proteins in chemoresistance is gradually being recognized by the scientific community. The latest research highlights the importance of elevated YTHDF1 in preserving the stem cell-like characteristics of cisplatin-resistant ovarian cancer cells. By promoting the translation of TRIM29 mRNA, YTHDF1 enhances the colony formation, spheroid formation capabilities, and invasiveness of cisplatin-resistant SKOV3/DDP and A2780/DDP cells239 (Fig. 5). This finding is significant for understanding the mechanisms of ovarian cancer therapy resistance and developing new therapeutic strategies.
In addition to the aforementioned m6A modification, m5C and m7G modifications also play significant roles in chemoresistance. In esophageal squamous cell carcinoma (ESCC), the m5C methyltransferase NSUN2 can upregulate the expression of TIGAR and GRB2, which increases the production of glutathione, reduces cell apoptosis, and promotes cell proliferation, collectively leading to chemoresistance in ESCC.552 Studies have also found that m5C reader proteins are associated with chemoresistance. In ovarian cancer patients, YBX1 modulates the expression of multiple downstream targets, including AKT, thereby promoting tumor proliferation and enhancing resistance to paclitaxel.302 Compared to non-resistant HCC cases, YBX1 expression is increased in chemotherapy-resistant HCC, indicating that YBX1 plays a key role in the observed chemoresistance in HCC.552 The m7G modification affects chemoresistance by regulating DNA damage repair, cancer migration, and invasion309,321,553,554 (Fig. 5). The m7G “writer” METTL1 suppresses the expression of S100A4 through m7G modification and upregulates miR-149-3p via a p53-dependent mechanism, thereby reducing the chemoresistance of CRC cells to cisplatin.548 Furthermore, the silencing of METTL1, which also methylates tRNAs at the variable loop of several tRNAs, has been shown to increase the sensitivity of cancer cells to 5-FU by increasing cell death555,556 (Fig. 5). These findings provide novel insights into the role of chemoresistance due to RNA modifications, which pave the way for developing new strategies to overcome resistance.
Non-coding RNAs in chemoresistance
LncRNAs
LncRNAs are frequently dysregulated in a diverse range of malignancies and engage in interactions with numerous RNAs and proteins, thereby exerting an impact on chemoresistance. LncRNAs regulate chemoresistance in cancer through a variety of molecular pathways including suppression of apoptosis, DNA damage response, multidrug efflux, EMT, as well as functioning as competitive endogenous RNA. When integrated with other regulatory mechanisms, these processes converge to form a highly intricate network of signaling that ultimately drives chemoresistance.557 LncRNAs can serve as molecular sponges for miRNAs, modulating miRNA activity and consequently activating signaling pathways associated with chemotherapeutic drug resistance, such as Wnt/β-catenin,558 PTEN/AKT,559 PTEN/PI3K560 and MAPK/ERK pathways. Additionally, the same lncRNA could regulate drug resistance in tumor cells through multiple mechanisms. For example, the lncRNA PVT1 promotes anti-apoptosis and chemotherapy resistance in gastric cancer by upregulating Bcl2 expression.561 Meanwhile, silencing lncRNA PVT1 with RNAi may hinder gastric cancer progression by increasing paclitaxel sensitivity.562 Additionally, PVT1 has been reported to act as a miRNA sponge, modulating miRNAs and their downstream signaling pathways to promote tumor drug resistance.563 For instance, VT1 may enhance cisplatin resistance in gastric cancer via the miR-3619-5p/TBL1XR1 axis and the miR-30a-5p/YAP1 pathway.564,565 Besides, lncRNAs can bind with RBPs to influence the DDR signaling pathway, regulating genes linked to cancer chemoresistance.566 Specifically, lncRNA SNHG12 enhances X-linked inhibitor of apoptosis protein (XIAP) transcription and stability by interacting with the RNA-binding protein Hu antigen R (HuR), promoting tumor growth and cisplatin resistance in NSCLC cells.325 LncRNA can also interact with other DDR-related protein to affect chemoresistance, including che-1, RBMX (RAN-binding motif protein X chromosome), PARP-1, YB-1 (Y-box protein 1).567,568,569 They also regulate efflux transporter activity, as seen with lncRNA linc00518 sponging miR-199a to affect ABCC1 expression, causing doxorubicin resistance in breast cancer cells.90 Recent studies indicate that lncRNAs are involved in the regulation of cancer-related stemness.570,571 Another study indicated that lncRNA NEAT1 was overexpressed in TNBC tissues and cell lines, and the silencing of NEAT1 was able to decrease stem cell populations, such as ALDH+, CD44+/CD24−, and SOX2+, and enhances the chemosensitivity of TNBC cells, including cisplatin and taxol.572,573 In addition, LncRNA SNHG14 contributed to trastuzumab tumorigenesis and resistance in breast cancer through the regulation of PABPC1 expression by H3K27 acetylation.574 Another lncRNA, HOTAIR, has been positively correlated with tamoxifen resistance, as it recruits the histone methyltransferase EZH2, which leads to tamoxifen resistance in breast cancer.575,576,577 Moreover, lncRNAs can perform a variety of functions in response to diverse environmental stimuli through dynamic structural changes. LncRNA LINC01956 undergoes dynamic structural remodeling that enhances the recruitment of O6-methylguanine DNA methyltransferase (MGMT) mRNA expression, which promotes DNA damage repair and tumor proliferation.578 LncRNAs can not only be regarded as potential biomarkers for the early diagnosis of cancer patients, but their expression profiles may also assist in predicting the sensitivity of cancer cells to different chemotherapy drugs, thereby alleviating chemoresistance (Fig. 5). the identification of lncRNAs that are uniquely expressed in chemoresistant cells can offer novel therapeutic alternatives for circumventing chemoresistance and cancer recurrence. Nevertheless, further research is requisite to validate the roles of the relevant lncRNAs.
miRNAs
miRNAs assume a crucial and pivotal role in the context of chemoresistance. A growing body of evidence have suggested that in various types of tumors, the expression levels of specific miRNAs are intricately associated with resistance to chemotherapeutic drugs. For instance, in prostate cancer, the upregulation of miR-200b-3p, miR-375, and miR-34b-3p may be associated with resistance to paclitaxel.579 Studies have revealed that miRNAs have the capacity to modulate the responsiveness of tumor cells to chemotherapeutic agents by engaging specific signaling cascades. An illustrative example is miR-99b-3p, which specifically targets the PPP2CA gene, consequently augmenting the phosphorylation of the AKT/mTOR signaling pathway. This modulation of the signaling cascade can substantially influence cell migration and potentially lead to the development of resistance to paclitaxel in breast cancer cells.580 Likewise, in HCC, miR-122 and miR-181a may facilitate resistance to sorafenib by modulating the RAS/RAF/ERK signaling pathway.581 MiR-31-5p, via extracellular vesicles (EVs), modulates the Hippo signaling pathway across diverse cell types within the tumor microenvironment, thereby contributing to the chemoresistance of pancreatic cancer cells to gemcitabine.582 MiRNAs can also influence the expression of target genes by binding to mRNA, thereby inhibiting translation or promoting degradation, which in turn affects the responsiveness of tumor cells to chemotherapeutic agents. Specifically, miR-223 downregulates the expression of FBXW7, resulting in resistance to doxorubicin in CRC583 (Fig. 5). Moreover, miR-223 suppresses the expression of FOXO3, contributing to the resistance of prostate cancer to docetaxel treatment and, through related mechanisms, also inducing cisplatin resistance in pancreatic cancer.584 Similarly, miR-21 downregulates the human DNA MutS homolog 2 (hMSH2) in CRC cells, thereby inducing resistance to 5-FU.
Moreover, miRNAs are capable of exerting an impact on the efficacy of chemotherapy by regulating the generation of tumor stem cells. In HCC, miR-2117 is found to be downregulated, and this downregulation serves to facilitate the self-renewal of liver tumor stem cells as well as tumorigenesis, ultimately culminating in cisplatin resistance.585 In NSCLC, miR-3163 targets the EXO1/Polη/Polι axis, inhibiting the growth of cancer stem-like cells in NSCLC and inducing apoptosis, thereby increasing the resistance of these cells to cisplatin.586 In NSCLC, miR-3163 targets the EXO1/Polη/Polι axis, inhibiting cancer stem-like cell growth and inducing apoptosis, which increases resistance to cisplatin. miRNAs can regulate gene transcription and epigenetics, affecting tumor cell response to chemotherapy. miRNA-381 may target multidrug resistance proteins (ABCG2, ABCC3, ABCC5) and stemness factors, enhancing glioblastoma sensitivity to temozolomide,587 while reducing the molecular level of miRNA-318 can lead to drug resistance in glioblastoma. miRNAs influence intracellular processes like apoptosis and DNA repair, affecting tumor cell drug resistance. For instance, increased miR-21 inhibits apoptosis in glioblastoma, causing sunitinib resistance.588 In NSCLC, miR-223 targets FBXW7, which then boosts autophagy and EMT, ultimately leading to resistance to both cisplatin and doxorubicin589 (Fig. 5). These research findings highlight the intricate and multifunctional role of miRNAs for chemoresistance and offer promising targets for the creation of innovative therapeutics, which holds significant implications for future cancer treatment strategies.
CircRNAs
Accumulating evidence has underscored the essential regulatory function of circRNAs in the carcinogenesis and progression of tumors. CircRNAs also play a role in tumor chemoresistance. CircRNAs can function as miRNA sponges and regulate chemosensitivity via the competitive endogenous RNA (ceRNA) mechanism. For instance, in osteosarcoma, circ_0004674 can sponge miR-142-5p, upregulate the anti-apoptotic protein MCL1 of the Bcl-2 family, leading to osteosarcoma progression and doxorubicin resistance.590 In laryngeal squamous cell carcinoma (LSCC), circPARD3 sponges miR-145-5p to activate the PRKCI-Akt-mTOR pathway and inhibit autophagy, promoting tumor cell proliferation, migration, invasion, and cisplatin resistance.591 Moreover, circRNAs regulate the sensitivity to chemotherapy drugs by directly interacting with proteins. In bladder cancer, circPTK2 binds with PABPC1, enhancing its ability to stabilize SETDB1 mRNA, promoting SETDB1-mediated EMT, significantly increasing in vitro migration and invasion capabilities, as well as gemcitabine resistance.592 CircIPO7 binds with the cytoplasmic Y-box binding protein-1 (YBX1), activating AKT phosphorylation to promote YBX1 nuclear translocation, increasing nasopharyngeal carcinoma’s resistance to cisplatin treatment.593 CircRNAs also affect the polarization of tumor-associated macrophages, thereby regulating chemoresistance. In ovarian cancer, circITGB6 directly interacts with IGF2BP2 and FGF9 mRNA, stabilizing FGF9 mRNA and inducing TAM polarization towards the M2 phenotype, increasing ovarian cancer’s resistance to cisplatin594 (Fig. 5). CircRNAs promote cell proliferation and induce tumor chemoresistance by activating related signaling pathways. For example, circTRIM1 enhances MARCKS translocation and activates the PI3K/AKT/mTOR signaling pathway, promoting chemoresistance to doxorubicin in TNBC.595 CircRNAs also regulate chemosensitivity by affecting intracellular molecular processes, such as apoptosis, autophagy, and DNA damage repair. The novel protein encoded by circATG4B can promote autophagy, inducing oxaliplatin resistance in CRC.596 CircPDIA3 can inhibit proptosis, thereby promoting oxaliplatin resistance in CRC597 (Fig. 5). In summary, circRNAs play a complex and crucial role in tumor chemoresistance, affecting the sensitivity of tumor cells to chemotherapy drugs through various mechanisms, including miRNA sponge function, direct interaction with proteins, affecting tumor-associated macrophage polarization, and regulating intracellular molecular processes. These discoveries offer an important theoretical foundation for the formulation of new cancer treatment strategies centered around circRNAs. They are anticipated to create new paths for future tumor treatment.
piRNAs
The aberrant expression of piRNA has been reported to promote chemoresistance across various cancer types.598,599,600,601,602 Numerous studies have shown that piRNAs and PIWILs are linked to key cancer traits, such as cell proliferation, evasion of cell death, metastasis, invasion, cell cycle regulation, all of which are linked with chemotherapy resistance393 (Fig. 5). For example, piR-1919609 and PIWIL2 was observed to be significantly upregulated in platinum-resistant ovarian cancer tissues. This overexpression is significantly linked to a poor prognosis adverse prognosis and a reduced recurrence-free survival period in ovarian cancer patients. Further investigations revealed that piR-1919609 primarily confers resistance to DDP by promoting cancer cell proliferation and inhibiting apoptosis.598 Similarly, studies have indicated that piR-39980 facilitates cellular motility and invasiveness by the stimulation of MMP-2, while concurrently inhibiting apoptosis via the negative regulation of SERPINB1.601,602 This dual mechanism plays a role in the development of chemoresistance in neuroblastoma and osteosarcoma cells. Additionally, a piRNA-like small RNA, piRNA-Ls, has been shown to induce chemoresistance to DDP-based therapy by suppressing apoptosis in lung squamous cell carcinoma.600 A recent study found that piR-FTH1 is frequently downregulated in six human cancer cell lines, with high Fth1 expression linked to doxorubicin resistance. Introducing external piR-FTH1 reduced Fth1 mRNA via HIWI2 and HILI, increasing TNBC cells’ doxorubicin sensitivity by 20-fold.599 In summary, piRNAs have been shown to regulate the expression of tumorigenic genes and cancer-suppressing genes at the transcriptional or post-transcriptional level by interacting with PIWI, thereby influencing the development of chemoresistance in various cancers. In summary, piRNAs interact with PIWI to regulate cancer-inducing genes and tumor-suppressing genes, affecting cancer chemoresistance (Fig. 5). They also recruit epigenetic writers, like histone-modifying enzymes and methyltransferases, to modulate DNA and m6A methylation and histone modifications, influencing key signaling pathways associated with therapeutic resistance. The crosstalk between these mechanisms will be further discussed.
Immunotherapy
Immunotherapy has emerged as a truly groundbreaking advancement in cancer treatment, for it triggers the patient’s immune system to identify and assault tumor cells. Immune checkpoint inhibitors, such as drugs targeting PD-1, PD-L1, and CTLA-4, block the immune evasion mechanisms of tumor cells, thereby enhancing the anti-tumor activity of immune cells.84,603,604,605,606 Additionally, technologies like adoptive cell transfer immunotherapy offer a variety of treatment options.607 Despite these advancements, resistance to immunotherapy has emerged as a significant focus of contemporary research. Epigenetic modifications are pivotal in this context, contributing to the tumor’s resistance to immune checkpoint inhibitors and other immunotherapies by modulating immune cell function, influencing the tumor microenvironment, and regulating gene expression.608,609 Acquiring a comprehensive understanding of these complex epigenetic regulatory networks is crucial for developing novel cancer treatment strategies. For instance, integrating epigenetic therapy with immunotherapy holds promise for enhancing the therapeutic response rates in cancer patients and potentially prolonging their survival, thereby offering increased hope for patient outcomes. In the subsequent text, we will provide a detailed examination of the specific effects of the aforementioned epigenetic modifications on resistance to immunotherapy.
Histone modifications in immunotherapy resistance
The intricate relationship between histone modification and immunotherapy resistance has been gradually explored. Recent studies have shown that histone acetylation could regulate the expression of immune checkpoints, helping tumors evade immune system surveillance and influencing the infiltration of immune cells in the tumor microenvironment. For example, HDACi can increase the levels of PD-1 ligands in melanoma, thereby enhancing the immunotherapeutic effects of PD-1 blockers.610 Moreover, HDACi can upregulate PD-L1 mRNA and protein expression in a time-dependent manner in TNBC cells and significantly enhance the in vivo response to PD-1/CTLA-4 blockade in the triple-negative 4T1 breast cancer mouse model.610 HDACi enhance the immunotherapy effect in TNBC by regulating tumor growth. As an HDAC inhibitor, SAHA effectively inhibits tumor growth and significantly extends overall survival in immunocompetent GBM intracranial xenograft mouse models. Additionally, SAHA suppresses the activity of regulatory T cells (Treg) by targeting the c-Myc/CCL1 signaling pathway in glioma stem cells, thereby enhancing the efficacy of PD-L1 blockade therapy611 (Fig. 6).
Histone methylation, like acetylation, plays a crucial role in the regulation of PD-L1 expression. Research has found that the histone demethylase LSD1 is a regulator of PD-L1 expression, exhibiting varying regulatory patterns across different types of cancer.612 In CRC cells, the expression of LSD1 can impair the TCF1+PD-1 precursor cell subset of CD8+ T cells in the tumor microenvironment, thereby increasing the body’s resistance to PD-1 therapy.613,614 Furthermore, high expression of LSD1 in tumors can also promote the expression of T-cell exhaustion markers, including PD-1, CTLA4, TIM3, and TIGIT, indicating that LSD1 expression contributes to T-cell exhaustion and leads to immune evasion. In clinical treatment, combination therapy with LSD1 inhibitors and PD-L1 has successfully helped many patients overcome resistance to PD-1 blocking antibodies.615,616 On the other hand, high expression of PRMT1 or PRMT5 is negatively correlated with the immune activation of CD8+ T cells and natural killer (NK) cells, but positively correlated with the infiltration of myeloid-derived suppressor cells (MDSCs) and cancer-associated fibroblasts (CAFs), suggesting an immunosuppressive microenvironment.617 Therefore, targeting PRMT1 and PRMT5 is expected to become a new approach to overcoming resistance to immunotherapy (Fig. 6).
Histone lactylation is also reported to be linked to immunotherapy resistance. H3K9la has been identified as a specific modification site in HNSCC. It is widely recognized that IL-11 transcriptionally activates immune checkpoint genes via JAK2/STAT3 signaling in CD8+T cells. In a recent study, H3K9la positively correlates with IL-11 expression and unfavorable immunotherapy responses in patients.618 Thus, targeting H3K9la may be favorable to activate CD8+ T cells and reduce immunotherapy resistance (Fig. 6). These research findings underscore the importance of histone modifications in tumor immune evasion and imply that aiming at histone modifications may be a promising new approach to overcome resistance to immunotherapy.
DNA methylation in immunotherapy resistance
DNA methylation predominantly manifests its influence on conferring resistance to immunotherapy through its regulatory mechanisms governing tumor immune evasion. Extensive studies have demonstrated that DNA methylation can exert an impact on the escape of tumor cells from the immune surveillance through modulating the transcription of immune checkpoint molecules. In esophageal malignancy (EC), excessive hypomethylation of well-recognized biomarkers, such as PD-L1 and HER2, has given rise to their overexpression within the tumor microenvironment, thereby endowing it with immunosuppressive characteristics.619,620 DNA methylation indeed has the capacity to influence the transcription of PD-L1, thereby fostering resistance to immunotherapy. In the context of glioblastoma, the EZH2/H3K27Me3/DNMT1 complex orchestrates the methylation of the AP-2α gene, inhibiting its transcriptional activity. This diminished expression of AP-2α is associated with elevated levels of PD-L1 in high-grade glioma tissues. Elevated expression of PD-L1 facilitates the evasion of tumor cells from T-cell-mediated immune surveillance.621 In melanoma, genome-wide hypomethylation induces the upregulation of PD-L1 and the secretion of inhibitory cytokines, concomitant with EMT alterations that collectively foster an immunosuppressive microenvironment.622,623 DNA methylation may also affect the expression of tumor-associated antigens, leading to immune evasion. Hypermethylation of the transporter 1 ATP-binding cassette subfamily B member (TAP1) has been shown to suppress TAP1 expression in immunoreactive cancer stem cells (CSCs) within murine models of breast carcinoma, these CSCs manifest a downregulation in the expression of transporter associated with antigen processing genes and co-stimulatory molecules, thereby reducing their sensitivity to T-cell-mediated immune surveillance and potentially culminating in immunotherapy resistance.624,625 Additionally, the methylation of chemokines can also lead to resistance to immunotherapy. In a murine model of hepatocellular carcinoma, the hypermethylation of CXCL9 and CXCL10 promoter leads to reduced expression of these chemokines. The downregulation of CXCL9 and CXCL10 reduces the count of T cells that infiltrate the tumor, thereby increasing the risk of resistance to immunotherapy.626 A study has developed an index of methylation-based epigenetic silencing (iMES) signature. They found that high iMES is linked to VEGF pathway silencing, endothelial cell attenuation, immune modulation, EZH2 induction, BAP1/SETD2 depletion, and resistance to immune checkpoint inhibition (ICI).627 Besides, the DNA methylation of related molecules may also affect the infiltration of immune cells, leading to resistance to immunotherapy. Hypermethylation of ACAP1 is negatively correlated with the infiltration level of tumor-infiltrating lymphocytes, thereby reducing the sensitivity to immunotherapy628 (Fig. 6). A thorough elucidation of these methylation-mediated mechanisms is essential for formulating approaches to surmount immunotherapy resistance and augment efficacy for cancer immunotherapy in the future.
RNA modifications in immunotherapy resistance
Recent studies have underscored the pivotal function of RNA modification regulators in regulating the response to immune checkpoint blockade therapy. Numerous investigations into m6A modification have elucidated its significant association with resistance to immune checkpoint inhibitors, particularly those targeting the PD-1/PD-L1 axis.222,629,630 The overexpression of METTL3 in thyroid cancer cells can enhance the efficacy of anti-PD-1 therapy. In a recent study, it was found that the downregulation of METTL3 promotes the demethylation of CD70 mRNA, leading to an increase in the number of regulatory T cells (Tregs) with immunosuppressive functions and terminally exhausted T cells, thereby enhancing the resistance to immune therapy.631 In addition, FTO is also a regulator of resistance to immunotherapy. Under metabolic stress, melanoma cells increase the level of FTO through autophagy and NF-κB signaling pathways, resulting in resistance to interferon-γ (IFN-γ) and anti-PD-1 treatment. In melanoma cells, knocking down the FTO gene can accelerate the RNA degradation rate of intrinsic pro-tumor genes including PD-1, CXCR4 and SOX10 through YTHDF2. Therefore, inhibiting FTO can reduce the resistance to immune therapy632 (Fig. 6).
Targeting m5C represents a novel approach in the field of mRNA-based therapeutics. m5C-modified mRNA can reduce immune recognition by inhibiting Toll-like receptor 3 (TLR3) in the endoplasmic reticulum, thereby promoting immune evasion.633 In immunologically “cold” tumors, the activation of the cGAS/STING axis (cyclic GMP-AMP synthase/STING axis) not only promotes tumor formation but also enhances resistance to PD-1 immunotherapy. NSUN2, known as an eraser of m5C, can suppress the cGAS/STING signaling pathway when overexpressed, thereby reducing the immunotherapy resistance of tumors.634 NSUN2 methylates ICAM-1 mRNA and promotes its translation, inhibiting the polarization of M2-type macrophages, which in turn reduces tumor metastasis and enhances the effectiveness of immunotherapy. Additionally, targeting the expression of NSUN2 may improve the immunotherapy outcomes in HNSCC.635,636 The knockout of METTL1 (m7G writer) significantly improves the efficacy of anti-PD-1 treatment for intrahepatic cholangiocarcinoma, indicating that modulating mRNA methylation can enhance the effects of immunotherapy556 (Fig. 6). Furthermore, the knockout of YBX1 reverses resistance to immunotherapy by blocking PD-L1 expression and activating T cells in the tumor microenvironment, providing a new strategy for immunotherapy.637
Non-coding RNAs in immunotherapy resistance
LncRNAs
LncRNAs are crucial in immunotherapy resistance, exerting an impact on a multitude of mechanisms and pathways. They are involved in the regulation of immune checkpoint molecules such as PD - L1, thereby facilitating tumor cells’ ability to evade the immune system. For example, lncRNA SNHG29 boosts PD-L1 expression by activating YAP, while LINC00460 acts as a molecular adsorbent for miR-186-3p, which results in an elevation for MYC, CD47, and PD-L1, consequently strengthening immune evasion in CRC cells.638,639 LncRNA MIR155HG modulates PD-L1 expression via m6A modification, aiding HCC’s immune escape.640 Additionally, lncRNAs can influence immune evasion by altering antigen expression. Cancer-associated fibroblast-derived EVs containing lncRNA RP11-161H23.5 reduce HLA-A expression in pancreatic carcinoma cells, facilitating tumor proliferation and immune evasion641 (Fig. 6).
Besides, lncRNAs exert a significant influence on various immune cells within the tumor microenvironment, such as T cells, myeloid-derived suppressor cells (MDSCs), and natural killer (NK) cells, thereby impacting their responsiveness to immunotherapy. Moreover, they are actively involved in multiple signaling pathways that contribute to the enhancement of regulatory T cells, ultimately creating an immunosuppressive milieu that favors tumor growth. For example, lnc-EGFR aids liver cancer cell growth by promoting Treg cell differentiation and suppressing CTL activity, facilitating immune escape.642 In breast cancer, increased lncRNA SNHG1 in CD4+ T cells boosts IDO expression by binding to miR-448, promoting Treg cell maturation and immune evasion.643 During the process of tumor immune escape, cytotoxic T lymphocytes (CTLs) may become dysfunctional or exhausted,644 and lncRNAs play a significant regulatory role in this process. For instance, lncRNA NEAT1 can impede the proliferation of CTLs by suppressing the cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway, thereby promoting immune escape.645 Similarly, lncRNAs influence other innate immune cells, contributing to an immunosuppressive microenvironment and facilitating immune escape. For instance, LINC00963 can hinder the differentiation and maturation of dendritic cells by binding to miR-612, which assists gastric cancer cells in evading the immune system.646 LncRNAs also participate in the formation of resistance to immunotherapy by affecting various molecular pathways within tumor cells, such as regulating cell cycle, apoptosis, metabolism, and epigenetic modifications. For example, lncRNA TYMSOS promotes the growth and metastasis of breast cancer cells and leads to immune escape through the CBX3/ULBP3 or SYVN1/ULBP3 axis.647 Furthermore, lncRNAs partake in the formation of resistance to immunotherapy by modulating diverse molecular pathways within tumor cells, including those related to cell cycle regulation, apoptosis, metabolism, and epigenetic modifications. For instance, lncRNA TYMSOS enhances breast cancer growth and metastasis, facilitating immune evasion via the CBX3/ULBP3 or SYVN1/ULBP3 axis648 (Fig. 6). These studies underscore the intricate role of lncRNAs in immunotherapy resistance and point to potential molecular targets for novel treatment strategies. The aim of these strategies is to surmount therapeutic resistance and enhance clinical responses. Further studies should concentrate on delving deeper into precise mechanisms of lncRNAs to further enhance cancer therapy.
miRNAs
miRNAs are crucial in immunotherapy resistance, modulating immune responses and affecting tumor interactions. In HCC, hypoxic tumor-derived exosomal miR-1290 promotes the polarization of M2 macrophages, triggers apoptosis in CD8+ T cells, and enhances EMT in HCC cells. These effects collectively aid tumor cells in evading the immune system.649 In breast cancer, the downregulation of miR-299-3p leads to an upregulation of CD47 expression. This, in turn, inhibits the activation and infiltration of macrophages and promotes tumor growth, ultimately resulting in immune escape within breast cancer.650 Particularly, in TNBC, exosome-carried miR-20a-5p is internalized into CD8+ T cells, causing T-cell dysfunction and further conferring resistance to PD-1 therapy in TNBC.651 Moreover, miRNAs can control immune checkpoint molecules like PD-L1, aiding tumor cells in evading immune attacks. In NSCLC, an increased level of miR-142-5p suppresses PTEN while elevating the levels of PI3K, p-Akt, and PD-L1, thereby facilitating tumor immune escape.652 miRNAs also assist tumor cells in evading immune detection, which allows for their unrestrained growth. In CRC, exosome-derived miR-372-5p targets the PTEN/AKT/NF-κB pathway to induce immune escape653 (Fig. 6). Collectively, studies indicate that miRNAs play a multifaceted role in tumor immune escape by exerting an impact on both the processes of tumor cells and the functions of immune cells. As such, they represent potential targets for future tumor immunotherapy.
CircRNAs
CircRNAs contribute to tumor immune evasion by regulating immune responses. They enable tumor cells to elude immune detection, and one of the ways they achieve this through modulating transcription of PD-L1 via miRNA sponging. As for gastric cancer, circ_0136666 upregulates PRKDC by sponging miR-375-3p, which activates the PD-L1 phosphorylation pathway, preventing PD-L1 degradation and aiding immune evasion.654 In esophageal cancer, circ-VIM promotes immune evasion by increasing PD-L1 expression through miR-124 sponging.655 Similarly, in gastric cancer, hsa_circ_0001479 boosts DEK expression by sponging miR-133a-5p. This, in turn, activates the Wnt/β-catenin pathway, which not only promotes cell growth but also reduces the infiltration of CD8+ T cells, thus facilitating immune evasion.656 In ovarian cancer, circ-NFIX promotes tumor progression and immune evasion by sponging miR-647 and activating IL-6R/JAK1/STAT3 signaling.657 CircRNAs can also influence immune cell function in the tumor microenvironment. HCC cells release exosomal circCCAR1, which is absorbed by CD8+ T cells, stabilizing PD-1 protein and causing T-cell dysfunction, leading to immune evasion.629 In NSCLC, circIGF2BP3 enhances tumor immune evasion by promoting PD-L1 deubiquitination, inhibiting CD8+ T-cell responses.658 The human tumor virus-encoded circE7 hampers T-cell function by reducing LGALS9 transcription, aiding immune evasion in HNSCC.659 Additionally, circRNAs contribute to innate immunity; for instance, AML cell-derived exosomal circ_001264 fosters immune evasion by promoting M2 macrophage polarization and PD-L1 expression660 (Fig. 6). Additionally, exosome circUHRF1 released from cancer cells induces natural killer cell exhaustion, potentially giving rise to resistance to PD-1 therapy in HCC.661 CircRNAs can influence immunotherapy sensitivity by affecting tumor cell glycolysis (Fig. 6), as seen with bladder cancer circFAM13B, which inhibits glycolysis via the IGF2BP1/PKM2 pathway, thereby enhancing immunotherapy sensitivity.662
Overall, circRNAs contribute to tumor immune evasion by sponging miRNAs to regulate PD-L1 expression, impacting signaling pathways and immune cell functions, and modulating immune responses in the tumor microenvironment. These insights offer potential targets and a theoretical framework for future developments.
piRNAs
Recent investigations have shed light on the fact that piRNAs may play a role in contributing to immunotherapy resistance. Specifically, the piRNA piR-hsa-30937, found in small extracellular vesicles from pancreatic neuroendocrine neoplasms, can be released into the microenvironment and enhance CD276 expression in macrophages via the PTEN/AKT pathway, leading CD276+ TAMs to inhibit T-cell antitumor immunity663 (Fig. 6). These findings further emphasize the complexity of the mechanisms underlying immunotherapy resistance and highlight the importance of continued exploration into the roles of various non-coding RNAs like piRNAs. An in-depth understanding of these mechanisms contributes to novel strategies to overcome immunotherapy resistance and improve the effectiveness of cancer treatment in the future.
