
Abstract
The ephrin receptors (EphRs) are the largest family of receptor tyrosine kinases (RTKs) that are abundantly expressed in the developing brain and play important roles at different stages of neurogenesis ranging from neural stem cell (NSC) fate specification to neural migration, morphogenesis, and circuit assembly. Defects in EphR signalling have been associated with several pathologies including neurodevelopmental disorders (NDDs), intellectual disability (ID), and neurodegenerative diseases (NDs). Here, we review our current understanding of the complex and dynamic role of EphRs in the brain and discuss how deregulation of these receptors contributes to disease, highlighting their potential as valuable druggable targets.
Similar content being viewed by others

Introduction
The first ephrin receptor (EphR), was discovered in 1987 as a new class of receptor tyrosine kinases (RTKs) [1]. Since then, 13 additional EphRs have been discovered leading to their classification into A- and B-type receptors. Currently, there are 9 EphA and 5 EphB receptors each requiring binding to their corresponding ephrin ligands to initiate signal transduction [2, 3]. The ligands are also grouped into A- and B-type classes. Although EphA receptors typically bind to ephrinA ligands, and EphB receptors bind to ephrinB ligands, cross-talk between the classes can occur [4], adding to the complexity of EphR signalling.
EphRs are abundantly expressed during brain development and play important roles in cellular communication, in particular short-range communication [3, 5, 6], in which the transfer of information between cells in close proximity occurs. Since both EphRs and ephrins are membrane-bound, direct cell-cell contact is often required to induce signalling by these receptors [2]. Eph receptors and ligands can also be expressed on the same cell (in cis) [7, 8], adding another level of complexity to understanding their mechanisms of action.
EphRs are important in modulating a spectrum of processes during corticogenesis, including proliferation, apoptosis, cell adhesion, cell division, and cell fate specification [7, 9,10,11,12]. In adulthood, however, EphRs are shown to regulate alternate processes ranging from synaptic remodelling, epithelial differentiation, immune function, and the self-renewal of stem cells [13,14,15,16,17,18]. Given that dysregulation of these pathways are associated with several neurodevelopmental disorders (NDDs) [19,20,21,22], a better understanding of signal transduction by EphRs in the developing and adult brain may lay the foundation for EphR targeted therapies. EphR structure and regulation, and their role in cancers, cardiovascular disease, and NDDs have been extensively reviewed [19,20,21,22]. Thus, this review article provides a brief synopsis on EphR, and is mainly centred on brain diseases, with particular focus on neurogenesis and neural stem cells (NSCs).
EphR structure and activation
EphRs are comprised of 7 domains (Fig. 1) [19, 23] with specificity of receptor-to-ligand binding being regulated by a cysteine-rich domain (CRD) and an epidermal growth factor-like (EGF) domain [24]. The CRD is tethered by a ligand-binding domain (LBD), leading to receptor dimerization and clustering [25]. The PSD-95, Dlg1, ZO-1 (PDZ) binding domain located at the C-terminal end, is required for anchoring the receptors from membrane to cytoskeleton, and is of particular importance for cell-cell communication [26]. Two fibronectin (FN) 3 domains and a sterile alpha motif (SAM) domain function to control protein-protein interactions, facilitating assembly of protein complexes required for signal transduction [21, 27]. Importantly, a kinase domain present in all RTKs, initiates the phosphorylation of tyrosine residues and is crucial for EphR activation [13]. At the N-terminus of EphRs, a juxta membrane (JM) region maintains the neighbouring kinase domain in an inactive form by inhibiting access to ATP [5]. This inhibition is removed following ligand binding, receptor dimerization, and phosphorylation of key residues, including those within the JM region.

The ephrin A and B ligands, as well as different domains of EphR are illustrated. Forward and reverse signalling are highlighted in which the receptor-carrying cell, or the ligand carrying cell initiates signal transduction, respectively. Phosphorylation sites for the receptors and ligands are highlighted. Figure made with BioRender.
Ephrin ligands bind to EphRs through their receptor binding domain (RBD). EphrinA ligands lack a cytoplasmic domain and their RBD is attached to the cell surface through a glycosylphosphatidylinositol (GPI) linker. EphrinB ligands possess a transmembrane domain [8], and an intracellular PDZ binding motif (Fig. 1) [19, 23]. Dimerization of ephrin ligands with their receptors is the catalyst for phosphorylation of tyrosine kinase domain [28]. The EphR and ephrins can function bi-directionally, where the signal comes from either the cell that carries the receptor (forward signal), or the cell that carries the ligand (reverse signal) (Fig. 1) [29, 30]. This signalling is often dependent on SRC family of kinases (SFKs), which are non-receptor kinases known to phosphorylate EphRs [19, 23, 31]. For example, binding of ephrinB to the ectodomain of EphR induces phosphorylation of their tyrosine residue and initiates reverse signalling mediated by SFK members, Src and Fyn [32,33,34,35]. A better understanding of EphR phosphorylation/activation will be instrumental in developing readouts to assess their biology and therapeutic potential (e.g. high throughout compound screens). This knowledge assists in designing novel pharmacological approaches to activate or block select EphRs in a context dependent manner.
EphR gene regulation and downstream effectors
EphRs and ephrins operate through modulating members of the Rho GTPase family, including RhoA, Rac1, and Cdc42 (Fig. 2), which are known to control components of cytoskeleton dynamics [36,37,38,39]. GTPases go through cycles of inactive (bound by GDP) and active (bound by GTP) forms and upon activation, they bind to their downstream targets. EphRs regulate the GDP-GTP transitions through guanine nucleotide exchange factors (GEFs), and GTPase-activating proteins (GAPS) (Fig. 2) [36,37,38,39]. An emerging GEF is the Ephexin protein family that activates RhoA, and controls the activation of Cdc42 [40, 41]. Ephexins are able to bind EphA4’s kinase domain [40, 41], an interaction documented in neurons [42]. Interestingly, ephrinA-induced activation of EphA upregulates Ephexin activity, which then triggers increased activity of RhoA [43]. These findings suggest that RhoA activation by EphAs may require Ephexin. Interestingly, Ephexins have been linked to neurological disorders such as depression, epilepsy, and Alzheimer’s disease (AD) [40, 44,45,46].

SRC family of kinases regulate EphR signalling through phosphorylation. Once active, the EphRs then modulate the activity of the Rho family of GTPases. EphRs modulate the switch between inactive (GDP) and active (GTP) confirmations of the Rho family, or EphRs can cause reversion of the GTP state back to GDP. Influencing cell behaviour, shape, and movement is important for cell migration, axon guidance, synaptic plasticity, tissue morphogenesis, and cancer metastasis. Figure made with BioRender.
GEFs including Intersectin and Kalirin, which activate Rac1 and Cdc42 respectively, are implicated in EphB activity [38, 47, 48]. For example, EphB2 influences dendritic spine structuring in hippocampal neurons by binding to Intersectin and Kalirin, thus activating Rac1 and Cdc42 [38, 47, 48]. Intersectin can also specifically bind to inactive EphB2 [38, 47], while Kalirin can only interact with a previously active EphB2 in early-born hippocampal neurons [48]. In addition, ephrinB1 activity upregulates Kalirin in dendritic spines of hippocampal neurons, which leads to subsequent activation of the downstream Rac1 effector, p21-activated kinase (PAK) [48]. Thus, within hippocampal neurons, Rac1 activity appears to be regulated by Kalirin.
Rac1 and Cdc42 deregulation has been implicated in intellectual disability (ID) [49]. For example, mutations in the gene encoding oligophrenin-1 (OPHN1), a Rho-GAP that regulates Rho GTPases, results in X-linked intellectual disability (XLID) characterized by dendritic spine abnormalities and impaired synaptic function [49, 50]. Similarly, mutations in PAK3, an effector for both Rac1 and Cdc42, has been identified in families with non-syndromic XLID. These mutations disrupt the kinase activity of PAK3, leading to deficits in dendritic spine morphology and cognitive impairments [49, 51]. Additionally, mutations in ARHGEF6, a GEF for Rac1 and Cdc42, are associated with ID and are thought to impair the regulation of actin dynamics necessary for proper neuronal connectivity [49, 52]. The involvement of Rac1 and Cdc42 in the maintenance of dendritic spines highlights their significance in diverse cellular processes underlying learning and memory, and thus their dysregulation may underlie the pathogenesis of NDDs [49].
EphR gene expression is regulated by numerous transcription factors (TFs) (Fig. 3). Among these transcriptional regulators are the Homeobox (HOX)-containing TFs including HOXA1/A2/A9/A13, HOXB1, HOXD13, and LIM1 which regulate EphR expression in a tissue-specific manner. For example, in the developing mouse brain, HOXA1 and HOXB1 induce the expression of EphA2 [53], while HOXA2 alters EphA4 expression in rhombomeres [54], the segmented regions of the developing vertebrate hindbrain that contribute to the formation of cranial nerves and neural circuits. Researchers have demonstrated that EphA4 is also positively regulated by Twist1, a basic helix-loop-helix TF [55, 56], in the context of coronal suture development and by LIM1 [55, 57] in motor neurons. Furthermore, in developing mouse limbs, HOXA13 and HOXD13 regulate the expression of EphA7 [58].