Targeted therapy
Targeted therapy predominantly depends on advancements in tumor molecular biology and genomics to address specific molecules or signaling pathways that are integral to tumor initiation and progression. Its primary classifications include protein kinase inhibitors, agents targeting tumor metabolism, and inhibitors of DNA damage repair pathways.664 Epigenetic regulators frequently exhibit aberrant expression in tumors that have developed resistance to targeted therapy, highlighting their role in the emergence of resistance to such treatments.665 A wealth of research has demonstrated that epigenetic alterations could profoundly influence the targeted therapy resistance.666 These regulators facilitate therapy resistance by modulating tumor proliferation, migration, invasion, apoptosis, and other critical factors.666 Consequently, clinical trials are exploring the combination of small-molecule inhibitors targeting epigenetic modulators with targeted therapies as a promising strategy to surmount resistance to targeted treatments. (Table 1) In the following section, we will delve into the epigenetic factors that govern resistance to targeted therapies.
Histone modifications in targeted therapy resistance
Recent research indicates that histone modifications impact resistance to protein kinase inhibitors by regulating cancer cell migration, invasion, and apoptosis through the modulation of oncogene transcriptional activity.84,667,668,669,670 Specifically, HDAC11 enhances the self-renewal capacity of lung adenocarcinoma stem cells and contributes to resistance to EGFR inhibitors by promoting Sox2 expression.671 Additionally, the combined use of HDAC inhibitors and EGFR inhibitors shows potential as candidate drugs for cancer treatment by promoting apoptosis through the activation of caspase 3/7.672 In NSCLC, the expression level of EZH2 is negatively correlated with resistance to epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs). Upregulating or promoting EZH2 expression can enhance the sensitivity of NSCLC cells to EGFR-TKIs673 (Fig. 7). Furthermore, targeting euchromatic histone-lysine N-methyltransferase 2 (EHMT2) can reverse EGFR-TKI resistance in NSCLC by epigenetically regulating the PTEN/AKT signaling pathway674 (Fig. 7). In conclusion, the targeting of histone PTMs, including HDACs and EZH2, represents a promising avenue for addressing drug resistance in cancer therapy. This approach not only augments the efficacy of current therapeutic agents but also offers innovative pathways for the development of novel anticancer drugs.
DNA methylation in targeted therapy resistance
DNA methylation exerts a significant impact on cellular apoptosis, consequently influencing the sensitivity towards targeted therapy. Elevated methylation in the death-associated protein kinase (DAPK) can give rise to resistance against anti-EGFR agents in NSCLC and HNSCC cell lines. This might be attributed to the fact that hypermethylation of DAPK attenuates pro-apoptotic signaling, thereby undermining the efficacy of anti-EGFR drugs675 (Fig. 7). DNA methylation is also capable of influencing the expression of cellular targets, thereby resulting in resistance to targeted therapy. In breast cancer, high methylation of the ESR1 promoter mediated by DNMTs leads to reduced ERα expression, resulting in tamoxifen resistance.676,677 Moreover, methylation can also lead to tamoxifen resistance by regulating the promoter methylation of ERα-related genes. High methylation of upstream genes of ERα suppresses ERα expression, thereby promoting tamoxifen resistance, such as p21, WT1, and miR-27b.678,679,680 High methylation of downstream genes of ERα leads to reduced expression, further leading to ERα dysfunction and inducing tamoxifen resistance, such as NAT1, ELOVL2 and PRA.681,682 Apart from ERα, methylation-induced inactivation of the PI3K/AKT/mTOR signaling pathway can enhance tamoxifen sensitivity.683 PTEN, being a crucial protein in the PI3K/AKT/mTOR signaling pathway, experiences increased AKT phosphorylation due to its hypermethylation, which consequently leads to tamoxifen resistance.684 However, the methylation of ERβ has an effect contrary to that of ERα. Hypomethylation of ERβ leads to tamoxifen resistance. DNA methylation can also affect the sensitivity to targeted therapy drugs by altering DNA repair related genes.685 For example, O-6-methylguanine-DNA methyltransferase (MGMT) is a key DNA damage repair gene, and low methylation of the MGMT promoter leads to reduced sensitivity to TMZ in GBM.686 Additionally, DNA methylation can impact the transport of targeted drugs into cells, thereby affecting drug sensitivity. OSCP1 encodes a widely substrate-specific organic solute carrier protein, and its high methylation restricts the transport of imatinib into nasopharyngeal cancer cells, resulting in imatinib resistance.687 During targeted drug therapy, DNA methylation can also affect cellular viability, giving rise to resistance to targeted therapy. In NSCLC, increased methylation of WIF promotes the cellular viability of gefitinib-resistant cells, further increasing cellular resistance688 (Fig. 7). Some tumor entities, like liver cancer, inherently exhibit resistance to TRAIL. In HCC, hypomethylation of Ache can impede the apoptosis of HCC cells induced by the cytokine TRAIL, leading to therapeutic resistance689 (Fig. 7). In summary, DNA methylation exerts its influence on the sensitivity to targeted therapy through a myriad of means. Gaining a comprehensive understanding of these mechanisms is of paramount importance for surmounting resistance and enhancing the effectiveness of cancer treatment.
RNA modifications in targeted therapy resistance
Recent research indicates that RNA modifications exert a crucial influence on modulating resistance to targeted therapies by influencing cellular processes such as apoptosis, proliferation, tumor migration, and invasion.690 Recent researches have shown METTL14 reduces the expression of HNF3γ mRNA through m6A modification, with HNF3γ being a key regulator of sorafenib resistance. The expression of HNF3γ not only promotes the differentiation of HCC cells, but also that of LCSCs. Therefore, the upregulation of METTL14 can lead to sorafenib resistance in liver cancer.690 In addition, autophagy mediated by FOXO3 results in sorafenib resistance of HCC. The deletion of METTL3 inhibits the m6A modification of FOXO3 mRNA via YTHDF1, thereby facilitating sorafenib resistance and cancer proliferation in HCC.238 Inhibiting the demethylase activity of FTO with Rhein can sensitize resistant cells to TKIs, downregulate Bcl-2 and MER proto-oncogene, tyrosine kinase (MERTK), thus regulating the PI3K-AKT-mTOR pathway and the Bcl-2 family of proteins, which affects the invasion and apoptosis of cancer cells.691,692,693 Therefore, the FTO-m6A axis has become a new indicator for TKI resistance. It has been reported that meclofenamic acid (MA), an FTO inhibitor, can also reverse TKI resistance.691 The inhibition of TSUC7 associated with m6A result in erlotinib resistance of lung cancer by regulating the stemness of EMT features in a Notch signaling activation-dependent manner.694 In A375, METTL3 augments the m6A modification of EGFR, boosts the RAF/MEK/ERK signaling pathway, and induces apoptosis, thereby promoting resistance to the BRAF (V600E) kinase inhibitor PLX4032695 (Fig. 7).
Recent studies have indicated that m5C modification is associated with resistance to targeted therapy.696,697,698 NSCLC with epidermal growth factor receptor (EGFR) mutations, m5C hypermethylation and NSUN2 are related to the intrinsic gefitinib resistance and tumor recurrence. The overexpressed NSUN2 interacts with YBX1, promoting the translation of quiescin sulfhydryl oxidase 1 (QSOX1) mRNA, thereby enhancing the resistance of non-small-cell lung cancer to gefitinib.697 M7G also shows relevant effects in the resistance to targeted therapy. In HCC, the elevation of m7G levels and the upregulation of METTL1 can lead to the resistance of HCC to lenvatinib.309 In lung adenocarcinoma, dual-specificity phosphatase 5 (DUSP5), regulated by YTHDF1-mediated m6A modification, promotes EMT and the resistance to epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) through the transforming growth factor-β (TGF-β)/Smad signaling pathway.699 The latest research on the combined strategy of RNA modification and targeted therapy covers multiple aspects, ranging from basic mechanism studies to clinical applications, demonstrating the great potential of RNA modification in cancer treatment and its synergistic effects with other therapies.
Non-coding RNAs in targeted therapy resistance
LncRNAs
LncRNAs are of vital significance in altering the sensitivity of cancer cells to targeted drugs by influencing molecule expression as well as signaling pathways. In NSCLC, decreased levels of lncRNA H19 are found in drug-resistant cells. The silencing of H19 can augment the expression of PKM2, and then contributing to erlotinib resistance in NSCLC700 (Fig. 7). Additionally, lncRNA UCA1 can cause erlotinib resistance by activating the AKT/mTOR pathway and inducing EMT, and it can also promote gefitinib resistance by binding to EZH2 and silencing CDKN1A epigenetically.701 Studies have demonstrated that LINC00665 can modulate SERPINE1 and enhance p-AKt expression within the PI3K/AKt pathway, resulting in trastuzumab resistance in gastric cancer.702 Additionally, lncRNAs could boost receptor signaling, which in turn heightens resistance to targeted therapies. For example, lncRNA HOTAIR is elevated in breast cancer, affecting estrogen receptor transcription and increasing tamoxifen resistance.703 LncRNAs can also affect the sensitivity of tumor cells to targeted therapy drugs by regulating cell cycle and apoptosis. For example, lncRNA PRNCR1 promotes the proliferation of breast cancer and inhibits apoptosis by regulating the microRNA-377/CCND2/MEK/MAPK axis, leading to resistance to tamoxifen in breast cancer704 (Fig. 7). LncRNAs can also influence the epigenetic modifications of specific molecules, causing changes in their levels, which affect the proliferation of cancer cells and mediate their drug sensitivity. For example, cyclin D1 is a key oncogene that promotes the proliferation of cancer cells. Studies have shown that in breast cancer, lncRNA DILA1 directly binds to cyclin D1, preventing its phosphorylation at the Thr286 site, thereby inhibiting its degradation and increasing the proliferation of breast cancer cells and promoting resistance to tamoxifen.705 Additionally, lncRNAs can act as competing ceRNAs, binding to specific miRNAs and thus alleviating the inhibition of their target mRNAs, which enables them to regulate the sensitivity to targeted therapy. In lung adenocarcinoma, lncRNA MALAT1 acts as a ceRNA, suppressing the expression of miR-125 and promoting the expression of Rab25, leading to resistance to EGFR-TKI (erlotinib) in lung adenocarcinoma.706 LncRNAs, like UCA1, can be secreted through exosomes, contributing to drug resistance in recipient cells (Fig. 7). In summary, lncRNAs are of vital importance in influencing the sensitivity of cancer cells to targeted drugs and contribute to drug resistance through multiple aspects, including molecular expression, signaling pathways, cell cycle regulation, apoptosis modulation, and epigenetic alterations. This underlines their intricate role in drug resistance and presents potential targets for novel therapies aimed at overcoming or reversing such resistance.
miRNAs
MiRNAs exert influence on contributing to resistance against targeted therapy by modulating the drug responsiveness of tumor cells via diverse mechanisms. They affect pathways and molecules involved in cell growth, impacting pathways like PI3K/AKT/mTOR, RAS/RAF/ERK, and STAT/JAK (Fig. 7). For instance, in gliomas, miRNA-7 exerts an inhibitory effect on IRS-1 and IRS-2, which are pivotal regulators within the IGF-1R/Akt pathway.707 In breast cancer, miRNA-205 could enhance tamoxifen resistance and proliferation by targeting E2F1.708 In CRC, miR-100 and miR-125b together activate the Wnt/β-catenin pathway, boosting resistance to cetuximab.709 Moreover, miRNAs have a bearing on receptor regulation and the sensitivity to therapy. In NSCLC, reduced miRNA-145 boosts EGFR, promoting cell growth and erlotinib resistance.710 Conversely, miR-145 inhibits platelet-derived growth factor receptor, decreasing proliferation and increasing erlotinib and gefitinib sensitivity.711 In breast cancer, miR-335-5p and miR-335-3p work in tandem to suppress estrogen receptor α, thereby heightening tamoxifen resistance.712 miRNAs influence therapy sensitivity by regulating processes like apoptosis, cell cycle, and EMT. In NSCLC, knockdown of miR-200c-3p enhances EMT, boosting resistance to erlotinib and gefitinib.713 MiR-146a-5p promotes migration and angiogenesis, causing trastuzumab resistance in HER2+ breast cancer714 (Fig. 7). In summary, miRNAs influence targeted therapy sensitivity by affecting signaling pathways, intracellular processes, and receptor regulation, highlighting their crucial role in tumor drug resistance and offering potential targets for new therapies.
CircRNAs
CircRNAs are increasingly for their influence in cancer treatment resistance, especially against targeted therapies. They exert an impact on biological functions through multiple modalities, such as functioning as miRNA sponges, influencing protein translation, or engaging in interactions with RBPs, thereby having a profound effect on therapy sensitivity. For example, circRNA_102481 acts as a sponge for miR-30a-5p to modulate the expression of ROR1. This regulatory action promotes cell proliferation while simultaneously inhibiting apoptosis, ultimately resulting in resistance to erlotinib and gefitinib in NSCLC715 (Fig. 7). In lung cancer, circRABL2B interacts with YBX1 to suppress MUC5AC, which in turn inhibits integrin β4/pSrc/p53 signaling as well as tumor stem cell characteristics, thereby augmenting erlotinib sensitivity.716 Additionally, circRNAs can influence targeted therapy sensitivity by binding to proteins. For instance, circ_SIRT1 binds to EIF4A3, upregulating ATG12, which enhances autophagy and imatinib resistance in chronic myeloid leukemia (CML).717 Reports suggest that certain circRNAs can trigger signaling pathways causing resistance to targeted therapies. In breast cancer, increased circCDYL2 stabilizes GRB7, preventing its degradation and boosting its interaction with FAK, which sustains AKT and ERK1/2 pathway activity, leading to trastuzumab resistance.718 Conversely, the reduction of certain circRNAs is likely to cause treatment resistance. For instance, in osimertinib-resistant lung adenocarcinoma, where decreased circFBXW7 activates the Wnt/β-catenin signaling pathway, further promoting proliferation and resistance to osimertinib719 (Fig. 7).
Certain circRNAs exert an influence on treatment sensitivity by modifying tumor cell metabolism as well as cell death pathways. For instance, circHIF1A enhances glycolysis changes, leading to cetuximab resistance in CRC.720 CircRNAs can also regulate treatment sensitivity by modulating cell death pathways. In breast cancer, the newly identified circVDAC3 confers resistance to trastuzumab by preventing the ubiquitination of HSPB1, reducing ferroptosis721 (Fig. 7). Similarly, circ-BGN can promote resistance to trastuzumab in breast cancer by enhancing OTUB1-mediated SLC7A11 deubiquitylation and alleviating ferroptosis.722 CircRNAs are of pivotal significance in modulating the sensitivity and resistance to cancer treatment, thus presenting novel therapeutic strategies and potential biomarkers for further exploration in the field of oncology.
Crosstalk between different epigenetic systems
Building on the discussion of single epigenetic mechanisms and their roles in tumorigenesis and therapeutic resistance, we now shift our attention to the intricate interplay between these various forms of epigenetic regulation. This crosstalk is crucial, as it underlies the complex gene expression patterns that fuel cancer progression and shape the resistance to therapies. Comprehending the interplay and cooperation among various epigenetic modifications is crucial for unraveling the full spectrum of their contributions to cancer biology. Epigenetic modifications, such as DNA methylation, histone post-translational modifications, and non-coding RNAs, operate in a sophisticated network that orchestrates gene expression patterns, thereby influencing cellular processes and contributing to the hallmarks of cancer.23,723,724 The crosstalk between these epigenetic markers is of particular importance, since it may lead to the activation of oncogenes or the suppression of tumor suppressor genes, which in turn modulates cancer cell behavior and response to therapeutic interventions.725,726
The interplay between DNA methylation and histone modifications, for instance, has been shown to create a repressive chromatin environment capable of silencing tumor suppressor genes, thereby promoting oncogenic transformation.727 Non-coding RNAs, including miRNAs and lncRNAs, also participate in this crosstalk by regulating the expression of genes involved in the epigenetic machinery, further shaping the epigenetic landscape of cancer cells.55 Moreover, the tumor microenvironment (TME) is replete with cellular and molecular components that can influence epigenetic marks on cancer cells, thereby affecting their phenotype and therapy response. The communication between cancer cells and the TME can lead to changes in the epigenome that reinforce the malignant properties of cancer cells and their resistance to treatment.
In the forthcoming sections, we will explore the intricate relationships between different epigenetic modifications and their collective impact on cancer progression and therapy resistance. By examining the latest research, we aim to provide insights into how these epigenetic interactions can be targeted to improve cancer treatment outcomes.
Histone modifications and DNA methylation
As we began to unravel the vast and intricate network of epigenetic modifications, we decided to find the entry point according to the levels of gene expression and gene regulation.728 It became apparent that the histone modifications are exactly the crucial key to unlocking this door, as they regulate genes at the chromatin level with global implications, and histone modification can serve as an anchor for other types of epigenetic modification or modifying enzymes.158 The interactive relationship between histone modifications and DNA methylation constitutes a significant component of epigenetic crosstalk.105 First, histone-modifying enzymes can directly act on DNMTs to regulate methylation levels. For example, the methylation of DNMT1 mediated by SET7/9 leads to a decrease in its stability and subsequent degradation, thereby causing a reduction in methylation levels across the entire genome.729 Second, previous studies have indicated that histone modifications can regulate the catalytic activity of DNMTs by the recruitment or dissociation of them.730 For instance, DNMT3A, DNMT3B and DNMT3L process an ADD (ATRX-DNMT3-DNMT3L) domain that interacts with unmodified N-terminal tails of histone H3, but H3K4me1/me2/me3 iteratively weakens this interaction and inhibits DNMT3A/B catalytic activity.731,732,733 Similarly, H3K36me2 can recruit DNMT3A and shapes the intergenic DNA methylation landscape.730 The interaction between histone methylation modification and DNA methylation has been well summarized in recent reviews.734,735,736 Third, the methylated DNA and histone-modifying enzymes can form complexes through certain “intermediate” proteins. For example, the methylated DNA may be recognized and bound by proteins known as MBDs.190,737 MBD proteins attract other proteins to the site, such as HDACs and other chromatin remodeling proteins capable of modifying histones, thereby creating compact, transcriptionally inactive heterochromatin.32,189
In summary, the collaboration between histone PTMs and DNA methylation for silencing of gene expression has been well-documented,738,739 and Importantly, such crosstalk/collaboration between them has been shown to promote therapeutic resistance in cancer.740 Tumor cells can invoke these epigenetic tools to synergistically achieve drug resistance.740 For example, clinical evidence indicates that BRAFV600E-mutated CRC shows a strong correlation with high levels of DNA methylation.740 When BRAF-mutated CRC exhibits a CpG island hypermethylation phenotype (CIMP-H), it often presents with therapeutic resistance and poor clinical prognosis.741 It has been confirmed that treatment with 5-azacytidine significantly reduces the level of DNA methylation in tumor cells, but the transcriptional repression of key tumor suppressor genes was not relieved. The increase in the histone deacetylation and histone methylation (H3K4me3 and H3K27me3) around the hypomethylated regions suggested that tumor cells could mobilize histone modifications to continue suppressing genes after DNA methylation was inhibited by the 5-azacytidine.740 Their work suggested that compensatory epigenomic alterations predominantly triggered by H3K27me3-based silencing following treatment-induced DNA methylation reduction, laying the groundwork for a potential DNMT and EZH2 inhibitor combination therapy in BRAFV600E CRC.
In recent years, it has been reported that the dysregulation of both histone modifications and DNA methylation often occurs simultaneously in tumors.609,742 For instance, the overexpression of DNMTs and HDACs in breast cancer tissues is positively correlated with each other and is associated with poor prognosis.743 Therefore, the combined use of DNMTi and HDACi shows great potential in overcoming resistance to cancer therapy. Chang et al. obtained a series of DNMT1/HDAC dual inhibitors by fusing the key pharmacophores from DNMT1 inhibitors (DNMT1i) and HDAC inhibitors (HDACi). Among them, compound (R)-23a demonstrates significant DNMT1 and HDAC inhibition both in vivo and in cells (colorectal tumor) and largely phenocopied the synergistic effects of combined DNMT1i and HDACi in reactivating epigenetically silenced tumor suppressor genes (TSGs).744 Huang et al. designed a dual DNMT and HDAC inhibitor (termed DNMT/HDACi) 15a, which exhibits immunomodulatory functions by increasing the intracellular level of double-stranded RNA to activate the RIG-I/MAVS pathway and enhances the effectiveness of immune checkpoint blockade therapy.743
Histone modifications and m6A RNA modification
Recent studies and reviews have highlighted the intricate crosstalk between histone modifications and m6A RNA modifications.745,746,747 Histone modifications can affect the local enrichment of m6A modifications by recruiting or releasing m6A writers, erasers, and readers.748,749 For example, it has been disclosed that H3K36me3 exhibited a similar CDS and 3’UTR distribution pattern to m6A. H3K36me3 can be directly identified and bound by METTL14. Upon meeting RNA Pol II, METTL14 then recruits other components of m6A MTC and mediates the deposition of m6A on newly synthesized RNA. Thus, the recognition of H3K36me3 by METTL14 and the specific recognition of DRACH motifs by m6A MTC allow for the accurate and dynamic deposition of m6A on the transcriptome, uncovering the importance of METTL14 in the selective and precise deposition of m6A.748 Another study found that METTL3 and METTL14 primarily bind to the promoter regions of the genome in leukemia cells and are associated with the histone H3K4me3 modification.750 Similarly, the m6A modification can also coordinately regulate histone modifications by recruiting histone-modifying enzymes and related proteins.751 Li et al. reported that the repressive histone mark H3K9me2 could be specifically removed by the induction of m6A-modified transcripts. They observed a genome-wide correlation between m6A and occupancy of the H3K9me2 demethylase KDM3B, and found that the m6A reader YTHDC1 physically interacts with and recruits KDM3B to m6A-associated chromatin regions, promoting H3K9me2 demethylation and gene expression.751 Moreover, m6A regulators can regulate histone modification by destabilizing histone modifying enzymes mRNAs.747 It is reported that METTL14 not only alters H3K27me3 modification, but also regulates H3K27ac modification by destabilizing CBP and p300 mRNAs.747 It is not difficult to envision that histone modifications and m6A RNA modifications, along with their respective writers, readers, and erasers, can engage in intricate crosstalk through mutual recognition and interaction. This complex network is anticipated to be progressively elucidated in forthcoming research.
In the past few years, it has been revealed histone modifications and m6A RNA modifications synergistically contribute to drug resistance in tumor, particularly in the framework of acquired resistance, through a complex crosstalk mechanism.752 For instance, KDM4C orchestrates ALKBH5 expression through elevating the chromatin openness, suppressing H3K9me3 and facilitating recruitment of MYB and Pol II. ALKBH5 is essential for sustaining leukemia stem cell characteristics, and mediates mRNA stability of receptor tyrosine kinase AXL by m6A modification (KDM4C-ALKBH5-AXL signaling axis).753 In glioblastoma with TMZ resistance, TMZ can induce a SOX4-dependent augmentation in chromatin openness at the region of METTL3 through enhancement of H3K27ac levels and the recruitment of RNA polymerase II. Furthermore, METTL3 decreasing perturbs the deposition of m6A on transcripts of genes, including EZH2, culminating in nonsense-mediated mRNA decay. This underscores the pivotal impaction for EZH2 in the modulating of METTL3 transcription through H3K27me3-independent manner.752
Histone modifications and ncRNAs
There exists a complex regulatory network among histone-modifying enzymes/histone modifications, lncRNAs, and miRNAs.754,755 In general, histone modifications are positioned “upstream” of lncRNAs and miRNAs, as they regulate gene expression at chromatin level, but the expression and localization of histone modifiers may also be regulated by lncRNAs and miRNAs.756 lncRNAs, especially those that have been well-studied for their oncogenic roles such as MALAT1, HOTAIR, H19 and so on, promote cancer development through various pathways, including interactions with histone modifications and miRNA.757,758 Among these three elements, miRNAs seem to be more inclined to a “downstream” position, acting as effector molecules, yet their effects can also entail changes in the levels of histone modifications and lncRNAs.754,759 In tumors, there is often a simultaneous dysregulation of histone modifications, lncRNAs and miRNAs, which have a synergistic effect on promoting tumor drug resistance.
Histone modifications or modifying enzymes can regulate the expression levels of lncRNAs through direct or indirect interacting, consequently affecting the levels of associated miRNAs, and ultimately leading to the induction or repression for resistance-associated pathways.760 For instance, as an oncogene frequently overexpressed in a variety of malignancies, KDM4C contributes to the progression of these cancers as well as their resistance to ionizing radiation and chemotherapy.120 KDM4C has been found to demethylate the MALAT1 promoter region to boost MALAT1 expression.761 MALAT1 has been proved to act as a sponge of a variety of miRNAs in many cancers. Take miR-328-3p as an example, the upregulation of MALAT1 led to its downregulation in AML cells, resulting in the overexpression of Cyclin D2 (CCND2), which elevates the fraction of cells arrested in the G1 phase and diminishes the responsiveness of HL-60/A to Ara-C.761,762 It is essential to acknowledge that histone variants could also impact expression of lncRNA. For instance, histone H1.3 exerts a suppressive effect on the expression of H19 and inhibits the proliferative capacity of ovarian cancer cells.763
Moreover, lncRNAs are able to regulate histone modifications in a variety of ways, as discussed in recent reviews. In brief, lncRNAs can serve as scaffolds to bring together multiple components of chromatin-modifying complexes. For instance, the lncRNA HOTAIR interacts with the PRC2 and the LSD1/CoREST/REST complex, facilitating the coordinated regulation of histone H3K27 methylation and H3K4 demethylation.764 It is reported that HOTAIR mediated the switching of histone H3 lysine 27 acetylation to methylation to promote EMT in gastric cancer.765 lncRNAs can guide histone modification enzymes to specific genomic loci, thereby influencing the local chromatin state and gene expression. For example, lncRNA CASC9 has been demonstrated to promote esophageal squamous cell carcinoma metastasis. Mechanistically, CASC9 can interact with the transcriptional coactivator CREB-binding protein (CBP) within the nucleus. This interaction leads to an increased presence of CBP and H3K27 acetylation at the LAMC2 promoter, thereby enhancing the expression of LAMC2.766 The lncRNA URRCC, whose expression is upregulated in renal cell carcinoma (RCC) samples and associated with poor prognosis, leading to promote RCC cells proliferation and invasion. Mechanistically, URRCC enhances the expression of EGFL7 via mediating histone H3 acetylation of EGFL7 promoter, activation of P-AKT signaling, and suppressing P-AKT downstream gene FOXO3.767
Compared to lncRNAs and miRNAs, other types of non-coding RNAs, such as circRNAs and piRNAs, are less frequently reported to interact with histone modifications. However, recent studies have also revealed the possibility of such crosstalk. Circ_0019435 recruited EZH2 by directly binding to suppress the expression of DKK1 and PTEN, thereby enhancing the progression of cervical cancer.768 In many cases, circRNA plays an anti-tumor role. Circ_SPECC1 was down-regulated in various gastric cancer cell lines. Circ_SPECC1 functions as a sponge to attach miR-526b and therefore regulates its target genes, including lysine demethylase 4A (KDM4A) and the downstream signaling target YAP1/KDM4A, and suppressing the invasion and growth of gastric cancer cells.769 In human bladder cancer, circXRN2, which is aberrantly downregulated in bladder cancer tissues and cell lines, suppresses tumor progression driven by H3K18 lactylation by activating the Hippo signaling pathway.770
Epigenetic agents in combination with cancer therapies
In the preceding discussion, we have systematically and comprehensively summarized the impact of epigenetics on cancer, demonstrating that epigenetic mechanisms contribute significantly to the onset and evolution of cancer. Targeting epigenetic pathways represents an innovative strategy for addressing the challenges posed by malignant tumors. Remarkably, within six-month in 2020, the FDA granted two approvals for Tazemetostat, an EZH2 inhibitor, heralding a groundbreaking new chapter in the field of epigenetic therapeutics for oncology.771 The utilization of epigenetic drugs in cancer therapy primarily focuses on the inhibition of aberrant DNA methylation, histone methylation, and histone acetylation.665 To date, several drugs targeting specific epigenetic mechanisms have been approved by FDA. These include histone deacetylase HDAC inhibitors such as vorinostat, romidepsin, and chidamide, as well as DNA methylation inhibitors like 5-azacytidine and decitabine and EZH2 inhibitor Tazemetostat above-metioned604 (Fig. 8). Furthermore, the combination of epigenetic drugs with other cancer treatment modalities, including radiotherapy, chemotherapy, and immunotherapy.666 Epigenetic drugs predominantly target regulatory mechanisms involved in the modification process to alter aberrant modification levels in cancer as described in Fig. 8. We will discuss the application of epigenetic agents in combination with other cancer therapies in clinical research subsequently.

Map of various inhibitors against epigenetic regulators. It delineates the principal epigenetic pathways, succinctly outlining their functions and interconnections. Additionally, it enumerates the key epigenetic regulators and their corresponding inhibitors for each pathway mentioned. DNMT DNA methyltransferase, TET ten-eleven translocation, HDAC histone deacetylase, KDM histone demethylase, KMT histone methyltransferase, KAT histone acetyltransferase, MBD methyl-CpG binding domain
Targeting histone modifications
Histone modification drugs are capable of rectifying dysregulated histone modification patterns within malignant cells to suppress the aberrant gene transcriptions associated with oncogenesis. Among the pharmacological agents targeting histone modifications, histone deacetylase inhibitors (HDACi) have been the most extensively investigated and clinically utilized. Notable examples of HDACi include vorinostat, trichostatin A (TSA), panobinostat, romidepsin, and mocetinostat.772 HDACi can directly induce alteration of the cancer epigenome leading to reactivation of epigenetically silenced genes and consequently cancer cell growth arrest, promotion of differentiation and induction of apoptosis.28 At present, HDACi are employed as a therapeutic option for hematological cancers, such as cutaneous T-cell lymphoma, peripheral T-cell lymphoma, multiple myeloma, and non-Hodgkin’s T-cell lymphoma.773,774Furthermore, HDACi have demonstrated potential efficacy in the treatment of various solid tumors, including breast cancer, lung cancer, and glioma.775,776 The preceding part describes a comprehensive picture on the epigenetic agents for overcoming resistance to cancer treatments (Fig. 8). At present, the combination of HDACi with radiotherapy, chemotherapy, immunity, and targeted therapy has also made progress in clinical research (Table 2). In conjunction with cisplatin, panobinostat is utilized alongside tamoxifen to address chemoresistance in breast cancer. Furthermore, the combination of Vorinostat with 5-FU, leucovorin, and oxaliplatin has demonstrated efficacy in overcoming chemoresistance in CRC. Additionally, HDACi serve as sensitizers for both conventional fractionated radiotherapy and fractionated stereotactic radiation therapy (NCT01378481 and NCT00821951). In the field of immunotherapy, panobinostat in conjunction with the CTLA4 inhibitor ipilimumab have been incorporated into clinical research studies. Besides, vorinostat combined with the anti-PD-1 monoclonal antibody in breast cancer has achieved phase II clinical trial (NCT00258349). Pembrolizumab, have been incorporated into clinical research studies. Furthermore, the combination of HDACi with the EGFR inhibitor lapatinib and the low-dose proteasome inhibitor bortezomib has demonstrated a potential effect in overcoming resistance to targeted therapies.
In addition to HDACi, EZH2 inhibitors are extensively utilized clinically. EZH2 inhibitors are categorized as disrupting the structural integrity of the Polycomb Repressive Complex 2 (PRC2) and targeting EZH2 methyltransferase activity, such as tazemetostat and valemetostat, function by directly reducing levels of trimethylated histone H3K27me3 through the inhibition methyltransferase activity of EZH2.777,778 Moreover, GSK126 the other kind of EZH2 inhibitor, which disrupts protein-protein interactions among PRC2 subunits, has achieved phase I clinical trials for the treatment of various cancers (NCT02082977). In overcoming targeted therapy, EZH2 inhibitors have also made breakthroughs. Tazemetostat, effectively inhibit prostate cancer progression when combined with enzalutamide, an androgen receptor inhibitor (NCT04179864). Part of EZH2 inhibitors is in clinical trials to verify the feasibility of combining with immune checkpoint inhibitors, such as tulmimetostat, XNW5004, CPI-1205 and SHR2554 (Table 2). Moreover, EZH2 inhibitors has been proven to be a booster radiotherapy (Table 2).
Over the past decade, substantial progress has been achieved in the domain of targeted drug research concerning histone readers. Notably, INCB054329, a BET domain inhibitor, is undergoing clinical trials for the treatment of Ewing’s sarcoma and advanced cancers (NCT03514407 and NCT02431260). Furthermore, the pharmaceutical research of BRD and BET domain protein inhibitors, specifically BMS-986158 and BMS-986378, have progressed to phase I trials in the context of pediatric cancer (NCT03936465). In general, the feasibility of targeting histone modifications in cancer therapy has been demonstrated, both as a standalone treatment and in combination with other therapeutic approaches.
Targeting DNA methylation
DNA methylation inhibitors have garnered considerable attention in oncological research owing to their potential to revise abnormal DNA methylation patterns linked to tumorigenesis.41,779,780 These pharmacological agents predominantly target DNMTs, which are categorized into cytosine analog oligonucleotide drugs, SAM competitors, and DNA binders. Notably, cytosine analogs have been utilized in clinical settings. These analogs are integrated into DNA during the process of DNA synthesis. During the catalytic activity of DNMTs, these cytosine analogs can form covalent bonds with DNMTs, thereby inhibiting DNMTs dissociating from chromatin,41 5-azacytidine (azacitidine) and 5-aza-2’-deoxycytidine (decitabine) have received clinical approval from the FDA for the treatment of myelodysplastic syndromes and acute leukemia. Furthermore, additional DNMT inhibitors are presented in the Table 2. DNMTs inhibitors play a role in reactivating excessively suppressed tumor suppressor genes, including p53 and p21, in cancer, thereby playing a crucial role in overcoming cancer treatment resistance (Fig. 8).515,781,782 Second-generation DNMT inhibitors with better pharmacokinetic properties, such as guadecitabine, have been developed. While DNMT inhibitors are established as a standard treatment for hematological malignancies, their efficacy in solid tumors remains limited. Consequently, current research is increasingly focused on their application as adjuvant therapies to enhance treatment outcomes.41,780,783 Notably, the integration of DNMT inhibitors with other therapeutic modalities, including radiotherapy, targeted therapy, chemotherapy, and immunotherapy, has demonstrated potential in enhancing the overall efficacy of cancer treatment in clinical trials (Table 2). For example, azacitidine, decitabine and guadecitabine combined with a variety of chemotherapy drugs, such as platinum, carboplatin and abraxane, have a significant effect on overcoming chemotherapy resistance in small cell lung cancer, acute leukemia and CRC (Table 2). Furthermore, the integration of DNMT inhibitors with widely used immune checkpoint inhibitors has progressed to clinical trial phases. Specifically, the combination of DNMT inhibitors with pembrolizumab, nivolumab, camrelizumab, and durvalumab is being investigated across a range of solid tumors (Table 2). The efficacy of DNMT inhibitors as adjuvant agents in cancer therapy has been substantiated through both preclinical and clinical studies.
Targeting ncRNAs
In the preceding section, we provided an overview of the aberrant expression of ncRNAs in cancer, delving into the mechanisms by which resistance to various cancer treatments can be overcome, which indicates that ncRNAs may play a crucial role in cancer therapy and in surmounting resistance to other therapeutic modalities. Presently, two principal strategies are employed to target ncRNAs for cancer treatment: the inhibition of overexpressed ncRNAs and the restoration of suppressed ncRNAs within cancerous cells.55 Therefore, it is necessary to discovery targeted inhibitors of ncRNAs and delivery systems to inhibit ncRNAs. There are six primary types of ncRNA inhibitors: (1) Antisense oligonucleotides (ASOs) bind to complementary RNA sequences, blocking their function and promoting degradation via RNase-H cleavage; (2) Antisense miRNAs (antagomirs) bind to complementary miRNAs, leading to their degradation and preventing them from interacting with target mRNAs. (3) Artificial miRNA sponge: synthetic RNA with multiple miRNA binding sites sequesters miRNA from its target mRNA; (4) Small molecules: disrupt any stage of RNA transcription; (5) siRNAs and shRNAs: synthetic double-stranded RNAs bind to complementary non-coding RNAs in AGO2, causing target RNA degradation; (6) CRISPR/Cas9 editing: uses Cas9 nuclease and guide RNA to accurately cut target nc RNA.784 Common delivery methods for ncRNA therapeutics include lipid nanoparticles, exosomes, antibodies, and peptides.785,786,787The FDA has approved this approach for certain diseases. For instance, vitravene (fomivirsen sodium), the first antisense nucleotide drug, is FDA-approved for cytomegalovirus retinitis. Nearly 20 RNA drugs are on the market, but none target tumors.788
ncRNA drugs face challenges in achieving clinical efficacy. In clinical trials for breast cancer and NSCLC, ASO drugs have not demonstrated significant anticancer efficacy.789,790 Currently, ncRNAs serve primarily as molecular markers in cancer treatment. Particularly, the FDA has approved the lncRNA PCA3 as a diagnostic marker for prostate cancer.791 Numerous ncRNAs are under investigation as cancer markers in clinical trials. In breast cancer chemotherapy, circulating miRNA types and levels in patients’ blood are monitored to aid in prognostication and treatment planning on an individual patient basis (NCT01722851).
The other approach for targeting ncRNAs in cancer that overexpression of ncRNAs, particularly using miRNA mimics to replace down-regulated tumor suppressor miRNAs, faces challenges such as off-target side effects. For instance, the miR-34 mimic MRX34 caused severe adverse events, including cytokine release syndrome, in five patients, halting a phase I cancer trial.792 Furthermore, off-target effects in non-neoplastic tissues pose a challenge to RNA drug therapy. The XIAP-targeting antisense oligonucleotide AEG35156, when taken up by glial cells or neurons, leads to a reduction in XIAP levels within these cells, which can result in peripheral chemotherapy-induced neuropathy in patients.793
Currently, ncRNAs emerge as viable predictors for cancer therapy in clinical trials, but integrating them with existing treatments poses challenges like tolerance, toxicity, and off-target effects. Preclinical studies show a strong link between ncRNAs and treatment resistance. Advances in single-cell sequencing and understanding tumor heterogeneity are expected to enhance ncRNA’s role in cancer treatment.
Targeting RNA modifications
Currently, there are no RNA modification inhibitors approved by the FDA for clinical application. Significant advancements have been achieved in the development of RNA modification therapeutics in recent years. Notably, FTO inhibitors are highly effective against cancer.794,795 Moreover, since the elucidation of the crystal structure of FTO in 2010, a range of inhibitors targeting its substrate binding sites has been developed, including anthraquinone, entacapone, meclofenamic acid (MA), MA2, as well as FB23 and FB23-2.795,796 The selectivity for FTO and cell membrane permeability improved with each generation.797,798 In preclinical investigations, FTO has demonstrated efficacy when used in conjunction with cancer therapeutic agents. MA enhances the sensitivity of leukemia cells to TKIs. Furthermore, entacapone combined with imatinib, a kind of TKIs, for leukemia treatment completed phase I clinical trials in 2022, highlighting its potential as a therapeutic enhancer in clinical settings (NCT04006769). In addition to FTO, many inhibitors of METTL3 have emerged in recent years, such as STM2457 STC-15, UZH1a (R-2a) STM2120. STC-15 for the treatment of AML has entered the clinical phase I trial stage in 2022 (NCT05584111). In thyroid cancer, the expression of METTL3 is relevant to anti-PD-1 therapy resistance.631 There is a paucity of studies on METTL3 inhibitor combined with other cancer treatments. METTL3 inhibitors may have the potential to enhance the efficacy of cancer therapies. This will be a new research direction for cancer treatment. It may be a novel avenue of oncology therapeutics. Furthermore, m6A reader inhibitors were demonstrated to function as radiosensitizers in 2023. The m6A reader inhibitor, DC-Y13-27, was found to directly bind to the YTHDF2 protein, thereby inhibiting its interaction with m6A-RNA in vitro, which alters the differentiation of myeloid-derived suppressor cells (MDSCs) and reduces their infiltration and suppressive activity. Consequently, DC-Y13-27 not only enhances the efficacy of radiotherapy but also augments the anti-tumor effects when radiation is combined with PD-L1 antibody therapy.
In conclusion, the challenge of achieving sustained cancer therapeutic doses with single- epigenetic agent arises from their low selectivity and broad spectrum of gene regulation. Consequently, optimizing pharmacokinetics and minimizing off-target toxicity are essential to establish a durable and tolerable therapeutic response in patients. The concept of utilizing low-dose epigenetic inhibitors to enhance therapeutic outcomes represents an innovative approach in the clinical application of these inhibitors. Over the past few years, a multitude of clinical trials have been undertaken to assess the specific approach. The integration of this therapeutic concept with the rapidly evolving techniques of single-cell sequencing is potential to become a pivotal strategy in overcoming resistance to cancer treatments.
Perspectives and challenges
In recent years, the domain of epigenetics has undergone substantial expansion, incorporating a diverse array of mechanisms including nucleosome positioning, chromatin remodeling, histone modifications, DNA methylation, RNA modifications, alternative RNA splicing, and non-coding RNAs (such as lncRNAs, miRNAs, circRNAs, piRNAs, among others), as well as pseudogenes (non-coding DNA).799 These elements can drive heritable alterations in gene function, culminating in phenotypic variations without modifications to the DNA sequence. It is crucial to acknowledge that the expression and activity of modifiers and regulators responsible for epigenetic modifications remain substantially affected by mutations within genetic code. This underscores the intricate interplay between genetic and epigenetic information.800 A comprehensive understanding of cellular life processes can only be attained by elucidating the mechanisms underlying the organization, transmission, and expression of both genetic and epigenetic information.
A considerable body of evidence indicates that widespread epigenetic dysregulation exists in tumors and is associated with resistance to cancer therapy. The intricate interplay between various epigenetic modifications is increasingly recognized as a key driver of therapeutic resistance in cancer. This epigenetic dysregulation not only influences the behavior of cancer cells but also reshapes the tumor microenvironment, thereby profoundly impacting the efficacy of cancer treatments. The complex network of epigenetic interactions can sculpt a landscape of resistance, enabling the tumor cells to adapt and survive under treatment. Understanding and targeting these epigenetic crosstalk pathways may offer novel therapeutic strategies to overcome resistance and enhance the responsiveness of tumors to treatment. However, a considerable challenge arises from the complexity and heterogeneity of epigenetic alterations within tumors. It is now recognized that within a single type of cancer, there can be a multitude of epigenetic disruptions, and due to the heterogeneity of cellular populations within the tumor, pinpointing the core drivers is exceedingly difficult. The interplay of various epigenetic modifications, such as DNA methylation, histone modifications, and non-coding RNAs, contributes to the phenotypic and functional diversity of cancer cells, ultimately fostering therapeutic resistance. Additionally, different therapeutic approaches can elicit distinct epigenetic landscape changes within tumors. For instance, there is substantial evidence that radiation therapy can modify histone modifications and chromatin structure within tumors, and it may also affect the levels of non-coding RNAs.801 However, it is rarely reported whether the original network of epigenetic crosstalk undergoes significant changes throughout the treatment process, whether the core of the original epigenetic crosstalk network is altered, and what impact such changes may have on therapy. This epigenetic heterogeneity, both between intertumor and intratumor, is a hallmark of human cancers and is increasingly recognized as a critical driver of resistance to cancer therapies.
The advancement of omics technologies such as multiplexed-scAEBS, scBS-Seq and single-cell ATAC-Seq have ushered in new opportunities for characterizing the epigenetic landscape of tumors and identifying resistance-associated factors.802 These technologies enable a more comprehensive view of the epigenome, which includes not only DNA methylation and histone modifications but also the complex interactions between different epigenetic regulators. The capacity to profile multiple epigenetic marks across a diverse array of tumors is crucial for unraveling the heterogeneity and pinpointing potential therapeutic targets. As research advances, there is an expanding trend towards integrating these omics data to uncover the complex network of epigenetic interactions that contribute to cancer progression and drug resistance. This approach offers promise for the development of innovative therapeutic strategies that target the epigenome, potentially overcoming resistance and improving treatment outcomes in cancer patients.
Currently, epigenetic medications often exhibit diminished efficacy in solid tumors, primarily due to the inherent heterogeneity and complex crosstalk within the epigenetic landscape.604,666,799 Consequently, they are not typically considered first-line treatments for most cancers. However, accumulating evidence suggests that epigenetic drugs can significantly contribute to the management of solid tumors by alleviating resistance to various cancer therapies. Despite their utility, these two therapeutic approaches encounter significant resistance when applied to solid tumors. Furthermore, technological progress has updated the traditional treatment methods. For instance, FLASH radiotherapy, an avant-garde technique of radiotherapy with a dose rate exceeding 40 Gy/s, exhibits a significant protective effect on mitochondria, outperforming traditional radiotherapy methods. FLASH therapy may exert a regulatory influence on histone modifications, notably histone lactylation, because of the protection of mitochondria with FLASH radiotherapy.803 Moreover, FLASH irradiation does not markedly compromise tumor vasculature and maintains the integrity of drug delivery systems. This preservation of vascular function, in turn, enhances the delivery of epigenetic therapeutics to solid tumors, potentially alleviating the radiotherapy resistance.804Nevertheless, the involvement of epigenetic mechanisms in drug resistance remains inadequately understood, and no established combination strategies have been developed for clinical implementation. Consequently, further investigation into epigenetic resistance within the contexts of endocrine therapy and differentiation therapy represents a prospective avenue for the development of epigenetic drugs in cancer treatment.
More interestingly, recent research has identified a noteworthy phenomenon wherein skin cells retain a long-term memory of radiotherapy via epigenetic inheritance, subsequently hindering wound healing.805 This raises questions about whether epigenetic inheritance similarly contributes to the retention of radiotherapy memory in tumor tissues. Furthermore, it prompts an inquiry into whether other therapeutic modalities induce long- lasting memory in tumors through epigenetic mechanisms. The implications of such long-term therapeutic memory for cancer treatment, whether advantageous or detrimental, warrant further investigation.
The advancement of epigenetics-based drugs in oncology can be advanced through two primary avenues. The first involves employing single-cell epigenetics technology to identify critical regulatory elements. The second entails integrating these insights with other therapeutic modalities to address and potentially surmount challenges related to drug resistance. In summary, the exploit of epigenetic insights contributes to epigenetics-based strategies, which are expected to display benefits in cancer therapy.
In conclusion, we have provided a comprehensive overview of the widespread epigenetic dysregulation in cancer and its association with therapeutic resistance in this review. We have focused on the molecular mechanisms by which epigenetic networks drive therapeutic resistance across different therapeutic contexts, with particular emphasis on the pivotal role of epigenetic crosstalk. Importantly, targeting epigenetic regulators/modifiers represents a promising strategy to overcome therapeutic resistance. We hope that this review will serve as a foundational and comprehensive reference for future research.
References
Koirala, M. & DiPaola, M. Overcoming cancer resistance: strategies and modalities for effective treatment. Biomedicines 12, 1801 (2024).
Tian, Y. et al. A protracted war against cancer drug resistance. Cancer Cell Int. 24, 326 (2024).
Vasan, N., Baselga, J. & Hyman, D. M. A view on drug resistance in cancer. Nature 575, 299–309 (2019).
Ponnusamy, L., Mahalingaiah, P. K. S. & Singh, K. P. Epigenetic reprogramming and potential application of epigenetic-modifying drugs in acquired chemotherapeutic resistance. Adv. Clin. Chem. 94, 219–259 (2020).
Han, T. S., Kim, D. S., Son, M. Y. & Cho, H. S. SMYD family in cancer: epigenetic regulation and molecular mechanisms of cancer proliferation, metastasis, and drug resistance. Exp. Mol. Med. 56, 2325–2336 (2024).
Sukocheva, O. A. et al. The crucial role of epigenetic regulation in breast cancer anti-estrogen resistance: Current findings and future perspectives. Semin. Cancer Biol. 82, 35–59 (2022).
Johnstone, S. E., Gladyshev, V. N., Aryee, M. J. & Bernstein, B. E. Epigenetic clocks, aging, and cancer. Science 378, 1276–1277 (2022).
Banerjee, R. et al. Epigenetic basis and targeting of cancer metastasis. Trends Cancer 8, 226–241 (2022).
Sun, L., Zhang, H. & Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 13, 877–919 (2022).
Ge, T. et al. Crosstalk between metabolic reprogramming and epigenetics in cancer: updates on mechanisms and therapeutic opportunities. Cancer Commun. 42, 1049–1082 (2022).
Jurkowska, R. Z. Role of epigenetic mechanisms in the pathogenesis of chronic respiratory diseases and response to inhaled exposures: From basic concepts to clinical applications. Pharmacol. Ther. 264, 108732 (2024).
Sun, C. et al. The interplay between histone modifications and nuclear lamina in genome regulation. J. Genet. Genomics 52, 24–38 (2025).
Skouras, P., Markouli, M., Papadatou, I. & Piperi, C. Targeting epigenetic mechanisms of resistance to chemotherapy in gliomas. Crit. Rev. Oncol. Hematol. 204, 104532 (2024).
Phillips, D. M. The presence of acetyl groups of histones. Biochem J. 87, 258–263 (1963).
Gupte, R., Liu, Z. & Kraus, W. L. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 31, 101–126 (2017).
Tjeertes, J. V., Miller, K. M. & Jackson, S. P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 28, 1878–1889 (2009).
Miller, K. M. & Jackson, S. P. Histone marks: repairing DNA breaks within the context of chromatin. Biochem. Soc. Trans. 40, 370–376 (2012).
Jackson, S. P. & Durocher, D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49, 795–807 (2013).
Kobza, K. et al. K4, K9 and K18 in human histone H3 are targets for biotinylation by biotinidase. FEBS J. 272, 4249–4259 (2005).
Zhu, D., Zhang, Y. & Wang, S. Histone citrullination: a new target for tumors. Mol. Cancer 20, 90 (2021).
Wang, X. et al. BRAFV600E restructures cellular lactylation to promote anaplastic thyroid cancer proliferation. Endocr. Relat. Cancer 30, e220344 (2023).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019).
Millan-Zambrano, G., Burton, A., Bannister, A. J. & Schneider, R. Histone post-translational modifications - cause and consequence of genome function. Nat. Rev. Genet 23, 563–580 (2022).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
Stoll, S., Wang, C. & Qiu, H. DNA methylation and histone modification in hypertension. Int. J. Mol. Sci. 19, 1174 (2018).
Zaib, S., Rana, N. & Khan, I. Histone modifications and their role in epigenetics of cancer. Curr. Med. Chem. 29, 2399–2411 (2022).
Strahl, B. D. & Allis, C. D. The language of covalent histone modifications. Nature 403, 41–45 (2000).
Li, Y. & Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 6, a026831 (2016).
Qu, J., Li, P. & Sun, Z. Histone lactylation regulates cancer progression by reshaping the tumor microenvironment. Front. Immunol. 14, 1284344 (2023).
Ilango, S. et al. Epigenetic alterations in cancer. Front. Biosci. 25, 1058–1109 (2020).
Wang, S. et al. Epigenetic regulation of hepatic lipid metabolism by DNA methylation. Adv. Sci. 10, e2206068 (2023).
Kaluscha, S. et al. Evidence that direct inhibition of transcription factor binding is the prevailing mode of gene and repeat repression by DNA methylation. Nat. Genet 54, 1895–1906 (2022).
Liu, F., Wang, Y., Gu, H. & Wang, X. Technologies and applications of single-cell DNA methylation sequencing. Theranostics 13, 2439–2454 (2023).
Nishiyama, A. & Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 37, 1012–1027 (2021).
Pfeifer, G. P., Szabo, P. E. & Song, J. Protein interactions at oxidized 5-Methylcytosine bases. J. Mol. Biol. 432, 1718–1730 (2020).
An, J. & Ko, M. Epigenetic modification of cytosines in hematopoietic differentiation and malignant transformation. Int. J. Mol. Sci. 24, 1727 (2023).
Xie, Q. et al. N(6)-methyladenine DNA modification in glioblastoma. Cell 175, 1228–1243.e1220 (2018).
Janulaitis, A., Klimasauskas, S., Petrusyte, M. & Butkus, V. Cytosine modification in DNA by BcnI methylase yields N4-methylcytosine. FEBS Lett. 161, 131–134 (1983).
Mattei, A. L., Bailly, N. & Meissner, A. DNA methylation: a historical perspective. Trends Genet. 38, 676–707 (2022).
Huang, W. et al. LncRNA-mediated DNA methylation: an emerging mechanism in cancer and beyond. J. Exp. Clin. Cancer Res. 41, 100 (2022).
Lee, A. V., Nestler, K. A. & Chiappinelli, K. B. Therapeutic targeting of DNA methylation alterations in cancer. Pharm. Ther. 258, 108640 (2024).
Cohn, W. E. Pseudouridine, a carbon-carbon linked ribonucleoside in ribonucleic acids: isolation, structure, and chemical characteristics. J. Biol. Chem. 235, 1488–1498 (1960).
Dubin, D. T. & Taylor, R. H. The methylation state of poly A-containing messenger RNA from cultured hamster cells. Nucleic Acids Res. 2, 1653–1668 (1975).
Perry, R. P., Kelley, D. E., Friderici, K. & Rottman, F. The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the 5’ terminus. Cell 4, 387–394 (1975).
Wei, C. M., Gershowitz, A. & Moss, B. Methylated nucleotides block 5’ terminus of HeLa cell messenger RNA. Cell 4, 379–386 (1975).
Liang, W. et al. mRNA modification orchestrates cancer stem cell fate decisions. Mol. Cancer 19, 38 (2020).
Ke, S. et al. m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 31, 990–1006 (2017).
Lin, S. et al. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 62, 335–345 (2016).
Liu, J. et al. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 20, 1074–1083 (2018).
Ma, J. Z. et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology 65, 529–543 (2017).
Zhang, S. et al. A review of current developments in RNA modifications in lung cancer. Cancer Cell Int 24, 347 (2024).
Yu, L. et al. The role of m5C RNA modification in cancer development and therapy. Heliyon 10, e38660 (2024).
Salmaninejad, A. et al. Genomic instability in cancer: molecular mechanisms and therapeutic potentials. Curr. Pharm. Des. 27, 3161–3169 (2021).
Sinkala, M. Mutational landscape of cancer-driver genes across human cancers. Sci. Rep. 13, 12742 (2023).
Nemeth, K., Bayraktar, R., Ferracin, M. & Calin, G. A. Non-coding RNAs in disease: from mechanisms to therapeutics. Nat. Rev. Genet. 25, 211–232 (2024).
Amaral, P. et al. The status of the human gene catalogue. Nature 622, 41–47 (2023).
Li, Y. Non-coding RNA performs its biological function by interacting with macromolecules. Int. J. Mol. Sci. 24, 16246 (2023).
Ashrafizadeh, M. et al. Non-coding RNA-based regulation of inflammation. Semin. Immunol. 59, 101606 (2022).
Slack, F. J. & Chinnaiyan, A. M. The role of non-coding RNAs in oncology. Cell 179, 1033–1055 (2019).
Panni, S., Lovering, R. C., Porras, P. & Orchard, S. Non-coding RNA regulatory networks. Biochim. Biophys. Acta Gene Regul. Mech. 1863, 194417 (2020).
Zhang, J. et al. Advances in the roles and mechanisms of mesenchymal stem cell derived microRNAs on periodontal tissue regeneration. Stem Cell Res. Ther. 15, 393 (2024).
Zhang, Q. et al. The epigenetic regulatory mechanism of PIWI/piRNAs in human cancers. Mol. Cancer 22, 45 (2023).
Perera, B. P. U. et al. PIWI-Interacting RNA (piRNA) and Epigenetic Editing in Environmental Health Sciences. Curr. Environ. Health Rep. 9, 650–660 (2022).
Fagan, S. G., Helm, M. & Prehn, J. H. M. tRNA-derived fragments: A new class of non-coding RNA with key roles in nervous system function and dysfunction. Prog. Neurobiol. 205, 102118 (2021).
Tang, X. et al. Review on circular RNAs and new insights into their roles in cancer. Comput. Struct. Biotechnol. J. 19, 910–928 (2021).
Yadav, M., Behera, D. K., Gupta, N. & Verma, V. K. Regulatory network of non-coding RNA in Helicobacter pylori: a systematic approach. Life Sci. 359, 123194 (2024).
He, S. et al. Structure of nucleosome-bound human BAF complex. Science 367, 875–881 (2020).
Klemm, S. L., Shipony, Z. & Greenleaf, W. J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet 20, 207–220 (2019).
de Miguel, F. J. et al. Mammalian SWI/SNF chromatin remodeling complexes promote tyrosine kinase inhibitor resistance in EGFR-mutant lung cancer. Cancer Cell 41, 1516–1534.e1519 (2023).
Xiang, K. et al. Chromatin remodeling in patient-derived colorectal cancer models. Adv. Sci. 11, e2303379 (2024).
Chen, L. & Huang, M. Oncometabolites in cancer: from cancer cells to the tumor microenvironment. Holist. Integr. Oncol. 3, 26 (2024).
Sinha, S., Molla, S. & Kundu, C. N. PARP1-modulated chromatin remodeling is a new target for cancer treatment. Med. Oncol. 38, 118 (2021).
Zou, M. et al. The critical function of the plastid rRNA methyltransferase, CMAL, in ribosome biogenesis and plant development. Nucleic Acids Res. 48, 3195–3210 (2020).