Various transcription factors that induce the expression of different EphRs are shown. PHF6 is highlighted as a master regulator that directly controls the expression of several EphR members including type-A and type-B receptors. Downstream known effectors are highlighted. Figure made with BioRender.
EphB2/B3 expression during cell migration of the intestinal epithelium is shown to be regulated by transcription factor 4 (TCF4) and ß-catenin [59]. EphB1 activity in retinal ganglion cells during the time of optic chiasm divergence is controlled by Zic family member 2 (Zic2) [55, 60]. Interestingly, HOXA9 regulates EphB4, influencing both endothelial cell migration and endothelial cell tube formation [61]. The regulation of EphB family members in the brain remains poorly understood, although TFs appear to be involved. As an example, EphB4 was shown to be regulated by the Valentino (Val) TF in boundary formation of the zebrafish hindbrain [55, 62].
PHF6: a new player in the regulation of EphA and EphB gene expression in the developing brain
The plant homeodomain finger-6 protein (PHF6) is a transcriptional regulator and an epigenetic modifier that is highly conserved in vertebrates with high expression during early fetal life, specifically during the early stages of corticogenesis [63,64,65]. Multiple mutations in the PHF6 gene on the X chromosome have been identified in the XLID, Börjeson-Forssman-Lehmann syndrome (BFLS). The most common recurrent PHF6 patient mutation, R342X, impairs the extended PHD2 (ePHD2) domain, whereby PHF6 is proposed to function as a truncated protein [66,67,68,69,70,71,72,73]. Mice harbouring the R342X mutation were shown to have phenotypic characteristic of BFLS [73]. The C99F-m is another point mutation (m) wherein cysteine-99 is replaced with phenylalanine (C99F) impairing the function of the PHD1 domain of PHF6 [65, 74]. Mice harbouring C99F mutations were also shown to display deficits in cognitive functions, emotionality, and social behaviour [65].
Most recently, several members of the EphR family including A and B subtypes are discovered to be direct transcriptional targets of PHF6 [74]. PHF6 directly binds to the EphA and EphB gene regulatory regions to alter their expression [74]. Strikingly, the expression of EphRs are significantly downregulated in BFLS mice, including in R342X and C99F-m [74]. Mechanistically, PHF6 is shown to alter NSC dynamics through regulation of EphA family members, in particular EphA4/7 [74]. Thus, alterations in the spatial and temporal patterns of eNSC fate commitment, which are essential for proper brain development, are tightly controlled by EphA members, suggesting their potential as viable therapeutic targets for the treatment of BFLS. Further investigations are required to establish the clinical manifestation of EphA4 upregulation in rescuing cellular, cognitive, and social deficits in BFLS (Fig. 4A, B) and other related XLID syndromes. In addition, subjecting BFLS mice brains to single cell analysis at different neurodevelopmental time points can assist in furthering our current understanding of disease and gaining novel mechanistic insights (Fig. 4C).

The therapeutic potential of upregulating EphA4 in BFLS mouse models can be explored using pharmacological agents following a high throughput screen (A) or genetic approaches to induce EphA4 expression (B). Detailed analysis of BFLS mouse brain and mouse behaviour can be assessed following EphA4 manipulation to establish the benefit of EphA4 targeting in preclinical models (B). Single cell analysis on BFLS mouse brains can further our knowledge on disease pathogenesis (C). Figure made with BioRender.
Regulation of EphR expression by micro-RNAs
In addition to TF regulation of EphR expression, micro-RNA (miRNA) have been shown to regulate EphR expression. miRNAs are a class of small non-coding RNA, typically ~20 nucleotides in length, that play an important role in the regulation of gene expression [75]. Deregulation of miRNA expression has been implicated in numerous diseases, including neurological disorders [76,77,78,79,80,81,82].
EphA2 is found to function downstream of the Ras-Raf-MAPK signalling pathway and is associated with different hallmarks of cancer including cell survival, proliferation, motility, and invasion [55, 83,84,85]. Interestingly, a study revealed that EphA2 is regulated by miR-26b in brain tumour cells, and elevated expression of miR-26b was shown to attenuate EphA2 expression and suppress glioma cell proliferation [86]. In the context of spinal cord injury, another miRNA, miR-93, was shown to degrade EphA4 protein expression. Infection of spinal cord neuronal cultures with miR-93 mimic decreased EphA4 protein expression and enabled neurite outgrowth [87]. Another example of EphR class-A regulation by miRNA includes miR-210-mediated downregulation of ephrinA3 in hypoxia conditions [88, 89]. Interestingly in the nervous system, ephrinA3 is also shown to be targeted by miR-4271 during dopaminergic neuronal differentiation, and this was shown to be correlated with the regulation of axonal growth [90].
In addition to the regulation of class-A EphRs, class-B EphRs can also be subject to the same mechanisms. miR-124 was demonstrated to repress ephrinB1 in NSCs, and downregulation of ephrinB1 was suggested to be required for miR-124-induced neuronal differentiation [12]. In yet another study, miR-204-mediated downregulation of EphB2 was demonstrated [91]. EphB2 is shown to regulate synaptic plasticity in hippocampal neurons with its expression levels significantly declined in the aged hippocampus. miR-204 was shown to target EphB2 and reduce the expression of the NMDA receptor subunit, NR1, in primary hippocampal neurons, a mechanism proposed to contribute to cognitive decline [91].
In summary, regulation of EphR expression can be summarized into transcriptional mechanisms mainly by TFs, and post-transcriptional mechanisms by miRNA in different model systems. Given that some of the transcriptional regulators outlined herein (e.g. PHF6) are also known epigenetic modifiers, it remains to be investigated whether epigenetic mechanisms are involved in the regulation of EphR expression.
Neural stem cells (NSCs) in brain health and disease
Neurogenesis, a process in which new neurons are generated from NSCs, is the corner stone of central nervous system (CNS) developmental. This process is comprised of proliferation and fate specification of NSCs, migration of newborn neurons, and their maturation, followed by neural circuit assembly [92, 93]. The ability of NSCs to self-renew and differentiate into different cell lineages that make up the brain is tightly regulated, and evidence is accumulating that deregulation of NSC behaviour can contribute to a number of NDDs. A better understanding of signalling mechanisms in the regulation of cell identity and NSC fate specification assists us to better understand NDDs.
Neurogenesis and its deregulation in neurodevelopmental disorders (NDD)
Neural development begins with specification of embryonic stem cells (ESCs) into embryonic neural stem cells (eNSCs), which then give rise to progenitors, neurons, and glial cells [94,95,96]. A progenitor cell is considered committed since it has a destined fate, while an NSC is uncommitted as it has the propensity to self-renew or differentiate. In the embryonic CNS, there are a population of progenitor cells known as neuroepithelial cells which are a highly polarized cell type with apical ends that line the ventricular surface, interfacing with cerebrospinal fluid (CSF). Similar to stem cells, neuroepithelial cells, are able to self-renew and undergo symmetric division to expand their progenitor pool during early CNS development [97]. In developing brain, neuroepithelial cells will differentiate into radial glial cells which will retain the polarity and localization of the neuroepithelial cells with their cell bodies located in the ventricular zone (VZ) and their processes spreading to the pial surface. Radial glial cells function as both scaffolding structures and as neural progenitors, and their long radial fibers will act as guides for migrating neurons during cortical development [98]. Radial glial cells will undergo asymmetric division, a characteristic not common in neuroepithelial cells, allowing them to self-renew while also giving rise to other cell types; including neurons and glial cells [99]. The switch from neuroepithelial to radial glial cells allows for the diversification of the cell types that make up the brain.
The formation of the cortical plate takes place during development and originates from a single layer of symmetrically dividing neuroepithelial cells [100, 101]. Specifically, these neuroepithelial cells will become apical radial glial cells and undergo further division to produce either basal progenitor cells or neurons [102, 103]. The migration of neurons from the VZ to the cortical plate is guided by the radial glial scaffold that extends from the VZ to the pial surface, passing through the intermediate zone (IZ), and directs newly born neurons to the developing cortex [104, 105]. Earlier-born neurons will form the deeper layers of the cortex, whereas later-born neurons will migrate further upwards to establish the upper cortical layers [106]. The subventricular zone (SVZ) acts in concert with the VZ wherein the intermediate progenitor cells housed here are prone to proliferate and increase the neuronal output necessary for cortical expansion, especially for neurons of the upper cortical layers [107]. Corticogenesis is completed when the progenitor cell population no longer undergoes continuous symmetric cell division or when progenitor cells are destined to generate glial cells, including astrocytes and oligodendrocytes [103].