Sharma, S. & Entian, K. D. Chemical Modifications of Ribosomal RNA. Methods Mol. Biol. 2533, 149–166 (2022).
Sharma, S. & Lafontaine, D. L. J. View from a bridge’: a new perspective on eukaryotic rRNA base modification. Trends Biochem. Sci. 40, 560–575 (2015).
Wang, W. et al. Loss of a single methylation in 23S rRNA delays 50S assembly at multiple late stages and impairs translation initiation and elongation. Proc. Natl Acad. Sci. USA 117, 15609–15619 (2020).
Rong, B. et al. Ribosome 18S m(6)A methyltransferase METTL5 promotes translation initiation and breast cancer cell growth. Cell Rep. 33, 108544 (2020).
Tseng, C. C. et al. Genetic variants in transcription factor binding sites in humans: triggered by natural selection and triggers of diseases. Int. J. Mol. Sci. 22, 4187 (2021).
Rosonina, E. A conserved role for transcription factor sumoylation in binding-site selection. Curr. Genet 65, 1307–1312 (2019).
Lambert, S. A. et al. The human transcription factors. Cell 175, 598–599 (2018).
Rimoldi, M. et al. DNA methylation patterns of transcription factor binding regions characterize their functional and evolutionary contexts. Genome Biol. 25, 146 (2024).
Chang, W. et al. Dynamic changes in whole genome DNA methylation, chromatin and gene expression during mouse lens differentiation. Epigenet. Chromatin 16, 4 (2023).
Audia, J. E. & Campbell, R. M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 8, a019521 (2016).
Bajbouj, K. et al. Histone modification in NSCLC: molecular mechanisms and therapeutic targets. Int. J. Mol. Sci. 22, 11701 (2021).
Cui, X., Dard, A., Reichheld, J. P. & Zhou, D. X. Multifaceted functions of histone deacetylases in stress response. Trends Plant Sci. 28, 1245–1256 (2023).
Ediriweera, M. K. Fatty acids as histone deacetylase inhibitors: old biochemistry tales in a new life sciences town. Drug Discov. Today 28, 103569 (2023).
Sheikh, B. N. & Akhtar, A. The many lives of KATs - detectors, integrators and modulators of the cellular environment. Nat. Rev. Genet 20, 7–23 (2019).
de Ruijter, A. J. et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 370, 737–749 (2003).
Jasim, S. A. et al. Histone Deacetylases (HDACs) Roles in Inflammation-mediated Diseases; Current Knowledge. Cell Biochem. Biophys. 83, 1375–1386 (2024).
Zhang, S. et al. Targeting histone modifiers in bladder cancer therapy - preclinical and clinical evidence. Nat. Rev. Urol. 21, 495–511 (2024).
Fraga, M. F. et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet 37, 391–400 (2005).
Miziak, P. et al. Acetylation of histone H3 in cancer progression and prognosis. Int. J. Mol. Sci. 25, 10982 (2024).
Li, Q. et al. PCAF inhibits hepatocellular carcinoma metastasis by inhibition of epithelial-mesenchymal transition by targeting Gli-1. Cancer Lett. 375, 190–198 (2016).
Liao, M. et al. LINC00887 promotes GCN5-dependent H3K27cr level and CRC metastasis via recruitment of YEATS2 and enhancing ETS1 expression. Cell Death Dis. 15, 711 (2024).
Mustachio, L. M. et al. Repression of GCN5 expression or activity attenuates c-MYC expression in non-small cell lung cancer. Am. J. Cancer Res. 9, 1830–1845 (2019).
Buchner, S. A. & Itin, P. Focal dermal hypoplasia syndrome in a male patient. Report of a case and histologic and immunohistochemical studies. Arch. Dermatol 128, 1078–1082 (1992).
Wisnieski, F. et al. Differential expression of histone deacetylase and acetyltransferase genes in gastric cancer and their modulation by trichostatin A. Tumour Biol. 35, 6373–6381 (2014).
Mitani, Y. et al. Histone H3 acetylation is associated with reduced p21(WAF1/CIP1) expression by gastric carcinoma. J. Pathol. 205, 65–73 (2005).
Ma, Y. et al. KAT7 promotes radioresistance through upregulating PI3K/AKT signaling in breast cancer. J. Radiat. Res. 64, 448–456 (2023).
Borbone, E. et al. Histone deacetylase inhibitors induce thyroid cancer-specific apoptosis through proteasome-dependent inhibition of TRAIL degradation. Oncogene 29, 105–116 (2010).
Duan, N. et al. Targeting the E2F1/Rb/HDAC1 axis with the small molecule HR488B effectively inhibits colorectal cancer growth. Cell Death Dis. 14, 801 (2023).
Tang, S. et al. Integrating the tumor-suppressive activity of Maspin with p53 in retuning the epithelial homeostasis: a working hypothesis and applicable prospects. Front. Oncol. 12, 1037794 (2022).
Sun, Y. et al. Therapeutic potential of tucidinostat, a subtype-selective HDAC inhibitor, in cancer treatment. Front. Pharm. 13, 932914 (2022).
Benedetti, R., Conte, M. & Altucci, L. Targeting Histone Deacetylases in Diseases: Where Are We? Antioxid. Redox Signal 23, 99–126 (2015).
Li, Y., Chen, X. & Lu, C. The interplay between DNA and histone methylation: molecular mechanisms and disease implications. EMBO Rep. 22, e51803 (2021).
Sutopo, N. C., Kim, J. H. & Cho, J. Y. Role of histone methylation in skin cancers: histone methylation-modifying enzymes as a new class of targets for skin cancer treatment. Biochim. Biophys. Acta Rev. Cancer 1878, 188865 (2023).
Copeland, R. A., Solomon, M. E. & Richon, V. M. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 8, 724–732 (2009).
Heightman, T. D. Chemical biology of lysine demethylases. Curr. Chem. Genomics 5, 62–71 (2011).
Beebe, D. W. et al. Is body focus restricted to self-evaluation? Body focus in the evaluation of self and others. Int. J. Eat. Disord. 20, 415–422 (1996).
Qin, J. et al. LINC00114 stimulates growth and glycolysis of esophageal cancer cells by recruiting EZH2 to enhance H3K27me3 of DLC1. Clin. Epigenet. 14, 51 (2022).
Varambally, S. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629 (2002).
Zhang, J. et al. Synthesis and biological evaluation of benzimidazole derivatives as the G9a Histone Methyltransferase inhibitors that induce autophagy and apoptosis of breast cancer cells. Bioorg. Chem. 72, 168–181 (2017).
Yin, C. et al. G9a promotes cell proliferation and suppresses autophagy in gastric cancer by directly activating mTOR. FASEB J. 33, 14036–14050 (2019).
Chae, Y. C. et al. FOXO1 degradation via G9a-mediated methylation promotes cell proliferation in colon cancer. Nucleic Acids Res. 47, 1692–1705 (2019).
Sun, L. et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 9, 287–300 (2006).
Sanchez-Carbayo, M. et al. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J. Clin. Oncol. 24, 778–789 (2006).
Kimchi, E. T. et al. Progression of Barrett’s metaplasia to adenocarcinoma is associated with the suppression of the transcriptional programs of epidermal differentiation. Cancer Res. 65, 3146–3154 (2005).
Wang, B. et al. Downregulation of KDM4A suppresses the survival of glioma cells by promoting autophagy. J. Mol. Neurosci. 60, 137–144 (2016).
Yang, Z. Q. et al. Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res. 60, 4735–4739 (2000).
Jie, X. et al. USP9X-mediated KDM4C deubiquitination promotes lung cancer radioresistance by epigenetically inducing TGF-beta2 transcription. Cell Death Differ. 28, 2095–2111 (2021).
Wang, Q., Wei, J., Su, P. & Gao, P. Histone demethylase JARID1C promotes breast cancer metastasis cells via down regulating BRMS1 expression. Biochem. Biophys. Res. Commun. 464, 659–666 (2015).
Bahl, S. & Seto, E. Regulation of histone deacetylase activities and functions by phosphorylation and its physiological relevance. Cell Mol. Life Sci. 78, 427–445 (2021).
Banerjee, T. & Chakravarti, D. A peek into the complex realm of histone phosphorylation. Mol. Cell Biol. 31, 4858–4873 (2011).
Pacaud, R. et al. Histone H3 phosphorylation in GBM: a new rational to guide the use of kinase inhibitors in anti-GBM therapy. Theranostics 5, 12–22 (2015).
Uguen, A. et al. Immunostaining of phospho-histone H3 and Ki-67 improves reproducibility of recurrence risk assessment of gastrointestinal stromal tumors. Virchows Arch. 467, 47–54 (2015).
Skaland, I. et al. Validating the prognostic value of proliferation measured by Phosphohistone H3 (PPH3) in invasive lymph node-negative breast cancer patients less than 71 years of age. Breast Cancer Res. Treat. 114, 39–45 (2009).
Takahashi, H. et al. Overexpression of phosphorylated histone H3 is an indicator of poor prognosis in gastric adenocarcinoma patients. Appl. Immunohistochem. Mol. Morphol. 14, 296–302 (2006).
Wang, Z. et al. Fructose-1,6-bisphosphatase 1 functions as a protein phosphatase to dephosphorylate histone H3 and suppresses PPARalpha-regulated gene transcription and tumour growth. Nat. Cell Biol. 24, 1655–1665 (2022).
Buetow, L. & Huang, D. T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 17, 626–642 (2016).
Komander, D. & Rape, M. The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 (2012).
Jeusset, L. M. & McManus, K. J. Developing targeted therapies that exploit aberrant histone ubiquitination in cancer. Cells 8, 165 (2019).
Jiang, Q. & Greenberg, R. A. Deciphering the BRCA1 tumor suppressor network. J. Biol. Chem. 290, 17724–17732 (2015).
Kalb, R. et al. BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep. 8, 999–1005 (2014).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Tarcic, O. et al. RNF20 and histone H2B ubiquitylation exert opposing effects in Basal-Like versus luminal breast cancer. Cell Death Differ. 24, 694–704 (2017).
Chernikova, S. B. et al. Deficiency in mammalian histone H2B ubiquitin ligase Bre1 (Rnf20/Rnf40) leads to replication stress and chromosomal instability. Cancer Res. 72, 2111–2119 (2012).
Shema, E. et al. The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev. 22, 2664–2676 (2008).
McCann, J. J. et al. USP22 functions as an oncogenic driver in prostate cancer by regulating cell proliferation and DNA repair. Cancer Res. 80, 430–443 (2020).
Zong, Z. et al. Alanyl-tRNA synthetase, AARS1, is a lactate sensor and lactyltransferase that lactylates p53 and contributes to tumorigenesis. Cell 187, 2375–2392.e2333 (2024).
Yang, J. et al. A positive feedback loop between inactive VHL-triggered histone lactylation and PDGFRbeta signaling drives clear cell renal cell carcinoma progression. Int. J. Biol. Sci. 18, 3470–3483 (2022).
Yu, J. et al. Histone lactylation drives oncogenesis by facilitating m(6)A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 22, 85 (2021).
Jiang, J. et al. Lactate modulates cellular metabolism through histone lactylation-mediated gene expression in non-small cell lung cancer. Front. Oncol. 11, 647559 (2021).
Zu, H. et al. SIRT2 functions as a histone delactylase and inhibits the proliferation and migration of neuroblastoma cells. Cell Discov. 8, 54 (2022).
Jin, J. et al. SIRT3-dependent delactylation of cyclin E2 prevents hepatocellular carcinoma growth. EMBO Rep. 24, e56052 (2023).
Teng, Y. et al. PAD2: a potential target for tumor therapy. Biochim. Biophys. Acta Rev. Cancer 1878, 188931 (2023).
Maeshima, K. et al. Is euchromatin really open in the cell? Trends Cell Biol. 34, 7–17 (2024).
Mohanan, S. et al. Potential role of peptidylarginine deiminase enzymes and protein citrullination in cancer pathogenesis. Biochem. Res. Int. 2012, 895343 (2012).
Chang, X. & Han, J. Expression of peptidylarginine deiminase type 4 (PAD4) in various tumors. Mol. Carcinog. 45, 183–196 (2006).
Chang, X. et al. Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer 9, 40 (2009).
Sabari, B. R., Zhang, D., Allis, C. D. & Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 18, 90–101 (2017).
Xie, J. Y. et al. The mechanisms, regulations, and functions of histone lysine crotonylation. Cell Death Discov. 10, 66 (2024).
Ntorla, A. & Burgoyne, J. R. The regulation and function of histone crotonylation. Front. Cell Dev. Biol. 9, 624914 (2021).
Martinez-Moreno, J. M. et al. The contribution of histone crotonylation to tissue health and disease: focus on kidney health. Front Pharm. 11, 393 (2020).
Li, K. & Wang, Z. Histone crotonylation-centric gene regulation. Epigenet. Chromatin 14, 10 (2021).
Jiang, G. et al. Protein lysine crotonylation: past, present, perspective. Cell Death Dis. 12, 703 (2021).
Li, K., Han, J. & Wang, Z. Histone modifications centric-regulation in osteogenic differentiation. Cell Death Discov. 7, 91 (2021).
Song, H. et al. Histone post-translational modification and the DNA damage response. Genes Dis. 10, 1429–1444 (2023).
Yao, W., Hu, X. & Wang, X. Crossing epigenetic frontiers: the intersection of novel histone modifications and diseases. Sig. Transduct. Target Ther. 9, 232 (2024).
Huang, H., Wang, D. L. & Zhao, Y. Quantitative crotonylome analysis expands the roles of p300 in the regulation of lysine crotonylation pathway. Proteomics 18, e1700230 (2018).
Yang, S., Fan, X. & Yu, W. Regulatory mechanism of protein crotonylation and its relationship with cancer. Cells 13, 1812 (2024).
Gallego-Bartolome, J. DNA methylation in plants: mechanisms and tools for targeted manipulation. N. Phytol. 227, 38–44 (2020).
Zhao, L. Y. et al. Mapping the epigenetic modifications of DNA and RNA. Protein Cell 11, 792–808 (2020).
Li, E. & Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 6, a019133 (2014).
Yu, Y. et al. A review on recent advances in assays for DNMT1: a promising diagnostic biomarker for multiple human cancers. Analyst 149, 1002–1021 (2024).
Li, D. et al. TET family of dioxygenases: crucial roles and underlying mechanisms. Cytogenet Genome Res. 146, 171–180 (2015).
Ginder, G. D. & Williams, D. C. Jr. Readers of DNA methylation, the MBD family as potential therapeutic targets. Pharm. Ther. 184, 98–111 (2018).
Bhootra, S. et al. DNA methylation and cancer: transcriptional regulation, prognostic, and therapeutic perspective. Med Oncol. 40, 71 (2023).
Liu, H. et al. Downregulation of FOXO3a by DNMT1 promotes breast cancer stem cell properties and tumorigenesis. Cell Death Differ. 27, 966–983 (2020).
Wu, X. Y. et al. DNMT1 promotes cell proliferation via methylating hMLH1 and hMSH2 promoters in EGFR-mutated non-small cell lung cancer. J. Biochem. 168, 151–157 (2020).
Zhai, P. et al. DNMT1-mediated NR3C1 DNA methylation enables transcription activation of connexin40 and augments angiogenesis during colorectal cancer progression. Gene 892, 147887 (2024).
Ley, T. J. et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 363, 2424–2433 (2010).
Narayanan, D. et al. Effect of DNMT3A variant allele frequency and double mutation on clinicopathologic features of patients with de novo AML. Blood Adv. 5, 2539–2549 (2021).
Deng, Q. F. et al. DNMT3A governs tyrosine kinase inhibitors responses through IAPs and in a cell senescence-dependent manner in non-small cell lung cancer. Am. J. Cancer Res. 13, 3517–3530 (2023).
Zhou, Y. et al. DNMT3A facilitates colorectal cancer progression via regulating DAB2IP mediated MEK/ERK activation. Biochim. Biophys. Acta Mol. Basis Dis. 1868, 166353 (2022).
Feng, M. et al. Terpenoids from quinoa reverse drug resistance of colon cancer by upregulating miR-495-3p. J. Sci. Food Agric 104, 8916–8927 (2024).
Rolls, W., Wilson, M. D. & Sproul, D. Using human disease mutations to understand de novo DNA methyltransferase function. Biochem. Soc. Trans. 52, 2059–2075 (2024).
He, D. et al. DNMT3A/3B overexpression might be correlated with poor patient survival, hypermethylation and low expression of ESR1/PGR in endometrioid carcinoma: an analysis of The Cancer Genome Atlas. Chin. Med J. 132, 161–170 (2019).
Liu, P. et al. Emerging role of different DNA methyltransferases in the pathogenesis of cancer. Front. Pharm. 13, 958146 (2022).
Pappalardi, M. B. et al. Discovery of a first-in-class reversible DNMT1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nat. Cancer 2, 1002–1017 (2021).
Peralta-Arrieta, I. et al. DNMT3B modulates the expression of cancer-related genes and downregulates the expression of the gene VAV3 via methylation. Am. J. Cancer Res. 7, 77–87 (2017).
Lopez-Moyado, I. F., Ko, M., Hogan, P. G. & Rao, A. TET enzymes in the immune system: from DNA demethylation to immunotherapy, inflammation, and cancer. Annu. Rev. Immunol. 42, 455–488 (2024).
Rasmussen, K. D. & Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 30, 733–750 (2016).
Salmeron-Barcenas, E. G. et al. TET enzymes and 5hmC levels in carcinogenesis and progression of breast cancer: potential therapeutic targets. Int. J. Mol. Sci. 25, 272 (2023).
Yue, X. & Rao, A. TET family dioxygenases and the TET activator vitamin C in immune responses and cancer. Blood 136, 1394–1401 (2020).
Jiang, S. Tet2 at the interface between cancer and immunity. Commun. Biol. 3, 667 (2020).
Xu, Q. et al. Loss of TET reprograms Wnt signaling through impaired demethylation to promote lung cancer development. Proc. Natl Acad. Sci. USA. 119, e2107599119 (2022).
Han, X. et al. TET1 promotes cisplatin-resistance via demethylating the vimentin promoter in ovarian cancer. Cell Biol. Int. 41, 405–414 (2017).
Laranjeira, A. B. A. et al. Upregulation of TET2 and resistance to DNA methyltransferase (DNMT) inhibitors in DNMT1-deleted cancer cells. Diseases 12, 163 (2024).
Du, Q., Luu, P. L., Stirzaker, C. & Clark, S. J. Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics 7, 1051–1073 (2015).
Jeltsch, A., Broche, J., Lungu, C. & Bashtrykov, P. Biotechnological applications of MBD domain proteins for DNA methylation analysis. J. Mol. Biol. 432, 1816–1823 (2020).
Liu, Z. et al. Hypoxia-induced suppression of alternative splicing of MBD2 promotes breast cancer metastasis via activation of FZD1. Cancer Res. 81, 1265–1278 (2021).
Pontes, T. B. et al. Reduced mRNA expression levels of MBD2 and MBD3 in gastric carcinogenesis. Tumour Biol. 35, 3447–3453 (2014).
Yan, W. et al. MBD3 promotes hepatocellular carcinoma progression and metastasis through negative regulation of tumour suppressor TFPI2. Br. J. Cancer 127, 612–623 (2022).
Derrien, A. C. et al. Germline MBD4 mutations and predisposition to uveal melanoma. J. Natl Cancer Inst. 113, 80–87 (2021).
Tsuboyama, N., Szczepanski, A. P., Zhao, Z. & Wang, L. MBD5 and MBD6 stabilize the BAP1 complex and promote BAP1-dependent cancer. Genome Biol. 23, 206 (2022).
Alarcón, C. R. et al. N6-methyladenosine marks primary microRNAs for processing. Nature 519, 482–485 (2015).
Zhou, C. et al. FTO fuels diabetes-induced vascular endothelial dysfunction associated with inflammation by erasing m6A methylation of TNIP1. J. Clin. Invest 133, e160517 (2023).
Zheng, W. et al. Multiple functions and mechanisms underlying the role of METTL3 in human cancers. Front. Oncol. 9, 1403 (2019).
Vu, L. P. et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 23, 1369–1376 (2017).
Cheng, M. et al. The m(6)A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-kappaB/MYC signaling network. Oncogene 38, 3667–3680 (2019).
Zeng, C., Huang, W., Li, Y. & Weng, H. Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J. Hematol. Oncol. 13, 117 (2020).
Lin, C. et al. METTL3 enhances pancreatic ductal adenocarcinoma progression and gemcitabine resistance through modifying DDX23 mRNA N6 adenosine methylation. Cell Death Dis. 14, 221 (2023).
Qu, J. et al. RNA demethylase ALKBH5 in cancer: from mechanisms to therapeutic potential. J. Hematol. Oncol. 15, 8 (2022).
Schumann, U., Shafik, A. & Preiss, T. METTL3 gains R/W access to the epitranscriptome. Mol. Cell 62, 323–324 (2016).
Shi, B. et al. The role, mechanism, and application of RNA methyltransferase METTL14 in gastrointestinal cancer. Mol. Cancer 21, 163 (2022).
Miyake, K. et al. A cancer-associated METTL14 mutation induces aberrant m6A modification, affecting tumor growth. Cell Rep. 42, 112688 (2023).
Hou, Y. et al. METTL14 modulates glycolysis to inhibit colorectal tumorigenesis in p53-wild-type cells. EMBO Rep. 24, e56325 (2023).
Chen, X. et al. METTL14-mediated N6-methyladenosine modification of SOX4 mRNA inhibits tumor metastasis in colorectal cancer. Mol. Cancer 19, 106 (2020).
Yang, X. et al. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol. Cancer 19, 46 (2020).
Fan, H. N. et al. METTL14-mediated m(6)A modification of circORC5 suppresses gastric cancer progression by regulating miR-30c-2-3p/AKT1S1 axis. Mol. Cancer 21, 51 (2022).
Gong, D. et al. The m6A-suppressed P2RX6 activation promotes renal cancer cells migration and invasion through ATP-induced Ca2+ influx modulating ERK1/2 phosphorylation and MMP9 signaling pathway. J. Exp. Clin. Cancer Res. 38, 233 (2019).
Zhang, C. et al. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. 8, 4766–4781 (2019).
Weng, H. et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell 22, 191–205.e199 (2018).
Wang, M. et al. Upregulation of METTL14 mediates the elevation of PERP mRNA N(6) adenosine methylation promoting the growth and metastasis of pancreatic cancer. Mol. Cancer 19, 130 (2020).
Yankova, E. et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597–601 (2021).
Sun, T. et al. LNC942 promoting METTL14-mediated m(6)A methylation in breast cancer cell proliferation and progression. Oncogene 39, 5358–5372 (2020).
Iles, M. M. et al. A variant in FTO shows association with melanoma risk not due to BMI. Nat. Genet 45, 428–432.e421 (2013).
Guan, Q. et al. Functions, mechanisms, and therapeutic implications of METTL14 in human cancer. J. Hematol. Oncol. 15, 13 (2022).
Zhou, Y. et al. N6-methyladenosine demethylase FTO promotes growth and metastasis of gastric cancer via m6A modification of caveolin-1 and metabolic regulation of mitochondrial dynamics. Cell Death Dis. 13, 72 (2022).
Li, J. et al. The m6A demethylase FTO promotes the growth of lung cancer cells by regulating the m6A level of USP7 mRNA. Biochem. Biophys. Res. Commun. 512, 479–485 (2019).
Li, Z. et al. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell 31, 127–141 (2017).
Yang, S. et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 10, 2782 (2019).
Huang, H. et al. FTO-dependent N (6)-methyladenosine modifications inhibit ovarian cancer stem cell self-renewal by blocking cAMP signaling. Cancer Res. 80, 3200–3214 (2020).
Sun, D. et al. Fat mass and obesity-associated protein regulates lipogenesis via m(6) A modification in fatty acid synthase mRNA. Cell Biol. Int. 45, 334–344 (2021).
Jeschke, J. et al. Downregulation of the FTO m(6)A RNA demethylase promotes EMT-mediated progression of epithelial tumors and sensitivity to Wnt inhibitors. Nat. Cancer 2, 611–628 (2021).
Yuan, Y. et al. ALKBH5 suppresses tumor progression via an m6A-dependent epigenetic silencing of pre-miR-181b-1/YAP signaling axis in osteosarcoma. Cell Death Dis. 12, 60 (2021).
Zhang, S. et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31, 591–606.e596 (2017).
Liu, X. et al. M(6)A demethylase ALKBH5 regulates FOXO1 mRNA stability and chemoresistance in triple-negative breast cancer. Redox Biol. 69, 102993 (2024).
Hao, L. et al. ALKBH5-mediated m(6)A demethylation of FOXM1 mRNA promotes progression of uveal melanoma. Aging 13, 4045–4062 (2021).
Yu, H. et al. ALKBH5 inhibited cell proliferation and sensitized bladder cancer cells to cisplatin by m6A-CK2alpha-mediated glycolysis. Mol. Ther. Nucleic Acids 23, 27–41 (2021).
Zhu, H. et al. ALKBH5 inhibited autophagy of epithelial ovarian cancer through miR-7 and BCL-2. J. Exp. Clin. Cancer Res. 38, 163 (2019).
Guo, X. et al. RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A-YTHDF2-dependent manner. Mol. Cancer 19, 91 (2020).
Tang, B. et al. m(6)A demethylase ALKBH5 inhibits pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation and mediating Wnt signaling. Mol. Cancer 19, 3 (2020).
He, Y. et al. ALKBH5-mediated m(6)A demethylation of KCNK15-AS1 inhibits pancreatic cancer progression via regulating KCNK15 and PTEN/AKT signaling. Cell Death Dis. 12, 1121 (2021).
Zhao, X. et al. Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomark. 21, 859–868 (2018).
Liu, L. et al. N6-methyladenosine-related genomic targets are altered in breast cancer tissue and associated with poor survival. J. Cancer 10, 5447–5459 (2019).
Han, D. et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 566, 270–274 (2019).
Lin, Z. et al. RNA m(6) A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. EMBO J. 39, e103181 (2020).
Hao, L. et al. m6A-YTHDF1-mediated TRIM29 upregulation facilitates the stem cell-like phenotype of cisplatin-resistant ovarian cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 1868, 118878 (2021).
Zhang, X. et al. N6-methyladenosine reader YTHDF1 promotes stemness and therapeutic resistance in hepatocellular carcinoma by enhancing NOTCH1 expression. Cancer Res. 84, 827–840 (2024).
Xu, Y. et al. YTH domain proteins: a family of m(6)A readers in cancer progression. Front. Oncol. 11, 629560 (2021).
Bai, Y. et al. YTHDF1 regulates tumorigenicity and cancer stem cell-like activity in human colorectal carcinoma. Front. Oncol. 9, 332 (2019).
Shi, Y. et al. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat. Commun. 10, 4892 (2019).
Chen, M. et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 67, 2254–2270 (2018).
Sheng, H. et al. YTH domain family 2 promotes lung cancer cell growth by facilitating 6-phosphogluconate dehydrogenase mRNA translation. Carcinogenesis 41, 541–550 (2020).
Yang, H. et al. Hypoxia inducible lncRNA-CBSLR modulates ferroptosis through m6A-YTHDF2-dependent modulation of CBS in gastric cancer. J. Adv. Res. 37, 91–106 (2022).
Yang, N. et al. Genetic variants in m6A modification genes are associated with esophageal squamous-cell carcinoma in the Chinese population. Carcinogenesis 41, 761–768 (2020).
Zhao, Y. et al. YTHDF3 facilitates eIF2AK2 and eIF3A recruitment on mRNAs to regulate translational processes in oxaliplatin-resistant colorectal cancer. ACS Chem. Biol. 17, 1778–1788 (2022).
Chen, S. et al. YTHDF3-mediated m6A modification of NKD1 regulates hepatocellular carcinoma invasion and metastasis by activating the WNT/beta-catenin signaling axis. Exp. Cell Res. 442, 114192 (2024).
Liao, J. et al. Insight into the structure, physiological function, and role in cancer of m6A readers-YTH domain-containing proteins. Cell Death Discov. 8, 137 (2022).
Jin, H. et al. N(6)-methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBioMedicine 47, 195–207 (2019).
Munshi, P. K., Govekar, D. P., Jadhav, A. P. & Telang, V. G. Diffuse venous synovial haemangioma arising from the knee joint. Indian J. Med. Sci. 49, 142–143 (1995).
Chen, L. et al. N6-methyladenosine reader YTHDF family in biological processes: Structures, roles, and mechanisms. Front. Immunol. 14, 1162607 (2023).
Zheng, Y. F. et al. Urinary nucleosides as biological markers for patients with colorectal cancer. World J. Gastroenterol. 11, 3871–3876 (2005).
Gao, Y. et al. Integrated analyses of m(1)A regulator-mediated modification patterns in tumor microenvironment-infiltrating immune cells in colon cancer. Oncoimmunology 10, 1936758 (2021).
Monshaugen, I. et al. Depletion of the m1A writer TRMT6/TRMT61A reduces proliferation and resistance against cellular stress in bladder cancer. Front. Oncol. 13, 1334112 (2023).
Li, J. et al. Differential analysis of RNA methylation regulators in gastric cancer based on TCGA data set and construction of a prognostic model. J. Gastrointest. Oncol. 12, 1384–1397 (2021).
Zhao, Y. et al. m1A regulated genes modulate PI3K/AKT/mTOR and ErbB pathways in gastrointestinal cancer. Transl. Oncol. 12, 1323–1333 (2019).
Wang, Y. et al. N(1)-methyladenosine methylation in tRNA drives liver tumourigenesis by regulating cholesterol metabolism. Nat. Commun. 12, 6314 (2021).
Zhang, Y., Chen, Y. & Wen, W. Four types of adenine-related RNA modification writers -mediated molecular subtypes contribute to predicting clinical outcomes and treatment options in bladder cancer. Front. Immunol. 14, 1152806 (2023).
Chen, W. et al. ALKBH1-mediated m(1) A demethylation of METTL3 mRNA promotes the metastasis of colorectal cancer by downregulating SMAD7 expression. Mol. Oncol. 17, 344–364 (2023).
Li, H. et al. ALKBH1 promotes lung cancer by regulating m6A RNA demethylation. Biochem. Pharm. 189, 114284 (2021).
Orsolic, I., Carrier, A. & Esteller, M. Genetic and epigenetic defects of the RNA modification machinery in cancer. Trends Genet. 39, 74–88 (2023).
Cheng, W. et al. Demethylation of m1A assisted degradation of the signal probe for rapid electrochemical detection of ALKBH3 activity with practical applications. Talanta 240, 123151 (2022).
Liu, Y. et al. Research progress of N1-methyladenosine RNA modification in cancer. Cell Commun. Signal 22, 79 (2024).
Tasaki, M. et al. ALKBH3, a human AlkB homologue, contributes to cell survival in human non-small-cell lung cancer. Br. J. Cancer 104, 700–706 (2011).
Wu, Y. et al. RNA m(1)A methylation regulates glycolysis of cancer cells through modulating ATP5D. Proc. Natl Acad. Sci. USA 119, e2119038119 (2022).
Kogaki, T. et al. TP53 gene status is a critical determinant of phenotypes induced by ALKBH3 knockdown in non-small cell lung cancers. Biochem. Biophys. Res. Commun. 488, 285–290 (2017).
Chen, Z. et al. Transfer RNA demethylase ALKBH3 promotes cancer progression via induction of tRNA-derived small RNAs. Nucleic Acids Res. 47, 2533–2545 (2019).
Zheng, L. et al. Deciphering the vital roles and mechanism of m5C modification on RNA in cancers. Am. J. Cancer Res. 13, 6125–6146 (2023).
Chen, B. et al. m5C regulator-mediated modification patterns and tumor microenvironment infiltration characterization in colorectal cancer: One step closer to precision medicine. Front. Immunol. 13, 1049435 (2022).
Chen, X. et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat. Cell Biol. 21, 978–990 (2019).
Zhang, Q. et al. RNA m(5)C regulator-mediated modification patterns and the cross-talk between tumor microenvironment infiltration in gastric cancer. Front. Immunol. 13, 905057 (2022).
Xue, S. et al. Depletion of TRDMT1 affects 5-methylcytosine modification of mRNA and inhibits HEK293 cell proliferation and migration. Biochem. Biophys. Res. Commun. 520, 60–66 (2019).
Liao, H. et al. Human NOP2/NSUN1 regulates ribosome biogenesis through non-catalytic complex formation with box C/D snoRNPs. Nucleic Acids Res. 50, 10695–10716 (2022).
Hughes, R. O., Davis, H. J., Nease, L. A. & Piskounova, E. Decoding the role of tRNA modifications in cancer progression. Curr. Opin. Genet Dev. 88, 102238 (2024).
Jin, S. et al. RNA 5-methylcytosine regulator NSUN3 promotes tumor progression through regulating immune infiltration in head and neck squamous cell carcinoma. Oral. Dis. 30, 313–328 (2024).
Delaunay, S. et al. Mitochondrial RNA modifications shape metabolic plasticity in metastasis. Nature 607, 593–603 (2022).
Zhao, Z. et al. NSUN4 mediated RNA 5-methylcytosine promotes the malignant progression of glioma through improving the CDC42 mRNA stabilization. Cancer Lett. 597, 217059 (2024).
Wu, J. et al. NSUN4-mediated m5C modification of circERI3 promotes lung cancer development by altering mitochondrial energy metabolism. Cancer Lett. 605, 217266 (2024).
Gu, X. et al. RNA 5-methylcytosine writer NSUN5 promotes hepatocellular carcinoma cell proliferation via a ZBED3-dependent mechanism. Oncogene 43, 624-635 (2024).
Selmi, T. et al. Sequence- and structure-specific cytosine-5 mRNA methylation by NSUN6. Nucleic Acids Res 49, 1006–1022 (2021).
Yang, R. et al. The RNA methyltransferase NSUN6 suppresses pancreatic cancer development by regulating cell proliferation. EBioMedicine 63, 103195 (2021).
Fang, X. et al. Role of m(5) C RNA methylation regulators in colorectal cancer prognosis and immune microenvironment. J. Clin. Lab Anal. 36, e24303 (2022).
Huang, Y. et al. Exploration of potential roles of m5C-related regulators in colon adenocarcinoma prognosis. Front. Genet 13, 816173 (2022).
Shen, H. et al. TET-mediated 5-methylcytosine oxidation in tRNA promotes translation. J. Biol. Chem. 296, 100087 (2021).
Li, X. & Meng, Y. Expression and prognostic characteristics of m(5) C regulators in low-grade glioma. J. Cell Mol. Med. 25, 1383–1393 (2021).
Wu, J. et al. Comprehensive Analysis of m5C RNA Methylation Regulator Genes in Clear Cell Renal Cell Carcinoma. Int. J. Genomics 2021, 3803724 (2021).
Xu, Z. et al. Roles of m5C RNA modification patterns in biochemical recurrence and tumor microenvironment characterization of prostate adenocarcinoma. Front. Immunol. 13, 869759 (2022).
Yu, G. et al. Comprehensive analysis of m5C methylation regulatory genes and tumor microenvironment in prostate cancer. Front. Immunol. 13, 914577 (2022).
Gu, X. et al. Vital roles of m(5)C RNA modification in cancer and immune cell biology. Front. Immunol. 14, 1207371 (2023).
Li, X. Y. & Yang, X. T. Correlation between the RNA methylation genes and immune infiltration and prognosis of patients with hepatocellular carcinoma: a pan-cancer analysis. J. Inflamm. Res. 15, 3941–3956 (2022).
Xue, C., Zhao, Y., Li, G. & Li, L. Multi-omic analyses of the m5C regulator ALYREF reveal its essential roles in hepatocellular carcinoma. Front. Oncol. 11, 633415 (2021).
Nagy, Z. et al. An ALYREF-MYCN coactivator complex drives neuroblastoma tumorigenesis through effects on USP3 and MYCN stability. Nat. Commun. 12, 1881 (2021).
He, Y. et al. Role of m(5)C-related regulatory genes in the diagnosis and prognosis of hepatocellular carcinoma. Am. J. Transl. Res. 12, 912–922 (2020).
Yu, W. et al. YAP 5-methylcytosine modification increases its mRNA stability and promotes the transcription of exosome secretion-related genes in lung adenocarcinoma. Cancer Gene Ther. 30, 149–162 (2023).
Yan, J. et al. FOXC2-AS1 stabilizes FOXC2 mRNA via association with NSUN2 in gastric cancer cells. Hum. Cell 34, 1755–1764 (2021).
Han, H. et al. N(7)-methylguanosine tRNA modification promotes esophageal squamous cell carcinoma tumorigenesis via the RPTOR/ULK1/autophagy axis. Nat. Commun. 13, 1478 (2022).
Ying, X. et al. METTL1-m(7) G-EGFR/EFEMP1 axis promotes the bladder cancer development. Clin. Transl. Med. 11, e675 (2021).
Yin, H. et al. 5-Methylcytosine (m(5)C) modification in peripheral blood immune cells is a novel non-invasive biomarker for colorectal cancer diagnosis. Front. Immunol. 13, 967921 (2022).
Zhu, W. et al. Positive epigenetic regulation loop between AR and NSUN2 promotes prostate cancer progression. Clin. Transl. Med. 12, e1028 (2022).
Kang, Y. et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J. Natl Cancer Inst. 105, 1485–1495 (2013).
Wang, L. et al. Distinct roles of m(5)C RNA methyltransferase NSUN2 in major gynecologic cancers. Front. Oncol. 12, 786266 (2022).
Zheng, Q. et al. Genetic characteristics and prognostic implications of m1A regulators in pancreatic cancer. Biosci. Rep. 41, BSR20210337 (2021).
Chen, X. H. et al. Regulations of m(6)A and other RNA modifications and their roles in cancer. Front Med 18, 622–648 (2024).
Nian, Z. et al. RNA epigenetic modifications in digestive tract cancers: friends or foes. Pharmacol. Res. 206, 107280, (2024).
Zhang, M. et al. Roles of RNA methylation on tumor immunity and clinical implications. Front. Immunol. 12, 641507 (2021).
Zhang, X. et al. Internal m 6 A and m 7 G RNA modifications in hematopoietic system and acute myeloid leukemia. Chin. Med. J. 137, 1033–1043 (2024).
Huang, M. et al. METTL1-mediated m7G tRNA modification promotes lenvatinib resistance in hepatocellular carcinoma. Cancer Res. 83, 89–102 (2023).
Garcia-Vilchez, R. et al. METTL1 promotes tumorigenesis through tRNA-derived fragment biogenesis in prostate cancer. Mol. Cancer 22, 119 (2023).
Ma, J. et al. METTL1/WDR4-mediated m(7)G tRNA modifications and m(7)G codon usage promote mRNA translation and lung cancer progression. Mol. Ther. 29, 3422–3435 (2021).
Pandolfini, L. et al. METTL1 promotes let-7 MicroRNA processing via m7G methylation. Mol. Cell 74, 1278–1290.e1279 (2019).
Wang, Z. et al. METTL1/WDR4-mediated tRNA m(7)G modification and mRNA translation control promote oncogenesis and doxorubicin resistance. Oncogene 42, 1900–1912 (2023).
Lu, Y. et al. RNA-binding protein QKI promotes the progression of HCC by interacting with long non-coding RNA EGOT. Int. Immunopharmacol. 136, 112297 (2024).
Huang, H. et al. FOXP3-regulated lncRNA NONHSAT136151 promotes colorectal cancer progression by disrupting QKI interaction with target mRNAs. J. Cell Mol. Med. 28, e18068 (2024).
Zakutansky, P. M. et al. Isoform balance of the long noncoding RNA NEAT1 is regulated by the RNA-binding protein QKI, governs the glioma transcriptome, and impacts cell migration. J. Biol. Chem. 300, 107595 (2024).
Neumann, D. P. et al. The landscape of alternative polyadenylation during EMT and its regulation by the RNA-binding protein Quaking. RNA Biol. 21, 1–11 (2024).
Jemal, A., Siegel, R., Xu, J. & Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 60, 277–300 (2010).
Siegel, R. L. et al. Cancer statistics for Hispanics/Latinos, 2015. CA Cancer J. Clin. 65, 457–480 (2015).
Zhang, H. et al. QKI-6 suppresses cell proliferation, migration, and EMT in non-small cell lung cancer. Front. Oncol. 12, 897553 (2022).
Zhao, Z. et al. QKI shuttles internal m(7)G-modified transcripts into stress granules and modulates mRNA metabolism. Cell 186, 3208–3226.e3227 (2023).
Xie, J. et al. The m(7)G reader NCBP2 promotes pancreatic cancer progression by upregulating MAPK/ERK Signaling. Cancers 15, 5454 (2023).
Arora, R. et al. NCBP2 and TFRC are novel prognostic biomarkers in oral squamous cell carcinoma. Cancer Gene Ther. 30, 752–765 (2023).
Nojima, T. & Proudfoot, N. J. Mechanisms of lncRNA biogenesis as revealed by nascent transcriptomics. Nat. Rev. Mol. Cell Biol. 23, 389–406 (2022).
Tan, Y. T. et al. LncRNA-mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Commun. 41, 109–120 (2021).
Sur, S. & Ray, R. B. Emerging role of lncRNA ELDR in development and cancer. FEBS J. 289, 3011–3023 (2022).
Bhan, A., Soleimani, M. & Mandal, S. S. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 77, 3965–3981 (2017).
Li, Y. et al. Pan-cancer characterization of immune-related lncRNAs identifies potential oncogenic biomarkers. Nat. Commun. 11, 1000 (2020).
Esposito, R. et al. Hacking the cancer genome: profiling therapeutically actionable long non-coding RNAs using CRISPR-Cas9 screening. Cancer Cell 35, 545–557 (2019).
Luo, J., Langer, L. F. & Liu, J. A novel role of LncRNA in regulating tumor metabolism and angiogenesis under hypoxia. Cancer Commun. 39, 2 (2019).
Bartonicek, N., Maag, J. L. & Dinger, M. E. Long noncoding RNAs in cancer: mechanisms of action and technological advancements. Mol. Cancer 15, 43 (2016).
Sideris, N. et al. LncRNAs in breast cancer: a link to future approaches. Cancer Gene Ther. 29, 1866–1877 (2022).
Gupta, R. A. et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076 (2010).
Hu, H. et al. LncRNA HOTAIR promotes DNA damage repair and radioresistance by targeting ATR in colorectal cancer. Oncol. Res. 32, 1335–1346 (2024).
Ren, M. M. et al. Roles of HOTAIR in lung cancer susceptibility and prognosis. Mol. Genet Genom. Med. 8, e1299 (2020).
Ma, J. et al. Long non-coding RNA ANRIL promotes chemoresistance in triple-negative breast cancer via enhancing aerobic glycolysis. Life Sci. 306, 120810 (2022).
Peng, Z., Zhang, C. & Duan, C. Functions and mechanisms of long noncoding RNAs in lung cancer. Onco Targets Ther. 9, 4411–4424 (2016).
Bhat, A. A. et al. MALAT1: A key regulator in lung cancer pathogenesis and therapeutic targeting. Pathol. Res Pr. 253, 154991 (2024).
Esfandi, F. et al. The expression of CCAT2, UCA1, PANDA and GHET1 long non-coding RNAs in lung cancer. Rep. Biochem. Mol. Biol. 8, 36–41 (2019).
Wang, C., Tao, X. & Wei, J. Effects of LncRNA MEG3 on immunity and autophagy of non-small cell lung carcinoma through IDO signaling pathway. World J. Surg. Oncol. 19, 244 (2021).
Huang, Z. et al. The role of long noncoding RNAs in hepatocellular carcinoma. Mol. Cancer 19, 77 (2020).
Klec, C., Gutschner, T., Panzitt, K. & Pichler, M. Involvement of long non-coding RNA HULC (highly up-regulated in liver cancer) in pathogenesis and implications for therapeutic intervention. Expert Opin. Ther. Targets 23, 177–186 (2019).
Wang, X. et al. Long non-coding RNA DILC regulates liver cancer stem cells via IL-6/STAT3 axis. J. Hepatol. 64, 1283–1294 (2016).
Diener, C., Keller, A. & Meese, E. Emerging concepts of miRNA therapeutics: from cells to clinic. Trends Genet 38, 613–626 (2022).
Ferragut Cardoso, A. P. et al. miRNA dysregulation is an emerging modulator of genomic instability. Semin. Cancer Biol. 76, 120–131 (2021).
Lu, T. X. & Rothenberg, M. E. MicroRNA. J. Allergy Clin. Immunol. 141, 1202–1207 (2018).
Cai, Y., Yu, X., Hu, S. & Yu, J. A brief review on the mechanisms of miRNA regulation. Genomics Proteom. Bioinforma. 7, 147–154 (2009).
Saliminejad, K., Khorram Khorshid, H. R., Soleymani Fard, S. & Ghaffari, S. H. An overview of microRNAs: biology, functions, therapeutics, and analysis methods. J. Cell Physiol. 234, 5451–5465 (2019).
Mishra, S., Yadav, T. & Rani, V. Exploring miRNA based approaches in cancer diagnostics and therapeutics. Crit. Rev. Oncol. Hematol. 98, 12–23 (2016).
Kim, T. & Croce, C. M. MicroRNA: trends in clinical trials of cancer diagnosis and therapy strategies. Exp. Mol. Med. 55, 1314–1321 (2023).
Seo, Y., Rhim, J. & Kim, J. H. RNA-binding proteins and exoribonucleases modulating miRNA in cancer: the enemy within. Exp. Mol. Med. 56, 1080–1106 (2024).
Metcalf, G. A. D. MicroRNAs: circulating biomarkers for the early detection of imperceptible cancers via biosensor and machine-learning advances. Oncogene 43, 2135–2142 (2024).
Peng, Y. & Croce, C. M. The role of MicroRNAs in human cancer. Sig. Transduct. Target Ther. 1, 15004 (2016).
Calin, G. A. et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl Acad. Sci. USA 99, 15524–15529 (2002).
Pfeffer, S. R., Yang, C. H. & Pfeffer, L. M. The Role of miR-21 in Cancer. Drug Dev. Res. 76, 270–277 (2015).
Zhu, X. et al. miR-145 sensitizes ovarian cancer cells to paclitaxel by targeting Sp1 and Cdk6. Int. J. Cancer 135, 1286–1296 (2014).
Yu, Y. et al. miR-21 and miR-145 cooperation in regulation of colon cancer stem cells. Mol. Cancer 14, 98 (2015).
Liu, C., Rokavec, M., Huang, Z. & Hermeking, H. Curcumin activates a ROS/KEAP1/NRF2/miR-34a/b/c cascade to suppress colorectal cancer metastasis. Cell Death Differ. 30, 1771–1785 (2023).
Huang, W. T., Kuo, S. H., Kuo, Y. C. & Lin, C. W. miR-155-regulated mTOR and Toll-like receptor 5 in gastric diffuse large B-cell lymphoma. Cancer Med. 11, 555–570 (2022).
Liu, F. et al. MiR-155 inhibits proliferation and invasion by directly targeting PDCD4 in non-small cell lung cancer. Thorac. Cancer 8, 613–619 (2017).
Qin, W. et al. MicroRNA-155 is a novel suppressor of ovarian cancer-initiating cells that targets CLDN1. FEBS Lett. 587, 1434–1439 (2013).
Li, X., Li, L. & Wu, J. The members of the miR-148/152 family inhibit cancer stem cell-like properties in gastric cancer via negative regulation of ITGA5. J. Transl. Med. 21, 105 (2023).
Cao, D. et al. MiR-128 suppresses metastatic capacity by targeting metadherin in breast cancer cells. Biol. Res 53, 43 (2020).
Li, M. et al. A test of miR-128-3p and miR-33a-5p in serum exosome as biomarkers for auxiliary diagnosis of non-small cell lung cancer. J. Thorac. Dis. 15, 2616–2626 (2023).
Kumar, S. et al. Analysis of miR-375-3p, miR-197-3p, and miR-15a-5p expression and their clinical relevance as biomarkers in lung cancer. Technol. Cancer Res Treat. 21, 15330338221080981 (2022).
Hsu, M. T. & Coca-Prados, M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature 280, 339–340 (1979).
Wang, Y. & Liu, B. Circular RNA in diseased heart. Cells 9, 1240 (2020).
Jin, J. et al. Circular RNA in renal diseases. J. Cell Mol. Med. 24, 6523–6533 (2020).
Kristensen, L. S. et al. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet 20, 675–691 (2019).
Lei, M. et al. Translation and functional roles of circular RNAs in human cancer. Mol. Cancer 19, 30 (2020).
Zhang, Z. H. et al. The function and mechanisms of action of circular RNAs in Urologic Cancer. Mol. Cancer 22, 61 (2023).
Li, J. et al. Circular RNAs in cancer: biogenesis, function, and clinical significance. Trends Cancer 6, 319–336 (2020).
Kristensen, L. S., Hansen, T. B., Veno, M. T. & Kjems, J. Circular RNAs in cancer: opportunities and challenges in the field. Oncogene 37, 555–565 (2018).
Sharma, A. R. et al. Recent research progress on circular RNAs: biogenesis, properties, functions, and therapeutic potential. Mol. Ther. Nucleic Acids 25, 355–371 (2021).
Chen, L. & Shan, G. CircRNA in cancer: fundamental mechanism and clinical potential. Cancer Lett. 505, 49–57 (2021).
Han, D. et al. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology 66, 1151–1164 (2017).
Zhang, P. F. et al. Circular RNA circTRIM33-12 acts as the sponge of MicroRNA-191 to suppress hepatocellular carcinoma progression. Mol. Cancer 18, 105 (2019).
Wang, L. et al. Circular RNA circRHOT1 promotes hepatocellular carcinoma progression by initiation of NR2F6 expression. Mol. Cancer 18, 119 (2019).
Zhang, L. et al. Circular RNA CircCACTIN promotes gastric cancer progression by sponging MiR-331-3p and regulating TGFBR1 expression. Int. J. Biol. Sci. 15, 1091–1103 (2019).
Zhu, Z. et al. Circular RNA circNHSL1 promotes gastric cancer progression through the miR-1306-3p/SIX1/vimentin axis. Mol. Cancer 18, 126 (2019).
Dai, X. et al. Circular RNA circFGD4 suppresses gastric cancer progression via modulating miR-532-3p/APC/beta-catenin signalling pathway. Clin. Sci. 134, 1821–1839 (2020).
Chen, B. et al. circEPSTI1 as a prognostic marker and mediator of triple-negative breast cancer progression. Theranostics 8, 4003–4015 (2018).
Liang, G. et al. Autophagy-associated circRNA circCDYL augments autophagy and promotes breast cancer progression. Mol. Cancer 19, 65 (2020).
Wang, D. et al. Circular RNA HSDL2 promotes breast cancer progression via miR-7978 ZNF704 axis and regulating hippo signaling pathway. Breast Cancer Res. 26, 105 (2024).
Shangguan, H. et al. Circular RNA circSLC25A16 contributes to the glycolysis of non-small-cell lung cancer through epigenetic modification. Cell Death Dis. 11, 437 (2020).
Cheng, Z. et al. circTP63 functions as a ceRNA to promote lung squamous cell carcinoma progression by upregulating FOXM1. Nat. Commun. 10, 3200 (2019).
Qiu, M. et al. The circular RNA circPRKCI promotes tumor growth in lung adenocarcinoma. Cancer Res. 78, 2839–2851 (2018).
Shi, Y. et al. Circular RNA LPAR3 sponges microRNA-198 to facilitate esophageal cancer migration, invasion, and metastasis. Cancer Sci. 111, 2824–2836 (2020).
Zhang, Y. et al. hsa_circRNA6448-14 promotes carcinogenesis in esophageal squamous cell carcinoma. Aging 12, 15581–15602 (2020).
Wong, C. H. et al. CircFOXK2 promotes growth and metastasis of pancreatic ductal adenocarcinoma by complexing with RNA-binding proteins and sponging MiR-942. Cancer Res. 80, 2138–2149 (2020).
Guo, X. et al. Circular RNA circBFAR promotes the progression of pancreatic ductal adenocarcinoma via the miR-34b-5p/MET/Akt axis. Mol. Cancer 19, 83 (2020).
Goto, Y., Seino, Y., Note, S. & Imura, H. The dual effect of alloxan modulated by 3-O-methylglucose or somatostatin on insulin secretion in the isolated perfused rat pancreas. Horm. Metab. Res. 12, 140–143 (1980).
Liu, Y. et al. The emerging role of the piRNA/piwi complex in cancer. Mol. Cancer 18, 123 (2019).
Charbe, N. B. et al. Small interfering RNA for cancer treatment: overcoming hurdles in delivery. Acta Pharm. Sin. B 10, 2075–2109 (2020).
Moyano, M. & Stefani, G. piRNA involvement in genome stability and human cancer. J. Hematol. Oncol. 8, 38 (2015).
Raza, A. et al. Dynamic liquid biopsy components as predictive and prognostic biomarkers in colorectal cancer. J. Exp. Clin. Cancer Res. 41, 99 (2022).
Garcia-Borja, E. et al. Critical appraisal of the piRNA-PIWI axis in cancer and cancer stem cells. Biomark. Res. 12, 15 (2024).
Lin, Y., Zheng, J. & Lin, D. PIWI-interacting RNAs in human cancer. Semin. Cancer Biol. 75, 15–28 (2021).
Fonseca Cabral, G. et al. piRNAs in gastric cancer: a new approach towards translational research. Int. J. Mol. Sci. 21, 2126 (2020).
Jian, Z., Han, Y. & Li, H. Potential roles of PIWI-interacting RNAs in lung cancer. Front. Oncol. 12, 944403 (2022).
Su, J. F. et al. PIWI-interacting RNAs: Mitochondria-based biogenesis and functions in cancer. Genes Dis. 8, 603–622 (2021).
Guo, B., Li, D., Du, L. & Zhu, X. piRNAs: biogenesis and their potential roles in cancer. Cancer Metastasis Rev. 39, 567–575 (2020).
Zhou, J. et al. PIWI-interacting RNAs: critical roles and therapeutic targets in cancer. Cancer Lett. 562, 216189 (2023).
Cheng, J. et al. piRNA, the new non-coding RNA, is aberrantly expressed in human cancer cells. Clin. Chim. Acta 412, 1621–1625 (2011).
Martinez, V. D., Enfield, K. S. S., Rowbotham, D. A. & Lam, W. L. An atlas of gastric PIWI-interacting RNA transcriptomes and their utility for identifying signatures of gastric cancer recurrence. Gastric Cancer 19, 660–665 (2016).
Shaker, F. H. et al. piR-823 tale as emerging cancer-hallmark molecular marker in different cancer types: a step-toward ncRNA-precision. Naunyn. Schmiedebergs Arch. Pharmacol. 398, 47–68 (2024).
Cheng, J. et al. piR-823, a novel non-coding small RNA, demonstrates in vitro and in vivo tumor suppressive activity in human gastric cancer cells. Cancer Lett. 315, 12–17 (2012).
Liu, T. et al. Piwi-interacting RNA-651 promotes cell proliferation and migration and inhibits apoptosis in breast cancer by facilitating DNMT1-mediated PTEN promoter methylation. Cell Cycle 20, 1603–1616 (2021).
Huang, G. et al. Altered expression of piRNAs and their relation with clinicopathologic features of breast cancer. Clin. Transl. Oncol. 15, 563–568 (2013).
Fu, A. et al. PIWI-interacting RNA 021285 is involved in breast tumorigenesis possibly by remodeling the cancer epigenome. Carcinogenesis 36, 1094–1102 (2015).
Zhang, H. et al. The expression of stem cell protein Piwil2 and piR-932 in breast cancer. Surg. Oncol. 22, 217–223 (2013).
Tan, L. et al. PIWI-interacting RNA-36712 restrains breast cancer progression and chemoresistance by interaction with SEPW1 pseudogene SEPW1P RNA. Mol. Cancer 18, 9 (2019).
Yin, J. et al. piR-823 contributes to colorectal tumorigenesis by enhancing the transcriptional activity of HSF1. Cancer Sci. 108, 1746–1756 (2017).
Mai, D. et al. Serum piRNA-54265 is a New Biomarker for early detection and clinical surveillance of Human Colorectal Cancer. Theranostics 10, 8468–8478 (2020).
Wang, S. et al. piR-823 inhibits cell apoptosis via modulating mitophagy by binding to PINK1 in colorectal cancer. Cell Death Dis. 13, 465 (2022).
Iyer, D. N. et al. Small RNA profiling of piRNAs in colorectal cancer identifies consistent overexpression of piR-24000 that correlates clinically with an aggressive disease phenotype. Cancers 12, 188 (2020).
Li, S., Kouznetsova, V. L., Kesari, S. & Tsigelny, I. F. piRNA in machine-learning-based diagnostics of colorectal cancer. Molecules 29, 4311 (2024).
Shi, Z. D. et al. Tumor cell plasticity in targeted therapy-induced resistance: mechanisms and new strategies. Sig. Transduct. Target Ther. 8, 113 (2023).
Rezayatmand, H., Razmkhah, M. & Razeghian-Jahromi, I. Drug resistance in cancer therapy: the Pandora’s Box of cancer stem cells. Stem Cell Res. Ther. 13, 181 (2022).
Huang, R. & Zhou, P. K. DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Sig. Transduct. Target Ther. 6, 254 (2021).
Le Tourneau, C. et al. Tumour growth kinetics assessment: added value to RECIST in cancer patients treated with molecularly targeted agents. Br. J. Cancer 106, 854–857 (2012).
Niu, X. et al. Cancer plasticity in therapy resistance: Mechanisms and novel strategies. Drug Resist. Updat 76, 101114 (2024).
Lei, Z. N. et al. Understanding and targeting resistance mechanisms in cancer. MedComm 4, e265 (2023).
Russo, M. et al. Cancer drug-tolerant persister cells: from biological questions to clinical opportunities. Nat. Rev. Cancer 24, 694–717 (2024).
Hosea, R. et al. The two sides of chromosomal instability: drivers and brakes in cancer. Sig. Transduct. Target Ther. 9, 75 (2024).
Perez-Gonzalez, A., Bevant, K. & Blanpain, C. Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat. Cancer 4, 1063–1082 (2023).
Franca, G. S. et al. Cellular adaptation to cancer therapy along a resistance continuum. Nature 631, 876–883 (2024).
Huang, R. X. & Zhou, P. K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Sig. Transduct. Target Ther. 5, 60 (2020).
Bukowski, K., Kciuk, M. & Kontek, R. Mechanisms of multidrug resistance in cancer chemotherapy. Int J. Mol. Sci. 21, 3233 (2020).
Bai, R. et al. Mechanisms of cancer resistance to immunotherapy. Front. Oncol. 10, 1290 (2020).
McLaughlin, M. et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 20, 203–217 (2020).
Beckers, C., Pruschy, M. & Vetrugno, I. Tumor hypoxia and radiotherapy: a major driver of resistance even for novel radiotherapy modalities. Semin. Cancer Biol. 98, 19–30 (2024).
de Perrot, M., Wu, L., Wu, M. & Cho, B. C. J. Radiotherapy for the treatment of malignant pleural mesothelioma. Lancet Oncol. 18, e532–e542 (2017).