The study of NSC deregulation in NDDs has garnered interest in the past decade. Specifically, evidence is accumulating that deregulation of NSC fate specification (proliferation versus differentiation) contributes to various NDDs [108, 109]. Research utilizing genetic models of a number of NDDs, including 2q23.1/2q33.1 deletion syndromes [110] and 9q34 deletion syndrome [111], indicates that these conditions can profoundly alter NSCs. Similarly, knockdown (KD) of the genes implicated in each of the aforementioned syndromes (e.g. TCF4, EHMT1, SATB2, and MBD5), were shown to reprogram the NSC transcriptome [109,110,111]. Another evidence comes from research on MECP2 mutations associated with Rett syndrome, in which impairments in the balance between neural progenitor cell (NPC) proliferation and differentiation through Notch signalling was demonstrated [112]. Furthermore, mutations associated with autism spectrum disorder (ASD) and ID have been investigated, whereby genes involved in the regulation of the Wnt/β-catenin pathway, a key pathway involved in neurogenesis, was documented. These mutations affect key upstream ligands, such as WNT1 and WNT2 [113,114,115,116], as well as the downstream TF, TCF7L2 [117,118,119]. Most recently, research using an ID-ASD patient’s primary fibroblasts confirmed the impairment of the Wnt/β-catenin pathway [119]. In further support, Wnt signalling has been shown to be deregulated in haploinsufficiency of the chromatin remodeler, CHD8, associated with ASD [109, 120,121,122], and eliciting KD of CHD8 in the developing mouse brain led to a decrease in the NPC population [109, 122, 123]. In yet another independent line of inquiry, our work has most recently established deregulation of NSCs associated with PHF6 loss-of-function mutations in the XLID syndrome, BFLS.
In summary, the knowledge into NSC abnormalities associated with ID, has opened up new avenues of research to harness the therapeutic potential of NSCs for better treatment of NDDs. In this context, EphR can be considered attractive druggable targets as outlined in this review.
EphR regulation of neurogenesis: role of EphRs in the SVZ
The subgranular zone (SGZ) of the hippocampus and the subventricular zone (SVZ) of the lateral ventricles are two regions with the highest density of stem cells in both the embryonic and adult brain [124,125,126] (Fig. 5). Researchers have investigated the role of EphR and their ligands in these stem cell dense regions, particularly their influence on the proliferation of NPCs within the SVZ [16, 127,128,129]. For instance, ephrinA2 expression is detected in neuroblasts and progenitor cells, and EphA7 is expressed in quiescent ependymal cells [129]. The signalling via ephrinA2/EphA7 interaction appears to negatively regulate the self-renewal of adult NPCs [129].

The expression of different EphRs and ephrin ligands is shown across the subventricular zone (SVZ), the subgranular zone (SGZ), and the cortex (CX). The cell types that make up each region is shown. Figure made with BioRender.
Similarly, ephrinB ligands are widely expressed in the SVZ, and loss- and gain-of-function studies have revealed that ephrinB/EphB signalling impacts the number of dividing cells within the SVZ [16, 129, 130]. Specifically, genetic ablation of ephrinB3 in a mouse model appears to increase the number of proliferating stem/progenitor cells in the lateral ventricle of adult mice [130]. At the same time, EphB3 is shown to suppress progenitor cell proliferation in the SVZ [128]. In contrast another EphB member, EphB2, which is expressed in NSCs and progenitor cells [16], induces cell proliferation in the SVZ through downregulation of neurogenic notch homolog protein 1 (Notch1) [131]. Interestingly, studies on mice lacking the Notch1 ligand, Delta1, showed downregulated EphB2 and ephrinB2 expression in neural crest cells [132], suggesting a feedforward mechanism between stem cell-related pathways and EphR. Of note, EphB2 is also shown to enhance neurogenesis following cerebral infarction [133, 134].
EphA4 is an extensively studied receptor of the ephrin family in the contexts of axon guidance, NSC proliferation during development, and neuroblast migration to the olfactory bulbs [10, 135, 136]. High expression of EphA4 is detected in hippocampal endothelial cells, mature astrocytes, neurons, and NSCs [10, 135,136,137,138,139]. EphA4 has a diverse set of signalling mechanisms compared to other EphRs and is able to bind both A- and B-class ephrin ligands [140, 141]. Ablation of EphA4 led to the misalignment of neuroblasts and deficits in astrocyte organization in the subventricular zone/rostral migratory stream/olfactory bulb (SVZ/RMS/OB), indicating that EphA4 is an important regulatory factor for proper neuroblast migration and in the spatial organization, orientation, and regulation of the SVZ/RMS/OB [135].
In summary, recent findings highlight the critical roles of EphRs and their ligands in regulating neurogenesis in SVZ. The neurogenic defects observed in disorders such as Rett syndrome, ASD, BFLS, and cerebral infarction emphasizes the potential value of targeting EphR signalling pathways for better treatment of these disorders. In particular, among all EphRs, focus should be given to EphA4, EphA7, and EphB2 as outlined in Sections V.I.
EphR regulation of neurogenesis: role of EphRs in the hippocampus, SGZ
The distinct and potentially overlapping roles of EphA and EphB receptors in regulating neural stem/progenitor cells (NSPCs) is also apparent in the dentate gyrus (DG) of the hippocampus, specifically in SGZ, where high expression of EphB1 in hippocampal NSPCs has been observed [142]. When stimulated by ephrinB3, EphB regulates proliferation and migration of NPCs [142]. Further studies on ephrinA5 knockout (KO) mice, which show downregulation of early neuron proliferation and survival in the DG, suggests a commonality between some roles of EphA and B receptors in the hippocampus [143]. In particular, EphA4’s indirect regulation of hippocampal precursor cell self-renewal within the adult brain is through D-serine regulated NMDAR signalling; important in synaptic plasticity and memory formation [144]. Inhibition of EphA4 signalling has also been shown to enhance the proliferation of hippocampal precursor cells [144]. This is consistent with the results obtained in NSCs whereby knockdown of EphA4 using an siRNA approach enhanced NSC proliferation and self-renewal [74].
The roles of EphRs in synaptic plasticity and memory formation, discussed in Section VI, highlights their importance in SGZ. It is not surprising that deregulation of these pathways are implicated in NDs such as AD [19, 22, 145, 146]. Targeting EphR signalling could offer novel therapeutic avenues to address deficits in synaptic plasticity and memory, potentially benefiting patients with neurodegenerative and psychiatric conditions.
EphR regulation of neurogenesis: role of EphRs in the developing cortex
In the developing cortex, ephrinB1 has been shown to be highly expressed in NPCs and is required for conserving progenitor fate. Blockade of ephrinB1 resulted in NPC differentiation and cell cycle exit [11, 147]. Similarly, EphA4 is highly expressed in NPCs of the developing cortex and EphA4-depleted embryos exhibited decreased cortical wall thickness and reduced proliferation as assessed by BrdU assays [10]. These studies led to proposing a model whereby NPC proliferation and maintenance in the embryonic cortex could be positively directed by ephrinB1 binding to EphA4 [10, 11]. Other studies have reported that inhibition of ephrinA1 and EphA2/A3/A4 interaction resulted in reduced neuron numbers induced by a reduced propensity for differentiation [148]. In yet another study, adult mice with ephrinA2 KO exhibited an abnormal neocortical laminar structure with a significant reduction in neuron density that was similar to what was seen in the neocortex of ASD children [149]. From these findings, ephrinA2 was proposed to be an important factor for neuronal fate [149]. Whether EphA-mediated regulation of neuronal fate and density stems directly from deregulation of NSCs/NPCs self-renewal, reprograming of pro-differentiation mechanisms, or both, requires additional investigation. In conclusion, given that alterations in cell identity and NPC fate in developing cortex are associated with various NDDs, such as microcephaly, lissencephaly, and ASD [150,151,152], understanding the roles of ephrin/EphR signalling in these processes could further our knowledge on the mechanisms underlying the pathogenesis of these diseases.
EphR in neurodegeneration
EphR/ephrins have been studied in the context of memory consolidation and development of memory-related diseases such as AD [19, 22]. EphR/ephrins are expressed in brain regions that regulate memory including cortex, hippocampus, and amygdala [22, 153, 154]. They are also abundantly expressed in post-synaptic flanks of synapses where they contribute to synaptic transmission, dendritic spine morphology, neurotransmitter release, and post-synaptic neurotransmitter trafficking/re-uptake [15, 22, 155,156,157], thus playing important roles in memory formation. Importantly, the expression of EphA4/B2 was shown to be reduced in the hippocampal tissue of patients with early-stage AD as well as in mouse models of AD [158]. ß-amyloid plaques, which are hallmarks of AD, attenuate EphB2 expression by initiating proteasome degradation [22, 153, 159, 160]. Interestingly, upon reinstating EphB2 activity, there is a decrease in ß-amyloid-derived neurotoxicity [159, 160], following EphB-NMDA-type glutamate receptor interaction at synapses. NMDA is a key player in memory formation and is known to be blocked by ß-amyloid in AD [19, 161]. Researchers have shown that the expression of EphB2 in a murine AD model restored NMDA activity and improved memory consolidation [153].