Shishido, K., Reinders, A. & Asuthkar, S. Epigenetic regulation of radioresistance: insights from preclinical and clinical studies. Expert Opin. Investig. Drugs 31, 1359–1375 (2022).
Porrazzo, A. et al. DNA repair in tumor radioresistance: insights from fruit flies genetics. Cell Oncol. (Dordr.) 47, 717–732 (2024).
Macedo-Silva, C. et al. Epigenetic mechanisms underlying prostate cancer radioresistance. Clin. Epigenetics 13, 125 (2021).
Haase, S. et al. Epigenetic reprogramming in pediatric gliomas: from molecular mechanisms to therapeutic implications. Trends Cancer 10, 1147–1160 (2024).
Chen, Q. et al. Histone acetyltransferases CBP/p300 in tumorigenesis and CBP/p300 inhibitors as promising novel anticancer agents. Theranostics 12, 4935–4948 (2022).
Ling, R. et al. HDAC-an important target for improving tumor radiotherapy resistance. Front. Oncol. 13, 1193637 (2023).
Ampferl, R. et al. Glucose starvation impairs DNA repair in tumour cells selectively by blocking histone acetylation. Radiother. Oncol. 126, 465–470 (2018).
Zhou, Y. et al. MMP14 Contributes to HDAC Inhibition-Induced Radiosensitization of Glioblastoma. Int. J. Mol. Sci. 22, 10403 (2021).
Kwon, S. J. et al. Targeting BRG1 chromatin remodeler via its bromodomain for enhanced tumor cell radiosensitivity in vitro and in vivo. Mol. Cancer Ther. 14, 597–607 (2015).
Liu, H. Y. et al. Acetylation of MORC2 by NAT10 regulates cell-cycle checkpoint control and resistance to DNA-damaging chemotherapy and radiotherapy in breast cancer. Nucleic Acids Res. 48, 3638–3656 (2020).
Rath, B. H., Waung, I., Camphausen, K. & Tofilon, P. J. Inhibition of the histone H3K27 demethylase UTX enhances tumor cell radiosensitivity. Mol. Cancer Ther. 17, 1070–1078 (2018).
Aymard, F. et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 21, 366–374 (2014).
Tong, D., Tang, Y. & Zhong, P. The emerging roles of histone demethylases in cancers. Cancer Metastasis Rev. 43, 795–821 (2024).
Daugaard, M. et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 19, 803–810 (2012).
Pfister, S. X. et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 7, 2006–2018 (2014).
Carvalho, S. et al. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife 3, e02482 (2014).
Duan, L. et al. JMJD2 promotes acquired cisplatin resistance in non-small cell lung carcinoma cells. Oncogene 38, 5643–5657 (2019).
Wen, R. et al. DSTN hypomethylation promotes radiotherapy resistance of rectal cancer by activating the Wnt/beta-catenin signaling pathway. Int. J. Radiat. Oncol. Biol. Phys. 117, 198–210 (2023).
Wu, Q. et al. Oleandrin enhances radiotherapy sensitivity in lung cancer by inhibiting the ATM/ATR-mediated DNA damage response. Phytother. Res. 38, 4151–4167 (2024).
Liu, Z. G. et al. Relationship between methylation status of ERCC1 promoter and radiosensitivity in glioma cell lines. Cell Biol. Int. 33, 1111–1117 (2009).
Pabalan, N. et al. Potential of RASSF1A promoter methylation as biomarker for endometrial cancer: A systematic review and meta-analysis. Gynecol. Oncol. 146, 603–608 (2017).
Gong, C. et al. AKBA inhibits radiotherapy resistance in lung cancer by inhibiting maspin methylation and regulating the AKT/FOXO1/p21 axis. J. Radiat. Res. 64, 33–43 (2023).
Guillaume, J. C., Evenou, P., Charpentier, P. & Avril, M. F. Phakomatosis pigmentovascularis type IIa. Ann. Dermatol Venereol. 115, 1113–1115 (1988).
Wang, L. et al. Adenovirus-mediated delivery of siRNA targeting TM4SF4 attenuated liver cancer cell growth in vitro and in vivo. Acta Biochim. Biophys. Sin. 45, 213–219 (2013).
Roy, K., Wang, L., Makrigiorgos, G. M. & Price, B. D. Methylation of the ATM promoter in glioma cells alters ionizing radiation sensitivity. Biochem. Biophys. Res. Commun. 344, 821–826 (2006).
Xu, M. et al. Reprimo (RPRM) is a novel tumor suppressor in pituitary tumors and regulates survival, proliferation, and tumorigenicity. Endocrinology 153, 2963–2973 (2012).
Misiewicz-Krzeminska, I. et al. Post-transcriptional Modifications Contribute to the Upregulation of Cyclin D2 in Multiple Myeloma. Clin. Cancer Res. 22, 207–217 (2016).
Huang, K. H. et al. Methylation of RASSF1A, RASSF2A, and HIN-1 is associated with poor outcome after radiotherapy, but not surgery, in oral squamous cell carcinoma. Clin. Cancer Res. 15, 4174–4180 (2009).
Zhu, X., Wang, Y., Tan, L. & Fu, X. The pivotal role of DNA methylation in the radio-sensitivity of tumor radiotherapy. Cancer Med. 7, 3812–3819 (2018).
Deng, L., Yin, Q., Liu, S. & Luo, D. MicroRNA-613 enhances nasopharyngeal carcinoma cell radiosensitivity via the DNA methyltransferase 3B/Tissue inhibitor of matrix metalloproteinase-3/signal transducer and activator of transcription-1/forkhead box O-1 axis. Dis. Markers 2022, 5699275 (2022).
Zhao, Y. et al. Enhancer RNA promotes resistance to radiotherapy in bone-metastatic prostate cancer by m(6)A modification. Theranostics 13, 596–610 (2023).
Yin, J. et al. METTL3-mediated m6A modification of LINC00839 maintains glioma stem cells and radiation resistance by activating Wnt/beta-catenin signaling. Cell Death Dis. 14, 417 (2023).
Huang, W. et al. N6-methyladenosine methyltransferases: functions, regulation, and clinical potential. J. Hematol. Oncol. 14, 117 (2021).
Shao, Y. et al. ALKBH5/YTHDF2-mediated m6A modification of circAFF2 enhances radiosensitivity of colorectal cancer by inhibiting Cullin neddylation. Clin. Transl. Med. 13, e1318 (2023).
Visvanathan, A. et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene 37, 522–533 (2018).
Taketo, K. et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int. J. Oncol. 52, 621–629 (2018).
Wu, P. et al. N6-methyladenosine modification of circCUX1 confers radioresistance of hypopharyngeal squamous cell carcinoma through caspase1 pathway. Cell Death Dis. 12, 298 (2021).
Hao, C. C. et al. Up-regulation of VANGL1 by IGF2BPs and miR-29b-3p attenuates the detrimental effect of irradiation on lung adenocarcinoma. J. Exp. Clin. Cancer Res. 39, 256 (2020).
Liu, Y. & Da, M. Wilms tumor 1 associated protein promotes epithelial mesenchymal transition of gastric cancer cells by accelerating TGF-beta and enhances chemoradiotherapy resistance. J. Cancer Res. Clin. Oncol. 149, 3977–3988 (2023).
Kowalski-Chauvel, A. et al. The m6A RNA demethylase ALKBH5 promotes radioresistance and invasion capability of glioma stem cells. Cancers 13, 40 (2020).
Wang, S. P. et al. FDI-6 inhibits the expression and function of FOXM1 to sensitize BRCA-proficient triple-negative breast cancer cells to Olaparib by regulating cell cycle progression and DNA damage repair. Cell Death Dis. 12, 1138 (2021).
Dong, Y. et al. TRIM56 reduces radiosensitization of human glioblastoma by regulating FOXM1-mediated DNA repair. Mol. Neurobiol. 59, 5312–5325 (2022).
Zhuang, H. et al. The role of m6A methylation in therapy resistance in cancer. Mol. Cancer 22, 91 (2023).
Huang, W. M. et al. m6A demethylase FTO renders radioresistance of nasopharyngeal carcinoma via promoting OTUB1-mediated anti-ferroptosis. Transl. Oncol. 27, 101576 (2023).
Xu, X. et al. METTL3-mediated m6A mRNA contributes to the resistance of carbon-ion radiotherapy in non-small-cell lung cancer. Cancer Sci. 114, 105–114 (2023).
Zhou, S. et al. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting beta-catenin through mRNA demethylation. Mol. Carcinog. 57, 590–597 (2018).
Du, H. et al. YTHDF3 mediates HNF1alpha regulation of cervical cancer radio-resistance by promoting RAD51D translation in an m6A-dependent manner. FEBS J. 290, 1920–1935 (2023).
Yu, M. et al. NSUN6-mediated 5-methylcytosine modification of NDRG1 mRNA promotes radioresistance in cervical cancer. Mol. Cancer 23, 139 (2024).
Liao, J. et al. Methyltransferase 1 is required for nonhomologous end-joining repair and renders hepatocellular carcinoma resistant to radiotherapy. Hepatology 77, 1896–1910 (2023).
Liu, M. et al. Prognostic model and ceRNA network of m7G- and radiosensitivity-related genes in hepatocellular carcinoma. Heliyon 10, e29925 (2024).
He, J. et al. LncRNA as a multifunctional regulator in cancer multi-drug resistance. Mol. Biol. Rep. 48, 1–15 (2021).
Yazarlou, F., Martinez, I. & Lipovich, L. Radiotherapy and breast cancer: finally, an lncRNA perspective on radiosensitivity and radioresistance. Front. Oncol. 14, 1437542 (2024).
Shen, Y. et al. XIST: a meaningful long noncoding RNA in NSCLC process. Curr. Pharm. Des. 27, 1407–1417 (2021).
Du, R. et al. Knockdown of lncRNA X inactive specific transcript (XIST) radiosensitizes non-small cell lung cancer (NSCLC) cells through regulation of miR-16-5p/WEE1 G2 checkpoint kinase (WEE1) axis. Int J. Immunopathol. Pharm. 35, 2058738420966087 (2021).
Liu, A. M. et al. Long noncoding RNA FAM201A involves in radioresistance of non-small-cell lung cancer by enhancing EGFR expression via miR-370. Eur. Rev. Med. Pharm. Sci. 23, 5802–5814 (2019).
Pei, R. et al. JMJD6-BRD4 complex stimulates lncRNA HOTAIR transcription by binding to the promoter region of HOTAIR and induces radioresistance in liver cancer stem cells. J. Transl. Med. 21, 752 (2023).
Qian, L. et al. lncRNA HOTAIR promotes DNA repair and radioresistance of breast cancer via EZH2. DNA Cell Biol. 39, 1-8 (2020).
Zheng, J. et al. Linc-RA1 inhibits autophagy and promotes radioresistance by preventing H2Bub1/USP44 combination in glioma cells. Cell Death Dis. 11, 758 (2020).
Zhou, Y. et al. A novel long noncoding RNA SP100-AS1 induces radioresistance of colorectal cancer via sponging miR-622 and stabilizing ATG3. Cell Death Differ. 30, 111–124 (2023).
Li, J., Lei, C., Chen, B. & Zhu, Q. LncRNA FGD5-AS1 facilitates the radioresistance of breast cancer cells by enhancing MACC1 expression through competitively sponging miR-497-5p. Front. Oncol. 11, 671853 (2021).
Lei, C. et al. LncRNA DUXAP8 induces breast cancer radioresistance by modulating the PI3K/AKT/mTOR pathway and the EZH2-E-cadherin/RHOB pathway. Cancer Biol. Ther. 23, 1–13 (2022).
Bi, Z. et al. Nanoparticles (NPs)-meditated LncRNA AFAP1-AS1 silencing to block Wnt/beta-catenin signaling pathway for synergistic reversal of radioresistance and effective cancer radiotherapy. Adv. Sci. 7, 2000915 (2020).
Yao, P. A. et al. The feedback loop of ANKHD1/lncRNA MALAT1/YAP1 strengthens the radioresistance of CRC by activating YAP1/AKT signaling. Cell Death Dis. 13, 103 (2022).
Labbe, M. et al. Loss of miR-200c-3p promotes resistance to radiation therapy via the DNA repair pathway in prostate cancer. Cell Death Dis. 15, 751 (2024).
Chen, H. et al. MiR-450a-5p inhibits autophagy and enhances radiosensitivity by targeting dual-specificity phosphatase 10 in esophageal squamous cell carcinoma. Cancer Lett. 483, 114–126 (2020).
You, G. R. et al. MiR-630 promotes radioresistance by induction of anti-apoptotic effect via Nrf2-GPX2 molecular axis in head-neck cancer. Cells. 12, 2853 (2023).
Li, W., Xing, X., Shen, C. & Hu, C. Tumor cell-derived exosomal miR-193b-3p promotes tumor-associated macrophage activation to facilitate nasopharyngeal cancer cell invasion and radioresistances. Heliyon 10, e30808 (2024).
Anastasov, N. et al. Radiation resistance due to high expression of miR-21 and G2/M checkpoint arrest in breast cancer cells. Radiat. Oncol. 7, 206 (2012).
Ueda, H. et al. miR-6855-5p Enhances radioresistance and promotes migration of pancreatic cancer by inducing epithelial-mesenchymal transition via suppressing FOXA1: potential of plasma exosomal miR-6855-5p as an indicator of radiosensitivity in patients with pancreatic cancer. Ann. Surg. Oncol. 32, 720–735 (2024).
Kawamura, K., Qi, F. & Kobayashi, J. Potential relationship between the biological effects of low-dose irradiation and mitochondrial ROS production. J. Radiat. Res. 59, ii91–ii97 (2018).
Alahdal, M. & Elkord, E. Non-coding RNAs in cancer immunotherapy: Predictive biomarkers and targets. Clin. Transl. Med. 13, e1425 (2023).
Di, L., Zhao, X. & Ding, J. Knockdown of circ_0008344 contributes to radiosensitization in glioma via miR-433-3p/RNF2 axis. J Biosci. 46, 82 (2021).
Xue, C. et al. The functional roles of the circRNA/Wnt axis in cancer. Mol. Cancer 21, 108 (2022).
Guan, Y. et al. Circular RNA circPITX1 knockdown inhibits glycolysis to enhance radiosensitivity of glioma cells by miR-329-3p/NEK2 axis. Cancer Cell Int. 20, 80 (2020).
Jin, Y. et al. Circ_0086720 knockdown strengthens the radiosensitivity of non-small cell lung cancer via mediating the miR-375/SPIN1 axis. Neoplasma 68, 96–107 (2021).
Qian, Y. et al. circ-ZNF609: A potent circRNA in human cancers. J. Cell Mol. Med. 25, 10349–10361 (2021).
Gao, C. et al. Circ_0055625 knockdown inhibits tumorigenesis and improves radiosensitivity by regulating miR-338-3p/MSI1 axis in colon cancer. World J. Surg. Oncol. 19, 131 (2021).
Guo, Y. M. et al. Germline polymorphisms and length of survival of nasopharyngeal carcinoma: an exome-wide association study in multiple cohorts. Adv. Sci. 7, 1903727 (2020).
Ippolito, M. R. et al. Gene copy-number changes and chromosomal instability induced by aneuploidy confer resistance to chemotherapy. Dev. Cell 56, 2440–2454.e2446 (2021).
Chen, N. N. et al. Doxorubicin resistance in breast cancer is mediated via the activation of FABP5/PPARgamma and CaMKII signaling pathway. Front. Pharm. 14, 1150861 (2023).
Liu, Y. P. et al. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm 2, 315–340 (2021).
Hu, C. et al. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: mechanism and clinical application. Clin. Epigenetics 13, 166 (2021).
Li, G. H. et al. Super-enhancers: a new frontier for epigenetic modifiers in cancer chemoresistance. J. Exp. Clin. Cancer Res. 40, 174 (2021).
Reyes, M. E. et al. Epigenetic modulation of cytokine expression in gastric cancer: influence on angiogenesis, metastasis and chemoresistance. Front. Immunol. 15, 1347530 (2024).
Lumpp, T. et al. Role of epigenetics for the efficacy of cisplatin. Int. J. Mol. Sci. 25, 1130 (2024).
Roca, M. S. et al. HDAC class I inhibitor domatinostat sensitizes pancreatic cancer to chemotherapy by targeting cancer stem cell compartment via FOXM1 modulation. J. Exp. Clin. Cancer Res. 41, 83 (2022).
Bi, L. et al. HDAC11 regulates glycolysis through the LKB1/AMPK signaling pathway to maintain hepatocellular carcinoma stemness. Cancer Res. 81, 2015–2028 (2021).
Gardner, E. E. et al. Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell 31, 286–299 (2017).
Chang, J. W. et al. EZH2 is associated with poor prognosis in head-and-neck squamous cell carcinoma via regulating the epithelial-to-mesenchymal transition and chemosensitivity. Oral. Oncol. 52, 66–74 (2016).
Deng, X. et al. A KLF4/PiHL/EZH2/HMGA2 regulatory axis and its function in promoting oxaliplatin-resistance of colorectal cancer. Cell Death Dis. 12, 485 (2021).
Liu, C. W. et al. Histone methyltransferase G9a drives chemotherapy resistance by regulating the glutamate-cysteine ligase catalytic subunit in head and neck squamous cell carcinoma. Mol. Cancer Ther. 16, 1421–1434 (2017).
Staberg, M. et al. Targeting glioma stem-like cell survival and chemoresistance through inhibition of lysine-specific histone demethylase KDM2B. Mol. Oncol. 12, 406–420 (2018).
Mathur, R. et al. Inhibition of demethylase KDM6B sensitizes diffuse large B-cell lymphoma to chemotherapeutic drugs. Haematologica 102, 373–380 (2017).
Dalvi, M. P. et al. Taxane-platin-resistant lung cancers co-develop hypersensitivity to JumonjiC demethylase inhibitors. Cell Rep. 19, 1669–1684 (2017).
Dalvi, M. P. & Martinez, E. D. JumonjiC demethylase inhibitors show potential for targeting chemotherapy-resistant lung cancers. Mol. Cell Oncol. 4, e1345352 (2017).
Nishida, Y. et al. The novel BMI-1 inhibitor PTC596 downregulates MCL-1 and induces p53-independent mitochondrial apoptosis in acute myeloid leukemia progenitor cells. Blood Cancer J. 7, e527 (2017).
Snyder, N. A. & Silva, G. M. Deubiquitinating enzymes (DUBs): Regulation, homeostasis, and oxidative stress response. J. Biol. Chem. 297, 101077 (2021).
Wang, A. et al. USP22 induces cisplatin resistance in lung adenocarcinoma by regulating gammaH2AX-mediated DNA damage repair and Ku70/Bax-mediated apoptosis. Front Pharm. 8, 274 (2017).
Yue, Q. et al. Histone H3K9 lactylation confers temozolomide resistance in glioblastoma via LUC7L2-mediated MLH1 intron retention. Adv. Sci. 11, e2309290 (2024).
Shi, Z. D. et al. Targeting HNRNPU to overcome cisplatin resistance in bladder cancer. Mol. Cancer 21, 37 (2022).
Bunch, B. et al. TAp73 expression and P1 promoter methylation, a potential marker for chemoresponsiveness to cisplatin therapy and survival in muscle-invasive bladder cancer (MIBC). Cell Cycle 18, 2055–2066 (2019).
Wu, M. et al. Low doses of decitabine improve the chemotherapy efficacy against basal-like bladder cancer by targeting cancer stem cells. Oncogene 38, 5425–5439 (2019).
Zeller, C. et al. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene 31, 4567–4576 (2012).
Arnold, C. N., Goel, A. & Boland, C. R. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int. J. Cancer 106, 66–73 (2003).
Eyre, R. et al. Reversing paclitaxel resistance in ovarian cancer cells via inhibition of the ABCB1 expressing side population. Tumour Biol. 35, 9879–9892 (2014).
Al-Abdulla, R. et al. Epigenetic events involved in organic cation transporter 1-dependent impaired response of hepatocellular carcinoma to sorafenib. Br. J. Pharm. 176, 787–800 (2019).
Liu, K. et al. Pharmacoepitranscriptomic landscape revealing m6A modification could be a drug-effect biomarker for cancer treatment. Mol. Ther. Nucleic Acids 28, 464–476 (2022).
Li, F. et al. Regulation of cisplatin resistance in bladder cancer by epigenetic mechanisms. Drug Resist Updat 68, 100938 (2023).
Decau, J. et al. Effect of nitrogen fertilization and time of the harvest on the production and content of nitrates in fresh bean pods (Phaseolus vulgaris L. var. Coco nain)]. Ann. Nutr. Aliment 34, 947–954 (1980).
Sun, Y. et al. METTL3 promotes chemoresistance in small cell lung cancer by inducing mitophagy. J. Exp. Clin. Cancer Res. 42, 65 (2023).
Wang, J. et al. N6-methyladenosine reader hnRNPA2B1 recognizes and stabilizes NEAT1 to confer chemoresistance in gastric cancer. Cancer Commun. 44, 469–490 (2024).
Zhu, L. et al. Impaired autophagic degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in gastric cancer. Cell Death Dis. 10, 383 (2019).
Li, E. et al. METTL3 promotes homologous recombination repair and modulates chemotherapeutic response in breast cancer by regulating the EGF/RAD51 axis. Elife 11, e75231 (2022).
Pan, X. et al. METTL3 promotes adriamycin resistance in MCF-7 breast cancer cells by accelerating pri-microRNA-221-3p maturation in a m6A-dependent manner. Exp. Mol. Med. 53, 91–102 (2021).
Liu, Y. et al. Overexpressed methyltransferase-like 1 (METTL1) increased chemosensitivity of colon cancer cells to cisplatin by regulating miR-149-3p/S100A4/p53 axis. Aging 11, 12328–12344 (2019).
Ouyang, L. et al. LncRNA FOXD1-AS1 regulates pancreatic cancer stem cell properties and 5-FU resistance by regulating the miR-570-3p/SPP1 axis as a ceRNA. Cancer Cell Int. 24, 4 (2024).
Nie, S. et al. ALKBH5-HOXA10 loop-mediated JAK2 m6A demethylation and cisplatin resistance in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 40, 284 (2021).
Li, X. D. et al. Long noncoding RNA just proximal to X-inactive specific transcript facilitates aerobic glycolysis and temozolomide chemoresistance by promoting stability of PDK1 mRNA in an m6A-dependent manner in glioblastoma multiforme cells. Cancer Sci. 112, 4543–4552 (2021).
Li, Z., Lu, W., Yin, F. & Huang, A. YBX1 as a prognostic biomarker and potential therapeutic target in hepatocellular carcinoma: A comprehensive investigation through bioinformatics analysis and in vitro study. Transl. Oncol. 45, 101965 (2024).
He, M. et al. M(7)G modification of FTH1 and pri-miR-26a regulates ferroptosis and chemotherapy resistance in osteosarcoma. Oncogene 43, 341–353 (2024).
Chen, J. et al. Metabolic reprogramming driven by METTL1-mediated tRNA m7G modification promotes acquired anlotinib resistance in oral squamous cell carcinoma. Transl. Res. 268, 28–39 (2024).
Okamoto, M. et al. tRNA modifying enzymes, NSUN2 and METTL1, determine sensitivity to 5-fluorouracil in HeLa cells. PLoS Genet 10, e1004639 (2014).
Liu, H. et al. Targeting tumour-intrinsic N(7)-methylguanosine tRNA modification inhibits MDSC recruitment and improves anti-PD-1 efficacy. Gut 72, 1555–1567 (2023).
Singh, D., Assaraf, Y. G. & Gacche, R. N. Long non-coding RNA mediated drug resistance in breast cancer. Drug Resist Updat 63, 100851 (2022).
Han, P. et al. The lncRNA CRNDE promotes colorectal cancer cell proliferation and chemoresistance via miR-181a-5p-mediated regulation of Wnt/β-catenin signaling. Mol. Cancer 16, 9 (2017).
Xing, S. et al. Deregulation of lncRNA-AC078883.3 and microRNA-19a is involved in the development of chemoresistance to cisplatin via modulating signaling pathway of PTEN/AKT. J. Cell Physiol. 234, 22657–22665 (2019).
Zhang, W. et al. LncRNA HOTAIR promotes chemoresistance by facilitating epithelial to mesenchymal transition through miR-29b/PTEN/PI3K signaling in cervical cancer. Cells Tissues Organs 211, 16–29 (2022).
Du, P. et al. LncRNA PVT1 mediates antiapoptosis and 5-fluorouracil resistance via increasing Bcl2 expression in gastric cancer. J. Oncol. 2019, 9325407 (2019).
Naseri, B. et al. lncRNA PVT1 silencing inhibits gastric cancer cells’ progression via enhancing chemosensitivity to paclitaxel. Gene 932, 148900 (2025).
Qu, H. et al. LncRNA PVT1 influences breast cancer cells glycolysis through sponging miR-145-5p. Genes Genomics 45, 581–592 (2023).
Wu, C. et al. Long noncoding RNA plasmacytoma variant translocation 1 regulates cisplatin resistance via miR-3619-5p/TBL1XR1 axis in gastric cancer. Cancer Biother Radiopharm. 35, 741–752 (2020).
Yao, W., Guo, P., Mu, Q. & Wang, Y. Exosome-derived Circ-PVT1 contributes to cisplatin resistance by regulating autophagy, invasion, and apoptosis via miR-30a-5p/YAP1 axis in gastric cancer cells. Cancer Biother Radiopharm. 36, 347–359 (2021).
Alemi, F. et al. Interaction between lncRNAs and RNA-binding proteins (RBPs) influences DNA damage response in cancer chemoresistance. Mol. Biol. Rep. 51, 308 (2024).
Tang, L. et al. Long non-coding RNA MIR200CHG promotes breast cancer proliferation, invasion, and drug resistance by interacting with and stabilizing YB-1. NPJ Breast Cancer 7, 94 (2021).
Song, Y. et al. RBMX contributes to hepatocellular carcinoma progression and sorafenib resistance by specifically binding and stabilizing BLACAT1. Am. J. Cancer Res. 10, 3644–3665 (2020).
Bruno, T. et al. Che-1 phosphorylation by ATM/ATR and Chk2 kinases activates p53 transcription and the G2/M checkpoint. Cancer Cell 10, 473–486 (2006).
Xu, Z. et al. Long non-coding RNA CCAT2 promotes oncogenesis in triple-negative breast cancer by regulating stemness of cancer cells. Pharm. Res. 152, 104628 (2020).
Hu, J. et al. TRPS1 confers multidrug resistance of breast cancer cells by regulating BCRP expression. Front. Oncol. 10, 934 (2020).
Shin, V. Y. et al. Long non-coding RNA NEAT1 confers oncogenic role in triple-negative breast cancer through modulating chemoresistance and cancer stemness. Cell Death Dis. 10, 270 (2019).
Brown, J. M., Wasson, M. D. & Marcato, P. The missing Lnc: the potential of targeting triple-negative breast cancer and cancer stem cells by inhibiting long non-coding RNAs. Cells 9, 763 (2020).
Dong, H. et al. Long non-coding RNA SNHG14 induces trastuzumab resistance of breast cancer via regulating PABPC1 expression through H3K27 acetylation. J. Cell Mol. Med. 22, 4935–4947 (2018).
Wu, C. & Luo, J. Long non-coding RNA (lncRNA) urothelial carcinoma-associated 1 (UCA1) enhances tamoxifen resistance in breast cancer cells via inhibiting mTOR signaling pathway. Med. Sci. Monit. 22, 3860–3867 (2016).
Yang, E. et al. EPIC-0628 abrogates HOTAIR/EZH2 interaction and enhances the temozolomide efficacy via promoting ATF3 expression and inhibiting DNA damage repair in glioblastoma. Cancer Lett. 588, 216812 (2024).
Khan, M. I. & Ahmad, A. LncRNA SNHG6 sponges miR-101 and induces tamoxifen resistance in breast cancer cells through induction of EMT. Front. Oncol. 12, 1015428 (2022).
Liao, X. et al. Dynamic structural remodeling of LINC01956 enhances temozolomide resistance in MGMT-methylated glioblastoma. Sci. Transl. Med. 16, eado1573 (2024).
Ding, L. et al. Role of noncoding RNA in drug resistance of prostate cancer. Cell Death Dis. 12, 590 (2021).
Mao, H. et al. Drug-resistant exosome miR-99b-3p induces macrophage polarization and confers chemoresistance on sensitive cells by targeting PPP2CA. Int. Immunopharmacol. 142, 113168 (2024).
Al-Gazally, M. E. et al. The role and mechanism of action of microRNA-122 in cancer: focusing on the liver. Int. Immunopharmacol. 123, 110713 (2023).
Qin, C. et al. Extracellular vesicles miR-31-5p promotes pancreatic cancer chemoresistance via regulating LATS2-Hippo pathway and promoting SPARC secretion from pancreatic stellate cells. J. Extracell. Vesicles 13, e12488 (2024).
Ding, J. et al. MiR-223 promotes the doxorubicin resistance of colorectal cancer cells via regulating epithelial-mesenchymal transition by targeting FBXW7. Acta Biochim. Biophys. Sin. 50, 597–604 (2018).
Feng, Q., He, P. & Wang, Y. MicroRNA-223-3p regulates cell chemo-sensitivity by targeting FOXO3 in prostatic cancer. Gene 658, 152–158 (2018).
Xia, Q., Liu, G., Lin, W. & Zhang, J. microRNA-2117 Negatively Regulates Liver Cancer Stem Cells Expansion and Chemoresistance Via Targeting SOX2. Mol. Carcinog. 64, 33–43 (2024).
Mandal, T. et al. The EXO1/Poleta/Poliota axis as a promising target for miR-3163-mediated attenuation of cancer stem-like cells in non-small cell lung carcinoma. Br. J. Cancer 131, 1668–1682 (2024).
Wang, Z. et al. Targeting miR-381-NEFL axis sensitizes glioblastoma cells to temozolomide by regulating stemness factors and multidrug resistance factors. Oncotarget 6, 3147–3164 (2015).
Miroshnichenko, S. & Patutina, O. Enhanced inhibition of tumorigenesis using combinations of miRNA-targeted therapeutics. Front. Pharm. 10, 488 (2019).
Wang, H. et al. MiR-223 regulates autophagy associated with cisplatin resistance by targeting FBXW7 in human non-small cell lung cancer. Cancer Cell Int. 20, 258 (2020).
Ma, X. L. et al. Doxorubicin-induced novel circRNA_0004674 facilitates osteosarcoma progression and chemoresistance by upregulating MCL1 through miR-142-5p. Cell Death Discov. 7, 309 (2021).
Gao, W. et al. circPARD3 drives malignant progression and chemoresistance of laryngeal squamous cell carcinoma by inhibiting autophagy through the PRKCI-Akt-mTOR pathway. Mol. Cancer 19, 166 (2020).
Meng, X. et al. CircPTK2/PABPC1/SETDB1 axis promotes EMT-mediated tumor metastasis and gemcitabine resistance in bladder cancer. Cancer Lett. 554, 216023 (2023).
Hong, X. et al. CircIPO7 promotes nasopharyngeal carcinoma metastasis and cisplatin chemoresistance by facilitating YBX1 nuclear localization. Clin. Cancer Res. 28, 4521–4535 (2022).
Li, H. et al. CircITGB6 promotes ovarian cancer cisplatin resistance by resetting tumor-associated macrophage polarization toward the M2 phenotype. J. Immunother Cancer. 10, e004029 (2022).
Li, Y. et al. CircTRIM1 encodes TRIM1-269aa to promote chemoresistance and metastasis of TNBC via enhancing CaM-dependent MARCKS translocation and PI3K/AKT/mTOR activation. Mol. Cancer 23, 102 (2024).
Pan, Z. et al. A novel protein encoded by exosomal CircATG4B induces oxaliplatin resistance in colorectal cancer by promoting autophagy. Adv. Sci. 9, e2204513 (2022).
Lin, J. et al. CircPDIA3/miR-449a/XBP1 feedback loop curbs pyroptosis by inhibiting palmitoylation of the GSDME-C domain to induce chemoresistance of colorectal cancer. Drug Resist. Updat 76, 101097 (2024).
Yan, Y. et al. piR-1919609 is an ideal potential target for reversing platinum resistance in ovarian cancer. Technol. Cancer Res. Treat. 23, 15330338241249692 (2024).
Balaratnam, S., West, N. & Basu, S. A piRNA utilizes HILI and HIWI2 mediated pathway to down-regulate ferritin heavy chain 1 mRNA in human somatic cells. Nucleic Acids Res. 46, 10635–10648 (2018).
Wang, Y. et al. A piRNA-like small RNA induces chemoresistance to cisplatin-based therapy by inhibiting apoptosis in lung squamous cell carcinoma. Mol. Ther. Nucleic Acids 6, 269–278 (2017).
Roy, J., Das, B., Jain, N. & Mallick, B. PIWI-interacting RNA 39980 promotes tumor progression and reduces drug sensitivity in neuroblastoma cells. J. Cell Physiol. 235, 2286–2299 (2020).
Das, B., Jain, N. & Mallick, B. piR-39980 promotes cell proliferation, migration and invasion, and inhibits apoptosis via repression of SERPINB1 in human osteosarcoma. Biol. Cell 112, 73–91 (2020).
Cui, J. W. et al. Tumor immunotherapy resistance: revealing the mechanism of PD-1 / PD-L1-mediated tumor immune escape. Biomed. Pharmacother. 171, 116203 (2024).
Hogg, S. J., Beavis, P. A., Dawson, M. A. & Johnstone, R. W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov. 19, 776–800 (2020).
Cao, J. & Yan, Q. Cancer epigenetics, tumor immunity, and immunotherapy. Trends Cancer 6, 580–592 (2020).
Lian, B., Chen, X. & Shen, K. Inhibition of histone deacetylases attenuates tumor progression and improves immunotherapy in breast cancer. Front. Immunol. 14, 1164514 (2023).
Rosenberg, S. A. & Restifo, N. P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015).
Dai, E. et al. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol. Cancer 20, 171 (2021).
Yang, J. et al. Epigenetic regulation in the tumor microenvironment: molecular mechanisms and therapeutic targets. Sig. Transduct. Target Ther. 8, 210 (2023).
Woods, D. M. et al. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol. Res. 3, 1375–1385 (2015).
Sun, T. et al. Suberanilohydroxamic acid (SAHA), a HDAC inhibitor, suppresses the effect of Treg cells by targeting the c-Myc/CCL1 pathway in glioma stem cells and improves PD-L1 blockade therapy. J. Neurooncol. 168, 457–471 (2024).
Xu, S. et al. LSD1 silencing contributes to enhanced efficacy of anti-CD47/PD-L1 immunotherapy in cervical cancer. Cell Death Dis. 12, 282 (2021).
Juneja, V. R. et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 214, 895–904 (2017).
Liu, Y. et al. LSD1 inhibition sustains T cell invigoration with a durable response to PD-1 blockade. Nat. Commun. 12, 6831 (2021).
Tu, W. J. et al. Targeting nuclear LSD1 to reprogram cancer cells and reinvigorate exhausted T cells via a novel LSD1-EOMES switch. Front. Immunol. 11, 1228 (2020).
Bao, L. et al. Targeting LSD1 in tumor immunotherapy: rationale, challenges and potential. Front. Immunol. 14, 1214675 (2023).
Wang, J. et al. Pan-cancer analysis identifies protein arginine methyltransferases PRMT1 and PRMT5 and their related signatures as markers associated with prognosis, immune profile, and therapeutic response in lung adenocarcinoma. Heliyon 9, e22088 (2023).
Wang, R. et al. H3K9 lactylation in malignant cells facilitates CD8(+) T cell dysfunction and poor immunotherapy response. Cell Rep. 43, 114686 (2024).
Battaglin, F., Naseem, M., Puccini, A. & Lenz, H. J. Molecular biomarkers in gastro-esophageal cancer: recent developments, current trends and future directions. Cancer Cell Int. 18, 99 (2018).
Dong, P. et al. Tumor-intrinsic PD-L1 signaling in cancer initiation, development and treatment: beyond immune evasion. Front. Oncol. 8, 386 (2018).
Long, S. et al. Epigenetically modified AP-2alpha by DNA methyltransferase facilitates glioma immune evasion by upregulating PD-L1 expression. Cell Death Dis. 14, 365 (2023).
Emran, A. A. et al. Targeting DNA methylation and EZH2 activity to overcome melanoma resistance to immunotherapy. Trends Immunol. 40, 328–344 (2019).
Ma, W. et al. Advances in predictive biomarkers for melanoma immunotherapy. Holist. Integr. Oncol. 3, 48 (2024).
Sultan, M. et al. Epigenetic silencing of TAP1 in Aldefluor(+) breast cancer stem cells contributes to their enhanced immune evasion. Stem Cells 36, 641–654 (2018).
Galassi, C., Esteller, M., Vitale, I. & Galluzzi, L. Epigenetic control of immunoevasion in cancer stem cells. Trends Cancer 10, 1052–1071 (2024).
Hong, Y. K. et al. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell Immunol. 336, 66–74 (2019).
Lu, X. et al. Silencing of genes by promoter hypermethylation shapes tumor microenvironment and resistance to immunotherapy in clear-cell renal cell carcinomas. Cell Rep. Med. 4, 101287 (2023).
Yi, Q. et al. ACAP1 deficiency predicts inferior immunotherapy response in solid tumors. Cancers 14, 5951 (2022).
Hu, Z. et al. Exosome-derived circCCAR1 promotes CD8 + T-cell dysfunction and anti-PD1 resistance in hepatocellular carcinoma. Mol. Cancer 22, 55 (2023).
Wang, L. et al. Targeting N6-methyladenosine reader YTHDF1 with siRNA boosts antitumor immunity in NASH-HCC by inhibiting EZH2-IL-6 axis. J. Hepatol. 79, 1185–1200 (2023).
Ning, J. et al. METTL3 inhibition induced by M2 macrophage-derived extracellular vesicles drives anti-PD-1 therapy resistance via M6A-CD70-mediated immune suppression in thyroid cancer. Cell Death Differ. 30, 2265–2279 (2023).
Su, R. et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell 38, 79–96.e11 (2020).
Andries, O. et al. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control Rel. 217, 337–344 (2015).
Chen, T. et al. NSUN2 is a glucose sensor suppressing cGAS/STING to maintain tumorigenesis and immunotherapy resistance. Cell Metab. 35, 1782–1798 e1788 (2023).
Luo, Y. et al. NSun2 deficiency protects endothelium from inflammation via mRNA methylation of ICAM-1. Circ. Res. 118, 944–956 (2016).
Lu, L. et al. High tRNA transferase NSUN2 gene expression is associated with poor prognosis in head and neck squamous carcinoma. Cancer Invest. 36, 246–253 (2018).
Maurya, P. K. et al. Role of Y box protein-1 in cancer: as potential biomarker and novel therapeutic target. J. Cancer 8, 1900–1907 (2017).
Ni, W. et al. Targeting cholesterol biosynthesis promotes anti-tumor immunity by inhibiting long noncoding RNA SNHG29-mediated YAP activation. Mol. Ther. 29, 2995–3010 (2021).
Luo, Q. et al. LINC00460/miR-186-3p/MYC feedback loop facilitates colorectal cancer immune escape by enhancing CD47 and PD-L1 expressions. J. Exp. Clin. Cancer Res. 43, 225 (2024).
Qiu, J. et al. Hypoxia-responsive lncRNA MIR155HG promotes PD-L1 expression in hepatocellular carcinoma cells by enhancing HIF-1α mRNA stability. Int. Immunopharmacol. 136, 112415 (2024).
Yao, H. et al. Extracellular vesicle-packaged lncRNA from cancer-associated fibroblasts promotes immune evasion by downregulating HLA-A in pancreatic cancer. J. Extracell. Vesicles 13, e12484 (2024).
Jiang, R. et al. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat. Commun. 8, 15129 (2017).
Sun, J. et al. Upregulated expression of indoleamine 2, 3-dioxygenase in CHO cells induces apoptosis of competent T cells and increases proportion of Treg cells. J. Exp. Clin. Cancer Res. 30, 82 (2011).
Farhood, B., Najafi, M. & Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: a review. J. Cell Physiol. 234, 8509–8521 (2019).
Ma, F. et al. LncRNA NEAT1 interacted with DNMT1 to regulate malignant phenotype of cancer cell and cytotoxic T cell infiltration via epigenetic inhibition of p53, cGAS, and STING in lung cancer. Front. Genet. 11, 250 (2020).
Zhu, H. et al. Long noncoding RNA LINC00963 promotes CDC5L-mediated malignant progression in gastric cancer. Onco Targets Ther. 13, 12999–13013 (2020).
Zhang, K. J., Tan, X. L. & Guo, L. LncRNA TYMSOS facilitates breast cancer metastasis and immune escape through downregulating ULBP3. iScience 26, 107556 (2023).
Li, G. et al. LIMIT is an immunogenic lncRNA in cancer immunity and immunotherapy. Nat. Cell Biol. 23, 526–537 (2021).
Yang, Y. et al. Hypoxic tumour-derived exosomal miR-1290 exacerbates the suppression of CD8+ T cells by promoting M2 macrophage polarization. Immunology 173, 672–688 (2024).
Tong, S. et al. Restoration of miR-299-3p promotes macrophage phagocytosis and suppresses malignant phenotypes in breast cancer carcinogenesis via dual-targeting CD47 and ABCE1. Int. Immunopharmacol. 130, 111708 (2024).
Li, W. et al. Cancer cell-derived exosomal miR-20a-5p inhibits CD8(+) T-cell function and confers anti-programmed cell death 1 therapy resistance in triple-negative breast cancer. Cancer Sci. 115, 347–356 (2024).
Wan, J., Ling, X., Peng, B. & Ding, G. miR-142-5p regulates CD4+ T cells in human non-small cell lung cancer through PD-L1 expression via the PTEN pathway. Oncol. Rep. 40, 272–282 (2018).
Wu, Y. et al. Colorectal cancer cell-derived exosomal miRNA-372-5p induces immune escape from colorectal cancer via PTEN/AKT/NF-kappaB/PD-L1 pathway. Int. Immunopharmacol. 143, 113261 (2024).
Miao, Z. et al. Hsa_circ_0136666 stimulates gastric cancer progression and tumor immune escape by regulating the miR-375/PRKDC Axis and PD-L1 phosphorylation. Mol. Cancer 22, 205 (2023).
Gao, C. et al. CircRNA VIM silence synergizes with sevoflurane to inhibit immune escape and multiple oncogenic activities of esophageal cancer by simultaneously regulating miR-124/PD-L1 axis. Cell Biol. Toxicol. 38, 825–845 (2022).
Zang, J. et al. Hsa_circ_0001479 accelerates tumorigenesis of gastric cancer and mediates immune escape. Int. Immunopharmacol. 124, 110887 (2023).
Wang, R. et al. m6A-modified circNFIX promotes ovarian cancer progression and immune escape via activating IL-6R/JAK1/STAT3 signaling by sponging miR-647. Int. Immunopharmacol. 124, 110879 (2023).
Liu, Z. et al. N(6)-methyladenosine-modified circIGF2BP3 inhibits CD8(+) T-cell responses to facilitate tumor immune evasion by promoting the deubiquitination of PD-L1 in non-small cell lung cancer. Mol. Cancer 20, 105 (2021).
Ge, J. et al. Human papillomavirus-encoded circular RNA circE7 promotes immune evasion in head and neck squamous cell carcinoma. Nat. Commun. 15, 8609 (2024).
Du, A., Yang, Q., Sun, X. & Zhao, Q. Exosomal circRNA-001264 promotes AML immunosuppression through induction of M2-like macrophages and PD-L1 overexpression. Int. Immunopharmacol. 124, 110868 (2023).
Zhang, P. F. et al. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol. Cancer 19, 110 (2020).
Lv, J. et al. HNRNPL induced circFAM13B increased bladder cancer immunotherapy sensitivity via inhibiting glycolysis through IGF2BP1/PKM2 pathway. J. Exp. Clin. Cancer Res. 42, 41 (2023).
Zhong, Y. et al. Small extracellular vesicle piR-hsa-30937 derived from pancreatic neuroendocrine neoplasms upregulates CD276 in macrophages to promote immune evasion. Cancer Immunol. Res. 12, 840–853 (2024).
Kaur, R., Bhardwaj, A. & Gupta, S. Cancer treatment therapies: traditional to modern approaches to combat cancers. Mol. Biol. Rep. 50, 9663–9676 (2023).
Dawson, M. A. & Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27 (2012).
Garcia-Martinez, L. et al. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 12, 1786 (2021).
Ranieri, R. et al. Current status and future perspectives in targeted therapy of NPM1-mutated AML. Leukemia 36, 2351–2367 (2022).
Zheng, J. et al. Targeting arginine methyltransferase PRMT5 for cancer therapy: updated progress and novel strategies. J. Med. Chem. 66, 8407–8427 (2023).
Diao, W. et al. Targeting histone demethylases as a potential cancer therapy (Review). Int. J. Oncol. 61, 103 (2022).
Xu, J. et al. Extracellular histones are major mediators of death in sepsis. Nat. Med. 15, 1318–1321 (2009).
Bora-Singhal, N. et al. Novel HDAC11 inhibitors suppress lung adenocarcinoma stem cell self-renewal and overcome drug resistance by suppressing Sox2. Sci. Rep. 10, 4722 (2020).
Parag-Sharma, K. et al. Synergistic efficacy of combined EGFR and HDAC inhibitors overcomes tolerance to EGFR monotherapy in salivary mucoepidermoid carcinoma. Oral. Oncol. 115, 105166 (2021).
Quan, C. et al. Loss of histone lysine methyltransferase EZH2 confers resistance to tyrosine kinase inhibitors in non-small cell lung cancer. Cancer Lett. 495, 41–52 (2020).
Wang, L. et al. Targeting EHMT2 reverses EGFR-TKI resistance in NSCLC by epigenetically regulating the PTEN/AKT signaling pathway. Cell Death Dis. 9, 129 (2018).
Ogawa, T. et al. Methylation of death-associated protein kinase is associated with cetuximab and erlotinib resistance. Cell Cycle 11, 1656–1663 (2012).
Jimenez-Garduno, A. M. et al. IL-1beta induced methylation of the estrogen receptor ERalpha gene correlates with EMT and chemoresistance in breast cancer cells. Biochem. Biophys. Res. Commun. 490, 780–785 (2017).
Kim, M. R. et al. TET2 directs mammary luminal cell differentiation and endocrine response. Nat. Commun. 11, 4642 (2020).
Redeuilh, G., Attia, A., Mester, J. & Sabbah, M. Transcriptional activation by the oestrogen receptor alpha is modulated through inhibition of cyclin-dependent kinases. Oncogene 21, 5773–5782 (2002).
Wang, L., Zhang, X. & Wang, Z. Y. The Wilms’ tumor suppressor WT1 regulates expression of members of the epidermal growth factor receptor (EGFR) and estrogen receptor in acquired tamoxifen resistance. Anticancer Res. 30, 3637–3642 (2010).
Mobini, K. et al. Aryl hydrocarbon-estrogen alpha receptor-dependent expression of miR-206, miR-27b, and miR-133a suppress cell proliferation and migration in MCF-7 cells. J. Biochem. Mol. Toxicol. 33, e22304 (2019).
Zhang, X. et al. High N-acetyltransferase 1 expression is associated with estrogen receptor expression in breast tumors, but is not under direct regulation by estradiol, 5alpha-androstane-3beta,17beta-diol, or dihydrotestosterone in breast cancer cells. J. Pharm. Exp. Ther. 365, 84–93 (2018).
Rickard, D. J. et al. Estrogen receptor isoform-specific induction of progesterone receptors in human osteoblasts. J. Bone Min. Res. 17, 580–592 (2002).
Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 20, 256–267 (2012).
Fan, Y. et al. PTEN promoter methylation predicts 10-year prognosis in hormone receptor-positive early breast cancer patients who received adjuvant tamoxifen endocrine therapy. Breast Cancer Res. Treat. 192, 33–42 (2022).
Chang, H. G. et al. Tamoxifen-resistant breast cancers show less frequent methylation of the estrogen receptor beta but not the estrogen receptor alpha gene. J. Mol. Med. 83, 132–139 (2005).
Xipell, E. et al. Endoplasmic reticulum stress-inducing drugs sensitize glioma cells to temozolomide through downregulation of MGMT, MPG, and Rad51. Neuro Oncol. 18, 1109–1119 (2016).
Nie, X. et al. Cloning, expression, and mutation analysis of NOR1, a novel human gene down-regulated in HNE1 nasopharyngeal carcinoma cell line. J. Cancer Res. Clin. Oncol. 129, 410–414 (2003).
Shen, Z. et al. The status of WIF1 methylation in cell-free DNA is associated with the insusceptibility for gefitinib in the treatment of lung cancer. J. Cancer Res. Clin. Oncol. 147, 2239–2248 (2021).
Park, S. S. et al. Ibulocydine sensitizes human hepatocellular carcinoma cells to TRAIL-induced apoptosis via calpain-mediated Bax cleavage. Int. J. Biochem. Cell Biol. 83, 47–55 (2017).
Zhou, T. et al. m6A RNA methylation-mediated HNF3gamma reduction renders hepatocellular carcinoma dedifferentiation and sorafenib resistance. Sig. Transduct. Target Ther. 5, 296 (2020).
Yan, F. et al. A dynamic N(6)-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. 28, 1062–1076 (2018).
Bu, T. et al. Organic anion transporters and PI3K-AKT-mTOR pathway mediate the synergistic anticancer effect of pemetrexed and rhein. J. Cell Physiol. 235, 3309–3319 (2020).
Sarno, J. et al. Dasatinib overcomes glucocorticoid resistance in B-cell acute lymphoblastic leukemia. Nat. Commun. 14, 2935 (2023).
Li, K. et al. M6A associated TSUC7 inhibition contributed to Erlotinib resistance in lung adenocarcinoma through a notch signaling activation dependent way. J. Exp. Clin. Cancer Res. 40, 325 (2021).
Bhattarai, P. Y. et al. METTL3 induces PLX4032 resistance in melanoma by promoting m(6)A-dependent EGFR translation. Cancer Lett. 522, 44–56 (2021).
Liu, W. W. et al. RNA modifications in cellular metabolism: implications for metabolism-targeted therapy and immunotherapy. Sig. Transduct. Target Ther. 9, 70 (2024).
Wang, Y. et al. Aberrant m5C hypermethylation mediates intrinsic resistance to gefitinib through NSUN2/YBX1/QSOX1 axis in EGFR-mutant non-small-cell lung cancer. Mol. Cancer 22, 81 (2023).
Qiu, L., Jing, Q., Li, Y. & Han, J. RNA modification: mechanisms and therapeutic targets. Mol. Biomed. 4, 25 (2023).
Fan, W. et al. DUSP5 regulated by YTHDF1-mediated m6A modification promotes epithelial-mesenchymal transition and EGFR-TKI resistance via the TGF-beta/Smad signaling pathway in lung adenocarcinoma. Cancer Cell Int. 24, 208 (2024).
Chen, C. et al. LncRNA H19 downregulation confers erlotinib resistance through upregulation of PKM2 and phosphorylation of AKT in EGFR-mutant lung cancers. Cancer Lett. 486, 58–70 (2020).
Xu, T. et al. LncRNA UCA1 induces acquired resistance to gefitinib by epigenetically silencing CDKN1A expression in non-small-cell lung cancer. Front. Oncol. 10, 656 (2020).
Wang, B. et al. Targeting LINC00665/miR-199b-5p/SERPINE1 axis to inhibit trastuzumab resistance and tumorigenesis of gastric cancer via PI3K/AKt pathway. Noncoding RNA Res. 10, 153–162 (2025).
Xue, X. et al. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene 35, 2746–2755 (2016).
Ouyang, J. et al. LncRNA PRNCR1 promotes breast cancer proliferation and inhibits apoptosis by modulating microRNA-377/CCND2/MEK/MAPK Axis. Arch. Med. Res. 52, 471–482 (2021).
Shi, Q. et al. LncRNA DILA1 inhibits Cyclin D1 degradation and contributes to tamoxifen resistance in breast cancer. Nat. Commun. 11, 5513 (2020).
Luo, J. et al. LncRNA MALAT-1 modulates EGFR-TKI resistance in lung adenocarcinoma cells by downregulating miR-125. Discov. Oncol. 15, 379 (2024).
Alamdari-Palangi, V., Amini, R. & Karami, H. MiRNA-7 enhances erlotinib sensitivity of glioblastoma cells by blocking the IRS-1 and IRS-2 expression. J. Pharm. Pharm. 72, 531–538 (2020).
Zhao, Y., Jin, L. J. & Zhang, X. Y. Exosomal miRNA-205 promotes breast cancer chemoresistance and tumorigenesis through E2F1. Aging 13, 18498–18514 (2021).
Lu, Y. et al. lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/beta-catenin signaling. Nat. Med. 23, 1331–1341 (2017).
Amri, J., Molaee, N., Baazm, M. & Karami, H. Targeting epidermal growth factor receptor by MiRNA-145 inhibits cell growth and sensitizes NSCLC cells to erlotinib. Asian Pac. J. Cancer Prev. 20, 2781–2787 (2019).
Chauhan, S. J., Thyagarajan, A. & Sahu, R. P. Effects of miRNA-149-5p and platelet-activating factor-receptor signaling on the growth and targeted therapy response on lung cancer cells. Int. J. Mol. Sci. 23, 6772 (2022).
Martin, E. C. et al. MicroRNA-335-5p and -3p synergize to inhibit estrogen receptor alpha expression and promote tamoxifen resistance. FEBS Lett. 591, 382–392 (2017).
Wang, H. Y. et al. MiR-200c-3p suppression is associated with development of acquired resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in EGFR mutant non-small cell lung cancer via a mediating epithelial-to-mesenchymal transition (EMT) process. Cancer Biomark. 28, 351–363 (2020).
Cabello, P. et al. miR-146a-5p promotes angiogenesis and confers trastuzumab resistance in HER2+ breast cancer. Cancers 15, 2138 (2023).
Yang, B. et al. Tumor-derived exosomal circRNA_102481 contributes to EGFR-TKIs resistance via the miR-30a-5p/ROR1 axis in non-small cell lung cancer. Aging 13, 13264–13286 (2021).
Lu, L. et al. EIF4a3-regulated circRABL2B regulates cell stemness and drug sensitivity of lung cancer via YBX1-dependent downregulation of MUC5AC expression. Int. J. Biol. Sci. 19, 2725–2739 (2023).
Zhang, R. et al. circ_SIRT1 upregulates ATG12 to facilitate Imatinib resistance in CML through interacting with EIF4A3. Gene 893, 147917 (2024).
Ling, Y. et al. circCDYL2 promotes trastuzumab resistance via sustaining HER2 downstream signaling in breast cancer. Mol. Cancer 21, 8 (2022).
Li, K. et al. Enhancement of TKI sensitivity in lung adenocarcinoma through m6A-dependent translational repression of Wnt signaling by circ-FBXW7. Mol. Cancer 22, 103 (2023).
Geng, Y. et al. CircHIF1A induces cetuximab resistance in colorectal cancer by promoting HIF1alpha-mediated glycometabolism alteration. Biol. Direct 19, 36 (2024).
Zou, Y. et al. crVDAC3 alleviates ferroptosis by impeding HSPB1 ubiquitination and confers trastuzumab deruxtecan resistance in HER2-low breast cancer. Drug Resist. Updat 77, 101126 (2024).
Wang, S. et al. A novel circular RNA confers trastuzumab resistance in human epidermal growth factor receptor 2-positive breast cancer through regulating ferroptosis. Environ. Toxicol. 37, 1597–1607 (2022).
Moore, L. D., Le, T. & Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38 (2013).
Hombach, S. & Kretz, M. Non-coding RNAs: classification, biology and functioning. Adv. Exp. Med. Biol. 937, 3–17 (2016).
Lanzetti, L. Oncometabolites at the crossroads of genetic, epigenetic and ecological alterations in cancer. Cell Death Differ. 31, 1582–1594 (2024).
Sadida, H. Q. et al. Epigenetic modifications: key players in cancer heterogeneity and drug resistance. Transl. Oncol. 39, 101821 (2024).
Pan, J. et al. Histone modifications and DNA methylation in psoriasis: a cellular perspective. Clin. Rev. Allergy Immunol. 68, 6 (2025).
Allis, C. D. & Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500 (2016).
Pradhan, S., Chin, H. G., Estève, P. O. & Jacobsen, S. E. SET7/9 mediated methylation of non-histone proteins in mammalian cells. Epigenetics 4, 383–387 (2009).
Weinberg, D. N. et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286 (2019).
Zhang, Y. et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 38, 4246–4253 (2010).
Ooi, S. K. et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717 (2007).
Noh, K. M., Allis, C. D. & Li, H. Reading between the Lines: “ADD”-ing Histone and DNA Methylation Marks toward a New Epigenetic “Sum”. ACS Chem. Biol. 11, 554–563 (2016).
Tibben, B. M. & Rothbart, S. B. Mechanisms of DNA methylation regulatory function and crosstalk with histone lysine methylation. J. Mol. Biol. 436, 168394 (2024).
Castillo-Aguilera, O. et al. DNA methylation targeting: the DNMT/HMT crosstalk challenge. Biomolecules 7, 3 (2017).
Du, J., Johnson, L. M., Jacobsen, S. E. & Patel, D. J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 16, 519–532 (2015).
Dai, L., Johnson-Buck, A. & Walter, N. G. Mechanistic model for epigenetic maintenance by methyl-CpG-binding domain proteins. bioRxiv, https://www.biorxiv.org/content/10.1101/2024.09.22.614380v1 (2024).
Vaissière, T., Sawan, C. & Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 659, 40–48 (2008).
Kondo, Y. Epigenetic cross-talk between DNA methylation and histone modifications in human cancers. Yonsei Med. J. 50, 455–463 (2009).
Lee, H. M. et al. Epigenome reprogramming through H3K27 and H3K4 trimethylation as a resistance mechanism to DNA methylation inhibition in BRAFV600E-mutated colorectal cancer. Clin. Cancer Res. 30, 5166–5179 (2024).
Kopetz, S. et al. Molecular profiling of BRAF-V600E-mutant metastatic colorectal cancer in the phase 3 BEACON CRC trial. Nat. Med. 30, 3261–3271 (2024).
Xu, X. et al. Metabolic reprogramming and epigenetic modifications in cancer: from the impacts and mechanisms to the treatment potential. Exp. Mol. Med. 55, 1357–1370 (2023).
Huang, W. et al. Dual inhibitors of DNMT and HDAC induce viral mimicry to induce antitumour immunity in breast cancer. Cell Death Discov. 10, 143 (2024).
Chang, Y. et al. Development of a first-in-class DNMT1/HDAC inhibitor with improved therapeutic potential and potentiated antitumor immunity. J. Med. Chem. 67, 16480–16504 (2024).