EphA4 can bind to both ephrinAs and ephrinBs, and appears to have a neuroprotective role in neurodegenerative diseases including AD and Parkinson’s [145, 146]. EphA4 appears to form a heterodimer with the platelet-derived growth factor receptor beta (PDGFRß), both of which are co-expressed in a human embryonic stem cell line [162]. PDGFRß, the receptor for the platelet-derived growth factors BB (PDGF-BB), is also expressed in neuronal and glial cells [163, 164]. Recently, combined treatment of the ephrinA1 ligand and PDGF-BB enhanced neurogenesis within the hippocampus, suggesting a synergy between EphA4 and PDGFRßs in regulation of neurogenesis [165].
EphR in brain tumour stem cells
EphRs and ephrins have been studied in the context of glioblastoma (GB) cancer cell invasion, migration, tumorigenicity, and maintenance of the brain tumour stem cell (BTSC) pool [166,167,168,169]. EphA2 was shown to promote BTSC derived xenografts, and its downregulation suppressed tumourigensis [166]. EphA3 expression is also elevated in GB cell lines and functions to preserve the stem cell-like properties of BTSCs [167]. A significant decrease in tumorigenicity of recurrent GB (rGB) cells following EphA2 and EphA3 inhibition has previously been reported to lead to inhibition of the AKT/ERK pathways [170]. Co-blockade of EphA2 and EphA3 enhanced differentiation of BTSCs, attenuated the expression of stem cell markers, and reduced the tumorigenicity of rGB [170].
B-class Eph receptors and ligands, are also implicated in several key processes within GB and contribute to tumourigenesis and poor prognosis [171,172,173,174,175]. Increased expression of EphB2 is detected in human U87 glioma cell line [173]. Human glioma samples exhibit elevated ephrinB3 [172], ephrinB2, and EphB4 expression [171]. Enhanced ephrinB1 signalling in U87 cells induced an increase in migration and invasiveness [173] and its blocking attenuated migration and invasion [172]. HIF-2a stabilization of EphB2 during hypoxia is shown to induce phosphorylation of paxillin, and this pathway appears to regulate GB invasion [175]. The protein reelin, which binds to EphB receptors, has been identified as a significant contributor to GB and cancer stem cell (CSC) migration [176]. Reelin is a glycoprotein involved in regulating neuronal migration and positioning during brain development, and binds the extracellular domain of EphB, inducing receptor clustering and activation in neurons, consequently controlling neuronal migration during nervous system development [177, 178]. Overall, EphBs and ephrinBs appear to facilitate the communication between tumour cells and their microenvironment, promoting tumour growth, invasion, and resistance to therapy [172,173,174,175]. Thus, targeting EphB and ephrinB signalling pathways may be an effective strategy to combat GB tumours.
Conclusion and future directions
EphR signalling plays important roles in the embryonic and adult brain, and tightly regulates NSC fate and neurogenesis [3, 5,6,7, 9]. In SGZ, EphB and their ephrinB ligands promote neurogenesis and migration of NPCs [127, 142]. In developing cortex, ephrinB1 maintains progenitor fate [11, 147], while EphA4 regulates the proliferation and maintenance of NPCs [10]. EphrinA and B ligands can have opposing roles on cell proliferation in SVZ, depending on the availability of the specific ligand and receptor interactions involved [16, 127, 129,130,131, 165]. These differing roles of EphRs and their ligands highlights the importance of detailing their mechanisms of action in a context dependent manner.
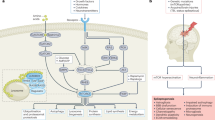
Disruptions in EphR signalling have been linked to several brain diseases (Fig. 6) [19, 22, 166,167,168,169]. In the rare XLID syndrome, BFLS, members of the EphA family including EphA4 suppress NSC self-renewal and proliferation [74], proposing a model whereby these receptors may be involved in fate specification of NSCs consequently promoting the generation of new neurons and their migration. In support of this model, defects in neuron density and migration are documented upon downregulation of PHF6, the gene causing BFLS [64, 74]. Deregulation of neurogenesis has also been reported in several other ID syndromes. Among well-studied XLID-associated proteins is the Fragile Mental Retardation Protein (FMRP), linked to Fragile X Syndrome (FXS), the most common form of inherited ID. Research utilizing FMR1 KO mice (the gene encoding the FRMP protein) has shown that absence of FMRP leads to abnormalities in synaptogenesis, synaptic structure, and function, mirroring the phenotype observed in patients with FXS [179]. Furthermore, FMR1 KO mice exhibit learning and memory deficits consistent with FXS characteristics, highlighting the critical role of FMRP in cognitive development and functioning [179]. In a number of studies, FXS has been characterized by excessive mGluR5 activity in neurons, outlining it as a disorder of NSC proliferation and differentiation [180,181,182,183,184]. Similarly, investigations into RNF12/RLIM E3 ubiquitin ligase, an XLID gene, in neural development using ESC models found that RNF12’s catalytic activity is essential for proper stem cell maintenance and neural differentiation, and these processes are disrupted by mutations associated with XLID [185, 186]. Mutations in RNF12/RLIM accelerates the expression of neural lineage markers and neurite outgrowth [185]. These characteristics have been observed in other types of IDs where abnormal neural specialization and irregular dendritic spine arborization have been observed [187,188,189], suggesting that abnormal neural development and differentiation are key mechanisms in XLID.

The involvement of different EphRs and ephrins in brain disorders are highlighted. Figure made with BioRender.
Beyond the study of XLID, diverse roles for EphRs are documented in glioma, ASD, AD, and neuronal injury. In gliomas, EphA2, EphA3, EphB2/B4, and ephrinB1/B2/B3 regulate different hallmarks of cancer including cell invasion, migration, and tumorigenicity [86, 166, 167, 170,171,172,173,174,175,176]. In ASD, ephrinA2 controls neocortical organization [149]. In spinal cord injury, inducing EphA4 promotes neuronal growth and recovery [87]. In AD, EphA4 and EphB2, regulate synaptic plasticity and memory consolidation [153, 158,159,160, 165]. It is therefore clear that EphR may take on different roles depending on the microenvironment and niche factors within the brain, their interacting network, and a cell’s transcriptional state.
Evidence is accumulating that stem cells and EphRs are closely associated in the regulation of multiple developmental processes. For example, HOX family members regulate EphR expression (Fig. 3). Interestingly, select HOX TF members regulate stem cell fate [190], neurogenesis, and development [53, 54, 58, 61]. In addition to HOX TF, another transcriptional regulator, PHF6, tightly regulates EphR expression, and restricts NSC self-renewal [74]. In further support, recent studies have highlighted a link between mutations in EphA7 [191] and ephrinB2 [192] in patients with cognitive impairments. Such interactions further highlights the significance of EphR in regulation of neurogenesis and their deregulation in ID patients [74]. Future research should focus on exploring the specific mechanisms by which impaired EphR signalling influences developmental delay. In addition, the intersection between stem cell regulation and EphR signalling during development merits in-depth investigation. It would be important to determine whether and how stem-cell-identity TFs modulate EphR expression during development. In particular, in conditions such as NDDs, where aberrant stem cell function and EphR dysregulation are closely associated, elucidating common regulatory mechanisms could lead to the development of novel druggable targets. Multi-omics analyses including single-cell RNA sequencing (scRNA-seq), the assay for transposase-accessible chromatin with sequencing (ATAC-seq) and mapping membrane protein interactions using platforms such as Mammalian Membrane Two-Hybrid assay (MaMTH) will assist in identifying key regulatory elements in the context of disease. Such global screens can lead to generation of new hypotheses and may also be employed to address several unanswered questions, including on potential cross talk of EphR and epigenetic modifiers, the cross talk of TFs and/or miRNAs in regulation of stem cell identity, and unraveling required binding partners that are found in complex with EphR and thus essential for signal transduction. In conclusion, the emerging role of EphRs in the regulation of NSC fate and deregulation of these signalling pathways in NDDs has opened up new avenues of investiagtion to tackle existing clinical challenges in NDD treatment.
References
Hirai H, Maru Y, Hagiwara K, Nishida J, Takaku F. A novel putative tyrosine kinase receptor encoded by the Eph gene. Science. 1987;238:1717–20.
Committee EN. Unified nomenclature for Eph family receptors and their ligands, the ephrins. Cell. 1997;90:403–4.
Darling TK, Lamb TJ. Emerging roles for Eph receptors and ephrin ligands in immunity. Front Immunol. 2019;10:1473.
Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52.
Lisabeth EM, Falivelli G, Pasquale EB. Eph receptor signaling and ephrins. Cold Spring Harb Perspect Biol. 2013;5:a009159.
Barquilla A, Pasquale EB. Eph receptors and ephrins: therapeutic opportunities. Annu Rev Pharmacol Toxicol. 2015;55:465–87.
Gerstmann K, Zimmer G. The role of the Eph/ephrin family during cortical development and cerebral malformations. Med Res Archiv. 2018;6:3.
Klein R. Eph/ephrin signalling during development. Development. 2012;139:4105–9.
Wilkinson DG. Regulation of cell differentiation by Eph receptor and ephrin signaling. Cell Adhes Migr. 2014;8:339–48.
North HA, Zhao X, Kolk SM, Clifford MA, Ziskind DM, Donoghue MJ. Promotion of proliferation in the developing cerebral cortex by EphA4 forward signaling. Development. 2009;136:2467–76.
Qiu R, Wang X, Davy A, Wu C, Murai K, Zhang H, et al. Regulation of neural progenitor cell state by ephrin-B. J Cell Biol. 2008;181:973–83.
Arvanitis DN, Jungas T, Behar A, Davy A. Ephrin-B1 reverse signaling controls a posttranscriptional feedback mechanism via miR-124. Mol Cell Biol. 2010;30:2508–17.
Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3:475–86.
Yamaguchi Y, Pasquale EB. Eph receptors in the adult brain. Curr Opin Neurobiol. 2004;14:288–96.
Tremblay ME, Riad M, Bouvier D, Murai KK, Pasquale EB, Descarries L, et al. Localization of EphA4 in axon terminals and dendritic spines of adult rat hippocampus. J Comp Neurol. 2007;501:691–702.
Conover JC, Doetsch F, Garcia-Verdugo J-M, Gale NW, Yancopoulos GD, Alvarez-Buylla A. Disruption of Eph/ephrin signaling affects migration and proliferation in the adult subventricular zone. Nat Neurosci. 2000;3:1091–7.
Sakamoto A, Ishibashi-Ueda H, Sugamoto Y, Higashikata T, Miyamoto S, Kawashiri M-A, et al. Expression and function of ephrin-B1 and its cognate receptor EphB2 in human atherosclerosis: from an aspect of chemotaxis. Clin Sci. 2008;114:643–50.
Durbin L, Brennan C, Shiomi K, Cooke J, Barrios A, Shanmugalingam S, et al. Eph signaling is required for segmentation and differentiation of the somites. Genes Dev. 1998;12:3096–109.
Taylor H, Campbell J, Nobes CD. Ephs and ephrins. Curr Biol. 2017;27:R90–5.
Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–80.
Liang L-Y, Patel O, Janes PW, Murphy JM, Lucet IS. Eph receptor signalling: from catalytic to non-catalytic functions. Oncogene. 2019;38:6567–84.
Lamprecht R, LeDoux J. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54.
Dravis C. Ephs, ephrins, and bidirectional signaling. Nat Educ. 2010;3:22.
Seiradake E, Harlos K, Sutton G, Aricescu AR, Jones EY. An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly. Nat Struct Mol Biol. 2010;17:398–402.
Sahoo AR, Buck M. Structural and functional insights into the transmembrane domain association of eph receptors. Int J Mol Sci. 2021;22:8593.
Liu X, Fuentes EJ. Emerging themes in PDZ domain signaling: structure, function, and inhibition. Int Rev Cell Mol Biol. 2019;343:129–218.
Campbell ID, Spitzfaden C. Building proteins with fibronectin type III modules. Structure. 1994;2:333–7.
Binns KL, Taylor PP, Sicheri F, Pawson T, Holland SJ. Phosphorylation of tyrosine residues in the kinase domain and juxtamembrane region regulates the biological and catalytic activities of Eph receptors. Mol Cell Biol. 2000;20:4791–805.
Klein R. Bidirectional modulation of synaptic functions by Eph/ephrin signaling. Nat Neurosci. 2009;12:15–20.
Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–75.
Wybenga-Groot LE, Baskin B, Ong SH, Tong J, Pawson T, Sicheri F. Structural basis for autoinhibition of the Ephb2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell. 2001;106:745–57.
Aoto J, Chen L. Bidirectional ephrin/Eph signaling in synaptic functions. Brain Res. 2007;1184:72–80.
Palmer A, Zimmer M, Erdmann KS, Eulenburg V, Porthin A, Heumann R, et al. EphrinB phosphorylation and reverse signaling: regulation by Src kinases and PTP-BL phosphatase. Mol Cell. 2002;9:725–37.
Georgakopoulos A, Litterst C, Ghersi E, Baki L, Xu C, Serban G, et al. Metalloproteinase/Presenilin1 processing of ephrinB regulates EphB‐induced Src phosphorylation and signaling. EMBO J. 2006;25:1242–52.
Knöll B, Drescher U. Src family kinases are involved in EphA receptor-mediated retinal axon guidance. J Neurosci. 2004;24:6248–57.
Ségaliny AI, Tellez-Gabriel M, Heymann M-F, Heymann D. Receptor tyrosine kinases: characterisation, mechanism of action and therapeutic interests for bone cancers. J Bone Oncol. 2015;4:1–12.
Locke C. Effects of localized EphB2 stimulation on dendritic filopodia of hippocampal neurons. PhD thesis, University of Massachusetts Amherst (2018).
Noren NK, Pasquale EB. Eph receptor–ephrin bidirectional signals that target Ras and Rho proteins. Cell Signal. 2004;16:655–66.
Herath NI, Boyd AW. The role of Eph receptors and ephrin ligands in colorectal cancer. Int J Cancer. 2010;126:2003–11.
Kim K, Lee S-A, Park D. Emerging roles of ephexins in physiology and disease. Cells. 2019;8:87.
Shamah SM, Lin MZ, Goldberg JL, Estrach S, Sahin M, Hu L, et al. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell. 2001;105:233–44.
Sahin M, Greer PL, Lin MZ, Poucher H, Eberhart J, Schmidt S, et al. Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse. Neuron. 2005;46:191–204.
Ogita H, Kunimoto S, Kamioka Y, Sawa H, Masuda M, Mochizuki N. EphA4-mediated Rho activation via Vsm-RhoGEF expressed specifically in vascular smooth muscle cells. Circ. Res. 2003;93:23–31.
Zhang J-C, Yao W, Qu Y, Nakamura M, Dong C, Yang C, et al. Increased EphA4-ephexin1 signaling in the medial prefrontal cortex plays a role in depression-like phenotype. Sci Rep. 2017;7:7133.
Veeramah KR, Johnstone L, Karafet TM, Wolf D, Sprissler R, Salogiannis J, et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia. 2013;54:1270–81.
Sell GL, Schaffer TB, Margolis SS. Reducing expression of synapse-restricting protein Ephexin5 ameliorates Alzheimer’s-like impairment in mice. J Clin Investig. 2017;127:1646–50.
Irie F, Yamaguchi Y. EphB receptors regulate dendritic spine development via intersectin, Cdc42 and N-WASP. Nat Neurosci. 2002;5:1117–8.
Penzes P, Beeser A, Chernoff J, Schiller MR, Eipper BA, Mains RE, et al. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37:263–74.
Govek E-E, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49.
Billuart P, Bienvenu T, Ronce N, Des Portes V, Vinet MC, Zemni R, et al. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature. 1998;392:923–6.
Bienvenu T, Des Portes V, McDonell N, Carrié A, Zemni R, Couvert P, et al. Missense mutation in PAK3, R67C, causes X‐linked nonspecific mental retardation. Am J Med Genet. 2000;93:294–8.
Kutsche K, Yntema H, Brandt A, Jantke I, Gerd Nothwang H, Orth U, et al. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat Genet. 2000;26:247–50.
Chen J, Ruley HE. An enhancer element in the EphA2 (Eck) gene sufficient for rhombomere-specific expression is activated by HOXA1 and HOXB1 homeobox proteins. J Biol Chem. 1998;273:24670–5.
Theil T, Frain M, Gilardi-Hebenstreit P, Flenniken A, Charnay P, Wilkinson DG. Segmental expression of the EphA4 (Sek-1) receptor tyrosine kinase in the hindbrain is under direct transcriptional control of Krox-20. Development. 1998;125:443–52.
Arvanitis DN, Davy A. Regulation and misregulation of Eph/ephrin expression. Cell Adhes Migr. 2012;6:131–7.
Ting M-C, Wu NL, Roybal PG, Sun J, Liu L, Yen Y, et al. EphA4 as an effector of Twist1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development. 2009;136:855–64.
Kania A, Jessell TM. Topographic motor projections in the limb imposed by LIM homeodomain protein regulation of ephrin-A: EphA interactions. Neuron. 2003;38:581–96.
Salsi V, Zappavigna V. Hoxd13 and Hoxa13 directly control the expression of the EphA7 Ephrin tyrosine kinase receptor in developing limbs. J Biol Chem. 2006;281:1992–9.
Batlle E, Henderson JT, Beghtel H, van den Born MM, Sancho E, Huls G, et al. β-Catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell. 2002;111:251–63.