Xu, Z. et al. Crosstalk between histone and m(6)A modifications and emerging roles of m(6)A RNA methylation. Front. Genet. 13, 908289 (2022).
Zhao, Y., Chen, Y., Jin, M. & Wang, J. The crosstalk between m(6)A RNA methylation and other epigenetic regulators: a novel perspective in epigenetic remodeling. Theranostics 11, 4549–4566 (2021).
Wang, Y. et al. N(6)-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat. Neurosci. 21, 195–206 (2018).
Huang, H. et al. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature 567, 414–419 (2019).
Huang, H., Weng, H. & Chen, J. The biogenesis and precise control of RNA m(6)A methylation. Trends Genet. 36, 44–52 (2020).
Barbieri, I. et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552, 126–131 (2017).
Li, Y. et al. N(6)-Methyladenosine co-transcriptionally directs the demethylation of histone H3K9me2. Nat. Genet. 52, 870–877 (2020).
Li, F. et al. Interplay of m(6) A and histone modifications contributes to temozolomide resistance in glioblastoma. Clin. Transl. Med. 11, e553 (2021).
Wang, J. et al. Leukemogenic chromatin alterations promote AML leukemia stem cells via a KDM4C-ALKBH5-AXL signaling axis. Cell Stem Cell 27, 81–97.e88 (2020).
Szczepanek, J. & Tretyn, A. MicroRNA-mediated regulation of histone-modifying enzymes in cancer: mechanisms and therapeutic implications. Biomolecules 13, 1590 (2023).
Nalbant, E. & Akkaya-Ulum, Y. Z. Exploring regulatory mechanisms on miRNAs and their implications in inflammation-related diseases. Clin. Exp. Med. 24, 142 (2024).
Bure, I. V., Nemtsova, M. V. & Kuznetsova, E. B. Histone modifications and non-coding RNAs: mutual epigenetic regulation and role in pathogenesis. Int. J. Mol. Sci. 23, 5801 (2022).
Paraskevopoulou, M. D. & Hatzigeorgiou, A. G. Analyzing MiRNA-LncRNA Interactions. Methods Mol. Biol. 1402, 271–286 (2016).
Nadhan, R., Isidoro, C., Song, Y. S. & Dhanasekaran, D. N. LncRNAs and the cancer epigenome: Mechanisms and therapeutic potential. Cancer Lett. 605, 217297 (2024).
Song, Y. X. et al. Non-coding RNAs participate in the regulatory network of CLDN4 via ceRNA mediated miRNA evasion. Nat. Commun. 8, 289 (2017).
Saleh, R. O. et al. lncRNA-microRNA axis in cancer drug resistance: particular focus on signaling pathways. Med Oncol. 41, 52 (2024).
Xue, L., Li, C., Ren, J. & Wang, Y. KDM4C contributes to cytarabine resistance in acute myeloid leukemia via regulating the miR-328-3p/CCND2 axis through MALAT1. Ther. Adv. Chronic Dis. 12, 2040622321997259 (2021).
Sharma, S. Unraveling the role of long non-coding RNAs in therapeutic resistance in acute myeloid leukemia: New prospects & challenges. Noncoding RNA Res. 9, 1203–1221 (2024).
Medrzycki, M. et al. Histone h1.3 suppresses h19 noncoding RNA expression and cell growth of ovarian cancer cells. Cancer Res. 74, 6463–6473 (2014).
Zhu, C., Wang, X., Wang, Y. & Wang, K. Functions and underlying mechanisms of lncRNA HOTAIR in cancer chemotherapy resistance. Cell Death Discov. 8, 383 (2022).
Song, Y. et al. Long non-coding RNA HOTAIR mediates the switching of histone H3 lysine 27 acetylation to methylation to promote epithelial-to-mesenchymal transition in gastric cancer. Int. J. Oncol. 54, 77–86 (2019).
Liang, Y. et al. LncRNA CASC9 promotes esophageal squamous cell carcinoma metastasis through upregulating LAMC2 expression by interacting with the CREB-binding protein. Cell Death Differ. 25, 1980–1995 (2018).
Zhai, W. et al. A positive feed-forward loop between LncRNA-URRCC and EGFL7/P-AKT/FOXO3 signaling promotes proliferation and metastasis of clear cell renal cell carcinoma. Mol. Cancer 18, 81 (2019).
Wang, Q. et al. Circ_0019435 exerts its functions in the cellular process of cervical cancer via epigenetically silencing DKK1 and PTEN. Reprod. Sci. 28, 2989–2999 (2021).
Chen, L. H., Wang, L. P. & Ma, X. Q. Circ_SPECC1 enhances the inhibition of miR-526b on downstream KDM4A/YAP1 pathway to regulate the growth and invasion of gastric cancer cells. Biochem. Biophys. Res Commun. 517, 253–259 (2019).
Xie, B. et al. CircXRN2 suppresses tumor progression driven by histone lactylation through activating the Hippo pathway in human bladder cancer. Mol. Cancer 22, 151 (2023).
Sun, M. Breast cancer study vetoed. Science 239, 252 (1988).
Parveen, R., Harihar, D. & Chatterji, B. P. Recent histone deacetylase inhibitors in cancer therapy. Cancer 129, 3372–3380 (2023).
Wang, P., Wang, Z. & Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 19, 5 (2020).
Petrella, A. et al. Histone deacetylase inhibitors in the treatment of hematological malignancies. Mini Rev. Med. Chem. 11, 519–527 (2011).
Zucchetti, B., Shimada, A. K., Katz, A. & Curigliano, G. The role of histone deacetylase inhibitors in metastatic breast cancer. Breast 43, 130–134 (2019).
Bass, A. K. A. et al. Comprehensive review for anticancer hybridized multitargeting HDAC inhibitors. Eur. J. Med. Chem. 209, 112904 (2021).
Hou, Y. et al. Transient EZH2 suppression by Tazemetostat during in vitro expansion maintains T-cell stemness and improves adoptive T-cell therapy. Cancer Immunol. Res. 13, 47-65 (2024).
Hsu, C., Konner, J. A. & Gounder, M. M. Epigenetic therapy in a rare ovarian cancer - a double-edged sword. N. Engl. J. Med. 391, 770–772 (2024).
Cristalli, C. & Scotlandi, K. Targeting DNA methylation machinery in pediatric solid tumors. Cells 13, 1209 (2024).
Zhou, S. et al. Targeting tumor endothelial cells with methyltransferase inhibitors: mechanisms of action and the potential of combination therapy. Pharm. Ther. 247, 108434 (2023).
Singh, M. et al. Current paradigms in epigenetic anticancer therapeutics and future challenges. Semin. Cancer Biol. 83, 422–440 (2022).
Chatterjee, B. et al. The phytochemical brazilin suppress DNMT1 expression by recruiting p53 to its promoter resulting in the epigenetic restoration of p21 in MCF7cells. Phytomedicine 95, 153885 (2022).
Xu, P., Hu, G., Luo, C. & Liang, Z. DNA methyltransferase inhibitors: an updated patent review (2012-2015). Expert Opin. Ther. Pat. 26, 1017–1030 (2016).
Roy, L. et al. Noncoding RNA as an influential epigenetic modulator with promising roles in cancer therapeutics. Drug Discov. Today 28, 103690 (2023).
Zhou, H., Hao, X., Zhang, P. & He, S. Noncoding RNA mutations in cancer. Wiley Interdiscip. Rev. RNA 14, e1812 (2023).
Aprile, M., Costa, V., Cimmino, A. & Calin, G. A. Emerging role of oncogenic long noncoding RNA as cancer biomarkers. Int. J. Cancer 152, 822–834 (2023).
Yan, H. & Bu, P. Non-coding RNA in cancer. Essays Biochem. 65, 625–639 (2021).
Liu, Y. et al. Noncoding RNAs regulate alternative splicing in Cancer. J. Exp. Clin. Cancer Res. 40, 11 (2021).
Oza, A. M. et al. Phase II study of CGP 69846A (ISIS 5132) in recurrent epithelial ovarian cancer: an NCIC clinical trials group study (NCIC IND.116). Gynecol. Oncol. 89, 129–133 (2003).
Paz-Ares, L. et al. Phase III study of gemcitabine and cisplatin with or without aprinocarsen, a protein kinase C-alpha antisense oligonucleotide, in patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 24, 1428–1434 (2006).
Kohaar, I., Petrovics, G. & Srivastava, S. A rich array of prostate cancer molecular biomarkers: opportunities and challenges. Int. J. Mol. Sci. 20, 1813 (2019).
Hong, D. S. et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 122, 1630–1637 (2020).
LaCasse, E. C. Pulling the plug on a cancer cell by eliminating XIAP with AEG35156. Cancer Lett. 332, 215–224 (2013).
Han, Z. et al. Crystal structure of the FTO protein reveals basis for its substrate specificity. Nature 464, 1205–1209 (2010).
Zhou, L. L., Xu, H., Huang, Y. & Yang, C. G. Targeting the RNA demethylase FTO for cancer therapy. RSC Chem. Biol. 2, 1352–1369 (2021).
Lovegrove, B. Teenage hypercalcaemia. Nurs 65, 1390–1392 (1969).
Wei, H. et al. The role of FTO in tumors and its research progress. Curr. Med. Chem. 29, 924–933 (2022).
Lan, Q. et al. The emerging roles of RNA m(6)A methylation and demethylation as critical regulators of tumorigenesis, drug sensitivity, and resistance. Cancer Res. 81, 3431–3440 (2021).
Guo, L., Lee, Y. T., Zhou, Y. & Huang, Y. Targeting epigenetic regulatory machinery to overcome cancer therapy resistance. Semin. Cancer Biol. 83, 487–502 (2022).
Kan, R. L., Chen, J. & Sallam, T. Crosstalk between epitranscriptomic and epigenetic mechanisms in gene regulation. Trends Genet. 38, 182–193 (2022).
Loe, A. K. H., Zhu, L. & Kim, T. H. Chromatin and noncoding RNA-mediated mechanisms of gastric tumorigenesis. Exp. Mol. Med. 55, 22–31 (2023).
Nam, A. S., Chaligne, R. & Landau, D. A. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat. Rev. Genet. 22, 3–18 (2021).
Kim, Y. E. et al. Effects of ultra-high doserate FLASH irradiation on the tumor microenvironment in lewis lung carcinoma: role of myosin light chain. Int. J. Radiat. Oncol. Biol. Phys. 109, 1440–1453 (2021).
Ma, Y. et al. Current views on mechanisms of the FLASH effect in cancer radiotherapy. Natl Sci. Rev. 11, nwae350 (2024).
Bian, X. et al. Epigenetic memory of radiotherapy in dermal fibroblasts impairs wound repair capacity in cancer survivors. Nat. Commun. 15, 9286 (2024).
Ye, P. et al. Histone deacetylase 2 regulates doxorubicin (Dox) sensitivity of colorectal cancer cells by targeting ABCB1 transcription. Cancer Chemother. Pharm. 77, 613–621 (2016).
Zhou, Y. et al. Deubiquitinase USP4 suppresses antitumor immunity by inhibiting IRF3 activation and tumor cell-intrinsic interferon response in colorectal cancer. Cancer Lett. 589, 216836 (2024).
Klaus, A. & Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 8, 387–398 (2008).
Pereira, F. et al. KDM6B/JMJD3 histone demethylase is induced by vitamin D and modulates its effects in colon cancer cells. Hum. Mol. Genet. 20, 4655–4665 (2011).
Watanabe, H. et al. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 8, 15 (2008).
Wan, L., Li, X., Shen, H. & Bai, X. Quantitative analysis of EZH2 expression and its correlations with lung cancer patients’ clinical pathological characteristics. Clin. Transl. Oncol. 15, 132–138 (2013).
Wang, R. et al. Effects of SMYD2-mediated EML4-ALK methylation on the signaling pathway and growth in non-small-cell lung cancer cells. Cancer Sci. 108, 1203–1209 (2017).
Takawa, M. et al. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 72, 3217–3227 (2012).
Iderzorig, T. et al. Comparison of EMT mediated tyrosine kinase inhibitor resistance in NSCLC. Biochem. Biophys. Res. Commun. 496, 770–777 (2018).
Zhang, P. et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 16, 864–875 (2014).
Wang, B. et al. Long noncoding RNA LINC02582 acts downstream of miR-200c to promote radioresistance through CHK1 in breast cancer cells. Cell Death Dis. 10, 764 (2019).
Cheng, C. et al. CRISPR/Cas9 library screening uncovered methylated PKP2 as a critical driver of lung cancer radioresistance by stabilizing beta-catenin. Oncogene 40, 2842–2857 (2021).
Hayami, S. et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 128, 574–586 (2011).
Radhakrishnan, R. et al. Histone deacetylase 10 regulates DNA mismatch repair and may involve the deacetylation of MutS homolog 2. J. Biol. Chem. 290, 22795–22804 (2015).
Kolas, N. K. et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318, 1637–1640 (2007).
Wang, Y. et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138, 660–672 (2009).
Jeong, K. W. et al. Establishment of active chromatin structure at enhancer elements by mixed-lineage leukemia 1 to initiate estrogen-dependent gene expression. Nucleic Acids Res. 42, 2245–2256 (2014).
Matkar, S. et al. An epigenetic pathway regulates sensitivity of breast cancer cells to HER2 inhibition via FOXO/c-Myc Axis. Cancer Cell 28, 472–485 (2015).
Dong, C. et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 23, 316–331 (2013).
Zhang, J. et al. The transcriptional landscape of lncRNAs reveals the oncogenic function of LINC00511 in ER-negative breast cancer. Cell Death Dis. 10, 599 (2019).
Kahl, P. et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 66, 11341–11347 (2006).
Schulte, J. H. et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 69, 2065–2071 (2009).
Bao, S. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006).
Tu, Y. et al. Smoothened promotes glioblastoma radiation resistance via activating USP3-mediated claspin deubiquitination. Clin. Cancer Res. 26, 1749–1762 (2020).
He, J., Nguyen, A. T. & Zhang, Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood 117, 3869–3880 (2011).
Shi, Y. et al. METTL14 inhibits hepatocellular carcinoma metastasis through regulating EGFR/PI3K/AKT signaling pathway in an m6A-dependent manner. Cancer Manag. Res. 12, 13173–13184 (2020).
Yang, P. et al. ALKBH5 holds prognostic values and inhibits the metastasis of colon cancer. Pathol. Oncol. Res. 26, 1615–1623 (2020).
Peng, F. et al. Oncogenic AURKA-enhanced N(6)-methyladenosine modification increases DROSHA mRNA stability to transactivate STC1 in breast cancer stem-like cells. Cell Res. 31, 345–361 (2021).
Zhang, J. et al. Knockdown of YTH N(6)-methyladenosine RNA binding protein 2 (YTHDF2) inhibits proliferation and promotes apoptosis in MGC-803 gastric cancer cells]. Xi Bao Yu Fen. Zi Mian Yi Xue Za Zhi 33, 1628–1634 (2017).
Tucillo, J. J. Dental casting alloys in our changing times. 2. Ceramic alloys. Dent. Tech. 27, 114–116 (1974).
Li, A., Omura, N., Hong, S. M. & Goggins, M. Pancreatic cancer DNMT1 expression and sensitivity to DNMT1 inhibitors. Cancer Biol. Ther. 9, 321–329 (2010).
Fu, Y. et al. The DNMT1-PAS1-PH20 axis drives breast cancer growth and metastasis. Sig. Transduct. Target Ther. 7, 81 (2022).
Na, F. et al. KMT2C deficiency promotes small cell lung cancer metastasis through DNMT3A-mediated epigenetic reprogramming. Nat. Cancer 3, 753–767 (2022).
Wang, J. et al. Interaction between DNMT3B and MYH11 via hypermethylation regulates gastric cancer progression. BMC Cancer 21, 914 (2021).
Wu, J. et al. TET1-mediated DNA hydroxymethylation activates inhibitors of the Wnt/beta-catenin signaling pathway to suppress EMT in pancreatic tumor cells. J. Exp. Clin. Cancer Res. 38, 348 (2019).
Ye, D. et al. DNMT3a-dermatopontin axis suppresses breast cancer malignancy via inactivating YAP. Cell Death Dis. 14, 106 (2023).
So, J. Y. et al. Induction of DNMT3B by PGE2 and IL6 at distant metastatic sites promotes epigenetic modification and breast cancer colonization. Cancer Res 80, 2612–2627 (2020).
Sun, M. et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl Acad. Sci. USA 110, 9920–9925 (2013).
Nepal, C. & Andersen, J. B. Alternative promoters in CpG depleted regions are prevalently associated with epigenetic misregulation of liver cancer transcriptomes. Nat. Commun. 14, 2712 (2023).
Yan, X. et al. DNMT3L inhibits hepatocellular carcinoma progression through DNA methylation of CDO1: insights from big data to basic research. J. Transl. Med. 22, 128 (2024).
Yang, G., Zeng, X., Wang, M. & Wu, A. The TET2/E-cadherin/beta-catenin regulatory loop confers growth and invasion in hepatocellular carcinoma cells. Exp. Cell Res. 363, 218–226 (2018).
Yao, W. et al. DNMT1-driven methylation of RORA facilitates esophageal squamous cell carcinoma progression under hypoxia through SLC2A3. J. Transl. Med. 22, 1167 (2024).
Liu, W. L. et al. MicroRNA-1269 promotes cell proliferation via the AKT signaling pathway by targeting RASSF9 in human gastric cancer. Cancer Cell Int. 19, 308 (2019).
Xiong, S. et al. Role of miR‑34 in gastric cancer: From bench to bedside (Review). Oncol. Rep. 42, 1635–1646 (2019).
Zhai, R. et al. miR-152 suppresses gastric cancer cell proliferation and motility by targeting CD151. Tumour Biol. 35, 11367–11373 (2014).
Zhao, Q. et al. miR-21 promotes EGF-induced pancreatic cancer cell proliferation by targeting Spry2. Cell Death Dis. 9, 1157 (2018).
Xu, C. & Qi, X. MiR-10b inhibits migration and invasion of pancreatic ductal adenocarcinoma via regulating E2F7. J. Clin. Lab. Anal. 34, e23442 (2020).
Xu, B. et al. Expression of miRNA-143 in pancreatic cancer and its clinical significance. Cancer Biother Radiopharm. 33, 373–379 (2018).
Liu, D. et al. MiR-135b-5p is an oncogene in pancreatic cancer to regulate GPRC5A expression by targeting transcription factor KLF4. Cell Death Discov. 8, 23 (2022).
Liang, G. et al. miR-196b-5p-mediated downregulation of TSPAN12 and GATA6 promotes tumor progression in non-small cell lung cancer. Proc. Natl Acad. Sci. USA 117, 4347–4357 (2020).
Meng, W. et al. miR-199a: a tumor suppressor with noncoding RNA network and therapeutic candidate in lung cancer. Int. J. Mol. Sci. 23, 8518 (2022).
Long, Z. & Wang, Y. miR-195-5p suppresses lung cancer cell proliferation, migration, and invasion via FOXK1. Technol. Cancer Res. Treat. 19, 1533033820922587 (2020).
Jia, T. et al. The function of miR-637 in non-small cell lung cancer progression and prognosis. Pulmonology 29, 111–118 (2023).
Yuan, X. et al. Breast cancer exosomes contribute to pre-metastatic niche formation and promote bone metastasis of tumor cells. Theranostics 11, 1429–1445 (2021).
Ding, Y. et al. miR-145 inhibits proliferation and migration of breast cancer cells by directly or indirectly regulating TGF-beta1 expression. Int. J. Oncol. 50, 1701–1710 (2017).
Zhang, L. et al. MiR-34b/c-5p and the neurokinin-1 receptor regulate breast cancer cell proliferation and apoptosis. Cell Prolif. 52, e12527 (2019).
Xia, M. et al. miR-10b-5p promotes tumor growth by regulating cell metabolism in liver cancer via targeting SLC38A2. Cancer Biol. Ther. 25, 2315651 (2024).
Shi, X. et al. microRNA-93-5p promotes hepatocellular carcinoma progression via a microRNA-93-5p/MAP3K2/c-Jun positive feedback circuit. Oncogene 39, 5768–5781 (2020).
Li, Y., Li, D., Yang, Y. & Wang, J. miR-15a-5p regulates liver cancer cell migration, apoptosis and cell cycle progression by targeting transcription factor E2F3. Crit. Rev. Eukaryot. Gene Expr. 32, 1–10 (2022).
Wang, J. et al. Targeted inhibition of the expression of both MCM5 and MCM7 by miRNA-214 impedes DNA replication and tumorigenesis in hepatocellular carcinoma cells. Cancer Lett. 539, 215677 (2022).
Heinemann, A. et al. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 72, 460–471 (2012).
Arnold, J. et al. miR-488-5p and its role in melanoma. Exp. Mol. Pathol. 112, 104348 (2020).
Zhang, X. et al. circRNA_0005529 facilitates growth and metastasis of gastric cancer via regulating miR-527/Sp1 axis. BMC Mol. Cell Biol. 22, 6 (2021).
Zhao, Q. et al. Hypoxia-induced circRNF13 promotes the progression and glycolysis of pancreatic cancer. Exp. Mol. Med. 54, 1940–1954 (2022).
He, Z. et al. Autophagy-associated circRNA circATG7 facilitates autophagy and promotes pancreatic cancer progression. Cell Death Dis. 13, 233 (2022).
Rong, Z. et al. CircRREB1 mediates metabolic reprogramming and stemness maintenance to facilitate pancreatic ductal adenocarcinoma progression. Cancer Res. 84, 4246–4263 (2024).
Jiang, M. et al. CircLIFRSA/miR-1305/PTEN axis attenuates malignant cellular processes in non-small cell lung cancer by regulating AKT phosphorylation. Mol. Cancer 23, 208 (2024).
Zheng, X. et al. The circRNA circSEPT9 mediated by E2F1 and EIF4A3 facilitates the carcinogenesis and development of triple-negative breast cancer. Mol. Cancer 19, 73 (2020).
Cao, L. et al. Circular RNA circRNF20 promotes breast cancer tumorigenesis and Warburg effect through miR-487a/HIF-1alpha/HK2. Cell Death Dis. 11, 145 (2020).
Fu, B. et al. Circular RNA circBCBM1 promotes breast cancer brain metastasis by modulating miR-125a/BRD4 axis. Int. J. Biol. Sci. 17, 3104–3117 (2021).
Liu, B. et al. Cytoskeleton remodeling mediated by circRNA-YBX1 phase separation suppresses the metastasis of liver cancer. Proc. Natl Acad. Sci. USA 120, e2220296120 (2023).
Gu, Y. et al. Circular RNA circIPO11 drives self-renewal of liver cancer initiating cells via Hedgehog signaling. Mol. Cancer 20, 132 (2021).
Bao, L. et al. LncRNA RUNX1-IT1 is downregulated in gastric cancer and suppresses the maturation of miR-20a by binding to its precursor. Histol. Histopathol. 38, 1321–1326 (2023).
Liu, J. et al. LncRNA H19 promoted the epithelial to mesenchymal transition and metastasis in gastric cancer via activating wnt/beta-catenin signaling. Dig. Dis. 40, 436–447 (2022).
Shi, Y. & Sun, H. Down-regulation of lncRNA LINC00152 suppresses gastric cancer cell migration and invasion through inhibition of the ERK/MAPK signaling pathway. Onco. Targets Ther. 13, 2115–2124 (2020).
Yang, A. et al. LncRNA UCA1 promotes development of gastric cancer via the miR-145/MYO6 axis. Cell Mol. Biol. Lett. 26, 33 (2021).
Liu, C. et al. LncRNA-CCAT5-mediated crosstalk between Wnt/beta-Catenin and STAT3 signaling suggests novel therapeutic approaches for metastatic gastric cancer with high Wnt activity. Cancer Commun. 44, 76–100 (2024).
Wang, J. et al. LINC00941 promotes pancreatic cancer malignancy by interacting with ANXA2 and suppressing NEDD4L-mediated degradation of ANXA2. Cell Death Dis. 13, 718 (2022).
Zhu, H. et al. LncRNA CYTOR promotes pancreatic cancer cell proliferation and migration by sponging miR-205-5p. Pancreatology 20, 1139–1148 (2020).
Kim, K. et al. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 32, 1616–1625 (2013).
Wu, G. et al. LncRNA BCAN-AS1 stabilizes c-Myc via N(6)-methyladenosine-mediated binding with SNIP1 to promote pancreatic cancer. Cell Death Differ. 30, 2213–2230 (2023).
Wei, F. et al. LncRNA-NEAT1 inhibits the occurrence and development of pancreatic cancer through spongy miR-146b-5p/traf6. Biotechnol. Genet Eng. Rev. 40, 1094–1112 (2024).
Karger, A. et al. ADPGK-AS1 long noncoding RNA switches macrophage metabolic and phenotypic state to promote lung cancer growth. EMBO J. 42, e111620 (2023).
Chen, Y. L. et al. LncRNA SLCO4A1-AS1 suppresses lung cancer progression by sequestering the TOX4-NTSR1 signaling axis. J. Biomed. Sci. 30, 80 (2023).
Hua, Q. et al. LINC01123, a c-Myc-activated long non-coding RNA, promotes proliferation and aerobic glycolysis of non-small cell lung cancer through miR-199a-5p/c-Myc axis. J. Hematol. Oncol. 12, 91 (2019).
Zhao, J. et al. Long noncoding RNA HOTAIR promotes breast cancer development through the lncRNA HOTAIR/miR-1/GOLPH3 axis. Clin. Transl. Oncol. 25, 3420–3430 (2023).
Liu, S. et al. A novel lncRNA ROPM-mediated lipid metabolism governs breast cancer stem cell properties. J. Hematol. Oncol. 14, 178 (2021).
Zhou, T. et al. LncRNA SPINT1-AS1 promotes breast cancer proliferation and metastasis by sponging let-7 a/b/i-5p. Pathol. Res. Pr. 217, 153268 (2021).
Xiu, B. et al. LINC02273 drives breast cancer metastasis by epigenetically increasing AGR2 transcription. Mol. Cancer 18, 187 (2019).
Liang, Y. et al. LncRNA PRBC induces autophagy to promote breast cancer progression through modulating PABPC1-mediated mRNA stabilization. Oncogene 43, 1019–1032 (2024).
Wang, C. et al. Long noncoding RNA HULC accelerates the growth of human liver cancer stem cells by upregulating CyclinD1 through miR675-PKM2 pathway via autophagy. Stem Cell Res. Ther. 11, 8 (2020).
Yuan, K. et al. Long noncoding RNA TLNC1 promotes the growth and metastasis of liver cancer via inhibition of p53 signaling. Mol. Cancer 21, 105 (2022).
Lu, L. et al. Exosomal lncRNA TUG1 from cancer-associated fibroblasts promotes liver cancer cell migration, invasion, and glycolysis by regulating the miR-524-5p/SIX1 axis. Cell Mol. Biol. Lett. 27, 17 (2022).
Li, H. et al. LncRNA MINCR promotes the development of liver cancer by regulating microRNA-107/beta-catenin. J. BUON 25, 972–980 (2020).
Shi, S. et al. PIWIL1 promotes gastric cancer via a piRNA-independent mechanism. Proc. Natl Acad. Sci. USA 117, 22390–22401 (2020).
Xu, L. et al. New insights into the function and mechanisms of piRNA PMLCPIR in promoting PM(2.5)-induced lung cancer. J. Adv. Res. S2090–1232(24)00372–2, https://doi.org/10.1016/j.jare.2024.08.029 (2024). Online ahead of print.
Yao, J. et al. piR-651 and its function in 95-D lung cancer cells. Biomed. Rep. 4, 546–550 (2016).
Peng, L. et al. piR-55490 inhibits the growth of lung carcinoma by suppressing mTOR signaling. Tumour Biol. 37, 2749–2756 (2016).
Liu, Y. et al. piR-hsa-211106 inhibits the progression of lung adenocarcinoma through pyruvate carboxylase and enhances chemotherapy sensitivity. Front. Oncol. 11, 651915 (2021).
Xie, J. et al. PIWIL1 interacting RNA piR-017061 inhibits pancreatic cancer growth via regulating EFNA5. Hum. Cell 34, 550–563 (2021).
Zhao, Q. et al. IL11 signaling mediates piR-2158 suppression of cell stemness and angiogenesis in breast cancer. Theranostics 13, 2337–2349 (2023).
Wu, L. et al. PIWI-interacting RNA-YBX1 inhibits proliferation and metastasis by the MAPK signaling pathway via YBX1 in triple-negative breast cancer. Cell Death Discov. 10, 7 (2024).
Ding, X. et al. piRNA-823 is involved in cancer stem cell regulation through altering DNA methylation in association with luminal breast cancer. Front. Cell Dev. Biol. 9, 641052 (2021).
Wu, Y. J. et al. PIWIL1 interacting RNA piR-017724 inhibits proliferation, invasion, and migration, and inhibits the development of HCC by silencing PLIN3. Front. Oncol. 13, 1203821 (2023).
Acknowledgements
This work is supported by the National Natural Science Foundation of China (82473574 and 82404200) and Projects from Sichuan Province (25QNJJ4600 and 2024ZYD0126).
Author information
Authors and Affiliations
Contributions
J.S., P.Y., H.B. drafted the manuscript, drew the figures and summarized the tables. C.C. contributed to the preparation of tables and specific sections of the manuscript. S.Z. conceived the manuscript. O.T., and W.Q.D provided valuable discussion and revised the manuscript. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, J., Yang, P., Chen, C. et al. Targeting epigenetic regulators as a promising avenue to overcome cancer therapy resistance. Sig Transduct Target Ther 10, 219 (2025). https://doi.org/10.1038/s41392-025-02266-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-025-02266-z