García-Frigola C, Carreres MI, Vegar C, Mason C, Herrera E. Zic2 promotes axonal divergence at the optic chiasm midline by EphB1-dependent and-independent mechanisms. Development. 2008;135:1833–41.
Bruhl T, Urbich C, Aicher D, Acker-Palmer A, Zeiher AM, Dimmeler S. Homeobox A9 transcriptionally regulates the EphB4 receptor to modulate endothelial cell migration and tube formation. Circ. Res. 2004;94:743–51.
Cooke J, Moens C, Roth L, Durbin L, Shiomi K, Brennan C, et al. Eph signalling functions downstream of Val to regulate cell sorting and boundary formation in the caudal hindbrain. Development. 2001;128:571–80.
Voss AK, Gamble R, Collin C, Shoubridge C, Corbett M, Gecz J, et al. Protein and gene expression analysis of Phf6, the gene mutated in the Borjeson-Forssman-Lehmann Syndrome of intellectual disability and obesity. Gene Expr Patterns. 2007;7:858–71.
Zhang C, Mejia LA, Huang J, Valnegri P, Bennett EJ, Anckar J, et al. The X-linked intellectual disability protein PHF6 associates with the PAF1 complex and regulates neuronal migration in the mammalian brain. Neuron. 2013;78:986–93.
Cheng C, Deng PY, Ikeuchi Y, Yuede C, Li D, Rensing N, et al. Characterization of a mouse model of Borjeson-Forssman-Lehmann syndrome. Cell Rep. 2018;25:1404–14.e1406.
Chao MM, Todd MA, Kontny U, Neas K, Sullivan MJ, Hunter AG, et al. T-cell acute lymphoblastic leukemia in association with Borjeson-Forssman-Lehmann syndrome due to a mutation in PHF6. Pediatr Blood Cancer. 2010;55:722–4.
Crawford J, Lower KM, Hennekam RC, Van Esch H, Megarbane A, Lynch SA, et al. Mutation screening in Borjeson-Forssman-Lehmann syndrome: identification of a novel de novo PHF6 mutation in a female patient. J Med Genet. 2006;43:238–43.
Gecz J, Turner G, Nelson J, Partington M. The Borjeson-Forssman-Lehman syndrome (BFLS, MIM #301900). Eur J Hum Genet. 2006;14:1233–7.
Jahani-Asl A, Cheng C, Zhang C, Bonni A. Pathogenesis of Borjeson-Forssman-Lehmann syndrome: insights from PHF6 function. Neurobiol Dis. 2016;96:227–35.
Lower KM, Solders G, Bondeson ML, Nelson J, Brun A, Crawford J, et al. 1024C> T (R342X) is a recurrent PHF6 mutation also found in the original Borjeson-Forssman-Lehmann syndrome family. Eur J Hum Genet. 2004;12:787–9.
Lower KM, Turner G, Kerr BA, Mathews KD, Shaw MA, Gedeon AK, et al. Mutations in PHF6 are associated with Borjeson-Forssman-Lehmann syndrome. Nat Genet. 2002;32:661–5.
Todd MA, Ivanochko D, Picketts DJ. PHF6 degrees of separation: the multifaceted roles of a chromatin adaptor protein. Genes. 2015;6:325–52.
Ahmed R, Sarwar S, Hu J, Cardin V, Qiu LR, Zapata G, et al. Transgenic mice with an R342X mutation in Phf6 display clinical features of Börjeson–Forssman–Lehmann Syndrome. Hum Mol Genet. 2021;30:575–94.
Rasool D, Burban A, Sharanek A, Madrigal A, Hu J, Yan K, et al. PHF6-mediated transcriptional control of NSC via Ephrin receptors is impaired in the intellectual disability syndrome BFLS. EMBO Rep. 2024;0:1–26.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66.
Calin GA, Liu C-G, Sevignani C, Ferracin M, Felli N, Dumitru CD, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci. 2004;101:11755–60.
Iorio MV, Ferracin M, Liu C-G, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–70.
Karaca E, Aykut A, Ertürk B, Durmaz B, Güler A, Büke B, et al. MicroRNA expression profile in the prenatal amniotic fluid samples of pregnant women with Down syndrome. Balk Med J. 2018;35:163–6.
Li Q, Song X-W, Zou J, Wang G-K, Kremneva E, Li X-Q, et al. Attenuation of microRNA-1 derepresses the cytoskeleton regulatory protein twinfilin-1 to provoke cardiac hypertrophy. J Cell Sci. 2010;123:2444–52.
Maciotta S, Meregalli M, Torrente Y. The involvement of microRNAs in neurodegenerative diseases. Front Cell Neurosci. 2013;7:265.
Zhang Q-S, Liu W, Lu G-X. miR-200a-3p promotes β-Amyloid-induced neuronal apoptosis through down-regulation of SIRT1 in Alzheimer’s disease. J Biosci. 2017;42:397–404.
Walker-Daniels J, Hess AR, Hendrix MJ, Kinch MS. Differential regulation of EphA2 in normal and malignant cells. Am J Pathol. 2003;162:1037–42.
Menges C, McCance D. Constitutive activation of the Raf–MAPK pathway causes negative feedback inhibition of Ras–PI3K–AKT and cellular arrest through the EphA2 receptor. Oncogene. 2008;27:2934–40.
Macrae M, Neve RM, Rodriguez-Viciana P, Haqq C, Yeh J, Chen C, et al. A conditional feedback loop regulates Ras activity through EphA2. Cancer Cell. 2005;8:111–8.
Wu N, Zhao X, Liu M, Liu H, Yao W, Zhang Y, et al. Role of microRNA-26b in glioma development and its mediated regulation on EphA2. PloS ONE. 2011;6:e16264.
Chen X, Yang H, Zhou X, Zhang L, Lu X. MiR-93 targeting EphA4 promotes neurite outgrowth from spinal cord neurons. J Mol Neurosci. 2016;58:517–24.
Fasanaro P, D’Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G, et al. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem. 2008;283:15878–83.
Zhang Y, Fei M, Xue G, Zhou Q, Jia Y, Li L, et al. Elevated levels of hypoxia‐inducible microRNA‐210 in pre‐eclampsia: new insights into molecular mechanisms for the disease. J Cell Mol Med. 2012;16:249–59.
Wang T, Chen J, Tang C-X, Zhou X-Y, Gao D-S. Inverse expression levels of EphrinA3 and EphrinA5 contribute to dopaminergic differentiation of human SH-SY5Y cells. J Mol Neurosci. 2016;59:483–92.
Danka Mohammed CP, Rhee H, Phee BK, Kim K, Kim HJ, Lee H, et al. miR‐204 downregulates EphB2 in aging mouse hippocampal neurons. Aging Cell. 2016;15:380–8.
Ming G-l, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223–50.
Zeiss CJ. Comparative milestones in rodent and human postnatal central nervous system development. Toxicol Pathol. 2021;49:1368–73.
Kamelska-Sadowska AM, Wojtkiewicz J, Kowalski IM. Review of the current knowledge on the role of stem cell transplantation in neurorehabilitation. BioMed Res Int. 2019;2019:3290894.
Gaspard N, Vanderhaeghen P. Mechanisms of neural specification from embryonic stem cells. Curr Opin Neurobiol. 2010;20:37–43.
Guillemot F. Cell fate specification in the mammalian telencephalon. Prog Neurobiol. 2007;83:37–52.
Götz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–88.
Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–20.
Malatesta P, Hartfuss E, Götz M. Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development. 2000;127:5253–63.
Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995;18:383–8.
Rakic P. Radial versus tangential migration of neuronal clones in the developing cerebral cortex. Proc Natl Acad Sci. 1995;92:11323–7.
Noctor SC, Martínez-Cerdeño V, Kriegstein AR. Contribution of intermediate progenitor cells to cortical histogenesis. Arch Neurol. 2007;64:639–42.
Stepien BK, Vaid S, Huttner WB. Length of the neurogenic period—a key determinant for the generation of upper-layer neurons during neocortex development and evolution. Front Cell Develop. Biol. 2021;9:676911.
Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145:61–83.
Hatten ME. Central nervous system neuronal migration. Annu Rev Neurosci. 1999;22:511–39.
Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–37.
Haubensak W, Attardo A, Denk W, Huttner WB. Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci. 2004;101:3196–201.
Ernst C. Proliferation and differentiation deficits are a major convergence point for neurodevelopmental disorders. Trends Neurosci. 2016;39:290–9.
Sacco R, Cacci E, Novarino G. Neural stem cells in neuropsychiatric disorders. Curr Opin Neurobiol. 2018;48:131–8.
Gigek C, Chen E, Ota V, Maussion G, Peng H, Vaillancourt K, et al. A molecular model for neurodevelopmental disorders. Transl Psychiatry. 2015;5:e565–65.
Chen ES, Gigek CO, Rosenfeld JA, Maussion G, Chen GG, Vaillancourt K, et al. Molecular convergence of neurodevelopmental disorders. Am J Hum Genet. 2014;95:490–508.
Li H, Zhong X, Chau KF, Santistevan NJ, Guo W, Kong G, et al. Cell cycle-linked MeCP2 phosphorylation modulates adult neurogenesis involving the Notch signalling pathway. Nat Commun. 2014;5:5601.
Lin P-I, Chien Y-L, Wu Y-Y, Chen C-H, Gau SS-F, Huang Y-S, et al. The WNT2 gene polymorphism associated with speech delay inherent to autism. Res Develop Disabil. 2012;33:1533–40.
Marui T, Funatogawa I, Koishi S, Yamamoto K, Matsumoto H, Hashimoto O, et al. Association between autism and variants in the wingless-type MMTV integration site family member 2 (WNT2) gene. Int J Neuropsychopharmacol. 2010;13:443–9.
Martin P, Yang X, Robin N, Lam E, Rabinowitz J, Erdman C, et al. A rare WNT1 missense variant overrepresented in ASD leads to increased Wnt signal pathway activation. Transl Psychiatry. 2013;3:e301–01.
Wassink TH, Piven J, Vieland VJ, Huang J, Swiderski RE, Pietila J, et al. Evidence supporting WNT2 as an autism susceptibility gene. Am J Med Genet. 2001;105:406–13.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–15.
Iossifov I, O’roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21.
El Khouri E, Ghoumid J, Haye D, Giuliano F, Drevillon L, Briand-Suleau A, et al. Wnt/β-catenin pathway and cell adhesion deregulation in CSDE1-related intellectual disability and autism spectrum disorders. Mol Psychiatry. 2021;26:3572–85.
Sakamoto I, Kishida S, Fukui A, Kishida M, Yamamoto H, Hino S-I, et al. A novel β-catenin-binding protein inhibits β-catenin-dependent Tcf activation and axis formation. J Biol Chem. 2000;275:32871–8.
Kobayashi M, Kishida S, Fukui A, Michiue T, Miyamoto Y, Okamoto T, et al. Nuclear localization of Duplin, a β-catenin-binding protein, is essential for its inhibitory activity on the Wnt signaling pathway. J Biol Chem. 2002;277:5816–22.
Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, Nott A, et al. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci. 2016;19:1477–88.
Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158:263–76.
Obernier K, Alvarez-Buylla A. Neural stem cells: origin, heterogeneity and regulation in the adult mammalian brain. Development. 2019;146:dev156059.
Ming G-l, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702.
Urbán N, Guillemot F. Neurogenesis in the embryonic and adult brain: same regulators, different roles. Front Cell Neurosci. 2014;8:396.
Genander M, Frisén J. Ephrins and Eph receptors in stem cells and cancer. Curr Opin Cell Biol. 2010;22:611–6.
Theus MH, Ricard J, Bethea JR, Liebl DJ. EphB3 limits the expansion of neural progenitor cells in the subventricular zone by regulating p53 during homeostasis and following traumatic brain injury. Stem Cells. 2010;28:1231–42.
Holmberg J, Armulik A, Senti K-A, Edoff K, Spalding K, Momma S, et al. Ephrin-A2 reverse signaling negatively regulates neural progenitor proliferation and neurogenesis. Genes Dev. 2005;19:462–71.
Ricard J, Salinas J, Garcia L, Liebl DJ. EphrinB3 regulates cell proliferation and survival in adult neurogenesis. Mol Cell Neurosci. 2006;31:713–22.
Katakowski M, Zhang Z, decarvalho AC, Chopp M. EphB2 induces proliferation and promotes a neuronal fate in adult subventricular neural precursor cells. Neurosci Lett. 2005;385:204–9.
De Bellard ME, Ching W, Gossler A, Bronner-Fraser M. Disruption of segmental neural crest migration and ephrin expression in delta-1 null mice. Develop Biol. 2002;249:121–30.
Xing S, He Y, Ling L, Hou Q, Yu J, Zeng J, et al. Blockade of EphB2 enhances neurogenesis in the subventricular zone and improves neurological function after cerebral cortical infarction in hypertensive rats. Brain Res. 2008;1230:237–46.
He Y, Zeng J, Yu J, He M, Cui C, Zhao Z, et al. EphB2-Fc promotes activation of endogenous neural stem cells after cerebral cortex infarction: experimental with hypertensive rats. Zhonghua Yi Xue Za Zhi. 2005;85:2395–9.
Todd KL, Baker KL, Eastman MB, Kolling FW, Trausch AG, Nelson CE, et al. EphA4 regulates neuroblast and astrocyte organization in a neurogenic niche. J Neurosci. 2017;37:3331–41.
Goldshmit Y, Galea MP, Wise G, Bartlett PF, Turnley AM. Axonal regeneration and lack of astrocytic gliosis in EphA4-deficient mice. J Neurosci. 2004;24:10064–73.
Deininger K, Eder M, Kramer ER, Zieglgänsberger W, Dodt H-U, Dornmair K, et al. The Rab5 guanylate exchange factor Rin1 regulates endocytosis of the EphA4 receptor in mature excitatory neurons. Proc Natl Acad Sci. 2008;105:12539–44.
Tremblay MÈ, Riad M, Chierzi S, Murai KK, Pasquale EB, Doucet G. Developmental course of EphA4 cellular and subcellular localization in the postnatal rat hippocampus. J Comp Neurol. 2009;512:798–813.
Goldshmit Y, Galea MP, Bartlett PF, Turnley AM. EphA4 regulates central nervous system vascular formation. J Comp Neurol. 2006;497:864–75.
Bowden TA, Aricescu AR, Nettleship JE, Siebold C, Rahman-Huq N, Owens RJ, et al. Structural plasticity of eph receptor A4 facilitates cross-class ephrin signaling. Structure. 2009;17:1386–97.
Qin H, Noberini R, Huan X, Shi J, Pasquale EB, Song J. Structural characterization of the EphA4-Ephrin-B2 complex reveals new features enabling Eph-ephrin binding promiscuity. J Biol Chem. 2010;285:644–54.
Chumley MJ, Catchpole T, Silvany RE, Kernie SG, Henkemeyer M. EphB receptors regulate stem/progenitor cell proliferation, migration, and polarity during hippocampal neurogenesis. J Neurosci. 2007;27:13481–90.
Hara Y, Nomura T, Yoshizaki K, Frisén J, Osumi N. Impaired hippocampal neurogenesis and vascular formation in ephrin‐A5‐deficient mice. Stem Cells. 2010;28:974–83.
Zhao J, Taylor CJ, Newcombe EA, Spanevello MD, O’Keeffe I, Cooper LT, et al. EphA4 regulates hippocampal neural precursor proliferation in the adult mouse brain by d-serine modulation of N-Methyl-d-Aspartate receptor signaling. Cereb Cortex. 2019;29:4381–97.
Jing X, Miwa H, Sawada T, Nakanishi I, Kondo T, Miyajima M, et al. Ephrin-A1-mediated dopaminergic neurogenesis and angiogenesis in a rat model of Parkinson’s disease. PloS ONE. 2012;7:e32019.
Willi R, Winter C, Wieske F, Kempf A, Yee B, Schwab M, et al. Loss of EphA4 impairs short‐term spatial recognition memory performance and locomotor habituation. Genes Brain Behav. 2012;11:1020–31.
Laussu J, Khuong A, Gautrais J, Davy A. Beyond boundaries—Eph: ephrin signaling in neurogenesis. Cell Adhes Migr. 2014;8:349–59.
Aoki M, Yamashita T, Tohyama M. EphA receptors direct the differentiation of mammalian neural precursor cells through a mitogen-activated protein kinase-dependent pathway. J Biol Chem. 2004;279:32643–50.
Homman-Ludiye J, Kwan WC, De Souza MJ, Rodger J, Bourne JA. Ephrin-A2 regulates excitatory neuron differentiation and interneuron migration in the developing neocortex. Sci Rep. 2017;7:11813.
Ben-Reuven L, Reiner O. Dynamics of cortical progenitors and production of subcerebral neurons are altered in embryos of a maternal inflammation model for autism. Mol Psychiatry. 2021;26:1535–50.
Xu D, Zhang F, Wang Y, Sun Y, Xu Z. Microcephaly-associated protein WDR62 regulates neurogenesis through JNK1 in the developing neocortex. Cell Rep. 2014;6:104–16.
Des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, et al. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61.
Grunwald IC, Korte M, Wolfer D, Wilkinson GA, Unsicker K, Lipp H-P, et al. Kinase-independent requirement of EphB2 receptors in hippocampal synaptic plasticity. Neuron. 2001;32:1027–40.
Martone ME, Holash JA, Bayardo A, Pasquale EB, Ellisman MH. Immunolocalization of the receptor tyrosine kinase EphA4 in the adult rat central nervous system. Brain Res. 1997;771:238–50.
Kayser MS, Nolt MJ, Dalva MB. EphB receptors couple dendritic filopodia motility to synapse formation. Neuron. 2008;59:56–69.
Contractor A, Rogers C, Maron C, Henkemeyer M, Swanson GT, Heinemann SF. Trans-synaptic Eph receptor-ephrin signaling in hippocampal mossy fiber LTP. Science. 2002;296:1864–9.
Bourgin C, Murai KK, Richter M, Pasquale EB. The EphA4 receptor regulates dendritic spine remodeling by affecting β1-integrin signaling pathways. J Cell Biol. 2007;178:1295–307.
Simón, de Maturana AM, R.L, Ricobaraza A, Escribano L, Schiapparelli L, et al. Early changes in hippocampal Eph receptors precede the onset of memory decline in mouse models of Alzheimer’s disease. J Alzheimer’s Dis. 2009;17:773–86.
Cissé M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, et al. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011;469:47–52.
Mroczko B, Groblewska M, Litman-Zawadzka A, Kornhuber J, Lewczuk P. Cellular receptors of amyloid β oligomers (AβOs) in Alzheimer’s disease. Int J Mol Sci. 2018;19:1884.
Liu J, Chang L, Song Y, Li H, Wu Y. The role of NMDA receptors in Alzheimer’s disease. Front Neurosci. 2019;13:43.
Chen Q, Sawada T, Sakaguchi K, Han F. Direct interaction of receptor tyrosine kinases, EphA4 and PDGFRβ, plays an important role in the proliferation of neural stem cells. J Neurorestoratol. 2017;5:133–41.
Sil S, Periyasamy P, Thangaraj A, Chivero ET, Buch S. PDGF/PDGFR axis in the neural systems. Mol Asp Med. 2018;62:63–74.
Ishii Y, Hamashima T, Yamamoto S, Sasahara M. Pathogenetic significance and possibility as a therapeutic target of platelet derived growth factor. Pathol Int. 2017;67:235–46.
Chen Q, Song H, Liu C, Xu J, Wei C, Wang W, et al. The interaction of EphA4 with PDGFRβ regulates proliferation and neuronal differentiation of neural progenitor cells in vitro and promotes neurogenesis in vivo. Front Aging Neurosci. 2020;12:7.
Binda E, Visioli A, Giani F, Lamorte G, Copetti M, Pitter KL, et al. The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell. 2012;22:765–80.
Day BW, Stringer BW, Al-Ejeh F, Ting MJ, Wilson J, Ensbey KS, et al. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell. 2013;23:238–48.
Nakada M, Anderson EM, Demuth T, Nakada S, Reavie LB, Drake KL, et al. The phosphorylation of ephrin‐B2 ligand promotes glioma cell migration and invasion. Int J Cancer. 2010;126:1155–65.
Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005;3:541–51.
Qazi MA, Vora P, Venugopal C, Adams J, Singh M, Hu A, et al. Cotargeting ephrin receptor tyrosine kinases A2 and A3 in cancer stem cells reduces growth of recurrent glioblastoma. Cancer Res. 2018;78:5023–37.
Tu Y, He S, Fu J, Li G, Xu R, Lu H, et al. Expression of EphrinB2 and EphB4 in glioma tissues correlated to the progression of glioma and the prognosis of glioblastoma patients. Clin Transl Oncol. 2012;14:214–20.
Nakada M, Drake KL, Nakada S, Niska JA, Berens ME. Ephrin-B3 ligand promotes glioma invasion through activation of Rac1. Cancer Res. 2006;66:8492–500.
Nakada M, Niska JA, Miyamori H, McDonough WS, Wu J, Sato H, et al. The phosphorylation of EphB2 receptor regulates migration and invasion of human glioma cells. Cancer Res. 2004;64:3179–85.
Zhu B, Li Y, Mao X. A review on the role of different ephrins in glioma. Eur J Pharmacol. 2022;917:174588.
Qiu W, Song S, Chen W, Zhang J, Yang H, Chen Y. Hypoxia-induced EPHB2 promotes invasive potential of glioblastoma. Int J Clin Exp Pathol. 2019;12:539.
Biamonte F, Sica G, Filippini A, D’Alessio A. Evidence of reelin signaling in GBM and its derived cancer stem cells. Brain Sci. 2021;11:745.
Bouché E, Romero-Ortega MI, Henkemeyer M, Catchpole T, Leemhuis J, Frotscher M, et al. Reelin induces EphB activation. Cell Res. 2013;23:473–90.
Sentürk A, Pfennig S, Weiss A, Burk K, Acker-Palmer A. Ephrin Bs are essential components of the Reelin pathway to regulate neuronal migration. Nature. 2011;472:356–60.
Qurashi A, Li X, Jin P. Fragile X mental retardation protein and stem cells. Results and Problems in Cell Differentiation vol. 54:157–64, Springer; 2012.
Jeon SJ, Kim J-W, Kim KC, Han SM, Go HS, Seo JE, et al. Translational regulation of NeuroD1 expression by FMRP: involvement in glutamatergic neuronal differentiation of cultured rat primary neural progenitor cells. Cell Mol Neurobiol. 2014;34:297–305.
Gong X, Zhang K, Wang Y, Wang J, Cui Y, Li S, et al. MicroRNA-130b targets Fmr1 and regulates embryonic neural progenitor cell proliferation and differentiation. Biochem Biophys Res Commun. 2013;439:493–500.
Scotto-Lomassese S, Nissant A, Mota T, Néant-Féry M, Oostra BA, Greer CA, et al. Fragile X mental retardation protein regulates new neuron differentiation in the adult olfactory bulb. J Neurosci. 2011;31:2205–15.
Callan MA, Cabernard C, Heck J, Luois S, Doe CQ, Zarnescu DC. Fragile X protein controls neural stem cell proliferation in the Drosophila brain. Hum Mol Genet. 2010;19:3068–79.
Castren M, Tervonen T, Kärkkäinen V, Heinonen S, Castrén E, Larsson K, et al. Altered differentiation of neural stem cells in fragile X syndrome. Proc Natl Acad Sci. 2005;102:17834–9.
Bustos F, Segarra-Fas A, Chaugule VK, Brandenburg L, Branigan E, Toth R, et al. RNF12 X-linked intellectual disability mutations disrupt E3 ligase activity and neural differentiation. Cell Rep. 2018;23:1599–611.
Zhang L, Huang H, Zhou F, Schimmel J, Pardo CG, Zhang T, et al. RNF12 controls embryonic stem cell fate and morphogenesis in zebrafish embryos by targeting Smad7 for degradation. Mol Cell. 2012;46:650–61.
Telias M, Ben-Yosef D. Modeling neurodevelopmental disorders using human pluripotent stem cells. Stem Cell Rev Rep. 2014;10:494–511.
Iwase S, Brookes E, Agarwal S, Badeaux AI, Ito H, Vallianatos CN, et al. A mouse model of X-linked intellectual disability associated with impaired removal of histone methylation. Cell Rep. 2016;14:1000–9.
Korb E, Herre M, Zucker-Scharff I, Gresack J, Allis CD, Darnell RB. Excess translation of epigenetic regulators contributes to fragile X syndrome and is alleviated by Brd4 inhibition. Cell. 2017;170:1209–23.e1220.
Sharanek A, Burban A, Hernandez-Corchado A, Madrigal A, Fatakdawala I, Najafabadi HS, et al. Transcriptional control of brain tumor stem cells by a carbohydrate binding protein. Cell Rep. 2021;36:109647.
Lévy J, Schell B, Nasser H, Rachid M, Ruaud L, Couque N, et al. EPHA7 haploinsufficiency is associated with a neurodevelopmental disorder. Clin Genet. 2021;100:396–404.
Lévy J, Haye D, Marziliano N, Casu G, Guimiot F, Dupont C, et al. EFNB2 haploinsufficien cy causes a syndromic neurodevelopmental disorder. Clin Genet. 2018;93:1141–7.
Acknowledgements
AJ-A holds a Canada research chair in Neurobiology of Disease and is supported by grants from NSERC (RGPIN-2016-00605) and CIHR (PJG-185800, 190379). We thank the members of the Jahani-Asl laboratory Behnaz Nateghi, Elham Mahmoudian, Ghazal Sadeghi, and Mohammad Al-Ayach for their critical review of this paper and insightful comments. All figures were generated using BioRender.
Author information
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rasool, D., Jahani-Asl, A. Master regulators of neurogenesis: the dynamic roles of Ephrin receptors across diverse cellular niches. Transl Psychiatry 14, 462 (2024). https://doi.org/10.1038/s41398-024-03168-4
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-024-03168-4
This article is cited by
-
Shaking into deficits: investigating behavioural and neuropathological outcomes associated with a novel preclinical model of infant abusive head trauma
Acta Neuropathologica Communications (2025)
-
Eph-ephrin signaling and its potential role in female reproductive tract development
Molecular Biology Reports (2025)
